
Bacteriophage Lysin Mediates the Binding of to Human Platelets through Interaction with Fibrinogen
The binding of bacteria to human platelets is a likely central mechanism in the pathogenesis of infective endocarditis. We have previously found that platelet binding by Streptococcus mitis SF100 is mediated by surface components encoded by a lysogenic bacteriophage, SM1. We now demonstrate that SM1-encoded lysin contributes to platelet binding via its direct interaction with fibrinogen. Far Western blotting of platelets revealed that fibrinogen was the major membrane-associated protein bound by lysin. Analysis of lysin binding with purified fibrinogen in vitro confirmed that these proteins could bind directly, and that this interaction was both saturable and inhibitable. Lysin bound both the Aα and Bβ chains of fibrinogen, but not the γ subunit. Binding of lysin to the Bβ chain was further localized to a region within the fibrinogen D fragment. Disruption of the SF100 lysin gene resulted in an 83±3.1% reduction (mean ± SD) in binding to immobilized fibrinogen by this mutant strain (PS1006). Preincubation of this isogenic mutant with purified lysin restored fibrinogen binding to wild type levels. When tested in a co-infection model of endocarditis, loss of lysin expression resulted in a significant reduction in virulence, as measured by achievable bacterial densities (CFU/g) within vegetations, kidneys, and spleens. These results indicate that bacteriophage-encoded lysin is a multifunctional protein, representing a new class of fibrinogen-binding proteins. Lysin appears to be cell wall-associated through its interaction with choline. Once on the bacterial surface, lysin can bind fibrinogen directly, which appears to be an important interaction for the pathogenesis of endocarditis.
Published in the journal:
. PLoS Pathog 6(8): e32767. doi:10.1371/journal.ppat.1001047
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001047
Summary
The binding of bacteria to human platelets is a likely central mechanism in the pathogenesis of infective endocarditis. We have previously found that platelet binding by Streptococcus mitis SF100 is mediated by surface components encoded by a lysogenic bacteriophage, SM1. We now demonstrate that SM1-encoded lysin contributes to platelet binding via its direct interaction with fibrinogen. Far Western blotting of platelets revealed that fibrinogen was the major membrane-associated protein bound by lysin. Analysis of lysin binding with purified fibrinogen in vitro confirmed that these proteins could bind directly, and that this interaction was both saturable and inhibitable. Lysin bound both the Aα and Bβ chains of fibrinogen, but not the γ subunit. Binding of lysin to the Bβ chain was further localized to a region within the fibrinogen D fragment. Disruption of the SF100 lysin gene resulted in an 83±3.1% reduction (mean ± SD) in binding to immobilized fibrinogen by this mutant strain (PS1006). Preincubation of this isogenic mutant with purified lysin restored fibrinogen binding to wild type levels. When tested in a co-infection model of endocarditis, loss of lysin expression resulted in a significant reduction in virulence, as measured by achievable bacterial densities (CFU/g) within vegetations, kidneys, and spleens. These results indicate that bacteriophage-encoded lysin is a multifunctional protein, representing a new class of fibrinogen-binding proteins. Lysin appears to be cell wall-associated through its interaction with choline. Once on the bacterial surface, lysin can bind fibrinogen directly, which appears to be an important interaction for the pathogenesis of endocarditis.
Introduction
The pathogenesis of infective endocarditis is a complex process, involving numerous host-pathogen interactions [1], [2]. A key interaction for disease establishment and progression is the binding of microbes to human components, including platelets, fibrinogen, fibrin, and fibronectin [3], [4], [5], [6], [7], [8]. Although this binding appears to be a central requirement for virulence, only a limited number of endocarditis-related adhesins has been identified [7], [8], [9].
Among the viridans group streptococci, Streptococcus mitis is a leading cause of endovascular infection [10], [11], [12], [13], [14]. Despite its increasing importance as a human pathogen, relatively little is known about the virulence determinants of this organism, particularly with regard to its interaction with platelets or other host components. Our previous studies identified two surface proteins (PblA and PblB) encoded by a lysogenic bacteriophage (SM1) that mediate the binding of S. mitis to human platelets, through their interaction with the membrane ganglioside GD3 [15], [16], [17]. Disruption of the genes encoding PblA and PblB results in a significant decrease in platelet binding in vitro, as well as a marked reduction in virulence, as measured by an animal model of endocarditis [16], [17].
Expression of these proteins on the bacterial surface is dependent upon the activities of phage holin and lysin, which permeabilize the cell envelope, thereby permitting the transport of PblA and PblB to the cell wall, where they attach to phosphocholine (PC) residues [16]. Of note, disruption of the gene encoding lysin (lys) resulted in a profound reduction in platelet binding, to levels that were significantly lower than those seen with either the parent strain, or a pblA/plbB double knock-out mutant [16]. These findings suggested that lysin mediates platelet binding in part through a mechanism independent of its role in the export of PblA and PblB.
For these reasons, we investigated the mechanisms by which lysin mediates binding to platelets, and whether this interaction contributes to the pathogenesis of streptococcal endocarditis. Our studies indicate that phage lysin can be localized on the bacterial surface through its interaction with PC residues. Surface-bound lysin can subsequently bind both free and platelet-associated fibrinogen, through its specific interaction with the Aα and Bβ chains of the protein. Loss of lysin expression is associated with reduced virulence in the setting of endocarditis, indicating that the binding of lysin to fibrinogen is an important factor in the pathogenesis of this infection.
Results
Characterization of lysinSM1 from bacteriophage SM1
Using the NCBI Conserved Domain Database (CDD) search system [18], bioinformatic analysis of the predicted amino acid sequence of lysinSM1 (Accession number Q9AF60), revealed that an amidase-5 domain (Pfam05382; amino acids 4–146) is present at the amino terminus, and a putative choline-binding domain is found at the carboxyl terminus (COG5263; amino acids 128–271; Fig. 1). The N-terminal domain of lysinSM1 (N-lysinSM1) exhibits 75% amino acid identity to the Pal lysin (accession number O03979) of the pneumococcal bacteriophage Dp-1, and 74% identity to the lysin (accession number Q8E0W3) of the prophage lambdaSa1 of Streptococcus agalactiae [19], [20]. The C terminus of lysinSM1 (C-lysinSM1) contains a choline-binding domain homologous to that found in the pneumococcal LytA autolytic enzyme (62% identity), which anchors the protein to PC residues present in LTA or teichoic acids [19].

To assess whether lysinSMl demonstrated its predicted biological activities, we first examined its binding to DEAE-cellulose, a property that is a hallmark of choline-binding proteins [21]. LysinSM1, N-lysinSM1, and C-lysinSM1 were expressed individually in Escherichia coli, and lysates from these strains were applied to a DEAE-cellulose column [16]. N-lysinSM1 failed to bind the matrix, with the protein being detected in the wash volumes (Fig. 2A). In contrast, lysinSM1 and C-lysinSM1 were only eluted with ™ buffer containing 2% choline chloride. Thus, lysinSM1 appears to be a choline-binding protein, with this interaction being mediated by the C terminus.

