
BosR (BB0647) Controls the RpoN-RpoS Regulatory Pathway and Virulence Expression in by a Novel DNA-Binding Mechanism
In Borrelia burgdorferi (Bb), the Lyme disease spirochete, the alternative σ factor σ54 (RpoN) directly activates transcription of another alternative σ factor, σS (RpoS) which, in turn, controls the expression of virulence-associated membrane lipoproteins. As is customary in σ54-dependent gene control, a putative NtrC-like enhancer-binding protein, Rrp2, is required to activate the RpoN-RpoS pathway. However, recently it was found that rpoS transcription in Bb also requires another regulator, BosR, which was previously designated as a Fur or PerR homolog. Given this unexpected requirement for a second activator to promote σ54-dependent gene transcription, and the fact that regulatory mechanisms among similar species of pathogenic bacteria can be strain-specific, we sought to confirm the regulatory role of BosR in a second virulent strain (strain 297) of Bb. Indeed, BosR displayed the same influence over lipoprotein expression and mammalian infectivity for strain Bb 297 that were previously noted for Bb strain B31. We subsequently found that recombinant BosR (rBosR) bound to the rpoS gene at three distinct sites, and that binding occurred despite the absence of consensus Fur or Per boxes. This led to the identification of a novel direct repeat sequence (TAAATTAAAT) critical for rBosR binding in vitro. Mutations in the repeat sequence markedly inhibited or abolished rBosR binding. Taken together, our studies provide new mechanistic insights into how BosR likely acts directly on rpoS as a positive transcriptional activator. Additional novelty is engendered by the facts that, although BosR is a Fur or PerR homolog and it contains zinc (like Fur and PerR), it has other unique features that clearly set it apart from these other regulators. Our findings also have broader implications regarding a previously unappreciated layer of control that can be involved in σ54–dependent gene regulation in bacteria.
Published in the journal:
. PLoS Pathog 7(2): e32767. doi:10.1371/journal.ppat.1001272
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001272
Summary
In Borrelia burgdorferi (Bb), the Lyme disease spirochete, the alternative σ factor σ54 (RpoN) directly activates transcription of another alternative σ factor, σS (RpoS) which, in turn, controls the expression of virulence-associated membrane lipoproteins. As is customary in σ54-dependent gene control, a putative NtrC-like enhancer-binding protein, Rrp2, is required to activate the RpoN-RpoS pathway. However, recently it was found that rpoS transcription in Bb also requires another regulator, BosR, which was previously designated as a Fur or PerR homolog. Given this unexpected requirement for a second activator to promote σ54-dependent gene transcription, and the fact that regulatory mechanisms among similar species of pathogenic bacteria can be strain-specific, we sought to confirm the regulatory role of BosR in a second virulent strain (strain 297) of Bb. Indeed, BosR displayed the same influence over lipoprotein expression and mammalian infectivity for strain Bb 297 that were previously noted for Bb strain B31. We subsequently found that recombinant BosR (rBosR) bound to the rpoS gene at three distinct sites, and that binding occurred despite the absence of consensus Fur or Per boxes. This led to the identification of a novel direct repeat sequence (TAAATTAAAT) critical for rBosR binding in vitro. Mutations in the repeat sequence markedly inhibited or abolished rBosR binding. Taken together, our studies provide new mechanistic insights into how BosR likely acts directly on rpoS as a positive transcriptional activator. Additional novelty is engendered by the facts that, although BosR is a Fur or PerR homolog and it contains zinc (like Fur and PerR), it has other unique features that clearly set it apart from these other regulators. Our findings also have broader implications regarding a previously unappreciated layer of control that can be involved in σ54–dependent gene regulation in bacteria.
Introduction
Bacterial gene expression is primarily controlled at the transcriptional level, which requires a central DNA-dependent RNA polymerase (RNAP) consisting of catalytic core (α2ββ'ω; E) and a dissociable σ factor [1]. Gene transcription occurs when the Eσ-promoter closed complex (CC) is converted to the open complex (OC). Among various σ factors, the alternative σ factor σ54(σN, RpoN) is employed by many bacteria to transcribe genes involved in a wide variety of cellular functions such as virulence, nitrogen metabolism, and stress responses [1]. Unlike other σ factors, the Eσ54 holoenzyme alone cannot melt the promoter. The Eσ54-CC (held by the interaction of Eσ54 with the unique -24/-12 promoter) remains in this conformation until the activator ATPase interacts with RNAP, which hydrolyzes ATP for promoter melting [2]–[4]. The activator ATPase, also known as the enhancer-binding protein (EBP), usually binds to an enhancer site located ∼100–150 bp upstream of the promoter. Typically, the EBP interacts with the RNAP via a DNA looping mechanism that is modulated by a DNA-bending protein such as integration host factor (IHF).
