
Herpesvirus Telomerase RNA (vTR) with a Mutated Template Sequence Abrogates Herpesvirus-Induced Lymphomagenesis
Telomerase reverse transcriptase (TERT) and telomerase RNA (TR) represent the enzymatically active components of telomerase. In the complex, TR provides the template for the addition of telomeric repeats to telomeres, a protective structure at the end of linear chromosomes. Human TR with a mutation in the template region has been previously shown to inhibit proliferation of cancer cells in vitro. In this report, we examined the effects of a mutation in the template of a virus encoded TR (vTR) on herpesvirus-induced tumorigenesis in vivo. For this purpose, we used the oncogenic avian herpesvirus Marek's disease virus (MDV) as a natural virus-host model for lymphomagenesis. We generated recombinant MDV in which the vTR template sequence was mutated from AATCCCAATC to ATATATATAT (vAU5) by two-step Red-mediated mutagenesis. Recombinant viruses harboring the template mutation replicated with kinetics comparable to parental and revertant viruses in vitro. However, mutation of the vTR template sequence completely abrogated virus-induced tumor formation in vivo, although the virus was able to undergo low-level lytic replication. To confirm that the absence of tumors was dependent on the presence of mutant vTR in the telomerase complex, a second mutation was introduced in vAU5 that targeted the P6.1 stem loop, a conserved region essential for vTR-TERT interaction. Absence of vTR-AU5 from the telomerase complex restored virus-induced lymphoma formation. To test if the attenuated vAU5 could be used as an effective vaccine against MDV, we performed vaccination-challenge studies and determined that vaccination with vAU5 completely protected chickens from lethal challenge with highly virulent MDV. Taken together, our results demonstrate 1) that mutation of the vTR template sequence can completely abrogate virus-induced tumorigenesis, likely by the inhibition of cancer cell proliferation, and 2) that this strategy could be used to generate novel vaccine candidates against virus-induced lymphoma.
Published in the journal:
. PLoS Pathog 7(10): e32767. doi:10.1371/journal.ppat.1002333
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002333
Summary
Telomerase reverse transcriptase (TERT) and telomerase RNA (TR) represent the enzymatically active components of telomerase. In the complex, TR provides the template for the addition of telomeric repeats to telomeres, a protective structure at the end of linear chromosomes. Human TR with a mutation in the template region has been previously shown to inhibit proliferation of cancer cells in vitro. In this report, we examined the effects of a mutation in the template of a virus encoded TR (vTR) on herpesvirus-induced tumorigenesis in vivo. For this purpose, we used the oncogenic avian herpesvirus Marek's disease virus (MDV) as a natural virus-host model for lymphomagenesis. We generated recombinant MDV in which the vTR template sequence was mutated from AATCCCAATC to ATATATATAT (vAU5) by two-step Red-mediated mutagenesis. Recombinant viruses harboring the template mutation replicated with kinetics comparable to parental and revertant viruses in vitro. However, mutation of the vTR template sequence completely abrogated virus-induced tumor formation in vivo, although the virus was able to undergo low-level lytic replication. To confirm that the absence of tumors was dependent on the presence of mutant vTR in the telomerase complex, a second mutation was introduced in vAU5 that targeted the P6.1 stem loop, a conserved region essential for vTR-TERT interaction. Absence of vTR-AU5 from the telomerase complex restored virus-induced lymphoma formation. To test if the attenuated vAU5 could be used as an effective vaccine against MDV, we performed vaccination-challenge studies and determined that vaccination with vAU5 completely protected chickens from lethal challenge with highly virulent MDV. Taken together, our results demonstrate 1) that mutation of the vTR template sequence can completely abrogate virus-induced tumorigenesis, likely by the inhibition of cancer cell proliferation, and 2) that this strategy could be used to generate novel vaccine candidates against virus-induced lymphoma.
Introduction
Telomerase is a multi-component ribonucleoprotein complex that governs the maintenance of telomeres, protein-associated hexameric sequence repeats at the end of linear chromosomes, and ensures chromosomal integrity and cellular survival [1], [2]. The telomerase complex consists of two core components, telomerase reverse transcriptase (TERT) and telomerase RNA (TR). In the complex, TR serves as the template for TERT, which catalyzes the addition of telomeric repeats (TTAGGG)n at chromosome ends [3]. Vertebrate TRs exhibit a universally conserved secondary structure comprised of four structural domains (Fig. 1): the pseudoknot (core) domain containing the template sequence in conserved region (CR) 1 (CR1), the CR4 and CR5 domains with a highly conserved stem-loop structure (CR4-5), the H/ACA box domain, and the CR7 domain [4]. CR1 encodes the template sequence that is utilized for the extension of the telomeric repeats, while the CR4-5 domain contributes to the processivity of telomerase and is essential for stable assembly with TERT. The H/ACA box and CR7 domains confer TR stability [4]–[6].

