
Vaccinia Virus–Encoded Ribonucleotide Reductase Subunits Are Differentially Required for Replication and Pathogenesis
Ribonucleotide reductases (RRs) are evolutionarily-conserved enzymes that catalyze the rate-limiting step during dNTP synthesis in mammals. RR consists of both large (R1) and small (R2) subunits, which are both required for catalysis by the R12R22 heterotetrameric complex. Poxviruses also encode RR proteins, but while the Orthopoxviruses infecting humans [e.g. vaccinia (VACV), variola, cowpox, and monkeypox viruses] encode both R1 and R2 subunits, the vast majority of Chordopoxviruses encode only R2 subunits. Using plaque morphology, growth curve, and mouse model studies, we investigated the requirement of VACV R1 (I4) and R2 (F4) subunits for replication and pathogenesis using a panel of mutant viruses in which one or more viral RR genes had been inactivated. Surprisingly, VACV F4, but not I4, was required for efficient replication in culture and virulence in mice. The growth defects of VACV strains lacking F4 could be complemented by genes encoding other Chordopoxvirus R2 subunits, suggesting conservation of function between poxvirus R2 proteins. Expression of F4 proteins encoding a point mutation predicted to inactivate RR activity but still allow for interaction with R1 subunits, caused a dominant negative phenotype in growth experiments in the presence or absence of I4. Co-immunoprecipitation studies showed that F4 (as well as other Chordopoxvirus R2 subunits) form hybrid complexes with cellular R1 subunits. Mutant F4 proteins that are unable to interact with host R1 subunits failed to rescue the replication defect of strains lacking F4, suggesting that F4-host R1 complex formation is critical for VACV replication. Our results suggest that poxvirus R2 subunits form functional complexes with host R1 subunits to provide sufficient dNTPs for viral replication. Our results also suggest that R2-deficient poxviruses may be selective oncolytic agents and our bioinformatic analyses provide insights into how poxvirus nucleotide metabolism proteins may have influenced the base composition of these pathogens.
Published in the journal:
. PLoS Pathog 6(7): e32767. doi:10.1371/journal.ppat.1000984
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000984
Summary
Ribonucleotide reductases (RRs) are evolutionarily-conserved enzymes that catalyze the rate-limiting step during dNTP synthesis in mammals. RR consists of both large (R1) and small (R2) subunits, which are both required for catalysis by the R12R22 heterotetrameric complex. Poxviruses also encode RR proteins, but while the Orthopoxviruses infecting humans [e.g. vaccinia (VACV), variola, cowpox, and monkeypox viruses] encode both R1 and R2 subunits, the vast majority of Chordopoxviruses encode only R2 subunits. Using plaque morphology, growth curve, and mouse model studies, we investigated the requirement of VACV R1 (I4) and R2 (F4) subunits for replication and pathogenesis using a panel of mutant viruses in which one or more viral RR genes had been inactivated. Surprisingly, VACV F4, but not I4, was required for efficient replication in culture and virulence in mice. The growth defects of VACV strains lacking F4 could be complemented by genes encoding other Chordopoxvirus R2 subunits, suggesting conservation of function between poxvirus R2 proteins. Expression of F4 proteins encoding a point mutation predicted to inactivate RR activity but still allow for interaction with R1 subunits, caused a dominant negative phenotype in growth experiments in the presence or absence of I4. Co-immunoprecipitation studies showed that F4 (as well as other Chordopoxvirus R2 subunits) form hybrid complexes with cellular R1 subunits. Mutant F4 proteins that are unable to interact with host R1 subunits failed to rescue the replication defect of strains lacking F4, suggesting that F4-host R1 complex formation is critical for VACV replication. Our results suggest that poxvirus R2 subunits form functional complexes with host R1 subunits to provide sufficient dNTPs for viral replication. Our results also suggest that R2-deficient poxviruses may be selective oncolytic agents and our bioinformatic analyses provide insights into how poxvirus nucleotide metabolism proteins may have influenced the base composition of these pathogens.
Introduction
Critical for the replication of all organisms and DNA viruses is the conversion of ribonucleotides to deoxynucleotides to serve as building blocks for genome synthesis and repair. Ribonucleotide reductase (RR) is a key enzyme involved in this process, catalyzing the reduction of rNDPs to dNDPs [1], [2]. RRs can be grouped into one of three classes, based on their requirement for oxygen and the mechanism by which a catalytically-important thiyl radical is generated [1]. Mammals typically encode class I RR proteins while class II and III proteins are found only in microorganisms [1], [3]. Class I RR enzymes are assembled from both large (R1; 80–100 kDa) and small (R2; 37–44 kDa) protein subunits, which associate to form enzymatically-active R12R22 tetrameric complexes [1]. These complexes require oxygen to generate a tyrosyl radical found within R2 subunits [1], [4], which is ultimately transferred to R1 subunits to generate a thiyl radical used in rNDP reduction. Transfer of the tyrosyl radical from R2 to R1 subunits is thought to occur through a “radical transfer pathway” that uses a series of at least eleven highly-conserved amino acid residues to promote long-range electron transfer [4], [5], [6], [7], [8]. Mutant proteins containing amino acid substitutions at either the tyrosine involved in radical formation [9] or any of the proposed transfer pathway residues [4], [6], [8], [10], [11] form inactive RR complexes, indicating that both radical formation and transfer are required for catalysis.
Mammalian cells encode a single R1 gene that is only transcribed during S-phase [12]. However, due to the long half-life (∼15 h) of R1 proteins, R1 levels remain essentially constant throughout the cell cycle [13]. The primary small subunit, R2, is also only expressed during S-phase [12], [14] however, this protein has a short half-life (∼3 h) and is rate-limiting for R1-R2 complex formation [13]. The short half-life of R2 is due to its polyubiquitination by the anaphase-promoting complex (APC)-Cdh1 ubiquitin ligase, which leads to its degradation during mitosis [15]. This degradation is dependent upon APC-Cdh1 recognition of a “KEN” box sequence in the N-terminus of R2 (Figure 1). Mammals also encode a second small subunit, p53R2, so named because its elevated expression in response to DNA damage is dependent upon the tumor suppressor p53 [16]. Although p53R2 is 80–90% identical to cellular R2 and can form active complexes with R1 [17], it lacks ∼33 N-terminal amino acid residues found in R2, including those containing the KEN box (Figure 1) [15]. The absence of the KEN box sequence likely explains why p53R2 levels are relatively constant throughout the cell cycle in the absence of DNA damage [18]. It has been hypothesized that p53R2 plays some role in supporting mitochondrial DNA synthesis and/or DNA repair outside of S-phase [17], [18], [19], [20]. Therefore, despite their similarity, R2 and p53R2 appear to be differentially regulated and probably serve different purposes during the cell cycle.

Many Chordopox-, herpes-, asfra - and iridoviruses, also encode their own class I RR proteins [21], [22], [23], [24], [25]. These enzymes are generally thought to support viral replication since ribonucleotide reduction is normally the rate-limiting step in de novo dNTP biogenesis [26]. Although most Chordopoxviruses encode RR proteins, many only encode one of the two RR subunits with a clear bias towards the conservation of R2 proteins (Table S1). Only the Suipox - and Orthopoxviruses contain both R1 and R2 genes. The latter group contains viruses of medical importance including variola virus, the causative agent of smallpox, as well as monkeypox and cowpox viruses, which are responsible for zoonoses in humans [27], [28]. Most understanding of poxvirus RR proteins comes from studies with another Orthopoxvirus, vaccinia virus (VACV).
Slabaugh and Mathews [29] were the first to show that RR activity increased in VACV-infected cells and subsequent studies identified the I4L [25], [30] and F4L [24] genes as those encoding the 87 kDa I4 (R1) and 37 kDa F4 (R2) proteins, respectively. Biochemical studies showed that VACV and cellular RR enzymes share many features, including a similar tertiary architecture, similar pH dependence, allosteric modulation of activity by nucleotides, and comparable specific activities on most rNDP substrates [31], [32], [33]. However, unlike cellular RR, the viral enzyme is less sensitive to allosteric modulation and shows little activity on UDP substrates, indicating that viral and cellular RR enzymes also differ in important ways [33]. The similarities between VACV and mammalian RR are not unexpected given that VACV (and other poxvirus) RR subunits typically share >70% sequence identity with their mouse and human homologs (Figure 1).
Previous studies have shown that inactivating VACV I4L does not affect plaque size and that I4L-deficient mutant strains replicate their DNA and produce viral particles to levels comparable to wild-type VACV in cell culture [34], [35]. Due to these observations, the I4L locus has been suggested to be an excellent site for insertion of foreign genes into VACV [36]. Furthermore, I4L mutants are only mildly attenuated in their virulence in mouse models, exhibiting an ∼10-fold increase in lethal dose 50 (LD50) values when compared to wild-type virus [34]. Paradoxically, another group reported that targeted inactivation of F4L attenuated VACV in mice by ∼1000-fold compared to wild-type virus [37], but the reason for this attenuation was unknown. Although these were separate studies using different strains of VACV, they suggested that the F4 subunit is more important for virus replication and pathogenesis than I4, despite the fact that both subunits are needed for RR activity [32].
We initiated our studies of VACV RR because VACV recombination appears to be catalyzed by poxviral DNA polymerases in vivo [38], and we wanted to determine if perturbing dNTP pools would affect this process. However, it soon became apparent that some of the mutant strains we generated exhibited previously uncharacterized replication defects. This prompted us to revisit how VACV RR affects viral replication and pathogenesis. To do this, we generated a panel of mutant strains containing mutations in the VACV RR genes. We also generated strains lacking a functional J2R (thymidine kinase or TK) gene, thus unable to access the parallel viral salvage pathway of dTTP biogenesis [39]. Our studies show that both the VACV R1 and R2 proteins can form a diversity of virus-virus and virus-host protein-protein interactions in vivo, but that the VACV R2 subunit is far more critical for VACV replication and pathogenicity than is the R1 subunit. Our model suggests that poxvirus R2 subunits form active complexes with host R1 proteins in order to ensure a sufficient dNTP supply to support viral replication. This model is substantiated by previous biochemical studies that found a chimeric RR enzyme consisting of VACV F4 and mouse R1 (MR1) to be more active than strictly viral or mouse RR complexes [33]. Our studies also provide insights into why poxviruses have often conserved their R2 but not their R1 genes (Table S1). To our knowledge this is the first report of a chimeric, virus-host RR forming in vivo. Our study provides further evidence that poxviruses recruit cellular enzymes, in addition to those previously identified such as topoisomerase II [40] and DNA ligase I [41], to support viral replication. Our bioinformatic analysis of other large DNA viruses suggests that recruitment of host RR subunits may represent a more widespread viral strategy to parasitize host nucleotide biosynthetic machinery.