We then examined whether purified lysinSM1 bound directly to PC residues of LTA purified from S. pneumoniae HS0001 and S. mitis SF100. Purified FLAGlysinSM1 was incubated with immobilized LTAs, and binding was assessed by ELISA with anti-FLAG antibody. As shown in Fig. 2B, lysinSM1 bound LTA from S. mitis SF100 and from S. pneumoniae HS0001, both of which contain PC. Of note, binding levels of lysinSM1 to these LTAs were comparable and concentration-dependent. In contrast, little or no binding to LTA was detected from strain HS0001-EA, which has no PC. C-lysinSM1 also bound LTA from SF100 and HS0001, whereas N-lysinSM1 did not (Figure S1). These results confirm that lysinSM1 interacts with the PC residues of LTA, and that binding is mediated by the predicted choline-binding domain within the C terminus.
As mentioned above, analysis of the predicted amino terminus of lysinSM1 indicated that it encodes an amidase with g-D-glutaminyl-L-lysin endopeptidase activity [20]. To assess its lytic activity, we tested the bactericidal properties of lysinSM1 in vitro (Fig. 2C). When compared with organisms treated with buffer alone, exposure of S. pneumoniae HS0001 to lysinSM1 resulted in a mean (± S.D.) reduction of 5.07±1.28 log10 CFU per ml (P<0.05). LysinSM1 was also active against the SM1 host strain SF100 and its isogenic variant, PS1006, though it only reduced mean titers by 0.8±0.04 (P<0.05) and 1.65±0.21 log10 CFU per ml (P<0.05), respectively. No bactericidal activity was seen when tested against Staphylococcus aureus, Streptococcus sanguinis, Streptococcus pyogenes, or E. coli. Of note, neither purified N-lysinSM1 nor C-lysinSM1 had bactericidal activity against strains HS0001 or SF100 (data not shown). Thus, lysinSM1 has lytic activity against PC positive strains, such as S. mitis and S. pneumoniae, though the latter species is considerably more sensitive to the enzyme. Moreover, lysinSM1 requires its choline-binding domain, in addition to its predicted amidase domain, for this activity.
Binding of recombinant lysinSM1 to human platelets
We have previously observed that disruption of the gene encoding lysinSM1 resulted in a significant reduction in platelet binding by S. mitis [16]. To assess whether lysinSM1 could interact directly with human platelets, we evaluated the binding of FLAGlysinSM1 to immobilized human platelets and to isolated platelet membranes (Fig. 3A). FLAGlysinSM1 was incubated with platelet monolayers or platelet membranes, and bound FLAGlysinSM1 was then detected with anti-FLAG antibody. When tested by this approach, we found that FLAGlysinSM1 strongly interacted with both whole platelets and platelet membranes in a concentration-dependent manner. In contrast, no binding of FLAGlysinSM1was seen to wells coated with only a casein-based blocking reagent (Western Blocking Reagent; Roche).
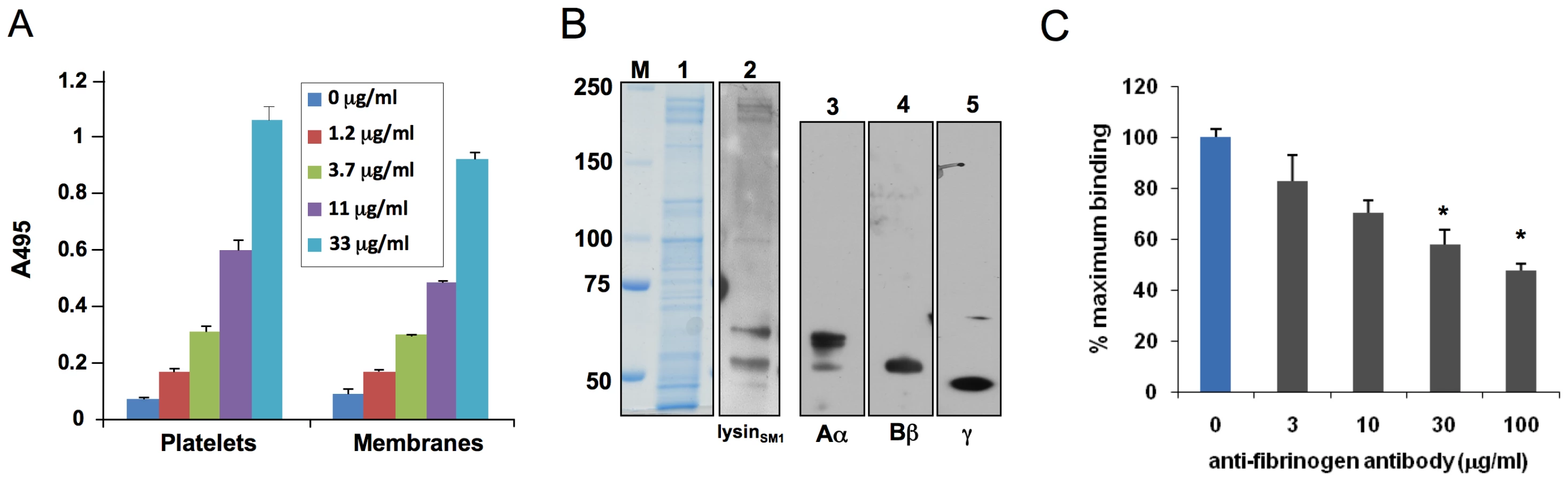
To identify the membrane receptor for lysinSM1, we assessed by far Western blotting the binding of FLAGlysinSM1 to platelet membranes that had undergone SDS-PAGE and transfer to nitrocellulose (Fig. 3B). Although the platelet membrane extracts contained numerous proteins, ranging in mass from 50 to 250 kDa, FLAGlysinSM1 bound only a small number of proteins. The highest levels of binding were seen with two proteins of MW 65 kDa and 55 kDa, which were similar to the molecular masses of the Aα and Bβ chains of human fibrinogen (64 and 56 kDa), respectively. To confirm that platelet membrane extracts contained fibrinogen, the preparations were probed with antibodies directed against the three major chains of fibrinogen (Aα, Bβ and γ). As shown Fig. 3B, each subunit of fibrinogen was present in the membrane extracts. To directly confirm that lysinSM1 bound fibrinogen on platelet membranes, we assessed whether lysinSM1 binding to immobilized platelet membranes was inhibited by anti-human fibrinogen IgG. As shown in Fig. 3C, pre-treatment of membranes with 30 or 100 µg/ml of anti-fibrinogen antibodies significantly reduced subsequent lysin binding. These results further indicate that fibrinogen is the principal component on platelet membranes and that lysinSM1 is bound to platelet membranes through it interaction with fibrinogen.