Telomerase activity is absent in most somatic cells, but commonly up-regulated in rapidly dividing cells including transformed cells [7]. Consistent with this observation, telomerase activity is significantly elevated in over 85% of human cancers and over 70% of immortalized human cell lines [8]. The absence of telomerase activity often leads to progressive telomere shortening resulting in cellular senescence and irreversible cell cycle arrest [9]. Several tumor-inducing viruses have evolved strategies to evade or subvert mechanisms controlling cellular senescence, mainly via the up-regulation of TERT, which is generally the limiting factor for telomerase activity [10]–[13]. It has been suggested that up-regulation of TERT expression and, consequently, increased telomerase activity ensures the proliferative potential of persistently infected cells.
One of the most efficient viruses with respect to induction of fatal tumors is Marek's disease virus (MDV). MDV is a lymphotropic herpesvirus that causes a well-described syndrome, Marek's disease (MD), in chickens. MD is characterized by neurological disorders, immune suppression, and malignant T cell lymphomas [14]. The rapid onset of lymphomas developing within 2 to 3 weeks post-infection (p.i.) and high tumor-induced mortalities of 90–100% in susceptible chickens make MDV-induced transformation an ideal model to study virus-induced tumorigenesis in a natural virus-host setting [15]. A number of MDV-encoded genes have been shown to be involved in MDV-induced transformation. The major MDV oncogene, meq, encodes a basic leucine zipper (bZIP) transcription factor similar to the cellular homologues c-Jun, c-Fos, and c-Myc. Meq dimerizes with other bZIP transcription factors and modulates expression of both cellular and viral genes [16], [17]. MDV also encodes other genes products and sequence elements, which perform auxiliary functions in transformation [18]. One such element is a TR homologue termed viral TR (vTR) that shares 88% sequence identity with chicken TR (chTR) [19]. The high sequence homology suggests vTR was likely acquired from the chicken genome during virus-host co-evolution. Compared to its cellular counterpart, chTR, interaction of vTR with TERT results in higher telomerase processivity [20], [21]. It was shown that vTR contributes to the rapid onset of lymphoma formation by serving as a template for TERT, but it also has functions that are independent of the telomerase complex. It is predominantly the telomerase-independent functions of vTR that are responsible for tumor progression and dissemination [21], [22].
In vitro experiments demonstrated that mutations in the template sequence within CR1 of human and mouse TR can result in telomere instabilities, aberrant chromosome separation and segregation, and ultimately apoptosis [23], [24]. TRs with a mutated template can induce unique checkpoint responses that are different from DNA damage or loss-of-telomerase responses, even at low mutant TR expression levels and in the presence of wild-type TR. In addition, pro-apoptotic effects were also shown for TRs harboring mutant template or oligonucleotides specifying mutant template sequences and such molecules are discussed as anti-tumor therapeutics in different types of cancers [23], [25], [26].
Here, we investigated the effect of a mutant vTR template sequence (AU5) on the tumor-promoting capacity of a highly oncogenic avian herpesvirus in its natural host. Mutation of the template sequence of MDV-encoded vTR completely abrogated virus-induced tumor formation in chickens. Introduction of a second mutation in the stem loop (CR4-5) region that abolishes a functional interaction of vTR with TERT restored lymphomagenesis, confirming that the abrogation of tumorigenesis shown for the mutant virus is dependent on telomerase activity through interaction of mutant vTR and TERT. In vaccination-challenge studies, the virus expressing mutant template protected chickens from lethal challenge with a very virulent MDV strain.
Results
Expression of mutant template sequence vTR significantly reduces proliferation of avian cancer cells in vitro
TRs harboring mutations in the template sequence were previously shown to result in telomere instabilities, aberrant chromosome separation and segregation, and ultimately apoptosis in mammalian cells in vitro [23]. Led by these previous observations, we hypothesized that expression of vTR encoding a mutated template sequence (AATCCCAATC to ATATATATAT), termed AU5, could have an effect on avian cancer cells that is similar to that described previously for mammalian cells [23], [24]. In order to test our hypothesis, we first screened a number of avian primary cells and permanent cancer cell lines to determine the optimal system that would provide sufficient levels of telomerase activity. We performed telomere repeat amplification protocol (TRAP) assays to detect telomerase activity in primary chicken embryo cell (CEC) cultures, the chicken fibroblast cell line DF-1 [27], and the quail cancer cell line QT35 [28]. CEC and DF-1 cells did not exhibit telomerase activity, while the QT35 cancer cell had high telomerase activity as evidenced by the presence of numerous TRAP products (Fig. 2A). A DF-1 cell line stably expressing TERT showed some telomerase activity, suggesting that TERT was the limiting factor for telomerase activity in this cell line. Based on the results, we used a previously established QT35 cancer cell line that allowed tetracycline-inducible expression [29] of vTR or vTR-AU5 (AU5). During the establishment of cell lines we observed that even un-induced AU5 cell lines replicated markedly slower. From many initial clones, only a single monoclonal AU5 cell line could be established, suggesting a strong selection against leaky AU5 expression. This effect has been previously observed during the development of mammalian cell lines expressing TR template mutants [23]. Therefore, polyclonal vTR and AU5 cell lines were used to determine the effect of AU5 expression on cancer cell proliferation. RT-qPCR analysis of polyclonal cells, confirmed leaky expression of the constructs and that vTR and AU5 expression could be increased by more than 300-fold upon induction with doxycycline after 5 days of treatment (Fig. 2B).