Results
Generation of VACV RR mutant strains
A series of mutant strains were generated in which one (ΔI4L; ΔF4L) or both (ΔI4L/ΔF4L) of the VACV RR genes were deleted from the viral genome (Figure 2A). We also constructed VACV encoding an insertional inactivation of the J2R (TK) gene in combination with ΔI4L and/or ΔF4L mutations generating ΔI4L/ΔF4L/ΔJ2R and ΔF4L/ΔJ2R strains. These strains provide insights into the relative biological importance of the de novo (RR-dependent) and salvage (TK-dependent) pathways in VACV replication. We also constructed VACV ΔF4L strains encoding a His6-tagged F4L gene, or a His6-tagged F4L gene encoding a Y300F amino acid substitution, inserted into the J2R locus. These viruses are referred to as VACV strains ΔF4L/ΔJ2RHisF4L and ΔF4L/ΔJ2RHisY300FF4L, respectively. The marker rescue strategies used to generate these mutant strains are depicted in Figure 2A.

PCR-based analysis confirmed the deletion or inactivation of the targeted loci in constructed VACV strains (data not shown). Western blotting confirmed the presence or absence of viral RR subunit expression in each of the isolates (Figure 2B). The ΔF4L/ΔJ2RHisF4L strain appeared to express elevated levels of F4 compared to wild-type virus, whereas the ΔF4L/ΔJ2RHisY300FF4L strain had slightly reduced F4 expression (Figure 2B). The former case is likely a result of the F4L gene being under the control of an early/late promoter present on the pSC66 transfer vector whereas the endogenous F4L promoter is activated only at early times during infection [42]. The lower F4 expression of the ΔF4L/ΔJ2RHisY300FF4L strain is likely due to its poor replication in culture (see below). These and other VACV strains are summarized in Table 1. See Text S1 for details of virus construction.

ΔF4L strains exhibit a small plaque phenotype
Plaque size and morphologies of the generated strains were analyzed on BSC-40 cells as an initial step to characterize their growth properties. The wild-type and ΔI4L strains exhibited similar plaque morphologies. These plaques typically had large central clearings and were accompanied by smaller secondary plaques that are formed when extracellular enveloped virus are released from infected cells and initiate new infections near primary plaque sites (Figure 3A). Upon measurement of primary plaque areas, no significant differences were found between wild-type and the ΔI4L strain (Figure 3B). In contrast, the ΔF4L, ΔF4L/ΔJ2R, and ΔI4L/ΔF4L/ΔJ2R strains all produced significantly smaller plaques (P<0.05) that were only 55–60% of the plaque size exhibited by wild-type virus (Figure 3B). In addition, the primary plaques produced by ΔF4L strains were typically devoid of nearby secondary plaques (Figure 3A). Incorporation of a His6-tagged form of F4L into the TK locus appeared to complement ΔF4L strain replication as the ΔF4L/ΔJ2RHisF4L strain displayed plaques characteristic of wild-type virus in terms of size and the presence of secondary plaques (Figure 3A and B). Strikingly, ΔF4L strains rescued with a His6-tagged F4L gene encoding the Y300F substitution produced plaques that were not only significantly smaller than wild-type virus [(P<0.05); Figure 3B], but were only 35–40% the size of plaques produced by any of the strains with F4L deleted and these differences were statistically significant (P<0.05). These results suggested that deletion of F4L has a more detrimental effect on plaque size than deletion of I4L. They further suggested that re-introduction of a His6-tagged F4L gene into the TK locus can rescue the small plaque phenotype of ΔF4L strains. However, this rescue effect is lost and the ΔF4L strain replication defect is exacerbated when the re-introduced F4 protein encodes the Y300F amino acid substitution.

Y300 represents a highly-conserved tyrosine residue found in essentially all mammalian small RR subunits (Figure 1). The homologous residue in mouse R2 (MR2; Y370) is required for the transfer of radicals from MR2 to MR1 subunits, which is necessary for catalysis [4]. A Y370F substitution abolishes catalysis but does not impede physical interaction of MR1 and MR2 subunits [4]. The same substitution of the homologous tyrosine residue in human p53R2 (Hp53R2) also abolishes RR activity of Human R1 (HR1)-Hp53R2 complexes [43]. Therefore, the Y300F substitution in F4 is predicted to inhibit catalysis while still allow for R1-R2 subunit interaction. These predicted properties of the Y300F F4 protein may explain the dominant negative-like phenotype exhibited by the ΔF4L/ΔJ2RHisY300FF4L strain.
ΔF4L strains exhibit impaired replication kinetics in culture
We also examined the growth kinetics of these mutant strains in HeLa cells. As previously reported [34], deleting I4L had little effect on viral replication, with the ΔI4L strain replicating to titers that were 2-fold lower than those produced by wild-type virus 48 h post-infection (Figure 3C). In contrast, large (>5-fold) differences between wild-type and ΔF4L strains were readily apparent by 18 h post-infection. This trend continued to the end of the experiment, with the wild-type strain producing ∼15–50-fold more virus than ΔF4L strains 48 h post-infection. This growth defect could be complemented with a His6-tagged F4L gene but not if this gene encoded the Y300F substitution (Figure 3D). In fact, the ΔF4L/ΔJ2RHisY300FF4L strain was unable to undergo productive replication in HeLa cells (Figure 3D). These results suggested that deletion of the F4L gene impairs VACV replication to a higher degree than deletion of I4L, and that concomitant deletion of F4L and J2R does not impede replication further (Figure 3C and 3D). Furthermore, the fact that one can rescue the ΔF4L growth defect with a His6-tagged form of F4L inserted at the TK locus, implies that the defect seen in a ΔF4L strain is not due to other possible idiosyncratic effects caused by deleting the F4L locus (Figure 3D). Finally, these studies further illustrate the dominant negative effect on virus growth imposed by a catalytically-inactive, Y300F-substituted F4 protein.
One explanation for the properties of virus encoding a Y300F-substituted F4 protein is that the mutant protein could be competing with cellular R2 proteins for binding to cellular and/or viral R1 subunits. However, the studies shown in Figure 3C and D suggested that deleting I4L does not result in significant replication defects and so the dominant negative phenotype was likely mediated by interaction with cellular R1 proteins. To rule out a role for I4 interaction in this dominant negative phenotype, the His6-tagged wild-type or Y300F-encoding F4L gene was inserted into the J2R locus of ΔI4L/ΔF4L strains. The ΔI4L/ΔF4L/ΔJ2RHisF4L strain produced plaques indistinguishable in size from those formed by wild-type virus (P>0.05; Figure 3B). However, deleting I4L had no further effects on the plating properties of the ΔI4L/ΔF4L/ΔJ2RHisY300FF4L strain. This strain still produced plaques that were significantly smaller than those produced by wild-type (P<0.05) or ΔF4L strains (P<0.05) and were not significantly different from ΔF4L/ΔJ2RHisY300FF4L virus plaques (P>0.05; Figure 3B). These observations implied that the plaque properties of ΔF4L strains are not influenced by the presence or absence of I4L.
We also tested the ability of other, His6-tagged Chordopoxvirus or host R2 proteins to rescue the small plaque phenotype of the ΔF4L strain. The R2 genes encoded by ECTV, MYXV and SFV R2 genes were all able to rescue the small plaque phenotype, but interestingly the Hp53R2 gene failed to rescue this phenotype (Figure 3B). These results implied that Chordopoxvirus R2 proteins have conserved a specific function and/or activity level that is not recapitulated by Hp53R2.
VACV DNA synthesis is impaired in cells infected with a ΔF4L strain
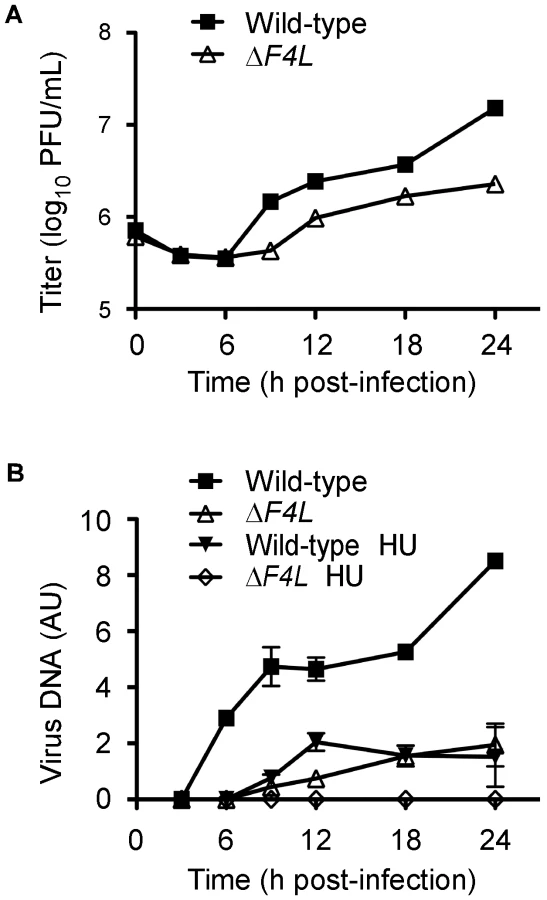
We hypothesized that the reduced replication of the ΔF4L strains was due to impaired genome replication. This is because RR plays a key role in dNTP biogenesis and our initial studies found that ΔF4L (Figure S1A), but not ΔI4L strains (Figure S1B), exhibited reduced late gene expression, which is a common consequence of defects in DNA replication. To test this hypothesis, BSC-40 cells were infected with wild-type or ΔF4L viruses and genome replication was measured in parallel with viral yields. The results of these experiments are shown in Figure 4. As in HeLa cells, the ΔF4L strain exhibited impaired replication kinetics in BSC-40 cells, generating only 15% of the total titer observed with the wild-type strain at 24 h post-infection (Figure 4A). This growth defect was associated with impaired DNA synthesis, with the ΔF4L strain exhibiting an ∼3 h delay in genome synthesis as well as an ∼5-fold reduction in DNA production at 24 h post-infection when compared to wild-type infections (Figure 4B). We also tested what effect the drug hydroxyurea (HU) would have on these strains, since previous studies have correlated HU resistance with changes in F4 expression [44]. Addition of 0.5 mM HU to ΔF4L strain-infected cultures completely blocked virus DNA synthesis. In contrast, wild-type virus still produced detectable amounts of genomic DNA, albeit with delayed kinetics, at levels comparable to what is seen in cells infected with the ΔF4L strain in the absence of HU (Figure 4B). These results suggested that the reduced yields observed with ΔF4L strains are at least partially due to impaired genome synthesis. Furthermore, because sensitivity to RR inhibitors is directly correlated to RR activity levels [45], the hypersensitivity of the ΔF4L strain to HU suggests that these effects on DNA replication are caused by a reduction in RR activity.