Binding of lysinSM1 to fibrinogen
Since fibrinogen is a key factor in the pathogenesis of infective endocarditis, and because it is a receptor for some bacterial adhesins [7], [22], [23], [24], [25], [26], we further investigated the interaction of this protein with lysinSM1. We first assessed the binding of the increasing concentrations of FLAGlysinSM1 to human fibrinogen (3 µg/ml) immobilized in microtiter wells. In control studies, no significant binding of fibrinogen by FLAG-tagged alkaline phosphatase (FLAGAP) was detected (Figure S2). In contrast, FLAGlysinSM1 showed significant binding to immobilized fibrinogen, which increased in direct proportion to the amount of protein applied (Fig. 4A). At concentrations above 125 µg/ml FLAGlysinSM1, binding reached a plateau, indicating that it was saturated. In addition, the binding of FLAGlysinSM1 to immobilized fibrinogen was effectively blocked by both unlabeled lysinSM1 and fibrinogen (Fig. 4B). Similar levels of lysin binding were seen with rat fibrinogen, the host used for our subsequent virulence assays (Figure S3).

For some bacteria, binding to fibrinogen is dependent on whether the protein is in solution or immobilized on a surface. For example, Group A and G streptococci can bind both soluble and immobilized forms of fibrinogen, whereas several oral streptococci appear to bind only immobilized fibrinogen [27], [28], [29]. To assess whether fibrinogen binding by lysinSM1 was phase-dependent, we reversed the binding conditions, such that FLAGlysinSM1, untagged lysinSM1, and FLAGAP (all at 10 µg/ml) were immobilized in microtiter wells, and probed with the increasing concentration of fibrinogen in solution. Under these conditions, fibrinogen was still found to bind lysinSM1 and FLAGlysinSM1 comparably, whereas no significant binding to FLAGAP was detected (Fig. 4C).
As noted above, we found that lysinSM1 bound two proteins associated with platelet membranes that corresponded to the Aα and Bβ chains of fibrinogen. To confirm that lysinSM1 binds specifically to these subunits, we assessed by far Western blotting the interaction of lysinSM1 with purified human fibrinogen (Fig. 4D). When separated by SDS-PAGE under reducing conditions, fibrinogen appeared as three bands, having the expected masses. When transferred to nitrocellulose and probed with FLAGlysinSM1, binding could be detected to the Aα and Bβ chains only, confirming the results seen with platelet membranes.
LysinSM1 binds fibrinogen fragment D but not fragment E
The fibrinogen molecule is comprised of two subunits, each containing three polypeptide chains (Aα, Bβ, and γ; Fig. 5A). Cleavage of fibrinogen with plasmin produces a series of fragments, most notably the E fragment containing the central part of the molecule, and the D fragment containing the terminal regions. To further identify the domains of fibrinogen bound by lysinSM1, we examined the interaction of FLAGlysinSM1 to the D and E fragments. When assessed by ELISA, FLAGlysinSM1 showed high levels of binding to immobilized fragment D, which were comparable to those seen with whole fibrinogen (Fig. 5B). In contrast, no significant binding to the E fragment was seen.

Purified fibrinogen fragment D contains three subunits, each representing a part of the three major chains (α chain fragment = 15 kDa, β chain fragment = 44.5 kDa, and γ chain fragment = 42 kDa) (Fig. 5A). To further identify the subdomains of fibrinogen bound by lysinSM1, purified fragment D was separated under reducing conditions and transferred to a nitrocellulose membrane. When assessed by far Western blotting, binding by FLAGlysinSM1, was limited to the Bβ chain component of fragment D with no binding detected to the Aα chain (Fig. 5C). These data indicate lysinSM1 binds a region contained within AA 134–461 of the Bβ chain. Of note, lysinSM1 bound the full-length Aα chain (Fig. 3B), but not its D or E fragments (Fig. 5C), suggesting that the lysinSM1 binding to the Aαchain requires the C terminus (AA 197–610).
LysinSM1 promotes the interaction of S. mitis SF100 with fibrinogen
To assess the impact of lysin expression on bacterial binding to fibrinogen, we compared the adherence of SF100 (WT) and PS1006 (Δlysin) to fibrinogen immobilized in microtiter wells. As shown in Fig. 6A, SF100 had high levels of binding to immobilized fibrinogen, which increased in proportion to the amount of fibrinogen in the wells. PS1006 showed markedly reduced levels of binding, as compared with the parent strain. For example, when tested with wells coated with 30 µg/ml of fibrinogen, PS1006 had only 18.8±4.7% (mean ± SD) of maximal binding, as compared with 89.7±12.8% for SF100 (P<0.05, unpaired t-test). Complementation of the lysin mutation in trans restored fibrinogen binding by PS1006 (Fig. 6B), thereby demonstrating that the loss of binding observed with lysin disruption was not due to polar or pleiotropic effects.

The above results suggested that the binding to immobilized fibrinogen by SF100 is mediated by lysinSM1 expressed on the bacterial surface. To confirm that lysin was sufficient to mediate fibrinogen binding, we next examined whether exogenous lysinSM1 could attach to the cell wall of PS1006 and restore binding. The PC-negative strain SK598 served as a negative control. Each strain was incubated with purified FLAGlysinSM1 at RT for 30 min. After washing to remove nonspecifically bound protein, cell wall bound FLAGlysinSM1 was extracted with 2% choline, and the amount of FLAGlysinSM1 recovered was assessed by Western blotting. As shown in Fig. 6C, exogenous FLAGlysinSM1 could readily be detected in the cell wall extracts of PS1006, whereas no binding of FLAGlysinSM1was observed with SK598.
We then assessed whether this interaction was sufficient to enhance the binding of PS1006 to fibrinogen (Fig. 6D). PS1006 was suspended in PBS containing a range of concentrations of purified lysinSM1 and then tested for its binding to immobilized fibrinogen, as described above. As expected, PS1006 incubated in PBS alone showed minimal levels of binding to fibrinogen. This was not due to a loss of PblA and PblB expression, since the pblA/pblB negative strain PS344 had levels of fibrinogen binding that were similar to those of the parent strain. Exposure of PS1006 to FLAGlysinSM1 increased fibrinogen binding in a concentration-dependent manner. Indeed, 10 µg per ml of FLAGlysinSM1 was sufficient to restore PS1006 binding to levels comparable to those seen with SF100.