To determine if AU5 inhibits cancer cell proliferation, we analyzed colony formation by measuring confluency over 31 days in the presence or absence of doxycycline. Constitutive or induced expression of wild-type vTR resulted in enhanced proliferation when compared cells harboring the vector control as described previously [21]. In contrast, cell lines harboring AU5 exhibited a significant growth defect when compared to vTR and control cell lines (Fig. 2C). Increased expression of AU5 following induction resulted in only slightly reduced cell proliferation when compared with non-induced cells, suggesting that expression of only low levels of AU5 are sufficient to reduce growth of the QT35 cancer cell line. We concluded that our results are consistent with those of TR over-expression in human and murine cancer cells [23], [24] and show that expression of the MDV vTR can help stabilize and/or promote growth, while mutation of the template sequence significantly impairs proliferation of the avian QT35 cancer cell line.
Mutation of the vTR template sequence abrogates MDV-induced lymphomagenesis
Since mutation of the template sequence of vTR resulted in decreased proliferation of QT35 cancer cells (Fig. 1C), we hypothesized that the mutation in the context of virus infection may have an effect on MDV replication and tumorigenesis in vivo. Therefore, we mutated the template sequence of vTR (AU5) in pRB-1B, an infectious bacterial artificial chromosome (BAC) clone of the highly oncogenic RB-1B MDV strain using two-step Red-mediated recombination [30], [31]. Two rounds of mutagenesis allowed the desired alteration of both copies of the diploid vTR gene within the MDV genome, and transfection of the recombinant BAC clone into CEC resulted in the reconstitution of the vTR template mutant virus (vAU5). Furthermore, a revertant clone (AU5rev) was generated in which the wild-type template sequence was restored in the mutant vAU5. Following virus reconstitution, we performed plaque size assays and multi-step growth kinetics in CEC that revealed that the growth properties of vAU5 were indistinguishable from those of parental (vRB-1B) and revertant (vAU5rev) viruses (Fig. 3A–D).

Next, we determined if expression of AU5 had an effect on MDV replication, disease, and tumor incidence in vivo. In two independent experiments, we infected 1-day-old P2a chickens with vRB-1B, vAU5, or vAU5rev and monitored virus levels in the blood using qPCR assays until 28 days post infection (dpi). vAU5 replication was significantly impaired when compared to parental and revertant viruses, indicating that the number of infected B and T cells is reduced (Fig. 4B and D). Consistent with the reduction of viremia, none of the chickens infected with vAU5 developed tumors in two independent experiments (0/10; 0/18) over the course of 13 weeks while parental (vRB-1B) or revertant (vAU5rev) viruses induced lymphomas in 92–100% of infected animals (Fig. 4A and C). We concluded from our data that expression of vTR harboring the AU5 mutation by MDV can completely abrogate virus-induced tumorigenesis in highly susceptible chickens, most likely by the elimination of MDV-infected and/or transformed cells by apoptosis.

Abrogation of MDV-induced lymphomagenesis caused by expression of mutant vTR is dependent on its interaction with TERT
We previously demonstrated that a mutation within the vTR P6.1 stem-loop can prevent incorporation of vTR into the telomerase complex and abolish enzymatic activity and telomere elongation [22]. To confirm that the absence of lymphoma in vAU5-infected animals was dependent on the presence of AU5 in the telomerase complex, we constructed mutant viruses in which the AU5 and P6.1 mutations were introduced into vTR either individually or together. Revertant viruses of each mutation were also generated. All constructed viruses replicated with kinetics comparable to those of parental and revertant viruses in vitro (Fig. 5). Upon infection of chickens with the recombinant viruses, qPCR analysis revealed that insertion of the P6.1 mutation into vAU5 (vAU5+P6.1mut) restored lytic virus replication to levels comparable to those of parental vRB-1B, while mutant virus only harboring the AU5 mutation (vAU5+P6.1rev) was significantly impaired in replication (Fig. 6A).


Like vAU5, vAU5+P6.1rev did not induce tumors in any of the infected chickens (0/19) (Fig. 6B). Two of the 19 chickens (11%) died over the course of the 13 week experiment, which was likely due to immunosuppression and generalized wasting, common characteristics of MD and observed in earlier reports using viruses that are unable to express vTR [21]. Viruses that contained the P6.1 and the AU5 mutation (vAU5+P6.1mut) induced lymphomas in 100% of infected animals. Furthermore, vP6.1, parental and complete revertant viruses caused tumors in all animals infected with the respective viruses. From the results we concluded that abrogation of lymphoma formation after infection with an MDV specifying the AU5 template mutation is indeed dependent on the interaction of template mutant vTR with TERT and has no effect if it is not incorporated into the telomerase complex.