ΔF4L strains are uniquely hypersensitive to cidofovir and HU
We hypothesized that the impaired genome replication of the ΔF4L strain was due to reduced dNTP pool sizes as a result of decreased RR activity. However, it is difficult to interpret the meaning of biochemical measurements of pool sizes because of uncertainties surrounding how dNTPs are distributed in infected cells. Instead, we tested whether VACV RR mutants exhibit an altered sensitivity to the antiviral drug cidofovir (CDV). CDV is converted by cellular kinases to the diphosphoryl derivative (CDVpp) [46] which is competitive with respect to dCTP [47] and inhibits VACV E9 DNA polymerase activity [48], [49]. Thus, CDV sensitivity can be used as an indirect probe for changes in dCTP pool sizes. Table 2 summarizes how RR mutations affect CDV sensitivity as assessed by plaque reduction assays and calculated 50% effective concentration (EC50) values. Wild-type and ΔF4L/ΔJ2RHisF4L strains exhibited similar mean EC50 values of 42.0 and 41.2 µM, respectively. The ΔI4L strain was significantly more sensitive than the aforementioned strains (P<0.05) having a mean EC50 value of 25.1 µM. However, loss of F4L (or F4L and J2R) resulted in greater hypersensitivities to CDV (P<0.05) with EC50 values ∼5–7-fold lower than wild-type values. The ΔF4L/ΔJ2RHisY300FF4L virus was even more sensitive to CDV (EC50 = 3.5 µM) than either wild-type (P<0.05) or ΔF4L (P<0.05) strains. As noted previously [50], [51], inactivation of J2R did not further alter VACV sensitivity to CDV (Table 2). The trends in CDV sensitivity closely mirrored those found in measurements of HU sensitivity using a plaque reduction assay (Table 2). The order of resistance to HU (from measurements of EC50) was wild-type ≥ΔF4L/ΔJ2RHisF4L>ΔI4L>ΔF4L>ΔF4L/ΔJ2RHisY300FF4L and seemed unaffected by the presence or absence of the J2R gene (Table 2). In order to determine if the hypersensitivities of ΔF4L and ΔF4L/ΔJ2RHisY300FF4L strains to CDV and HU were specific and not simply due to the reduced replicative abilities of these viruses, we performed a plaque reduction assay using phosphonoacetic acid (PAA). PAA is a pyrophosphate analog and DNA polymerase inhibitor that is noncompetitive with dNTPs [52]. Therefore, the efficacy of PAA in inhibiting virus replication would not be expected to be dependent upon RR activity or dNTP pool sizes. Consistent with this, RR mutant VACV strains were not hypersensitive to PAA when compared to wild-type virus (Table 2). These mutant strains were also not hypersensitive to isatin-β-thiosemicarbazone (IBT), which causes aberrant late viral mRNA biogenesis [53] (data not shown). Collectively, these data all point to a deficiency in dNTP pools as being the cause of the ΔF4L strain growth deficiency (Figure 3) and suggest that F4, and not I4, is the critical determinant of growth efficiency and drug sensitivity.

Viral and human RR subunits interact in VACV-infected cells
Our data suggested that F4 may form functional complexes with host R1 proteins to support viral replication. This hypothesis was strengthened by previous studies that found purified mouse and VACV RR subunits to form functional chimeric RR complexes in vitro [33]. To determine if virus-host RR interactions could occur in vivo, co-immunoprecipitation experiments were performed with VACV-infected HeLa cell lysates using antibodies against HR1, Human R2 (HR2) or Hp53R2 RR subunits. F4 co-immunoprecipitated with each of the host RR subunits (Figure 5A) and the efficiency of “pull-down” was the same in extracts prepared from cells infected with wild-type and ΔI4L strains (Figure 5B), suggesting that the presence of I4 does not significantly impede F4 interaction with host RR subunits. Interaction of F4 with cellular R2 subunits, while unexpected, may not be that surprising given that R2 subunits interact with one another in addition to interacting with homodimers of R1 [1]. We thought these interactions may be in part due to enhanced cellular RR subunit expression after infection. However, we were unable to observe induction of cellular RR expression by 24 h post-infection (Figure S2). To further confirm the immunoprecipitation results, VACV strains expressing either Flag-tagged HR1 (ΔJ2RFlagHR1) or Flag-tagged I4 (ΔI4L/ΔJ2RFlagI4L) were constructed and used in new immunoprecipitation experiments. Immunoprecipitation with anti-Flag antibodies confirmed the interaction of HR1 and I4 with F4 as well as with HR2 and Hp53R2 (Figure 6A). We typically observed weaker R2 bands in immunoprecipitations of Flag-tagged HR1 compared to Flag-tagged I4 despite similar amounts of these two proteins being immunoprecipitated (Figure 6A). This result was likely due to competition between the Flag-tagged HR1 protein and endogenous HR1, whereas Flag-tagged I4 is expressed in the ΔI4L background and thus does not have to compete for binding to R2 proteins with endogenous I4. We also prepared extracts from cells infected with ΔF4L/ΔJ2RHisY300FF4L or ΔF4L/ΔJ2RHisF4L viruses and observed that these His6-tagged proteins could also be co-immunoprecipitated with HR1 protein (Figure 6B). Reciprocal co-immunoprecipitation experiments confirmed an interaction between F4 and HR1 proteins (Figure S3).


Other Chordopoxvirus R2 proteins rescued the replication defect of VACV ΔF4L strains (Figure 3B). Therefore, we determined whether these proteins could also interact with HR1. ECTV, MYXV, and SFV R2 proteins all co-immunoprecipitated with HR1 (Figure 6B). Although there appeared to be differences in the efficiency of HR1 association, western blotting of lysates showed that this reflected differences in R2 expression levels (Figure 6B). These results confirm that RR subunits from poxviruses that infect a diversity of mammalian hosts have conserved the capacity to interact with HR1.
In uninfected cells, mammalian RR subunits show an exclusively cytoplasmic distribution [54], [55], [56]. Confocal microscopy studies with antibodies directed against endogenous (Figure S4A) or epitope-tagged (Figure S4B) RR subunits suggested that VACV infection did not alter host RR localization and VACV RR subunits were also found to exhibit a similar cytoplasmic distribution.
Requirement of C-terminal residues of F4 for interaction with HR1
The previous studies showed that F4 interacts with HR1 but did not prove whether such an interaction was essential for viral replication. Numerous structural and peptide-inhibition studies of class I RR proteins have identified a C-terminal peptide (boxed in Figure 1) in R2 subunits as critical for interaction with R1 proteins [11], [57], [58], [59], [60], [61]. Since this C-terminal peptide is well conserved in F4 (Figure 1), we speculated that HR1-F4 interactions were also dependent on this peptide. To test this hypothesis, we generated the VACV strain ΔF4L/ΔJ2RHisF4LΔR1BD, encoding a truncation mutant of F4 that lacks the C-terminal seven residues representing the putative R1-binding domain (R1BD). We also generated an R1BD mutant that also encodes the Y300F substitution, (ΔF4L/ΔJ2RHisY300FF4LΔR1BD). As shown in Figure 7A, His6-tagged F4 co-immunoprecipitated with HR1 in HeLa cell extracts. However, there was a clear reduction (by ∼90%) in co-immunoprecipitation of His6-tagged F4 proteins lacking the R1BD, despite comparable levels of these two forms of F4 in lysates and immunoprecipitates. Thus, F4 appears to have conserved the R1-binding peptide encoded by class I RRs.

We used plaque area measurements to determine if deleting the R1BD would alter VACV plating properties (Figure 7B). The control viruses exhibited the same relative plaque sizes noted previously (i.e. wild-type = ΔF4L/ΔJ2RHisF4L>ΔF4L>ΔF4L/ΔJ2RHisY300FF4L) and the differences were all significant (P<0.05). However, the ΔF4L/ΔJ2RHisF4LΔR1BD and ΔF4L/ΔJ2RHisY300FF4LΔR1BD strains produced plaques no different in size from those produced by ΔF4L strains (P>0.05). This suggested that the F4 R1BD was not only required for RR activity, but that the HR1-F4 interaction was also responsible for the dominant negative effects observed with strains encoding the Y300F-substituted F4 protein with an intact R1BD. We also confirmed in these studies that inactivation of J2R alone had no significant effect on plaque size (Figure 7B).
Correlation of ΔF4L strain replication with host RR subunit expression
Our results suggested that deleting the F4L gene renders VACV highly dependent upon the host cell for provision of a complementing RR activity. This leads to the prediction that the efficiency of growth of a ΔF4L virus will depend upon the level of cellular RR activity. To test this hypothesis, we used two pancreatic cancer cell lines that have been previously reported to exhibit high (PANC-1) and low (CAPAN-2) levels of RR subunit expression and activity [45], [62]. We prepared cell-free extracts from wild-type virus-infected (or mock-infected) PANC-1 and CAPAN-2 cells, and used western blots to measure the levels of RR proteins. This study confirmed that HR1, HR2, and Hp53R2 are expressed at lower levels in CAPAN-2 cells, relative to PANC-1 cells, and that this phenotype is unaffected by VACV infection (Figure 8A). We then seeded approximately equal numbers of PANC-1 and CAPAN-2 cells into culture dishes and infected them with wild-type and mutant strains. The total titers for each of these infections at 48 or 72 h post-infection are plotted in Figure 8B. Division of the mean titers obtained in PANC-1 cells by those obtained in CAPAN-2 cultures for each virus gave an estimate of the fold difference in replication efficiencies for each strain in these cells (Figure 8C). These data showed that PANC-1 cells support a relatively normal level of replication of most mutant VACV strains. For example, the wild-type virus grew only ∼3–6-fold better on PANC-1 cells than did ΔF4L, and ΔI4L/ΔF4L, and ΔF4L/ΔJ2R strains (Figure 8B). One exception to this rule is that the wild-type virus produced titers ∼16-fold higher than the ΔI4L/ΔF4L/ΔJ2R strain on PANC-1 cells (Figure 8B). This suggested that in certain cell types and in the absence of J2R and F4L, I4L may play some role in supporting VAC replication.