Borrelia burgdorferi (Bb), the Lyme disease spirochete [5]–[6], encodes three σ factors: the housekeeping σ70 (RpoD, BB0712), and two alternative σ factors, σ54 (RpoN, BB0450) and σS (RpoS, BB0771) [7]. Abundant evidence [8]–[11] has revealed that Bb RpoN directly binds to the -24/-12 site in rpoS promoter and thus activates rpoS which, in turn, modulates the differential expression of more than 100 genes involved in Bb virulence, stress adaptation, and many other functions. Studies have indicated that the Bb RpoS regulon is triggered by various environmental stimuli including temperature, pH, cell density, and unknown mammalian host factors [12]–[15]. The RpoN-RpoS pathway (or the Rrp2-RpoN-RpoS pathway) [8]–[11], [16]–[18] plays a central role in modulating the differential expression of Bb outer surface lipoproteins such as outer surface protein C (OspC) [14], [19]–[20] and decorin-binding protein A (DbpA) [21]–[23], which are critical for Bb to transmit from the arthropod tick vector to mammalian hosts and to maintain its natural life cycle. Activation of the RpoN-dependent rpoS gene requires the activation of Rrp2 (BB0763), a putative EBP [17]. Although Rrp2 was presumed to be the sole NtrC-like EBP in Bb, Rrp2 seems to be unconventional as an EBP in that it apparently does not bind specifically to the RpoN-dependent promoter of rpoS [8], [24]. The efficient translation of rpoS mRNA also requires the small RNA DsrA and an atypical RNA chaperone Hfq [25]–[26].
Emanating from our general interest in virulence expression in the Lyme disease spirochete, we previously showed that a manganese transporter, BB0219 (BmtA), is required for full virulence by Bb [27]. The implication that metal transport, and perhaps metal control over borrelial gene regulation, could influence Bb's virulence prompted us to expand our study to examine other molecules of Bb implicated in metal-sensing. To this end, BosR (BB0647) is a ferric uptake regulator (Fur)-like homologue in Bb [7], [28] that has been postulated to contribute to the regulation of oxidative stress responses [29]. In many bacteria, Fur globally regulates iron homeostasis and other functions [30]. Given the provocative finding that Bb seems not to accumulate iron [31], it remained tenuous as to whether BosR is involved in iron uptake by Bb. Nonetheless, BosR may influence cellular functions other than iron acquisition. Recently, we and others surprisingly found that expression of the RpoS regulon was significantly impaired in a mutant deficient in bosR, leading to the additional unexpected finding that BosR somehow functions as a second activator to promote σ54-dependent rpoS transcription, and that such control by BosR ultimately governs the expression of virulence-associated membrane lipoproteins and mammalian infectivity by Bb [32]–[35]. Although this discovery has represented a major advance in further understanding the regulatory control of virulence expression by the Lyme disease spirochete, the observation has engendered many new unanswered questions. Among them include whether BosR is indeed a global regulator common to more than the one virulent strain of Bb (examined in previous studies) [32]–[34], and how BosR may act mechanistically to exert its positive control over the RpoN-RpoS regulatory pathway in Bb. In this report, we provide further evidence for the direct involvement of BosR in the activation of rpoS, and thus the RpoS regulon, in a second virulent strain of Bb. We also present evidence that BosR functions as a DNA-binding protein, but it has many features that markedly distinguish it from either of its Fur or PerR homologs. Defining this novel involvement of BosR relative to its control over the RpoN-RpoS pathway is important for elucidating Bb's host adaptation and pathogenesis, and could lead to innovative strategies for thwarting Lyme disease. This study also expands our understanding of bacterial sigma factor regulatory networks, and establishes a new paradigm of an additional transcriptional activator that is absolutely required for σ54–dependent gene regulation in a bacterial pathogen.