Role of lysinSM1 in the pathogenesis of infective endocarditis
To assess the impact of lysinSM1 on pathogenesis, we compared the relative virulence of SF100, PS344 and PS1006 in a rat co-infection model of infective endocarditis [16], [55]. We first compared SF100 with PS344 to confirm previous results obtained in a rabbit model of infection [16]. As was observed with rabbits, disruption of pblA and pblB was also associated with attenuated virulence in rats, with PS344 having significantly reduced levels of bacteria within all tissues (Table 1). Disruption of lysinSM1 also produced a significant reduction in virulence. Rats co-infected with SF100 and PS1006 had significantly lower densities of the lysin mutant strain in vegetations (mean ± SD = 5.07±1.50 log10 CFU/g) as compared with the parent strain (6.91±1.35 log10 CFU/g; n = 8, P = 0.009). Densities of PS1006 were also significantly reduced in kidneys (P = 0.008) and spleens (P<0.001) as compared with SF100. We then examined the relative impact on virulence of abrogated PblA and PblB expression, versus loss of lysin (Table 1). In animals co-infected with PS344 and PS1006, titers of the latter mutant were significantly reduced in all tissues examined, as compared with the former. In particular, the mean densities of PS1006 within vegetations (6.59±1.45 log10 CFU/g) were significantly lower than those of PS344 (8.32±0.76; n = 8; P = 0.008), as were densities within kidneys (P = 0.027) and spleens (P = 0.006). We then re-analyzed these data by comparing the ratio of PS344 to PS1006 within tissues, with the CFU of each strain normalized to the number of CFU within the inoculum (competition index) (Figure S4). When assessed by this approach, the levels of the lysinSM1 mutant PS1006 remained significantly reduced in all tissues, as compared with PS344. Thus, lysinSM1 appears to be a significant virulence determinant in the setting of infective endocarditis. Moreover, its role in pathogenesis is not due solely to any effect it may have on PblA and PblB expression. Instead, it appears to have an impact upon the development of infective endocarditis independent of these other phage-encoded proteins.

Discussion
The binding of pathogenic bacteria to platelets is thought to play a key role in the pathogenesis of infective endocarditis. This interaction may be important both for the initial attachment of bacteria to the endocardial surface, and for the subsequent formation of vegetations. Numerous endocarditis-associated pathogens have been shown to bind platelets directly in vitro, through a variety of mechanisms [3], [4], [7], [8], [25], [30]. The ability to bind platelets in vitro has been linked to virulence for several of the most common endocarditis-associated species, including Staphylococcus aureus, Streptococcus gordonii, and Streptococcus sanguinis [5], [31], [32], [33]. Previous work from our laboratory has shown that platelet binding by S. mitis strain SF100 is mediated in part by two proteins (PblA and PblB) encoded by the lysogenic bacteriophage SM1 [16]. The functional localization of these proteins to the cell surface requires the phage lysin (lysinSM1), which permeabilizes the host organism, thereby permitting the transport of PblA and PblB from the cytoplasm to the bacterial surface, and their subsequent attachment to the cell wall [16]. During the course of these studies, we noted that disruption of the gene encoding lysinSM1 reduced platelet binding in vitro more profoundly than the loss of PblA and PblB localization, indicating that lysin had a role in platelet binding beyond facilitating PblA and PblB transport. It was unknown, however, whether lysin itself could directly mediate binding, or rather, the effects of lysin on bacterial permeability led to the surface expression of other proteins (either phage or bacterial) that could enhance platelet binding.
Our current results demonstrate that lysin can bind human platelets directly through its interaction with fibrinogen. Purified lysin was found to bind fibrinogen, regardless of whether the proteins were in solution or immobilized. The binding of lysin with fibrinogen also was saturable, consistent with a receptor-ligand interaction. Lysin binding was restricted to the D fragment of the Aα and Aβ chains, further indicating that this is a specific process. This interaction appears to be important for the binding of S. mitis to fibrinogen, since disruption of the gene encoding lysin markedly reduced fibrinogen binding by bacteria in vitro. The addition of exogenous purified lysin to these mutants restored binding to WT levels, confirming that lysin can directly mediate the interaction of S. mitis with fibrinogen.
A number of other bacterial proteins have been shown to bind fibrinogen, including the M protein and serum opacity factor of Streptococcus pyogenes, FbsA of Streptococcus agalactiae, SdrG of Staphylococcus epidermidis, and several proteins of Staphylococcus aureus (clumping factors A and B, fibronectin binding protein A) [4], [22], [25], [30], [34]. However, none of these proteins exhibit any primary sequence homology with lysinSM1. The staphylococcal autolysins Aaa and Aae do resemble lysinSM1, in that they appear to have both enzymatic and fibrinogen binding activities in vitro [35], [36]. A search against the SMART and Pfam databases indicates that collectively these proteins belong to the NlpC/P60 superfamily of proteins, containing their catalytic domain that are characteristic of this group of proteins. However, the predicted catalytic activity of lysinSM1 (amidase 5) is different from that autolysins Aaa and Aae. LysinSM1 has no sequence similarity to either the Aaa or Aae protein, and unlike these other proteins, it is a choline-binding protein. Thus, lysin appears to be a multi-functional protein that can mediate S. mitis binding to fibrinogen, in addition to its role in the transit of the PblA and PblB proteins to the cell surface.
LysinSM1 was also associated with increased virulence in a rat model of infective endocarditis. When animals were co-infected with the parent strain SF100 and the lysinSM1 mutant PS1006, densities of the lysin mutant were significantly reduced within vegetations, kidneys, and spleens, as compared with the parent strain. Moreover, the virulence of PS1006 was also attenuated, when compared with its pblA and pblB-deficient isogenic variant, PS344. These results indicate that, beyond its importance for PblA and PblB expression, lysin contributes to virulence through a mechanism beyond its role in the transport of these bacteriophage-encoded adhesins. It is possible that there are other, unrecognized phage-encoded virulence factors that require lysin for export or localization. However, in view of the ability of lysin to bind fibrinogen directly (both human and rat), and that fibrinogen binding has been associated with virulence for several other adhesins [37], [38], [39], [40], [41], it is likely that this interaction of lysin with fibrinogen contributes to the pathogenesis of infective endocarditis by S. mitis. Given that lysin-fibrinogen binding enhances bacterial adherence to platelets in vitro, and that bacterium-platelet binding has been linked to virulence, it is likely that lysin-mediated binding to platelets via fibrinogen is an important pathogenetic interaction. However, it is also possible that lysin mediates streptococcal binding to fibrinogen on other surfaces, such as damaged endothelium. Finally, it is conceivable that lysin contributes to virulence through other, as yet unidentified interactions.
In summary, lysin is a novel fibrinogen-binding protein encoded by a lysogenic bacteriophage of S. mitis. In addition to its expected role in cell wall degradation, lysin also appears to be an adhesin mediating the attachment of this organism to human platelets, through its interaction with cell wall PC, fibrinogen, and the platelet membrane receptor for fibrinogen, glycoprotein IIb/IIIa (Fig. 7). Lysin also appears to contribute significantly to virulence, which could explain the persistence of certain bacteriophages within their host organisms. Although induction of the phage lytic cycle extracts a toll on host viability, in vivo this may be more than offset by the enhanced virulence resulting from lysin expression. Since fibrinogen is also present within gingival crevicular fluid, lysin-fibrinogen binding may also contribute to the colonization of oral surfaces by S. mitis [42], [43]. Although we do not know the exact prevalence of lysinSM1 homologs in other organisms, recent studies of S. pneumoniae and Enterococcus faecalis indicate that lysogenic bacteriophages encoding homologs of PblA and PblB are often present within these species [44], [45]. Since lysins are required for the phage life cycle, these findings suggest that homologs of lysinSM1 may also be encoded by such prophages. If so, then lysin binding to fibrinogen could prove to be an important interaction for a range of Gram-positive pathogens.