A more notable feature of this experiment is that all virus tested grew better on PANC-1 cells compared to CAPAN-2 cells. The wild-type, ΔI4L, and ΔF4L/ΔJ2RHisF4L strains produced yields 6–8-fold higher on PANC-1 cells than CAPAN-2 cells 48 h post-infection and this difference was greatly exacerbated by deletion or mutation of F4L (Figure 8C). For example, the ΔF4L strain grew 18–30-fold better on PANC-1 cells and the ΔF4L/ΔJ2RHisY300FF4L strain yielded a 113-fold increase in titer on PANC-1 cells compared to CAPAN-2 cells. In fact, titering of input inocula indicated that the ΔF4L/ΔJ2RHisY300FF4L strain did not productively replicate in CAPAN-2 cells (data not shown). This suggested that the reduced RR activity of CAPAN-2 cells imposes a barrier to replication of this mutant. Collectively, these results suggested that the replication defects exhibited by ΔF4L and ΔF4L/ΔJ2RHisY300FF4L strains can be complemented in human cancer cell lines over-expressing cellular RR subunits. However, direct evidence for the linkage between cellular RR levels and mutant rescue requires further studies.
VACV RR subunits are differentially required for pathogenesis in mice
We used an animal model to determine if the apparent differential requirement for VACV RR subunits for replication in culture would be recapitulated in vivo. We infected groups of five NMRI mice with equal doses of wild-type, ΔI4L, ΔF4L, or ΔI4L/ΔF4L strains and tracked changes in animal body weight over 24 days. The wild-type and ΔI4L strains exhibited a similar degree of virulence, causing the death of 5/5 and 4/5 animals, respectively, within seven days of infection. In contrast, both ΔF4L and ΔI4L/ΔF4L strains were highly attenuated, with all animals displaying little to no signs of disease and surviving the infections (Figure 9A). There were small, transient drops in body weight for animals infected with the ΔF4L strain around days 5 and 7, otherwise these animals, and those infected with the ΔI4L/ΔF4L strain, showed no obvious signs of morbidity when compared to the mock-infected control group (Figure 9A). To obtain a more quantitative measurement of the pathogenic nature of these infections, we isolated lung tissues from mice infected with the aforementioned strains on day 5 post-infection. Wild-type and ΔI4L strains clearly had a replication advantage over ΔF4L and ΔI4L/ΔF4L strains with lung titers approximately 4 logs higher than the latter two strains (Figure 9B). These results indicate that VACV RR subunits are differentially required for virulence in mice.

Discussion
Acquisition of a suitable supply of dNTPs to support replication is a challenging feat for mammalian DNA viruses because most host cells exist predominantly in a terminally-differentiated and quiescent state [63]. The S-phase-specific nature of host R2 expression leaves quiescent cells with only p53R2-R1 complexes to maintain a low (∼2–3% the level of cycling cells [64]) level of RR activity to meet the demands of DNA repair and mitochondrial genome synthesis [16], [19], [65]. Since ribonucleotide reduction is the rate-limiting step in mammalian dNTP biogenesis [26], low RR activity may pose a barrier to productive infection. Therefore, DNA viruses must replicate only in cycling cells, induce host RR activity upon infection, and/or encode their own RR enzymes [63]. Many large DNA viruses, including herpes-, irido-, asfra - and poxviruses have evolved the later strategy.
It is clear that herpesvirus-encoded RR proteins are important because inactivation of viral RR genes leads to replication defects in vitro and in animals [66], [67], [68], [69]. Furthermore, inhibiting complex formation by herpes simplex virus (HSV) R1 and R2 proteins with a C-terminal R2 peptide mimic has been shown to prevent HSV replication in culture [70], [71], [72]. Interestingly, β-herpesviruses, only encode an R1 gene, although it is still required for virulence [63]. However, it is unlikely to play a catalytic role in dNTP biogenesis as it encodes mutations at key catalytic residues that would render this subunit inactive in RR complexes [73], [74], [75]. Recent evidence suggests that β-herpesviruses may induce host RR protein expression, possibly explaining why viral RR function was not conserved [75], [76]. What biological purpose is served by β-herpesvirus R1 proteins is unclear, although it has been suggested that these proteins might play some role in inhibiting apoptosis [77].
The increasing availability of virus genome sequences has revealed that differential conservation of viral RR genes is actually a widespread phenomenon among eukaryotic DNA viruses. However, in contrast to the case of β-herpesviruses, most DNA viruses that encode a single RR subunit encode R2 proteins while R1 is frequently absent. For example, iridoviruses in the Megalocytivirus genus only encode an R2 subunit while all other iridoviruses encode both RR subunits [78]. Furthermore, certain members of the Phycodnaviridae and Ascoviridae viral families also only encode R2 subunits [79]. This R2 bias is also seen in bacteriophage belonging to the Siphoviridae and Myoviridae families, suggesting that even prokaryotic viruses have biased conservation of RR genes [79].
Perhaps the most biased conservation of RR genes is found in poxviruses, with a clear favoring of R2 over R1 (Table S1). Even Orthopoxviruses, which typically encode both R1 and R2 genes, contain a member (horsepox virus) that encodes a fragmented R1 gene [80]. Many other Chordopoxviruses show no evidence of ever having encoded an R1 activity, including the Leporipoxviruses MYXV and SFV which we sequenced ten years ago [81], [82]. At that time, the close similarity of MYXV and SFV R2 subunits to mammalian R2 proteins, and absence of a viral R1 homolog, led us to suggest that Leporipoxvirus R2 subunits were likely forming chimeric complexes with host R1 proteins [82]. Subsequent biochemical studies of VACV F4 and I4 by Chimploy and Mathews [33] found that mixing purified F4 and I4 with MR1 and MR2 proteins, respectively, resulted in functional, chimeric RR enzymes. However, the two kinds of chimeric RRs did not display identical properties. Whereas the native complexes (i.e. I42F42 and MR12MR22) were about equally active, the I42MR22 enzyme exhibited ∼5-fold less activity and the MR12F42 enzyme showed up to 2-fold more activity than either native complex [33]. These observations may explain why Child et al. reported their ΔI4L strain to exhibit no observable replication defect in culture and only a small (∼10-fold) increase in LD50 for mice compared to wild-type VACV [34]. However, in an attempt to develop new vaccine strains, Lee et al. generated a F4L insertional inactivation mutant and reported significant increases of ∼1000-fold in the LD50 of this mutant in a similar mouse model used by Child et al. with their ΔI4L strain [34]. Collectively these independent bioinformatic, biochemical, and molecular genetic studies all suggested that poxvirus R2 subunits might be more important for viral replication than R1 subunits. However, the contributions of poxvirus R1 and R2 subunits to viral replication and pathogenesis had never been directly compared nor was it clear why R2 subunits may be more important to the poxvirus life cycle.
We examined this issue in detail by generating a panel of VACV RR mutant strains and analyzing their plaque, growth, and pathogenic properties. Our studies clearly show that these properties are far more affected in ΔF4L strains compared to ΔI4L strains (Figures 3 and 9). Combining F4L and I4L deficiencies caused no further inhibition of virus growth, suggesting that the phenotype is dominated by the integrity of the F4L locus. We also showed that inactivating VACV J2R in the ΔF4L background did not further impede replication (Figure 3), suggesting that the salvage pathway for dNTP production is either not required for replication in culture, or is sufficiently complemented by host TK enzymes. This result made it possible to use the J2R locus as a site for introducing ectopic copies of different recombinant R2 proteins. The ΔF4L strain's phenotype can be completely complemented by a gene encoding His6-tagged F4 and by genes encoding other Orthopoxvirus or Leporipoxvirus R2 proteins (Figure 3B). Why Hp53R2 failed to rescue this phenotype is unclear but several possibilities exist. For one, R1-p53R2 complexes exhibit only 40–60% the activity of R1-R2 complexes [17] and this reduced activity might not meet some activity threshold required for efficient viral replication. Secondly, significant fractions of Hp53R2 proteins are bound in inactive complexes by p53 and p21 proteins, and are only released after appropriate signaling pathways have been activated [83], [84]. Therefore, even if one over-expresses Hp53R2, it may not produce a sufficient level of “free” Hp53R2 that could complex with R1 proteins. Finally, Hp53R2 has recently been shown to inhibit MEK2, a kinase involved in the activation of the Ras-Raf-MAPK signaling pathway [85]. This inhibition could be detrimental as activation of the MAPK pathway is required for VACV replication [86]. These possibilities are currently being addressed. Attempts to generate a VACV strain over-expressing HR2 have thus far been unsuccessful and so it is unclear whether HR2 can complement a ΔF4L strain.
The hypothesis that F4 protein can compete with (or replace) cellular small subunits to form chimeric RR complexes in vivo is strongly supported by the dominant-negative phenotype exhibited by Y300F-substituted F4 protein (Figure 3). Viruses encoding these mutant proteins replicate very poorly and produce extremely small plaques. Furthermore, this phenotype is not altered by the presence or absence of I4 (Figure 3B). The genetic data are fully concordant with our immunoprecipitation experiments, which showed that F4 interacted with cellular RR subunits (Figure 5A) and that this interaction was unaffected by I4 (Figure 5B). We also found that other Chordopoxvirus R2 proteins co-immunoprecipitated with HR1 (Figure 6B), which is consistent with the ability of these proteins to rescue the replication defect of the ΔF4L strain. The ability of various poxvirus R2 subunits to interact with HR1 might be explained by the high degree of sequence conservation amongst mammalian RR subunits and the fact that Chordopoxvirus R2 subunits are typically >70% identical to mammalian subunits. The observation that poxvirus RR genes are generally more similar to cellular RR genes than other virus RR genes has led to the suggestion that poxviruses have acquired RR genes through horizontal transfer events with their host [87], [88]. Interestingly, other viral and bacterial pathogens have also likely acquired RR enzymes through host gene capture [89], [90], [91]. It would be of interest to determine if Chordopoxvirus R2 proteins exhibit a quantitative binding preference for R1 proteins isolated from their natural hosts (e.g. MR1 with ECTV R2 and rabbit R1 with MYXV and SFV R2), as that would be an expected consequence of evolutionary adaption to a particular host. Potential differences in binding affinities between poxvirus and host subunits may provide further insight into factors that contribute to poxvirus host range which remain poorly defined.
During infection, ∼8-fold more F4 than I4 subunits are synthesized [92]. In tissue culture, levels of mammalian R1 are constant during the cell cycle due to its long half-life [13], while R2 subunits are quickly degraded late in mitosis leading to a much shorter half-life [15]. Given the relatively reduced activity of R1-Hp53R2 complexes [17], it is possible that production of F4 in excess allows these subunits to form needed complexes with both viral and host R1 subunits. Interestingly, poxvirus R2 subunits, like Hp53R2, lack much of the N-terminal sequences found in HR2 including phosphorylation and ubiquitination sites that may regulate HR2 function and degradation (Figure 1) [15]. This may explain why F4 protein levels are stable for at least 12 h after infection [92]. It seems likely that adaptive changes during evolution has led to conservation of poxvirus R2 enzymatic function yet has resulted in a loss of regulatory sequences that may restrict viral subunit levels in the host.