Results
Inactivation of bosR in Bb strain 297
Previously, we [34] and others [32] found that the mutation of bosR in Bb strain B31 abolished RpoS, OspC and DbpA expression. However, there are some notable discrepancies between these two studies. Hyde et al. [32] found that the mutant exhibited defects in growth in vitro and in the expression of NapA (or Dps, implicated in protecting DNA from damage during starvation or oxidative stress), whereas we [34] showed that the bosR mutant had normal growth and NapA expression comparable to WT Bb. It is also well-documented that transcriptional regulators and gene control mechanisms can differ widely among bacterial pathogens of the same species [36]–[38], and variations in strain-specific genetic contents, gene expression profiling, and pathogenicity have been observed, in particular, for different strains of Bb [39]–[40]. Thus, to more broadly investigate the role of BosR in Bb pathogenesis and gene regulation, we generated bosR mutants in another virulent WT Bb strain (strain 297) via homologous recombination. As in strain B31 [34], bosR in strain 297 was predicted to be cotranscribed and form an operon with bb0646 and bb0648 (Fig. S1A). To verify this, RT-PCR using specific primers and Bb cDNA was performed. As shown in Fig. S1B, amplicons spanning the junction of bb0646/bosR (lane 3), bosR/bb0648 (lane 5), or bb0646 - bb0648 (lane 6) were generated, indicating the operonic nature of bb0646, bosR, and bb0648. Of note, the sequences of this operon and its flanking genes (bb0645 and bb0649) are identical between both strains 297 and B31 (data not shown). Thus, the same strategies used in the creation of the bosR mutant in B31 [34] were employed to inactivate bosR in Bb 297. When the suicide vector pOY24 was transformed into Bb 297, two kanamycin-resistant bosR mutant clones (OY08/A11 and OY08/F4) were obtained. To cis-complement the bosR mutation, the suicide plasmid pOY83 [34] containing the bb0649-bb0648-bosR-PflgB-aadA cassette was introduced into the bosR mutants. As a result, two streptomycin-resistant clones (OY33/A6 and OY33/F7) were isolated. The inactivation and complementation of bosR in these strains were confirmed using PCR (Fig. S1C). Moreover, RT-PCR and immunoblot analyses revealed that BosR expression was detected in both WT and the complemented strains, but not in the mutants (Fig. S1D and E).
To ensure that all mutants and complements retained the plasmids cp26, lp25 and lp28-1 that are essential for Bb virulence [41]–[42], PCR-based plasmid profiling was performed. As shown in Fig. S2A, the WT and bosR mutant OY08/A11 contained the same plasmid profiles. In addition, OY08/F4 and the complemented strains (OY33/A6 and OY33/F7) contained the same plasmid profiles as that of the WT strain (data not shown). The bosR mutant exhibited spirochetal morphology and movement identical to that of WT under dark-field microscopy. No discernable growth defect was observed when bosR was inactivated, and the mutants displayed similar growth patterns to that of WT (Fig. S2B).
BosR is essential for Bb to establish mammalian infection
The role of BosR in Bb strain 297 mammalian infectivity was assessed using the murine needle-challenge model of Lyme borreliosis [43]–[44]. All mice inoculated with WT or the complemented strains at a 104 inoculum became infected, and motile spirochetes were isolated from all tissues from these mice (Table 1). In contrast, the bosR mutants were not recovered from any mice inoculated with either 104 or 107 bacteria. These data establish that previous findings implicating BosR in Bb's infectivity and virulence [32], [34] were not unique to strain B31, and thus BosR appears to be essential for conferring virulence to other pathogenic strains of Bb.

BosR controls the expression of rpoS, ospC and dbpA
To determine whether the loss of Bb strain 297 virulence in the bosR mutant correlated with a loss in the expression of rpoS, ospC, and dbpA, as has been noted for strain B31 [32], [34], we further assessed the effect of the bosR mutation on gene expression. WT 297, the bosR mutants, and the cis-complemented strains were cultured at 37°C in BSK-H medium at pH 6.8, conditions under which the RpoS regulon is highly induced [10], [14]–[15], [25]. Cells were harvested at late-log phase and subjected to immunoblot and RT-PCR analyses. As shown in Fig. 1, when bosR was inactivated, the expression of rpoS, ospC and dbpA, was essentially abolished at both the protein (Fig. 1A) and mRNA (Fig. 1B) levels. When the bosR mutation was cis-complemented, gene expression was fully restored.

To further investigate the influence of BosR on gene expression, a bosR expression construct (pOY112) was created using a newly-developed lac-based inducible expression system [45]. In pOY112, bosR transcription was placed under the direct control of the IPTG-inducible PpQE30 promoter. Bb 297 transformed with pOY112 were cultivated in the presence of various amounts of IPTG. Late log-phase cells were harvested and analyzed by immunoblot. Relative to protein levels in cells grown in medium without IPTG, 50 µM of IPTG induced the production of BosR, as well as increased the levels of RpoS, OspC, and DbpA (Fig. 1C), suggesting that BosR activates expression of these genes. Gene expression in WT 297 containing the empty vector (grown under various concentrations of IPTG) was not altered (data not shown). We also complemented the bosR mutation in trans using the IPTG-inducible bosR expression construct pOY112. As shown in Fig. 1D, when BosR was expressed from pOY112 by IPTG, expression of RpoS, OspC and DbpA was consequently restored.