Materials and Methods
Ethics statement
Blood was obtained from healthy human volunteers, using a protocol approved by the Committee on Human Research at the University of California, San Francisco. All human studies were conducted according to the principles expressed in the Declaration of Helsinki. Written informed consent was obtained from all study participants prior to their participation. All procedures involving rats were approved by the Los Angeles Biomedical Research Institute animal use and care committee, following the National Institutes of Health guidelines for animal housing and care.
Reagents
N terminal Met-FLAG-alkaline phosphatase (FLAGAP) and purified rat fibrinogen were purchased from Sigma-Aldrich. Purified human fibrinogen and the fibrinogen fragment D and E (produced by cleavage of fibrinogen with plasmin) were obtained from Haematologic Technologies. Rabbit anti-human fibrinogen polyclonal IgG was purchased from Innovative Research.
Cloning and expression of lysinSM1
Genomic DNA was isolated from SF100, using Wizard Genomic DNA purification kits (Promega), according to the manufacturer's instructions. Polymerase chain reaction (PCR) was performed with the primers listed in Table S2. To clone lys gene into E. coli expression vector, PCR products were purified, digested, and ligated into pET28FLAG to express FLAG-tagged versions of full length lysinSM1 (amino acids [AA] 1–295), the amino terminus of lysinSM1 (AA 1–158; N-lysinSM1), or the carboxy terminus of lysinSM1 (AA 141–295; C - lysinSM1). Untagged lysinSM1, C-lysinSM1, and His-tagged N-lysinSM1 (HisN-lysinSM1) were cloned into pET22b(+) (Novagen). The plasmids were then introduced to E. coli BL21(DE3) by transformation. LysinSM1, FLAGlysinSM1, C-lysinSM1 and FLAGC-lysinSM1 were purified with DEAE-cellulose columns, as described previously [16]. FLAGN-lysinSM1 and HisN-lysinSM1 were purified by either Ni-NTA (Promega) or anti-FLAG M2 agarose affinity chromatography (Sigma-Aldrich), according to the manufacturers' instructions.
Deletion or complementation of the lysin gene (lys)
A gene replacement cassette was constructed by cloning the chromosomal regions flanking lys upstream and downstream of the cat gene in pC326 [16]. A 339 bp upstream segment was amplified using primers KO4F and KO4R, and then digested with XhoI and HindIII. A 513 bp downstream segment was amplified with primers KO6F and KO6R, and then digested with EcoRI. The upstream and downstream fragments were cloned sequentially into the corresponding sites of pC326. The resulting plasmid, pKO-lys, was introduced into SF100 by natural transformation as previously described [17]. In brief, overnight SF100 cultures were diluted 100-fold in fresh THB supplemented with 20% heat-inactivated horse serum, 200 ng/ml competence-stimulating peptide (CSP; DWRISETIRNLIFPRRK), and 1 µg/ml of plasmid. Transformation mixtures were incubated 4 h at 37°C and then plated on blood agar containing 5 µg chloramphenicol per ml. To complement in trans the lys mutation in PS1006, lys was amplified using primers 3206-XbaI and 5206-EcoRI and then cloned into the streptococcal expression vector pDE123. This plasmid was derived from pDC123 by replacing the chloramphenicol resistance marker with an erythromycin resistance marker [46]. The resulting plasmid, pDE-lys, was introduced into PS1006 by natural transformation.
Strains and growth conditions
The bacteria and plasmids used in this study are listed in Table S1. S. mitis strains were grown in Todd-Hewitt broth (Difco) supplemented with 0.5% yeast extract (THY). PS344 (ΔORF47-PblB::pVA891) and PS1006 (ΔlysinSM1) are isogenic variants of S. mitis SF100, which is an endocarditis-associated clinical isolate [16]. All three strains grow comparably well in vitro. S. pneumoniae strains were grown in either a chemically defined medium (CDM; JRH bioscience) [47] supplemented with 0.1% choline chloride, or THY. S. pneumoniae HS0001 is a nonencapsulated pneumococcal strain derived from the TIGR4 strain by deleting the capsule synthesis locus as described previously [48]. S. pneumoniae HS0001-EA is a PC-negative strain derived from HS0001 as described previously [49]. Escherichia coli DH5α and BL21(DE3) strains were grown at 37°C under aeration in Luria broth (LB; Difco). Appropriate concentrations of antibiotics were added to the media, if required.
Purification of lysinSM1 and C-lysinSM1 in DEAE-cellulose
Transformed E. coli BL21(DE3) cells were harvested by centrifugation, washed and suspended in 50 mM Tris-maleate (™) buffer (Sigma-Aldrich), pH 6.3. Cells were disrupted by treatment with B-PER lysis solution (Pierce, Rockford, IL) and the debris was removed by centrifugation at 4,000 rpm for 10 min at 4°C. Supernatants were loaded on a 2 ml DEAE-cellulose (Sigma-Aldrich) column equilibrated with 50 mM ™ buffer, pH 6.3. The column was washed with at least 3 volumes of 50 mM ™ buffer, pH 6.3, containing 1.5 M NaCl and 0.1% choline chloride, until no protein was detected in the eluent. The retained proteins were then eluted with 50 mM ™ buffer, pH 6.3, containing 1.5 M NaCl and 2% choline chloride. Recombinant protein was dialyzed against PBS and then stored at −70°C.
Bactericidal assay
Early log phage (A600 = 0.5) bacteria were harvested by centrifugation and suspended in PBS at approximately 108–109 CFU/ml. Bacteria samples were then incubated with or without 30 µg/ml of purified lysinSM1 at 37°C for 30 min. Samples were serially diluted in PBS and plated onto blood agar, to determine the number of surviving bacteria.
Isolation of platelet membranes
Platelet membranes were prepared by glycerol lysis and gradient centrifugation, as previously described [50]. In brief, isolated human platelets were lysed in 5 volumes of lysis buffer (8.5 mM Tris-Cl, 96.5 mM NaCl, 85.7 mM glucose, 1 mM EDTA, 10 mM EGTA [pH 7.4]) containing Complete Protease Inhibitor Cocktail (Roche). The sample was centrifuged (5,900× g, 10 min) to remove unlysed platelets, and the supernatant was applied to a sucrose step gradient (10 ml of 33% sucrose on 5 ml of 66% sucrose in buffer). After ultracentrifugation (90 min, 63,000× g, 4°C), the membranes were removed, dialyzed against PBS containing 10% glycerol, and stored at −70°C.