We hypothesized that the ability of Chordopoxvirus R2 proteins to interact with HR1 was due to the high degree of conservation of the C-terminal seven residues between poxvirus and mammalian R2 subunits (Figure 1). This C-terminal motif has been well-characterized in R1-R2 interaction studies of various class I RR enzymes [11], [57], [58], [59], [60], [61] and an oligopeptide mimic (7FTLDADF1) of mammalian R2 C-termini has been shown to inhibit RR activity [57]. Positions 1, 5, and 7 in this mimic are the most critical determinants of RR inhibition [57], and the residues at these positions are conserved in the C-terminus of F4 (FSLDVDF) suggesting that VACV and mammalian RR share a common R1-R2 subunit interaction mechanism. The large differences between C-terminal sequences of HSV (YAGAVVNDL) and mammalian R2 subunits likely explains why no evidence could be found for interaction of HSV RR proteins with host subunits [23] and why peptide mimics of the HSV R2 C-terminus are highly selective antivirals [71]. Previous studies have used the F4 heptapeptide to generate an affinity column for I4 purification [93]. Therefore, we thought it was likely that F4 interacted with R1 proteins in a similar manner as found with cellular R2 subunits. Indeed, interaction of F4 proteins lacking the putative R1BD with HR1 was clearly impaired (Figure 7A) and strains expressing these truncated proteins were unable to rescue the small plaque phenotype of the ΔF4L mutant (Figure 7B). However, deleting the R1BD from Y300F F4 did suppress the dominant negative phenotype (Figure 7B), which further implied that F4 functionally interacts with HR1 through the C-terminus of F4.
Collectively our data show that VACV F4 proteins (and likely other poxvirus R2 proteins) are required for efficient viral replication in culture as well as for pathogenesis (Figure 9). While our studies of CDV and HU sensitivities (Table 2) suggest a defect in RR activity and subsequent dNTP pool biogenesis as the underlying cause for the defect of ΔF4L strains, it is possible that these are only indirect consequences of inactivation of F4L and other functions of F4 are required for replication. However, the dominant negative phenotype of the Y300F-encoding strains in the presence or absence of I4 (Figure 3B), the requirement of the R1BD to produce this phenotype and interact with HR1, and the similar localization observed with viral and cellular RR subunits in infected cells (Figure S4) all support the simple conclusion that F4 must form complexes with host R1 proteins to facilitate dNTP biogenesis.
The critical importance of this interaction for VACV replication suggests that ΔF4L strains may act as selective oncolytic agents. A wide variety of human cancers exhibit elevated RR expression patterns and prolonged treatment of patients with RR inhibitors can lead to drug resistance as result of HR2 gene amplification [94], [95], [96]. For example, a recent study of patients with non-small lung cancer found that elevated host RR expression levels in patients' tumors were directly correlated with reduced response to chemotherapy and poorer prognoses [94]. Interestingly, hrR3, a HSV mutant strain with an inactivated viral R1 gene, replicates more efficiently in cancers with elevated host RR expression [97] and shows promise as an oncolytic agent in mouse models [98]. The enhanced replication of ΔF4L and ΔF4L/ΔJ2RHisY300FF4L strains in PANC-1 cells relative to CAPAN-2 pancreatic cancer cell lines (Figure 8C) correlates well with the higher levels of RR subunits in PANC-1 cells (Figure 8A) and documented differences in RR activities between these two cell lines [45]. While further evidence will be needed to prove that host RR proteins complement the ΔF4L strain replication defect in PANC-1 cells, our results build a strong circumstantial case for the dependence of these F4L mutant strains on host RR activity. Since VACV and other, non-Orthopoxviruses (e.g. Leporipoxviruses [99], [100], and Yatapoxviruses [101]) that encode R2 subunits have shown potential for use in cancer virotherapy, we suggest that deletion of R2 subunit genes from these viruses may create more selective oncolytic agents.
During our studies we noted that poxviruses that encode R2 and TK genes tend to have higher A+T base content in their genomes than poxviruses lacking these genes (Table S1). This strong correlation suggests that hybrid virus-host RR complexes and/or poxvirus TK proteins have contributed to the establishment of unique dNTP pools that have influenced viral genome composition during the co-evolution of poxviruses with their hosts. Recently an APC mimic has been identified in poxviruses that lack R2 and TK genes including Molluscipoxviruses, Parapoxviruses, and crocodilepox virus [102]. The APC mimic in orf virus, termed “PACR” (poxvirus APC/cyclosome regulator) inhibits APC activity and causes mammalian cells to accumulate in G2/M phases of the cell cycle [102]. Since APC targets mammalian TK [103] and R2 [15] proteins for degradation during mitosis, poxvirus APC mimics may serve to prevent degradation of these host nucleotide metabolism proteins during viral replication. This is supported by the finding that PACR inhibits host TK degradation during the cell cycle [102]. This reliance on host nucleotide metabolism proteins may explain why poxviruses encoding APC mimics have low A+T content in their genomes (Table S1). Strict reliance on host nucleotide metabolism machinery may also explain why GC-rich Mollusci - and Parapoxviruses have a rather limited host range when compared to AT-rich Orthopoxviruses which encode their own RR and TK [104] (Table S1). Therefore, poxviruses appear to have acquired different mechanisms to obtain dNTPs for replication, which may ultimately influence their genomic composition and host tropism.
Materials and Methods
Cell and virus culture
Cell and virus culture methods have been described elsewhere [105]. Wild-type VACV and its mutant derivatives were derived from strain Western Reserve (WR) originally acquired from the American Type Culture Collection. Non-transformed African Green Monkey kidney cells (BSC-40) were normally cultured in modified Eagle's medium (MEM) supplemented with 5% fetal bovine serum (FBS). HeLa human cervical adenocarcinoma and human embryonic lung (HEL) cells were cultured in Dulbeccos MEM (DMEM) supplemented with 10% FBS. PANC-1 and CAPAN-2 cells are human pancreatic epithelioid carcinoma and adenocarcinoma lines, respectively and were also cultured in DMEM supplemented with 10% FBS. All of the above cell lines were originally obtained from the American Type Culture Collection. A U20S human osteosarcoma cell line that expresses Cre recombinase was a kind gift from Dr. J. Bell (University of Ottawa). These cells were maintained in DMEM supplemented with 10% FBS. Cells were cultured in Opti-MEM media (Invitrogen; Carlsbad, CA) for experiments requiring transfections.
Materials
(S)-1-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine or cidofovir (CDV or HPMPC) was from Dr. K. Hostetler (University of California, San Diego). Hydroxyurea (HU) was obtained from Alfa Aesar (Ward Hill, MA). X-gal and X-glu substrates were obtained from Sigma Chemical Co. (St. Louis, MO) and Clontech (Palo Alto, CA), respectively. Phosphonoacetic acid (PAA) was from Sigma Chemical Co. Isatin-β-thiosemicarbazone (IBT) was from Pfaltz and Bauer (Waterbury, CT). Mycophenolic acid (MPA) and xanthine were obtained from Sigma Chemical Co. Hypoxanthine was obtained from ICN Biomedicals, Inc. (Aurora, OH). Compounds were diluted to their final concentration in MEM (CDV; HU; PAA; IBT) or in a 1∶1 mixture of MEM and 1.7% noble agar (X-gal; X-glu) immediately prior to use. Taq and PfuUltra DNA polymerases were obtained from Fermentas (Burlington, ON) and Stratagene (La Jolla, CA), respectively.
Antibodies, western blotting, and immunoprecipitation
Normal mouse and goat serum and goat polyclonal antibodies against human R1 (HR1), human R2 (HR2), and human p53R2 (Hp53R2) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Mouse monoclonal antibodies against HR1 and HR2 were from Millipore (Billerica, MA) and Santa Cruz Biotechnology, Inc., respectively. Mouse monoclonal antibodies against Flag and His6 (His) epitopes were from Sigma and Roche (Mississauga, ON), respectively. Rabbit anti-Flag epitope polyclonal antibodies were obtained from Sigma. A mouse monoclonal antibody was raised against bacterially-expressed, recombinant ECTV R2 antigen by ProSci (Poway, CA). The resulting antibody also recognizes VACV F4 and was used for western blotting. In some cases, a rabbit anti-F4 polyclonal antibody was also used for western blotting. The plasmid used to express recombinant ECTV R2 antigen and the rabbit anti-F4 antibody were kindly provided by Dr. M. Barry (University of Alberta). A rabbit anti-VACV I4 polyclonal antibody was obtained from Dr. C. Mathews (Oregon State University). Although this antibody recognizes VACV I4, it also cross-reacts with cellular R1 on western blots [92]. The mouse monoclonal antibody against VACV I3 has been described [40] and the mouse monoclonal antibody against cellular actin was from Sigma.
Protein extracts for western blots and immunoprecipitations were prepared from cell cultures by lysing cells on ice in a buffer containing 150 mM NaCl, 20 mM Tris (pH 8.0), 1 mM EDTA, and 0.5% NP-40 along with freshly-added phenylmethylsulfonyl fluoride (100 µg/mL) and protease inhibitor tablets (Roche). For western blots, 20–40 µg of total protein were subjected to SDS-PAGE and subsequently blotted with appropriate antibodies after transfer to nitrocellulose membranes. Membranes were scanned using an Odyssey scanner (Li-COR Biosciences).
Protein extracts for immunoprecipitations were recovered as described above 6–8 h post-infection from 107 HeLa cells infected with indicated strains at a multiplicity of infection (MOI) of 10. Extracts were then pre-cleared by incubation with normal mouse or goat serum along with protein G sepharose beads (GE Healthcare Life Sciences; Piscataway, NJ) for 30 min at 4°C with constant inversion. The samples were subsequently centrifuged (2,500 rpm, 1 min, 4°C) and supernatants were transferred to fresh tubes. These extracts were then incubated with the primary antibodies overnight at 4°C with constant inversion. Fresh protein G beads were then added to the extracts and incubated for 2 h at 4°C after which the beads were collected (2,500 rpm, 1 min, 4°C) and washed four times with lysis buffer. The resulting bead-protein complexes were resuspended in SDS-PAGE loading buffer, boiled for 15 min and subjected to SDS-PAGE. Western blotting was then performed as described above. Whole cell extracts (lysates) were also blotted with indicated antibodies and represented ∼5% of the input material used for immunoprecipitations.