These data, consistent with previous studies [32]–[34], further corroborate that BosR activates the expression of rpoS, ospC and dbpA. Of note, expression of rrp2, rpoN or bb0646 (the gene downstream from bosR in the bb0648-bosR-bb0646 operon) was not affected by the bosR mutation (Fig. 1B), implying that the phenotypes observed were not due to impairment in the expression of rrp2, rpoN, or bb0646. In addition, NapA levels were found to be similar in WT, the bosR mutant, and the complemented strains (Fig. 1A).
BosR controls ospC and dbpA expression through RpoS
Although our data suggested that the expression of both ospC and dbpA was activated by BosR, it remained unknown how BosR controlled the expression of these two lipoproteins. Given our finding that rpoS transcription was abolished in the bosR mutant, and the observations that expression of ospC and dbpA are directly regulated by RpoS through RpoS-specific promoters [46]–[48], we hypothesized that BosR likely regulated the expression of rpoS which, in turn, influences ospC and dbpA. To test this hypothesis, an IPTG-inducible rpoS expression construct was generated to render RpoS synthesis independent of BosR. This vector, pOY110, was then introduced into the bosR mutant OY08/A11. When RpoS was induced from pOY110 by IPTG, expression of OspC and DbpA was consequently rescued, although expression of BosR was absent (Fig. 2), suggesting that the controlled induction of RpoS could overcome the BosR deficiency and that BosR indirectly, rather than directly, controls OspC and DbpA expression.

Recombinant BosR purification and metal content analysis
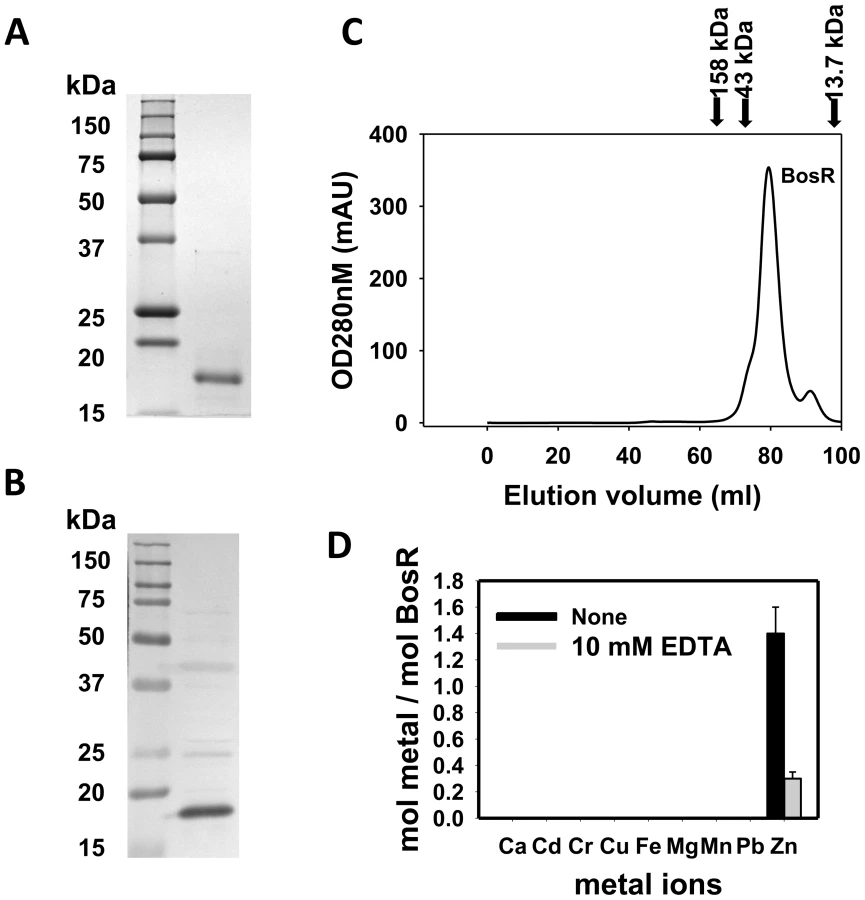
Recombinant BosR (rBosR) was hyper-expressed in E. coli and purified to apparent homogeneity. SDS-PAGE analysis indicated that BosR has an apparent molecular mass of ∼18.7 kDa (Fig. 3A), which is in agreement with the apparent mass of native BosR in Bb (Fig. 3B). Furthermore, when analyzed by size-exclusion chromatography, purified BosR eluted predominantly as a dimer with a molecular mass of ∼38 kDa (Fig. 3C). Although recombinant BosR has been obtained previously and Zn2+ was found to affect BosR's in vitro binding to DNA [28]–[29], it remained unclear whether BosR contains bound metal. Therefore, metal content analysis was carried out by inductively coupled plasma atomic emission spectrometry (ICP-AES) [27], [49]. rBosR did not contain detectable levels (<0.001 ppm) of metal ions such as Fe or Mn (Fig. 3D). Rather, it contained 1.4 mol of zinc per mol of protein. Moreover, in order to remove bound metal(s) from BosR, we also dialyzed the protein in the presence of 10 mM EDTA. However, 0.3 mol of zinc/mol of proteins remained in the demetallated BosR (Fig. 3D), suggesting that the recombinant protein bound zinc avidly. Of note, these properties are typical of the dimeric bacterial Fur protein [50] or the Bacillus subtilis H2O2 stress response regulator PerR [51].