Far western blot analysis
Samples were separated by electrophoresis through 4–12% NuPAGE Bis-Tris gels (Invitrogen) and transferred onto nitrocellulose membranes. The membrane were treated with a casein-based blocking solution (Western Blocking Reagent; Roche) at room temperature, and then incubated for 1 h with FLAGlysinSM1 (5 µg/ml) or purified human fibrinogen (1 µg/ml) suspended in PBS-0.05% Tween 20 (PBS-T). The membranes were then washed three times for 15 min in PBS-T, and bound probe proteins were detected with mouse anti-FLAG monoclonal antibody (Sigma-Aldrich) or rabbit anti-fibrinogen polyclonal IgG antibody.
Lysin binding to platelet monolayers or platelet membranes
Washed, fixed human platelets or purified platelet membranes were immobilized in 96 well microtiter plates as described previously [51]. To reduce non-specific adherence, the wells were then treated with the casein-based blocking reagent for 1 h at room temperature. The blocking solution was removed by aspiration, and the wells were incubated with 0 to 100 µg of FLAGlysinSM1 in PBS for 1 h, at RT, followed by washing to remove unbound protein. Bound FLAGlysinSM1 was detected by ELISA with anti-FLAG antibody. For some studies, the wells containing platelet membranes were pretreated with 0 to 100 µg/ml of rabbit anti-fibrinogen antibody for 30 min, followed by washing to remove unbound antibody. Binding by FLAGlysinSM1 (5 µg/ml) was then assessed as described above.
Binding of recombinant FLAGlysinSM1 to fibrinogen and fibrinogen fragments
Rat fibrinogen (10 µg/ml), human fibrinogen, or human fibrinogen D or E fragments (all 15 nM in PBS), were immobilized in 96-well microtiter dishes by overnight incubation at 4°C. The wells were washed twice with PBS and blocked with 300 µl of casein-based blocking solution for 1 h at room temperature. The plates were washed three times with PBS, and a range of FLAGlysinSM1 concentrations in PBS with Tween 20 (0.05%) were added. The plates were then incubated for 2 h at 37°C. Unbound protein was removed by washing with PBS, and plates were incubated with mouse anti-FLAG antibodies for 1 h at 37°C. Binding was assessed by ELISA, using HRP-conjugated rabbit anti-mouse IgG, for 1 h at 37°C. FLAGAP (25–100 µg/ml) served as a control for nonspecific binding.
To examine the binding of fibrinogen to immobilized FLAGlysinSM1, untagged lysinSM1, or FLAGAP (10 µg/ml in PBS) were immobilized in 96 well microtiter plates, followed by blocking of the wells with the casein blocking solution. The wells were incubated with a range of human fibrinogen for 1 h at room temperature, followed by washing. Bound fibrinogen was detected by ELISA, using anti-human fibrinogen IgG.
Assay for lysinSM1 binding to bacterial cell walls
Cultures of PS1006 and S. mitis SK598 in the early log phage of growth (A600 = 0.5) were harvested by centrifugation and suspended in PBS. The bacteria were incubated with purified FLAGlysinSM1 (0 to 10 µg/ml) for 30 min at room temperature. The samples were washed twice with PBS to remove unbound FLAGlysinSM1 and incubated with PBS-2% choline chloride to elute choline-binding proteins from the cell walls, as described previously [21]. Eluted cell wall proteins were harvested by centrifugation and loaded onto SDS-PAGE. Cell wall bound FLAGlysinSM1 was detected by western blotting with anti-FLAG antibody.
Assay for the binding of SF100 to immobilized fibrinogen
Overnight cultures of S. mitis SF100 or its isogenic mutants (PS1006 and PS344) were diluted 1∶10 in fresh THY broth, incubated for 1 h at 37°C, and then exposed to UV light (λ = 312 nm) for 3 min, to induce the expression of the lysogenic bacteriophage SM1. The cultures were then incubated at 37°C for an additional 2 h, followed by harvesting by centrifugation. The pellets were suspended in PBS, and adjusted to a concentration of 106 CFU/ml. One hundred microliters of each suspension were added to wells that had been coated overnight with 30 µg/well of fibrinogen in carbonate buffer. The plates were incubated at room temperature for 1 h, and the wells were washed three times with PBS to remove nonadherent bacteria. The wells were then treated with 50 µl of trypsin (2.5 mg/ml) for 30 min at 37°C to release the bound bacteria. The number of bound bacteria was determined by plating serial dilutions of the recovered bacteria onto blood agar.
Lipoteichoic acid (LTA) purification
LTA was prepared from S. pneumoniae HS0001 and S. mitis strains by organic solvent extraction and octyl-Sepharose chromatography, as previously described [52]. In brief, bacteria were cultured at 37°C for 10 h in CDM with 0.1% choline chloride (Fisher scientific Inc.). To purify PC negative LTA, S. pneumoniae HS0001-EA was cultured for 16 h in CDM supplemented with 2% ethanolamine. Pelleted bacteria were suspended in 0.05 M sodium acetate (pH 4.0) and lysed by sonication. After extraction from the lysate with a chloroform and methanol mixture (1∶0.9), the LTA was adsorbed onto an octyl-Sepharose CL-4B (Sigma-Aldrich) equilibrated in a mixture of 15% n-propanol and 0.05 M sodium acetate (pH 4.7). The absorbed LTA was then eluted with 35% n-propanol in 0.05 M sodium acetate (pH 4.7).
Analysis of LTA structure
Purified LTA was analyzed by matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry [52] (Figure S5). In brief, 1 µl of a sample (1 µg/ml) and 1 µl of matrix solution (0.5 M 2, 5-dihydroxybenzoic acid and 0.1% trifluoroacetic acid in methanol) were applied to a sample plate. After drying, the sample was analyzed with a mass spectrometer (Voyager Biospectrometry DE Pro workstation; PerSeptive Biosystems). Purified LTA showed three major peaks that corresponded to LTA with five, six, and seven repeating units, respectively. The mass difference between the major peaks was 1299 or 1100 amu, corresponding to an oligosaccharide repeating unit with two PC groups or two phosphoethanolamine groups [52]. In addition, PC expression by strains HS0001 and SF100 was directly assessed by western blotting with anti-PC antibody (TEPC-15; Sigma-Aldrich) [53] (Figure S6).