Plaque morphology and replication analyses
Plaque dimensions were measured on 60-mm-diameter dishes of confluent BSC-40 cells infected with ∼100 plaque-forming units (PFU) of the indicated strain. After 48 h of infection, triplicate plates were stained with crystal violet and scanned using an HP ScanJet 6300C scanner. The resulting image files were analyzed using ImageJ v1.04g software (National Institutes of Health, USA). Unpaired t-tests or one-way ANOVA tests were performed on mean plaque areas between wild-type and each of the various RR mutant strains using GraphPad Prism (San Diego, CA) software (version 4.0). In some cases two different RR mutant strains were also compared for differences in mean plaque areas. A P value of <0.05 was considered to be statistically significant.
Growth analyses were conducted in BSC-40, HeLa, PANC-1 and CAPAN-2 cell cultures using the indicated MOI and strains. Cells were harvested by scraping monolayers into the culture media at the indicated time points followed by three rounds of freeze-thawing. Virus stocks were titered on BSC-40 cells.
For viral genome replication analyses, at the indicated times post-infection, BSC-40 cells were harvested by scraping, collected by centrifugation (800 rpm, 10 min, 4°C) washed once with PBS, and resuspended in 500 µL of 10× saline-sodium citrate (SSC) loading buffer containing 1 M ammonium acetate [106]. The cells were then disrupted by three cycles of freeze-thaw and 50-µL aliquots of the lysates were applied to a Zeta probe membrane using a slot-blot apparatus (Bio-Rad, Richmond, CA). Samples were denatured with 1.5 M NaCl and 0.5 M NaOH and washed twice with 10× SSC loading buffer. The membrane was then hybridized with a 32P-labeled E9L gene probe. After the membrane was washed with SSC buffer and air dried, it was exposed to a phosphorimager screen, imaged using a Typhoon 8600 phosphorimager and the data were processed using ImageQuant software, (version 5.1) [40]. In some cases 0.5 mM HU was added to the media 1 h post-infection.
Plaque reduction assays
Plaque-reduction assays were performed as previously described [105]. Briefly, 35-mm-diameter dishes of confluent BSC-40 cells were inoculated with ∼100 PFU of the indicated virus strains, and 1 h after infection either drug-free medium or medium containing the indicated doses of CDV or HU was added to the cultures and the plates were incubated at 37°C for 48 h. Plates were then stained with crystal violet to visualize and count plaques. Mean 50% effective concentration (EC50) values and their 95% confidence intervals (CIs) were calculated using nonlinear regression analyses with GraphPad Prism software after three independent experiments had been performed. In cases where the 95% CIs of two different EC50 values did not overlap, these two EC50 values were considered to be statistically different (P<0.05).
Confocal microscopy
HeLa cells were grown on coverslips in 24-well plates and infected with the indicated virus strains at a MOI of 5 for 10 h. The cells were fixed for 30 min on ice with 4% paraformaldehyde in PBS. The fixed cells were blocked and permeabilized for 1 h at RT in PBS containing 0.1% Tween (PBS-T) as well as 10% BSA. The coverslips were then incubated with the primary antibodies diluted in PBS-T (1% BSA) for 2 h at RT, washed three times and then incubated with secondary antibodies conjugated to Alexa 488 or 594 (Invitrogen) for 1 h at RT. The cells were then counterstained with 10 ng/mL 4′,6′-diamidino-2-phenylindole (DAPI) in PBS-T for 15 min. The specimens were examined using a Zeiss 710 Laser-Scanning confocal microscope equipped with DAPI, Alexa 488, and Alexa 594 filters. Images were captured and processed using ZEN 2009 software and Adobe Photoshop (version 10.0.1).
Recombinant viruses
BSC-40 cells were grown to confluence and then infected for 1 h with the appropriate VACV strain (see below) at a MOI of 2 in 0.5 mL of PBS. The cells were then transfected with 2 µg of linearized plasmid DNA using Lipofectamine 2000 (Invitrogen). See Text S1 and for details regarding the primers and transfer vectors used to generate recombinant VACV strains. The cells were returned to the incubator for another 5 h, the transfection solution was replaced with 5 mL of fresh growth medium, and the cells were cultured for 24–48 h at 37°C. Virus progeny were released by freeze-thawing, and the virus titer was determined on BSC-40 cells. To identify recombinant virus, plaques were stained with X-gal or X-glu (both at 0.4 mg/mL) in solid growth media, or cultured in media containing 25 µg/mL MPA supplemented with xanthine (250 µg/mL) and hypoxanthine (15 µg/mL) for selection of yfp-gpt-encoding strains (see Text S1; [107]). The PCR was used to confirm insertions/deletions in the resulting recombinant viruses. The primers: 5′-GATGAATGTCCTGGATTGGA-3′ & 5′-ATTCCAAAGATCCGACGGTA-3′ were used to PCR amplify ∼700 bp of I4L sequence that should not be present in ΔI4L strains. The primers: 5′-ATGGAACCCATCCTTGCACC-3′ & 5′-ATCTTCTTGAGACATAACTC-3′ were used to amplify ∼930 bp of F4L sequence that should not be present in ΔF4L strains. Disruption of J2R sequence was detected with primers: 5′-TCCTCTCTAGCTACCACCGCAATAG-3′ & 5′-GTGCGGCTACTATAACTTTTTTCC-3′ that bind to regions of J2R flanking the insertion site of pSC66 vector [108] sequences (see below). Primers TGGATTCGTACAAATTGGATTCTAT & AATTGCTATTTCAGAGATGAGGTTC were used to amplify an ∼800 bp fragment from VACV DNA polymerase (E9L) sequence to serve as a positive control for amplification. In some cases western blotting was used to confirm the presence or absence of gene expression in the described VACV strains. Details of how each VACV strain was constructed are provided in Text S1 and the marker rescue strategies used in these studies are depicted in Figure 2A.
PfuUltra DNA polymerase (Stratagene) was used to PCR-amplify DNA for cloning whereas Taq DNA polymerase (Fermentas) was used for PCR diagnostic purposes. Plasmid constructs were verified by sequencing and all virus strains were plaque-purified a minimum of three times in BSC-40 cells or Cre recombinase-expressing U20S cells. All VACV strains were characterized by PCR (data not shown) and sometimes by western blotting (Figure 2B), although for brevity only characterization of the main viral strains discussed throughout this study is shown.
Animal studies
Female NMRI mice, 3 to 4 weeks of age, were obtained from Charles River Laboratories (Brussels, Belgium). Mice were utilized at 5 mice per infection or control group for morbidity studies. Mice were anesthetized using ketamine-xylazine and inoculated intransally (or mock-inoculated) with 4×104 PFU of virus diluted in 30 µL of saline. Animal body weights were recorded over the next 24 days or until the animals had to be euthanized because of more than 30% loss in body weight. To determine viral titers in lungs, two (wild-type infections) or five animals (ΔI4L, ΔF4L, and ΔI4L/ΔF4L infections) were euthanized on day 5. Lung samples were removed aseptically, weighed, homogenized in MEM, and frozen at −70°C until assayed by titrations on HEL cells. For mouse pathogenicity experiments, the ΔI4L/ΔF4L strain was generated with the pDGloxPKODEL vector. This allowed for removal of the yfp-gpt marker cassette from the I4L locus after passage in U20S cells expressing Cre recombinase. No differences were found in replication in culture between strains expressing the yfp-gpt cassette and those with this cassette deleted by Cre recombination (Figure S5). See Text S1 for further details.
Ethics statement
All animal work was approved by the K.U. Leuven Animal Care and Use Committee. All animal guidelines and policies were in accordance with the Belgian Royal Decree of 14 November 1993 concerning the protection of laboratory animals and the European Directive 86-609-EEC for the protection of vertebrate animals used for experimental and other scientific purposes.
Genbank gene ID numbers
-
VACV I4L (VACWR073) (Genbank ID: 3707606)
-
VACV F4L (VACWR043) (Genbank ID: 3707500)
-
VACV J2R (VACWR094) (Genbank ID: 3707550)
Genbank protein accession numbers
-
human R2 (HR2; Genbank accession: NP_001025.1)
-
mouse R2 (MR2; Genbank accession: NP_033130.1)
-
human p53R2 (Hp53R2; Genbank accession: BAD12267.1)
-
mouse p53R2 (Mp53R2; Genbank accession: Q6PEE3.1)
-
VACV F4 (Genbank accession: AAO89322.1),
-
ectromelia EVM028 (Genbank accession: NP_671546.1),
-
myxoma virus m015L (Genbank accession: NP_051729.1),
-
Shope fibroma gp015L (Genbank accession: NP_051904.1)
-
human R1 (HR1; Genbank accession: AAD37491)
-
mouse R1 (mR1; Genbank accession: AAH16450)
-
VACV I4 (Genbank accession: AAO89352)
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. NordlundP
ReichardP
2006
Ribonucleotide reductases.
Annu Rev Biochem
75
681
706
2. KolbergM
StrandKR
GraffP
AnderssonKK
2004
Structure, function, and mechanism of ribonucleotide reductases.
Biochim Biophys Acta
1699
1
34
3. TorrentsE
TrevisiolC
RotteC
HellmanU
MartinW
2006
Euglena gracilis ribonucleotide reductase: the eukaryote class II enzyme and the possible antiquity of eukaryote B12 dependence.
J Biol Chem
281
5604
5611
4. RovaU
AdraitA
PotschS
GraslundA
ThelanderL
1999
Evidence by mutagenesis that Tyr(370) of the mouse ribonucleotide reductase R2 protein is the connecting link in the intersubunit radical transfer pathway.
J Biol Chem
274
23746
23751
5. HimoF
SiegbahnPE
2003
Quantum chemical studies of radical-containing enzymes.
Chem Rev
103
2421
2456
6. StubbeJ
NoceraDG
YeeCS
ChangMC
2003
Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer?
Chem Rev
103
2167
2201
7. NordlundP
SjobergBM
EklundH
1990
Three-dimensional structure of the free radical protein of ribonucleotide reductase.
Nature
345
593
598
8. RovaU
GoodtzovaK
IngemarsonR
BehravanG
GraslundA
1995
Evidence by site-directed mutagenesis supports long-range electron transfer in mouse ribonucleotide reductase.
Biochemistry
34
4267
4275
9. LarssonA
SjobergBM
1986
Identification of the stable free radical tyrosine residue in ribonucleotide reductase.
EMBO J
5
2037
2040
10. EkbergM
SahlinM
ErikssonM
SjobergBM
1996
Two conserved tyrosine residues in protein R1 participate in an intermolecular electron transfer in ribonucleotide reductase.
J Biol Chem
271
20655
20659
11. ClimentI
SjobergBM
HuangCY
1992
Site-directed mutagenesis and deletion of the carboxyl terminus of Escherichia coli ribonucleotide reductase protein R2. Effects on catalytic activity and subunit interaction.