Vaccination with vTR template mutant virus confers protection against lethal MDV challenge
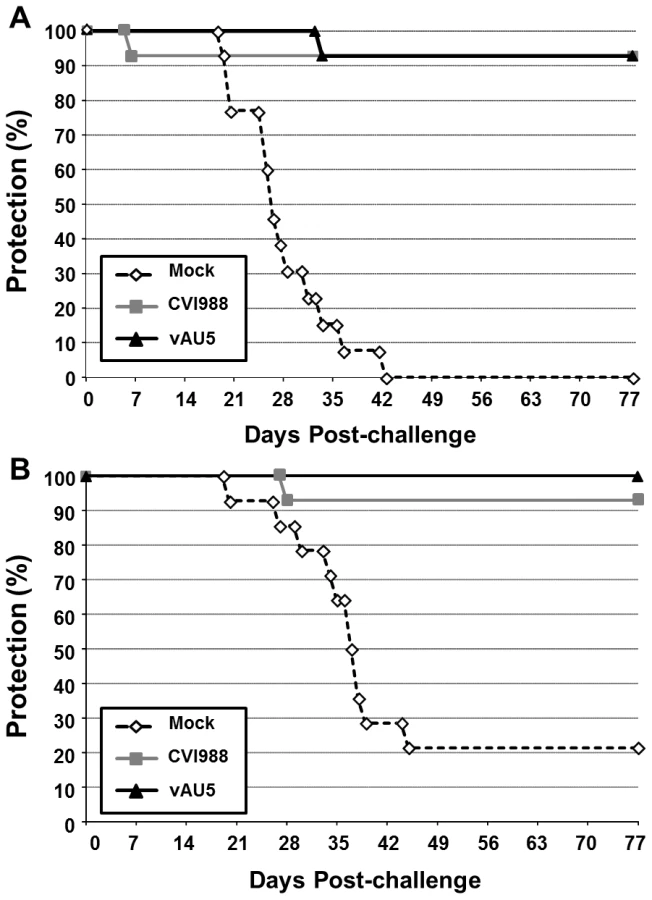
Since MDV harboring the template mutant vTR did not induce tumors but still replicated in chickens, we addressed the question whether vAU5 could induce a robust enough immune response to serve as a vaccine. Groups of 1-day-old P2a (highly susceptible to MD) and N2a (partially resistant to MD) chickens were inoculated with diluent, vAU5, or the widely used, commercial vaccine strain CVI988. Vaccinated chickens were challenged 10 days later with the very virulent RB-1B MDV strain. Chickens receiving the diluent developed tumors with expected frequencies of 100% in the P2a chickens and 79% in the N2a chickens after 13 weeks (Fig. 7A and B) [32]. vAU5 vaccinated N2a chickens were completely protected from lethal challenge, while 7% of the animals vaccinated with the commercial CVI988 vaccine strain developed disease with a protective index of 91%. In P2a chickens that are highly susceptible to MD, both vAU5 and CVI988 efficiently induced protection against challenge infection, as only 1 animal in each group developed disease. The protective index of vAU5 and CVI988 in P2a animals was 93% and 92%, respectively. These results suggest that mutation of the template region of vTR in a virulent MDV can serve as a strategy to induce protection against virus-induced lymphomas.
Discussion
We here report on effects of a mutation in the template sequence (CR1) of vTR encoded by MDV on virus replication and tumorigenesis in a natural virus-host model. Mutation of the vTR template sequence from AATCCCAATC to ATATATATAT (AU5) resulted in decreased proliferation of the QT35 avian cancer cell line (Fig. 2), as had been described for TR in mammalian cells [23], [24]. Introduction of the template sequence mutation in vTR in the context of the viral genome and infection of MD-susceptible chickens with mutant virus (vAU5) resulted in complete absence of tumors and low-level viral replication in vivo (Fig. 4). Secondary mutation of the vTR stem-loop sequence (P6.1), abolishing the interaction of mutant vTR with TERT, restored virus-induced tumorigenesis (Fig. 6), thus showing that vTR-TERT interaction and functional telomerase activity is required for the anti-tumorigenic effects of the mutant template sequence in a viral background. Vaccination with MDV harboring the vTR template mutation not only abrogated herpesvirus-induced tumorigenesis, but also protected chickens from a lethal challenge with a very virulent MDV strain.