Rat model of infective endocarditis
The relative virulence of SF100 and its isogenic variants was compared in a competition model of infective endocarditis, as described previously [16], [54]. In brief, Sprague-Dawley female rats (250 to 300 g each) were first anesthetized with ketamine (35 mg/kg) and xylazine (10 mg/kg). A sterile polyethylene catheter was surgically placed across the aortic valve of each animal, such that the tip was positioned in the left ventricle, to induce the formation of sterile vegetations (nonbacterial thrombotic endocarditis). The catheters were left in place throughout the study. Seven days post-catheterization, the animals were infected intravenously with an inoculum of 105 CFU containing a 1∶1 mixture of a) SF100 and PS344, b) SF100 and PS1006, or c) PS344 and PS1006. At 72 hr post-infection, the rats were euthanized with thiopental (100 mg IP). Animals were included in the final analysis only if the catheters were correctly positioned across the aortic valve at the time of sacrifice, and if macroscopic vegetations were visible. All cardiac vegetations, as well as samples of the kidneys and spleens, were harvested, weighed, homogenized in saline, serially diluted, and plated onto 8% Todd Hewitt agar (±2.5 µg/ml of chloramphenicol or 5 µg/ml of erythromycin) for quantitative culture. The plates were cultured for 48 h at 37°C, and bacterial densities were expressed as the log10 CFU per gram of tissue. Differences in means were compared for statistical significance by the paired t-test. The data were also analyzed by calculating a “competition index,” which was defined as the ratio of the paired strains within tissues for each animal, normalized by the ratio of organisms in the inoculum. The mean of the log10 normalized ratios was tested against the hypothesized ‘no effect’ mean value of 0, as described previously, using a paired t-test, with P<0.05 as the threshold for statistical significance [55].
Data analysis
Data expressed as means ± standard deviations were compared for statistical significance by the paired or unpaired t test, as indicated.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. KnoxKW
HunterN
1991 The role of oral bacteria in the pathogenesis of infective endocarditis. Aust Dent J 36 286 292
2. BashoreTM
CabellC
FowlerVJr
2006 Update on infective endocarditis. Curr Probl Cardiol 31 274 352
3. GaneshVK
RiveraJJ
SmedsE
KoYP
BowdenMG
2008 A structural model of the Staphylococcus aureus ClfA-fibrinogen interaction opens new avenues for the design of anti-staphylococcal therapeutics. PLoS Pathog 4 e1000226
4. McDevittD
NanavatyT
House-PompeoK
BellE
TurnerN
1997 Characterization of the interaction between the Staphylococcus aureus clumping factor (ClfA) and fibrinogen. Eur J Biochem 247 416 424
5. MiajlovicH
LoughmanA
BrennanM
CoxD
FosterTJ
2007 Both complement - and fibrinogen-dependent mechanisms contribute to platelet aggregation mediated by Staphylococcus aureus clumping factor B. Infect Immun 75 3335 3343
6. FitzgeraldJR
FosterTJ
CoxD
2006 The interaction of bacterial pathogens with platelets. Nat Rev Microbiol 4 445 457
7. LoughmanA
FitzgeraldJR
BrennanMP
HigginsJ
DownerR
2005 Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A. Mol Microbiol 57 804 818
8. O'BrienL
KerriganSW
KawG
HoganM
PenadesJ
2002 Multiple mechanisms for the activation of human platelet aggregation by Staphylococcus aureus: roles for the clumping factors ClfA and ClfB, the serine-aspartate repeat protein SdrE and protein A. Mol Microbiol 44 1033 1044
9. FordI
DouglasCW
1997 The role of platelets in infective endocarditis. Platelets 8 285 294
10. Bek-ThomsenM
TettelinH
HanceI
NelsonKE
KilianM
2008 Population diversity and dynamics of Streptococcus mitis, Streptococcus oralis, and Streptococcus infantis in the upper respiratory tracts of adults, determined by a nonculture strategy. Infect Immun 76 1889 1896
11. NgKH
LeeS
YipSF
QueTL
2005 A case of Streptococcus mitis endocarditis successfully treated by linezolid. Hong Kong Med J 11 411 413
12. HuangIF
ChiouCC
LiuYC
HsiehKS
2002 Endocarditis caused by penicillin-resistant Streptococcus mitis in a 12-year-old boy. J Microbiol Immunol Infect 35 129 132
13. HallGE
BaddourLM
2002 Apparent failure of endocarditis prophylaxis caused by penicillin-resistant Streptococcus mitis. Am J Med Sci 324 51 53
14. ChayakulP
HortiwakulR
YipintsoiT
IngviyaN
2002 Varidans streptococci in the oral flora of the patients at risk for infective endocarditis: species and penicillin susceptibilities. J Med Assoc Thai 85 825 830
15. MitchellJ
SullamPM
2009 Streptococcus mitis phage-encoded adhesins mediate attachment to {alpha}2-8-linked sialic acid residues on platelet membrane gangliosides. Infect Immun 77 3485 3490
16. MitchellJ
SibooIR
TakamatsuD
ChambersHF
SullamPM
2007 Mechanism of cell surface expression of the Streptococcus mitis platelet binding proteins PblA and PblB. Mol Microbiol 64 844 857
17. BensingBA
SibooIR
SullamPM
2001 Proteins PblA and PblB of Streptococcus mitis, which promote binding to human platelets, are encoded within a lysogenic bacteriophage. Infect Immun 69 6186 6192
18. Marchler-BauerA
PanchenkoAR
ShoemakerBA
ThiessenPA
GeerLY
2002 CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res 30 281 283
19. SheehanMM
GarciaJL
LopezR
GarciaP
1997 The lytic enzyme of the pneumococcal phage Dp-1: a chimeric lysin of intergeneric origin. Mol Microbiol 25 717 725
20. PritchardDG
DongS
KirkMC
CarteeRT
BakerJR
2007 LambdaSa1 and LambdaSa2 prophage lysins of Streptococcus agalactiae. Appl Environ Microbiol 73 7150 7154
21. CaubinJ
MartinH
RoaA
CosanoI
PozueloM
2001 Choline-binding domain as a novel affinity tag for purification of fusion proteins produced in Pichia pastoris. Biotechnol Bioeng 74 164 171
22. Ni EidhinD
PerkinsS
FrancoisP
VaudauxP
HookM
1998 Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol Microbiol 30 245 257
23. WalshEJ
MiajlovicH
GorkunOV
FosterTJ
2008 Identification of the Staphylococcus aureus MSCRAMM clumping factor B (ClfB) binding site in the alphaC-domain of human fibrinogen. Microbiology 154 550 558
24. GeorgeNP
WeiQ
ShinPK
KonstantopoulosK
RossJM