Biochemistry
31
4801
4807
12. BjorklundS
SkogS
TribukaitB
ThelanderL
1990
S-phase-specific expression of mammalian ribonucleotide reductase R1 and R2 subunit mRNAs.
Biochemistry
29
5452
5458
13. EngstromY
ErikssonS
JildevikI
SkogS
ThelanderL
1985
Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits.
J Biol Chem
260
9114
9116
14. ChabesAL
BjorklundS
ThelanderL
2004
S Phase-specific transcription of the mouse ribonucleotide reductase R2 gene requires both a proximal repressive E2F-binding site and an upstream promoter activating region.
J Biol Chem
279
10796
10807
15. ChabesAL
PflegerCM
KirschnerMW
ThelanderL
2003
Mouse ribonucleotide reductase R2 protein: a new target for anaphase-promoting complex-Cdh1-mediated proteolysis.
Proc Natl Acad Sci U S A
100
3925
3929
16. TanakaH
ArakawaH
YamaguchiT
ShiraishiK
FukudaS
2000
A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage.
Nature
404
42
49
17. GuittetO
HakanssonP
VoevodskayaN
FriddS
GraslundA
2001
Mammalian p53R2 protein forms an active ribonucleotide reductase in vitro with the R1 protein, which is expressed both in resting cells in response to DNA damage and in proliferating cells.
J Biol Chem
276
40647
40651
18. WangX
ZhenchukA
WimanKG
AlbertioniF
2009
Regulation of p53R2 and its role as potential target for cancer therapy.
Cancer Lett
276
1
7
19. BourdonA
MinaiL
SerreV
JaisJP
SarziE
2007
Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion.
Nat Genet
39
776
780
20. HakanssonP
HoferA
ThelanderL
2006
Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells.
J Biol Chem
281
7834
7841
21. TidonaCA
DaraiG
2000
Iridovirus homologues of cellular genes–implications for the molecular evolution of large DNA viruses.
Virus Genes
21
77
81
22. BoursnellM
ShawK
YanezRJ
VinuelaE
DixonL
1991
The sequences of the ribonucleotide reductase genes from African swine fever virus show considerable homology with those of the orthopoxvirus, vaccinia virus.
Virology
184
411
416
23. FrameMC
MarsdenHS
DutiaBM
1985
The ribonucleotide reductase induced by herpes simplex virus type 1 involves minimally a complex of two polypeptides (136K and 38K).
J Gen Virol
66(Pt 7)
1581
1587
24. SlabaughM
RosemanN
DavisR
MathewsC
1988
Vaccinia virus-encoded ribonucleotide reductase: sequence conservation of the gene for the small subunit and its amplification in hydroxyurea-resistant mutants.
J Virol
62
519
527
25. TengelsenLA
SlabaughMB
BiblerJK
HrubyDE
1988
Nucleotide sequence and molecular genetic analysis of the large subunit of ribonucleotide reductase encoded by vaccinia virus.
Virology
164
121
131
26. ElfordHL
FreeseM
PassamaniE
MorrisHP
1970
Ribonucleotide reductase and cell proliferation. I. Variations of ribonucleotide reductase activity with tumor growth rate in a series of rat hepatomas.
J Biol Chem
245
5228
5233
27. ParkerS
NuaraA
BullerRM
SchultzDA
2007
Human monkeypox: an emerging zoonotic disease.
Future Microbiol
2
17
34
28. VorouRM
PapavassiliouVG
PierroutsakosIN
2008
Cowpox virus infection: an emerging health threat.
Curr Opin Infect Dis
21
153
156
29. SlabaughMB
JohnsonTL
MathewsCK
1984
Vaccinia virus induces ribonucleotide reductase in primate cells.
J Virol
52
507
514
30. SchmittJF
StunnenbergHG
1988
Sequence and transcriptional analysis of the vaccinia virus HindIII I fragment.
J Virol
62
1889
1897
31. SlabaughMB
MathewsCK
1984
Vaccinia virus-induced ribonucleotide reductase can be distinguished from host cell activity.
J Virol
52
501
506
32. HendricksSP
MathewsCK
1998
Allosteric regulation of vaccinia virus ribonucleotide reductase, analyzed by simultaneous monitoring of its four activities.
J Biol Chem
273
29512
29518
33. ChimployK
MathewsCK
2001
Mouse ribonucleotide reductase control: influence of substrate binding upon interactions with allosteric effectors.
J Biol Chem
276
7093
7100
34. ChildSJ
PalumboGJ
BullerRM
HrubyDE
1990
Insertional inactivation of the large subunit of ribonucleotide reductase encoded by vaccinia virus is associated with reduced virulence in vivo.
Virology
174
625
629
35. RajagopalI
AhnBY
MossB
MathewsCK
1995
Roles of vaccinia virus ribonucleotide reductase and glutaredoxin in DNA precursor biosynthesis.
J Biol Chem
270
27415
27418
36. HowleyPM
SpehnerD
DrillienR
1996
A vaccinia virus transfer vector using a GUS reporter gene inserted into the I4L locus.
Gene
172
233
237
37. LeeMS
RoosJM
McGuiganLC
SmithKA
CormierN
1992
Molecular attenuation of vaccinia virus: mutant generation and animal characterization.
J Virol
66
2617
2630
38. GammonDB
EvansDH
2009
The 3′-to-5′ exonuclease activity of vaccinia virus DNA polymerase is essential and plays a role in promoting virus genetic recombination.
J Virol
83
4236
4250
39. BlackME
HrubyDE
1992
A single amino acid substitution abolishes feedback inhibition of vaccinia virus thymidine kinase.
J Biol Chem
267
9743
9748
40. LinYC
LiJ
IrwinCR
JenkinsH
DeLangeL
2008
Vaccinia virus DNA ligase recruits cellular topoisomerase II to sites of viral replication and assembly.
J Virol
82
5922
5932
41. ParanN
De SilvaFS
SenkevichTG
MossB
2009
Cellular DNA ligase I is recruited to cytoplasmic vaccinia virus factories and masks the role of the vaccinia ligase in viral DNA replication.
Cell Host Microbe
6
563
569
42. RosemanNA
SlabaughMB
1990
The vaccinia virus HindIII F fragment: nucleotide sequence of the left 6.2 kb.
Virology
178
410
418
43. XueL
ZhouB
LiuX
WangT
ShihJ
2006
Structurally dependent redox property of ribonucleotide reductase subunit p53R2.
Cancer Res
66
1900
1905
44. SlabaughMB
MathewsCK
1986
Hydroxyurea-resistant vaccinia virus: overproduction of ribonucleotide reductase.
J Virol
60
506
514
45. DuxburyMS
ItoH
BenoitE
ZinnerMJ
AshleySW
2004
Retrovirally mediated RNA interference targeting the M2 subunit of ribonucleotide reductase: A novel therapeutic strategy in pancreatic cancer.
Surgery
136
261
269
46. CihlarT
ChenMS
1996
Identification of enzymes catalyzing two-step phosphorylation of cidofovir and the effect of cytomegalovirus infection on their activities in host cells.
Mol Pharmacol
50
1502
1510
47. XiongX
SmithJL
KimC
HuangES
ChenMS
1996
Kinetic analysis of the interaction of cidofovir diphosphate with human cytomegalovirus DNA polymerase.
Biochem Pharmacol
51
1563
1567
48. MageeWC
HostetlerKY
EvansDH
2005
Mechanism of inhibition of vaccinia virus DNA polymerase by cidofovir diphosphate.
Antimicrob Agents Chemother
49
3153
3162
49. MageeWC
AldernKA
HostetlerKY
EvansDH
2008
Cidofovir and (S)-9-[3-hydroxy-(2-phosphonomethoxy)propyl]adenine are highly effective inhibitors of vaccinia virus DNA polymerase when incorporated into the template strand.
Antimicrob Agents Chemother
52
586
597
50. KernER
PrichardMN
QuenelleDC
KeithKA
TiwariKN
2009
Activities of certain 5-substituted 4′-thiopyrimidine nucleosides against orthopoxvirus infections.
Antimicrob Agents Chemother
53
572
579
51. PrichardMN
KeithKA
JohnsonMP
HardenEA
McBrayerA
2007
Selective phosphorylation of antiviral drugs by vaccinia virus thymidine kinase.
Antimicrob Agents Chemother
51
1795
1803
52. TaddieJA
TraktmanP
1993
Genetic characterization of the vaccinia virus DNA polymerase: cytosine arabinoside resistance requires a variable lesion conferring phosphonoacetate resistance in conjunction with an invariant mutation localized to the 3′-5′ exonuclease domain.
J Virol
67
4323
4336
53. CresawnSG
PrinsC
LatnerDR
ConditRC
2007
Mapping and phenotypic analysis of spontaneous isatin-beta-thiosemicarbazone resistant mutants of vaccinia virus.
Virology
363
319
332
54. EngstromY
RozellB
1988
Immunocytochemical evidence for the cytoplasmic localization and differential expression during the cell cycle of the M1 and M2 subunits of mammalian ribonucleotide reductase.
EMBO J
7
1615
1620
55. PontarinG
FijolekA
PizzoP
FerraroP
RampazzoC
2008
Ribonucleotide reduction is a cytosolic process in mammalian cells independently of DNA damage.
Proc Natl Acad Sci U S A
105
17801
17806
56. EngstromY
RozellB
HanssonHA
StemmeS
ThelanderL
1984
Localization of ribonucleotide reductase in mammalian cells.
EMBO J
3
863
867
57. FisherA
YangFD
RubinH
CoopermanBS
1993
R2 C-terminal peptide inhibition of mammalian and yeast ribonucleotide reductase.
J Med Chem
36
3859
3862
58. PenderBA
WuX
AxelsenPH
CoopermanBS
2001
Toward a rational design of peptide inhibitors of ribonucleotide reductase: structure-function and modeling studies.
J Med Chem
44
36
46
59. UhlinU
EklundH
1994
Structure of ribonucleotide reductase protein R1.
Nature
370
533
539
60. LiuzziM
DezielR
MossN
BeaulieuP
BonneauAM
1994
A potent peptidomimetic inhibitor of HSV ribonucleotide reductase with antiviral activity in vivo.
Nature
372
695
698
61. UppstenM
FarnegardhM
DomkinV
UhlinU
2006
The first holocomplex structure of ribonucleotide reductase gives new insight into its mechanism of action.
J Mol Biol
359
365
377
62. DuxburyMS
ItoH
ZinnerMJ
AshleySW
WhangEE
2004
RNA interference targeting the M2 subunit of ribonucleotide reductase enhances pancreatic adenocarcinoma chemosensitivity to gemcitabine.