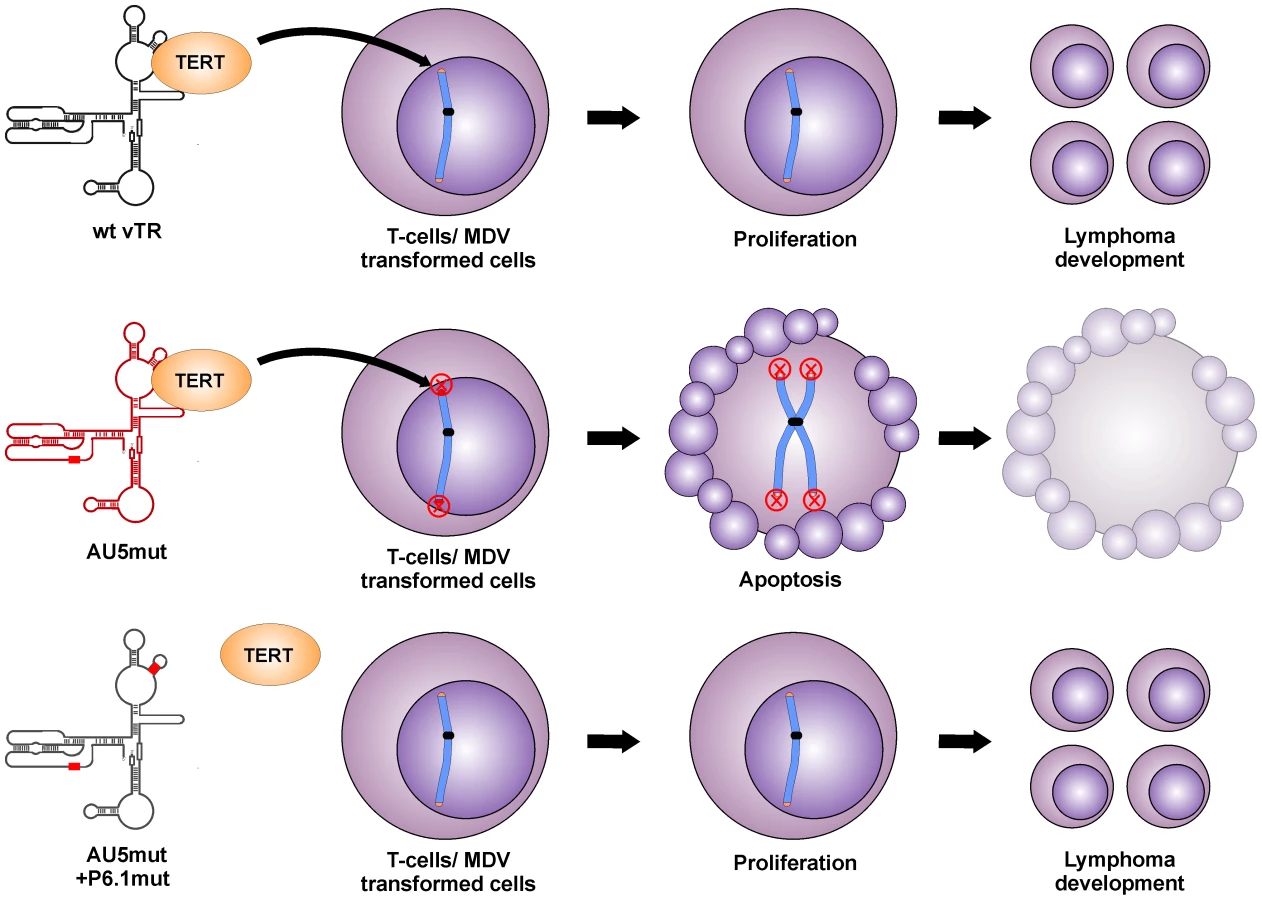
We surmise that the reduced proliferation of QT35 expressing vTR with a template sequence mutation, as well as the absence of tumors and greatly reduced lytic replication in chickens are both caused by the incorporation of mutant telomeric repeat sequences into host telomeres of infected cells, which eventually leads to telomere crisis and apoptosis (Fig. 8). This sequence of events has been shown previously in other mammalian systems in vitro, where even low levels of mutant TR induced a unique checkpoint response resulting in telomere instabilities, aberrant chromosome separation and segregation, and apoptosis [23], [24]. In addition, the pro-apoptotic effect of TRs harboring mutant templates or oligonucleotides specifying mutant template sequences has also been shown [23], [25], [26].
It is interesting to note that the QT35 cancer cell line was previously shown to maintain MDV in a latent state [33] and that the cells express MDV vTR at very low levels (Fig. 2B). Despite the expression of endogenous quail TR and MDV vTR, AU5 expression had a negative effect on the replication of QT35 cancer cells. Consequently, induced over-expression of wild-type vTR led to increased proliferation of QT35 cancer cells, further lending support to the interpretation that vTR performs an important function in the early maintenance of transformed cells [21]. Induced expression of the AU5 sequence leading to incorporation of mutant template sequences significantly reduced proliferation presumably by inducing apoptosis, again consistent with previous studies on mammalian TRs [23].
vTR was previously shown to contribute to MDV-induced lymphomagenesis. Deletion of vTR in the MDV genome resulted in significantly reduced tumor incidences but the mutation did not affect virus replication in vivo [21]. Mutation of the vTR template region (AU5) in MDV, however, completely abrogated tumorigenesis and reduced viral loads in infected animals, likely via inhibition of cancer cell replication through induction of apoptosis. vTR has at least two functions during lymphomagenesis, one that is dependent and one that is independent of telomerase activity. The telomerase-dependent function plays an important role in the early onset of disease but is dispensable for tumorigenesis. This conclusion is supported by studies showing that MDV harboring a mutated vTR incapable of interaction with TERT (P6.1mut) can still induce tumors in chickens, albeit resulting in a delayed onset of tumor formation [22]. vTR functions that are independent of its presence in the telomerase complex seem to be important for lymphomagenesis but are poorly understood. We utilized this previously published mutation to determine if the induction of apoptosis and abrogation of tumorigenesis is dependent on the incorporation of AU5 into the telomerase complex. While MDV harboring AU5 vTR are incapable of inducing tumors, mutation in the vTR-TERT interaction domain (vAU5+P6.1mut) in vAU5 completely restored tumorigenesis.
We therefore concluded that restored ability of vAU5+P6.1mut to induce tumors is presumably caused by the inability of the telomerase to incorporate mutant telomeric repeats at the ends of host chromosomes and cause telomere crisis and apoptosis; hence the pro-oncogenic functions of vTR that are independent of the telomerase complex prevail. Our results demonstrate that vAU5+P6.1mut can efficiently cause lymphoma, which confirms that vTR has tumor-promoting functions independent of the telomerase complex and that they are mediated by a vTR domain outside of the template region.