2006 Staphylococcus aureus adhesion via Spa, ClfA, and SdrCDE to immobilized platelets demonstrates shear-dependent behavior. Arterioscler Thromb Vasc Biol 26 2394 2400
25. FitzgeraldJR
LoughmanA
KeaneF
BrennanM
KnobelM
2006 Fibronectin-binding proteins of Staphylococcus aureus mediate activation of human platelets via fibrinogen and fibronectin bridges to integrin GPIIb/IIIa and IgG binding to the FcgammaRIIa receptor. Mol Microbiol 59 212 230
26. SimpsonKH
BowdenG
HookM
AnvariB
2003 Measurement of adhesive forces between individual Staphylococcus aureus MSCRAMMs and protein-coated surfaces by use of optical tweezers. J Bacteriol 185 2031 2035
27. LeeSY
KimKK
ChoeSJ
2001 Binding of oral streptococci to human fibrinogen. Oral Microbiol Immunol 16 88 93
28. ReuterswardA
MiornerH
WagnerM
KronvallG
1985 Variations in binding of mammalian fibrinogens to streptococci groups A, B, C, E, G and to Staphylococcus aureus. Acta Pathol Microbiol Immunol Scand B 93 77 82
29. KuuselaP
VartioT
VuentoM
MyhreEB
1985 Attachment of staphylococci and streptococci on fibronectin, fibronectin fragments, and fibrinogen bound to a solid phase. Infect Immun 50 77 81
30. PietrocolaG
SchubertA
VisaiL
TortiM
FitzgeraldJR
2005 FbsA, a fibrinogen-binding protein from Streptococcus agalactiae, mediates platelet aggregation. Blood 105 1052 1059
31. HerzbergMC
NobbsA
TaoL
KilicA
BeckmanE
2005 Oral streptococci and cardiovascular disease: searching for the platelet aggregation-associated protein gene and mechanisms of Streptococcus sanguis-induced thrombosis. J Periodontol 76 2101 2105
32. KerriganSW
DouglasI
WrayA
HeathJ
ByrneMF
2002 A role for glycoprotein Ib in Streptococcus sanguis-induced platelet aggregation. Blood 100 509 516
33. DouglasCW
BrownPR
PrestonFE
1990 Platelet aggregation by oral streptococci. FEMS Microbiol Lett 60 63 67
34. CourtneyHS
DaleJB
HastyDL
2002 Mapping the fibrinogen-binding domain of serum opacity factor of group A streptococci. Curr Microbiol 44 236 240
35. HeilmannC
HartleibJ
HussainMS
PetersG
2005 The multifunctional Staphylococcus aureus autolysin Aaa mediates adherence to immobilized fibrinogen and fibronectin. Infect Immun 73 4793 4802
36. HeilmannC
ThummG
ChhatwalGS
HartleibJ
UekotterA
2003 Identification and characterization of a novel autolysin (Aae) with adhesive properties from Staphylococcus epidermidis. Microbiology 149 2769 2778
37. BrouilletteE
LacasseP
ShkretaL
BelangerJ
GrondinG
2002 DNA immunization against the clumping factor A (ClfA) of Staphylococcus aureus. Vaccine 20 2348 2357
38. EntenzaJM
MoreillonP
SennMM
KormanecJ
DunmanPM
2005 Role of sigmaB in the expression of Staphylococcus aureus cell wall adhesins ClfA and FnbA and contribution to infectivity in a rat model of experimental endocarditis. Infect Immun 73 990 998
39. SullamPM
ValoneFH
MillsJ
1987 Mechanisms of platelet aggregation by viridans group streptococci. Infect Immun 55 1743 1750
40. DhawanVK
YeamanMR
CheungAL
KimE
SullamPM
1997 Phenotypic resistance to thrombin-induced platelet microbicidal protein in vitro is correlated with enhanced virulence in experimental endocarditis due to Staphylococcus aureus. Infect Immun 65 3293 3299
41. CheungAL
EberhardtKJ
ChungE
YeamanMR
SullamPM
1994 Diminished virulence of a sar-/agr - mutant of Staphylococcus aureus in the rabbit model of endocarditis. J Clin Invest 94 1815 1822
42. AC
SGR
IL
JO
2003 Bacteria-binding plasma proteins in pellicles formed on hydroxyapatite in vitro and on teeth in vivo. Oral Microbiology Immunology 18 203 207
43. NagataH
MurakamiY
InoshitaE
ShizukuishiS
TsunemitsuA
1990 Inhibitory effect of human plasma andsaliva on co-aggregation between Bacteroides gingivalis and Streptococcus mitis. J Dent REs 69 1476 1479
44. YasminA
KennyJG
ShankarJ
DarbyAC
HallN
2010 Comparative genomics and transduction potential of Enterococcus faecalis temperate bacteriophages. J Bacteriol 192 1122 1130
45. RomeroP
CroucherNJ
HillerNL
HuFZ
EhrlichGD
2009 Comparative genomic analysis of ten Streptococcus pneumoniae temperate bacteriophages. J Bacteriol 191 4854 4862
46. ChaffinDO
C.E.R
1998 Blue/white screening of recombinant plasmids in Gram-positive bacteria by interruption of alkaline phosphatase gene (phoZ) expression. Gene 219 91 99
47. van de RijnI
KesslerRE
1980 Growth characteristics of group A streptococci in a new chemically defined medium. Infect Immun 27 444 448
48. DillardJP
VanderseaMW
YotherJ
1995 Characterization of the cassette containing genes for type 3 capsular polysaccharide biosynthesis in Streptococcus pneumoniae. J Exp Med 181 973 983
49. YotherJ
LeopoldK
WhiteJ
FischerW
1998 Generation and properties of a Streptococcus pneumoniae mutant which does not require choline or analogs for growth. J Bacteriol 180 2093 2101
50. HarmonJT
GrecoNJ
JamiesonGA
1992 Isolation of human platelet plasma membranes by glycerol lysis. Methods Enzymol 215 32 36
51. SibooIR
ChambersHF
SullamPM
2005 Role of SraP, a Serine-Rich Surface Protein of Staphylococcus aureus, in binding to human platelets. Infect Immun 73 2273 2280
52. SeoHS
CarteeRT
PritchardDG
NahmMH
2008 A new model of pneumococcal lipoteichoic acid structure resolves biochemical, biosynthetic, and serologic inconsistencies of the current model. J Bacteriol 190 2379 2387
53. SeoHS
MichalekSM
NahmMH
2008 Lipoteichoic acid is important in innate immune responses to Gram-positive bacteria. Infect Immun 76 206 213
54. XiongYQ
BensingBA
BayerAS
ChambersHF
SullamPM
2008 Role of the serine-rich surface glycoprotein GspB of Streptococcus gordonii in the pathogenesis of infective endocarditis. Microb Pathog 45 297 301
55. GianfaldoniC
CensiniS
HilleringmannM
MoschioniM
FacciottiC
2007 Streptococcus pneumoniae pilus subunits protect mice against lethal challenge. Infect Immun 75 1059 1062
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 8
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- The Transcription Factor Rbf1 Is the Master Regulator for -Mating Type Controlled Pathogenic Development in
- PKC Signaling Regulates Drug Resistance of the Fungal Pathogen via Circuitry Comprised of Mkc1, Calcineurin, and Hsp90
- Contribution of Coagulases towards Disease and Protective Immunity
- Early Severe Inflammatory Responses to Uropathogenic Predispose to Chronic and Recurrent Urinary Tract Infection