Oncogene
23
1539
1548
63. LemboD
BruneW
2009
Tinkering with a viral ribonucleotide reductase.
Trends Biochem Sci
34
25
32
64. PontarinG
FerraroP
HakanssonP
ThelanderL
ReichardP
2007
p53R2-dependent ribonucleotide reduction provides deoxyribonucleotides in quiescent human fibroblasts in the absence of induced DNA damage.
J Biol Chem
282
16820
16828
65. KimuraT
TakedaS
SagiyaY
GotohM
NakamuraY
2003
Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools.
Nat Genet
34
440
445
66. de WindN
BernsA
GielkensA
KimmanT
1993
Ribonucleotide reductase-deficient mutants of pseudorabies virus are avirulent for pigs and induce partial protective immunity.
J Gen Virol
74(Pt 3)
351
359
67. CameronJM
McDougallI
MarsdenHS
PrestonVG
RyanDM
1988
Ribonucleotide reductase encoded by herpes simplex virus is a determinant of the pathogenicity of the virus in mice and a valid antiviral target.
J Gen Virol
69(Pt 10)
2607
2612
68. JacobsonJG
LeibDA
GoldsteinDJ
BogardCL
SchafferPA
1989
A herpes simplex virus ribonucleotide reductase deletion mutant is defective for productive acute and reactivatable latent infections of mice and for replication in mouse cells.
Virology
173
276
283
69. HeinemanTC
CohenJI
1994
Deletion of the varicella-zoster virus large subunit of ribonucleotide reductase impairs growth of virus in vitro.
J Virol
68
3317
3323
70. McClementsW
YamanakaG
GarskyV
PerryH
BacchettiS
1988
Oligopeptides inhibit the ribonucleotide reductase of herpes simplex virus by causing subunit separation.
Virology
162
270
273
71. DutiaBM
FrameMC
Subak-SharpeJH
ClarkWN
MarsdenHS
1986
Specific inhibition of herpesvirus ribonucleotide reductase by synthetic peptides.
Nature
321
439
441
72. CoopermanBS
2003
Oligopeptide inhibition of class I ribonucleotide reductases.
Biopolymers
71
117
131
73. PatroneM
PercivalleE
SecchiM
FiorinaL
Pedrali-NoyG
2003
The human cytomegalovirus UL45 gene product is a late, virion-associated protein and influences virus growth at low multiplicities of infection.
J Gen Virol
84
3359
3370
74. SunY
ConnerJ
1999
The U28 ORF of human herpesvirus-7 does not encode a functional ribonucleotide reductase R1 subunit.
J Gen Virol
80(Pt 10)
2713
2718
75. LemboD
DonalisioM
HoferA
CornagliaM
BruneW
2004
The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis.
J Virol
78
4278
4288
76. LemboD
GribaudoG
HoferA
RieraL
CornagliaM
2000
Expression of an altered ribonucleotide reductase activity associated with the replication of murine cytomegalovirus in quiescent fibroblasts.
J Virol
74
11557
11565
77. BruneW
MenardC
HeesemannJ
KoszinowskiUH
2001
A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism.
Science
291
303
305
78. WilliamsT
Barbosa-SolomieuV
ChincharVG
2005
A decade of advances in iridovirus research.
Adv Virus Res
65
173
248
79. LundinD
TorrentsE
FurrerE
Larsson BirganderP
SahlinM
PooleAM
SjöbergBM
2005
RNRdb: The ribonucleotide reductase database. version 1.0.
Dept of Molecular Biology & Functional Genetics, Stockholm University, Stockholm, Sweden http://rnrdb.molbio.su.se
80. TulmanER
DelhonG
AfonsoCL
LuZ
ZsakL
2006
Genome of horsepox virus.
J Virol
80
9244
9258
81. CameronC
Hota-MitchellS
ChenL
BarrettJ
CaoJX
1999
The complete DNA sequence of myxoma virus.
Virology
264
298
318
82. WillerDO
McFaddenG
EvansDH
1999
The complete genome sequence of shope (rabbit) fibroma virus.
Virology
264
319
343
83. XueL
ZhouB
LiuX
QiuW
JinZ
2003
Wild-type p53 regulates human ribonucleotide reductase by protein-protein interaction with p53R2 as well as hRRM2 subunits.
Cancer Res
63
980
986
84. XueL
ZhouB
LiuX
HeungY
ChauJ
2007
Ribonucleotide reductase small subunit p53R2 facilitates p21 induction of G1 arrest under UV irradiation.
Cancer Res
67
16
21
85. PiaoC
JinM
KimHB
LeeSM
AmatyaPN
2009
Ribonucleotide reductase small subunit p53R2 suppresses MEK-ERK activity by binding to ERK kinase 2.
Oncogene
28
2173
2184
86. AndradeAA
SilvaPN
PereiraAC
De SousaLP
FerreiraPC
2004
The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication.
Biochem J
381
437
446
87. BratkeKA
McLysaghtA
2008
Identification of multiple independent horizontal gene transfers into poxviruses using a comparative genomics approach.
BMC Evol Biol
8
67
88. FileeJ
PougetN
ChandlerM
2008
Phylogenetic evidence for extensive lateral acquisition of cellular genes by Nucleocytoplasmic large DNA viruses.
BMC Evol Biol
8
320
89. FileeJ
BaptesteE
SuskoE
KrischHM
2006
A selective barrier to horizontal gene transfer in the T4-type bacteriophages that has preserved a core genome with the viral replication and structural genes.
Mol Biol Evol
23
1688
1696
90. JordanA
TorrentsE
SalaI
HellmanU
GibertI
1999
Ribonucleotide reduction in Pseudomonas species: simultaneous presence of active enzymes from different classes.
J Bacteriol
181
3974
3980
91. HughesAL
FriedmanR
2003
Genome-wide survey for genes horizontally transferred from cellular organisms to baculoviruses.
Mol Biol Evol
20
979
987
92. HowellML
RosemanNA
SlabaughMB
MathewsCK
1993
Vaccinia virus ribonucleotide reductase. Correlation between deoxyribonucleotide supply and demand.
J Biol Chem
268
7155
7162
93. SlabaughMB
DavisRE
RosemanNA
MathewsCK
1993
Vaccinia virus ribonucleotide reductase expression and isolation of the recombinant large subunit.
J Biol Chem
268
17803
17810
94. SouglakosJ
BoukovinasI
TaronM
MendezP
MavroudisD
2008
Ribonucleotide reductase subunits M1 and M2 mRNA expression levels and clinical outcome of lung adenocarcinoma patients treated with docetaxel/gemcitabine.
Br J Cancer
98
1710
1715
95. WrightJA
AlamTG
McClartyGA
TaggerAY
ThelanderL
1987
Altered expression of ribonucleotide reductase and role of M2 gene amplification in hydroxyurea-resistant hamster, mouse, rat, and human cell lines.
Somat Cell Mol Genet
13
155
165
96. RosellR
FelipE
TaronM
MajoJ
MendezP
2004
Gene expression as a predictive marker of outcome in stage IIB-IIIA-IIIB non-small cell lung cancer after induction gemcitabine-based chemotherapy followed by resectional surgery.
Clin Cancer Res
10
4215s
4219s
97. YoonSS
CarrollNM
ChioccaEA
TanabeKK
1998
Cancer gene therapy using a replication-competent herpes simplex virus type 1 vector.
Ann Surg
228
366
374
98. SpearMA
SunF
ElingDJ
GilpinE
KippsTJ
2000
Cytotoxicity, apoptosis, and viral replication in tumor cells treated with oncolytic ribonucleotide reductase-defective herpes simplex type 1 virus (hrR3) combined with ionizing radiation.
Cancer Gene Ther
7
1051
1059
99. StanfordMM
McFaddenG
2007
Myxoma virus and oncolytic virotherapy: a new biologic weapon in the war against cancer.
Expert Opin Biol Ther
7
1415
1425
100. WooY
KellyKJ
StanfordMM
GalanisC
ChunYS
2008
Myxoma virus is oncolytic for human pancreatic adenocarcinoma cells.
Ann Surg Oncol
15
2329
2335
101. HuY
LeeJ
McCartJA
XuH
MossB
2001
Yaba-like disease virus: an alternative replicating poxvirus vector for cancer gene therapy.
J Virol
75
10300
10308
102. MoM
FlemingSB
MercerAA
2009
Cell cycle deregulation by a poxvirus partial mimic of anaphase-promoting complex subunit 11.
Proc Natl Acad Sci U S A
106
19527
19532
103. KePY
ChangZF
2004
Mitotic degradation of human thymidine kinase 1 is dependent on the anaphase-promoting complex/cyclosome-CDH1-mediated pathway.
Mol Cell Biol
24
514
526
104. McFaddenG
2005
Poxvirus tropism.
Nat Rev Microbiol
3
201
213
105. AndreiG
GammonDB
FitenP
De ClercqE
OpdenakkerG
2006
Cidofovir resistance in vaccinia virus is linked to diminished virulence in mice.
J Virol
80
9391
9401
106. DeMasiJ
TraktmanP
2000
Clustered charge-to-alanine mutagenesis of the vaccinia virus H5 gene: isolation of a dominant, temperature-sensitive mutant with a profound defect in morphogenesis.
J Virol
74
2393
2405
107. FalknerFG
MossB
1988
Escherichia coli gpt gene provides dominant selection for vaccinia virus open reading frame expression vectors.
J Virol
62
1849
1854
108. WasilenkoST
StewartTL
MeyersAF
BarryM
2003
Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis.
Proc Natl Acad Sci U S A
100
14345
14350
109. DvoracekB
ShorsT
2003
Construction of a novel set of transfer vectors to study vaccinia virus replication and foreign gene expression.
Plasmid
49
9
17
110. ChakrabartiS
SislerJR
MossB
1997
Compact, synthetic, vaccinia virus early/late promoter for protein expression.
Biotechniques
23
1094
1097
111. TulmanER
AfonsoCL
LuZ
ZsakL
KutishGF
2004
The genome of canarypox virus.
J Virol
78
353
366
112. UptonC
HoggD
PerrinD
BooneM
HarrisNL
2000
Viral genome organizer: a system for analyzing complete viral genomes.
Virus Res
70
55
64
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 7
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- RNA Virus Replication Complexes
- Functional Genetic Diversity among Complex Clinical Isolates: Delineation of Conserved Core and Lineage-Specific Transcriptomes during Intracellular Survival
- Extreme CD8 T Cell Requirements for Anti-Malarial Liver-Stage Immunity following Immunization with Radiation Attenuated Sporozoites
- A Systems Immunology Approach to Plasmacytoid Dendritic Cell Function in Cytopathic Virus Infections