The fact that vAU5 was unable to induce tumors in highly susceptible P2a chickens suggested that it could serve as a potential vaccine against MD. Vaccination with vAU5 protected chickens from lethal challenge infection with the very virulent MDV strain RB-1B at least as efficiently as the commercial vaccine strain CVI988/Rispens that is commonly used in the field [34]. However, the residual mortality observed in some chickens has to be clarified to ensure the safety of the vaccine candidate. A similar phenomenon of low levels of mortality in highly susceptible birds, similar to the one observed here, was also observed with MDV mutants in which the major oncoprotein of MDV, Meq, was absent. Infection with meq deletion viruses did not cause tumors [35], but severe lymphoid atrophy and immunosuppression was evident [36]. Likely, a combination of vTR template mutation with other modifications in the MDV genome targeting genes important for replication could therefore increase the safety of the vaccine and prevent lymphoid atrophy. Here, we suggest a new strategy that could be applied to the next generation of MD vaccines, which will certainly be needed because recently isolated MDV strains are capable of evading immune protection provided by current vaccines [15], [37], [38].
Materials and Methods
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of Cornell University (permit number 2002-0085 and 2008-0018). The animal care facilities and programs of Cornell University meet the requirements of the law (89–544, 91–579, 94–276) and NIH regulations on laboratory animals, and are in compliance with the Animal Welfare Act, PL 279. All experimental procedures were in compliance with approval of Cornell University's Institutional Animal Care and Use Committee (IACUC) and all efforts were made to minimize suffering.
Generation of mutant MDV
Recombinant viruses were generated by two-step Red-mediated recombination as previously described [30], [31]. Primers used for construction of template sequence (CR1) AU5 mutants (vAU5) and revertants (vAU5rev) are shown in Table 1. Primers used for construction of the P6.1 vTR-TERT interaction domain mutants and revertants have previously been published [22].

Propagation of MDV
CEC cultures were prepared from 10-day-old specific-pathogen-free (SPF) embryos using standard methods [39]. Recombinant viruses were reconstituted from purified BAC DNA in CEC cultures using CaPO4 transfection [40]. The loxP flanked mini-F sequences within the infectious clones were removed by co-transfection with a Cre recombinase expression vector (pCAGGS-NLS/Cre) as previously described and screened via analytical PCR [30]. Virus propagation, plaque area measurements and multi-step growth kinetics were also performed as described previously [41].
Animal studies
SPF P2a (MHC haplotype B19B19) or N2a (MHC haplotype B21B21) chickens were obtained from departmental flocks and housed in poultry isolation units. Chickens were inoculated with 1,000 or 2,000 plaque forming units (PFU) of virus by intra-abdominal injection and evaluated for symptoms of MD on a daily basis. Necropsies were performed on chickens showing clinical signs of MD, as well as all remaining chickens at the termination of the experiment.
Chicken blood DNA extraction and qPCR assays
DNA was extracted from whole blood of eight chickens for each group randomly selected prior to the experiment and MDV genomic copies were determined by qPCR assays [41]. Briefly, MDV DNA copy numbers were detected using primers and probe specific for the MDV infected cell protein 4 (ICP4) locus that were normalized to cellular genome copies of chicken inducible nitric oxide synthase (iNOS).
Cloning of vTR and AU5 Tet-on expression constructs and generation of stable cell lines
Tet-on constructs were generated by digestion of the pCMS-vTR and pCMS-vTR-AU5 constructs previously described [22] with EcoRI and XbaI. Resulting vTR or AU5 fragments were then cloned into the pcDNA4/TO/myc-his vector (Invitrogen, Carlsbad, CA) to generate pcDNA4/TO-vTR and pcDNA4/TO-AU5, respectively.
Inducible cell lines were generated based on QT35TR19, a previously described Tet-inducible QT35 cancer cell line (kindly provided by Karel A. Schat, Cornell University) and maintained as described previously [29]. To generate control, vTR, and AU5 expressing cell lines, QT35TR19 cells were transfected with pcDNA4/TO (empty vector), pcDNA4/TO-vTR, or pcDNA4/TO-AU5 using Lipofectamine2000 (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. Monoclonal and polyclonal cell lines were selected with 5 µg/ml blasticidin and 500 µg/ml zeocin (Invitrogen). All cell lines were used between passage 5 and 10 in cell proliferation and RT-qPCR assays.
Cell proliferation assays
Proliferation of Tet-inducible cell lines was evaluated as previously described [23]. Briefly, 2×103 cells of each cell line were seeded into 35 mm dishes in triplicate and maintained in media with or without 1 µg/ml doxycycline with 2/3 media changed every 4–5 days. After 31 days, cells were fixed with 90% ice-cold acetone and stained with 1% crystal violet in 50% methanol. Percent confluency was determined using NIH ImageJ software by calculating the total area on the plates covered by cell colonies over the total area of the plate. The average % confluency was determined from three independent experiments.
RT-qPCR assays for analysis of Tet-inducible vTR expression
One-thousand vTR, AU5, or empty vector Tet-inducible cells were treated with or without 1 µg/ml doxycycline in triplicate. After 3 and 5 days, total RNA was prepared using RNA STAT 60 as described previously [42]. Reverse transcription was performed using the ThermoScriptTM RT-PCR system (Invitrogen, Carlsbad, CA) with random hexamer oligonucleotides according to manufacturer's instructions.
Copies of vTR cDNA were determined by qPCR assays using the TaqMan Fast Universal Master Mix system (Applied Biosystems, Inc.) according to manufacturer's instructions and performed in an ABI Prism 7500 Fast Real-Time PCR System (Applied Biosystems, Inc.). Results were analyzed with the Sequence Detection Systems version V2.0.3 software using the comparative Ct method (2−ΔΔCt) of relative quantification. Primers and probe for the detection of MDV vTR and quail glyceraldehyde-3-phosphate dehydrogenase (GAPDH) that served as an endogenous control have been described previously [43], [44].
Statistical analysis
Significant differences in % confluency assays and MDV replication using qPCR assays were determined using Student's t test or Tukey-Kramer comparison of means.
Zdroje
1. BlackburnEH 2005 Telomeres and telomerase: their mechanisms of action and the effects of altering their functions. FEBS Lett 579 859 862
2. BlackburnEH 2005 Telomerase and Cancer: Kirk A. Landon—AACR prize for basic cancer research lecture. Mol Cancer Res 3 477 482
3. GreiderCWBlackburnEH 1987 The telomere terminal transferase of tetrahymena is a ribonucleoprotein Enzyme with 2 kinds of primer specificity. Cell 51 887 898
4. ChenJLBlascoMAGreiderCW 2000 Secondary structure of vertebrate telomerase RNA. Cell 100 503 514
5. ChenJLGreiderCW 2004 Telomerase RNA structure and function: implications for dyskeratosis congenita. Trends Biochem Sci 29 183 192
6. MitchellJRChengJCollinsK 1999 A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3′ end. Mol Cell Biol 19 567 576
7. KimNWPiatyszekMAProwseKRHarleyCBWestMD 1994 Specific association of human telomerase activity with immortal cells and cancer. Science 266 2011 2015
8. ShayJWBacchettiS 1997 A survey of telomerase activity in human cancer. Eur JCancer 33 787 791
9. BellonMNicotC 2008 Regulation of telomerase and telomeres: Human tumor viruses take control. J Natl Cancer Inst 100 98 108
10. HorikawaIBarrettJC 2003 Transcriptional regulation of the telomerase hTERT gene as a target for cellular and viral oncogenic mechanisms. Carcinogenesis 24 1167 1176
11. LiuHLuanFJuYShenHYGaoLF 2007 In vitro transfection of the hepatitis B virus PreS2 gene into the human hepatocarcinoma cell line HepG2 induces upregulation of human telomerase reverse transcriptase. Biochem Biophys Res Commun 355 379 384
12. ZhuZWWilsonATGopalakrishnaKBrownKELuxonBA 2010 Hepatitis C virus core protein enhances telomerase activity in Huh7 cells. J Med Virol 82 239 248
13. ZhuZWWilsonATGopalakrishnaKNBrownKLuxonBA 2008 Hepatitis C virus core protein upregulates telomerase activity in hepatoma cells. Hepatology 48 974A 974A
14. CalnekBW 2001 Pathogenesis of Marek's disease virus infection. HiraiK Marek's disease Berlin, Germany Springer 25 55
15. OsterriederNKamilJPSchumacherDTischerBKTrappS 2006 Marek's disease virus: from miasma to model. Nat Rev Microbiol 4 283 294
16. BrownACBaigentSJSmithLPChattooJPPetherbridgeLJ 2006 Interaction of MEQ protein and C-terminal-binding protein is critical for induction of lymphomas by Marek's disease virus. Proc Natl Acad Sci U S A 103 1687 1692
17. BrownACSmithLPKgosanaLBaigentSJNairV 2009 Homodimerization of the Meq viral oncoprotein is necessary for induction of T-cell lymphoma by Marek's disease virus. J Virol 83 11142 11151
18. JarosinskiKWTischerBKTrappSOsterriederN 2006 Marek's disease virus: lytic replication, oncogenesis and control. Expert Rev Vaccines 5 761 772
19. FragnetLBlascoMAKlapperWRasschaertD 2003 The RNA subunit of telomerase is encoded by Marek's disease virus. J Virol 77 5985 5996
20. FragnetLKutERasschaertD 2005 Comparative functional study of the viral telomerase RNA based on natural mutations. J Biol Chem 280 23502 23515
21. TrappSParcellsMSKamilJPSchumacherDTischerBK 2006 A virus-encoded telomerase RNA promotes malignant T cell lymphomagenesis. J Exp Med 203 1307 1317
22. KauferBBTrappSJarosinskiKWOsterriederN 2010 Herpesvirus telomerase RNA(vTR)-dependent lymphoma formation does not require interaction of vTR with telomerase reverse transcriptase (TERT). PloS Pathog 6 e1001073
23. KimMMRiveraMABotchkinaILShalabyRThorAD 2001 A low threshold level of expression of mutant-template telomerase RNA inhibits human tumor cell proliferation. Proc Natl Acad Sci U S A 98 7982 7987
24. LinJSmithDLBlackburnEH 2004 Mutant telomere sequences lead to impaired chromosome separation and a unique checkpoint response. Mol Biol Cell 15 1623 1634
25. DjojosubrotoMWChinACGoNSchaetzleinSMannsMP 2005 Telomerase antagonists GRN163 and GRN163L inhibit tumor growth and increase chemosensitivity of human hepatoma. Hepatology 42 1127 1136
26. GellertGCDikmenZGWrightWEGryaznovSShayJW 2006 Effects of a novel telomerase inhibitor, GRN163L, in human breast cancer. Breast Cancer Res Treat 96 73 81
27. HimlyMFosterDNBottoliIIacovoniJSVogtPK 1998 The DF-1 chicken fibroblast cell line: transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses. Virology 248 295 304
28. MoscoviciCMoscoviciMGJimenezHLaiMMCHaymanMJ 1977 Continuous tissue-culture cell lines derived from chemically-induced tumors of Japanese quail. Cell 11 95 103
29. LiXHJarosinskiKWSchatKA 2006 Expression of Marek's disease virus phosphorylated polypeptide pp38 produces splice variants and enhances metabolic activity. Vet Microbiol 117 154 168
30. JarosinskiKWMargulisNGKamilJPSpatzSJNairVK 2007 Horizontal transmission of Marek's disease virus requires U S2, the UL13 protein kinase, and gC J Virol 81 10575 10587
31. TischerBKvon EinemJKauferBOsterriederN 2006 Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. BioTechniques 40 191 197
32. SchatKACalnekBWFabricantJ 1982 Characterization of two highly oncogenic strains of Marek's disease virus. Avian Pathol 11 593 605
33. YamaguchiTKaplanSLWakenellPSchatKA 2000 Transactivation of latent Marek's disease herpesvirus genes in QT35, a quail fibroblast cell line, by herpesvirus of turkeys. J Virol 74 10176 10186
34. WitterRLKreagerKS 2004 Serotype 1 viruses modified by backpassage or insertional mutagenesis: approaching the threshold of vaccine efficacy in Marek's disease. Avian Dis 48 768 782
35. LeeLFLupianiBSilvaRFKungHJReddySM 2008 Recombinant Marek's disease virus (MDV) lacking the Meq oncogene confers protection against challenge with a very virulent plus strain of MDV. Vaccine 26 1887 1892
36. KatneniUSchmidtCKeelerCGolovanSReddyS Analysis of immune signaling in Marek's disease virus vaccinated chickens. SpatzSMoodyA The 5 th International Workshop on the Molecular Pathogenesis of Marek's Disease Virus and 1 st Symposium on Avian Herpesviruses 2010; Athens, GA United States Department of Agriculture, Agriculture Research Service 47 47
37. GimenoIM 2008 Marek's disease vaccines: A solution for today but a worry for tomorrow? Vaccine 26 C31 C41
38. WozniakowskiGSamorek-SalamonowiczEKozdrunW 2011 Molecular characteristics of Polish field strains of Marek's disease herpesvirus isolated from vaccinated chickens. Acta Vet Scand 53 10
39. SchatKASellersHS 2008 Cell-culture methods. Dufour-ZavalaLSwayneDEGlissonJRPearsonJEReedWM A laboratory manual for the identification and characterization of avian pathogens: American Association of Avian Pathologists, Jacksonville, FL 195 203
40. SchumacherDTischerBKFuchsWOsterriederN 2000 Reconstitution of Marek's disease virus serotype 1 (MDV-1) from DNA cloned as a bacterial artificial chromosome and characterization of a glycoprotein B-negative MDV-1 mutant. J Virol 74 11088 11098
41. JarosinskiKWOsterriederNNairVKSchatKA 2005 Attenuation of Marek's disease virus by deletion of open reading frame RLORF4 but not RLORF5a. J Virol 79 11647 11659
42. JarosinskiKWO'ConnellPHSchatKA 2003 Impact of deletions within the Bam HI-L fragment of attenuated Marek's disease virus on vIL-8 expression and the newly identified transcript of open reading frame LORF4. Virus Genes 26 255 269
43. ChbabNEgererAVeigaIJarosinskiKWOsterriederN 2010 Viral control of vTR expression is critical for efficient formation and dissemination of lymphoma induced by Marek's disease virus (MDV). Vet Res 41
44. PiepenbrinkMSLiXHO'ConnellPHSchatKA 2009 Marek's disease virus phosphorylated polypeptide pp38 alters transcription rates of mitochondrial electron transport and oxidative phosphorylation genes. Virus Genes 39 102 112
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 10
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Regulates Cell Stress Response and Apoptosis
- The SARS-Coronavirus-Host Interactome: Identification of Cyclophilins as Target for Pan-Coronavirus Inhibitors
- Biochemical and Structural Insights into the Mechanisms of SARS Coronavirus RNA Ribose 2′-O-Methylation by nsp16/nsp10 Protein Complex
- Evolutionarily Divergent, Unstable Filamentous Actin Is Essential for Gliding Motility in Apicomplexan Parasites