BosR directly impacts rpoS expression
In silico analysis predicted that Bb BosR contains an N-terminal winged helix-turn-helix DNA binding domain and a C-terminal dimerization domain. Three-dimensional (3D) protein modeling using the Swiss-model program (http://swissmodel.expasy.org/) indicated that the structure of the DNA-binding domain of BosR is quite similar to the Vibrio cholerae Fur protein [52] and the B. subtilis PerR protein [51] (Fig. S3), suggesting that, consistent with previous reports [28]–[29], BosR may be a DNA-binding protein. Moreover, our aforementioned data revealed that BosR impacted rpoS expression at the transcription level. Thus, EMSAs were performed to examine potential interactions between BosR and the rpoS promoter. Consistent with previous studies [28]–[29], BosR bound to the promoter of Bb napA (from −336 to +48, relative to the ATG start codon) (Fig. 4A). However, BosR did not bind to the ospC or dbpBA promoters under our tested conditions (Fig. 4B and C), providing support that BosR likely does not impact ospC and dbpA directly. Although BosR did not bind to the probe ZM126 that encompasses the rpoS promoter from −67 to −8 (Fig. 4D), BosR, in a dose-dependent manner, bound to the rpoS promoter (PrpoS) encompassing 277 bp of the rpoS upstream region and 245 bp of the rpoS encoding region (Fig. 5A). Of note, binding of rBosR generated multiple shifted bands, suggesting the possible existence of multiple BosR binding sites (BSs) in the probe. As an initial approach to identify the BosR binding sequence, DNase I footprinting assays were performed. As shown in Fig. 5B, three BosR BSs were recognized in the PrpoS DNA. Specifically, BosR BS1, BS2, or BS3 spanned regions of −193 to −137, −120 to −46, or −29 to +43 (relative to the ATG start codon, where A is +1), respectively (Fig. 5C).


To corroborate the DNase I-footprinting data, EMSA employing synthesized double-stranded (ds) DNA oligonucleotides representing different BosR BSs were performed. As shown in Fig. 6B and C, BosR bound strongly to both ZM132 (representing BS1) and ZM127 (representing BS2). Moreover, the binding of BosR to labeled BS1 (ZM132) or BS2 (ZM127) was inhibited by the addition of a 200-fold excess of unlabeled DNA, but not inhibited by the addition of a 200-fold excess of non-specific competitor ZM126 DNA (Fig. 6D and E), suggesting that BosR binds to BS1 (ZM132) or BS2 (ZM127) specifically. Binding to both probes also was abrogated by the addition of α-BosR antibody, but not by the addition of control rat serum (Fig. 6D and E), indicating that the DNA shift was indeed caused by BosR. Of note, BosR, like Fur or PerR, putatively comprises an N-terminal DNA binding motif domain and a C-terminal domain involved in protein dimerization. Both domains are essential for Fur/PerR recognizing and binding to its target DNA as a homodimer [51]–[52]. Therefore, interaction with either domain of BosR by antibody may also interrupt protein binding to DNA and prevent DNA-BosR complex formation. Similarly, we also examined the binding of BosR to BS3 using EMSA. As shown in Fig. 7, although BosR did not bind to probe ZM160 that corresponds to the 5′ sequence of BS3, the protein bound to probe ZM161 (encompassing rpoS from +4 to +63).


BosR exhibits different affinities for the three binding sites
EMSAs employing target DNA sequences (representing the three BosR binding sites) exposed to increasing concentrations of rBosR were used as means of inferring BosR binding affinities for the three binding sites. As shown in Fig. 6 and 7, concentrations of 50, 20, or 200 nM of rBosR induced shifts by ZM132 (BS1), ZM127 (BS2), or ZM161 (BS3), respectively, suggesting that BosR has an affinity for these DNA targets in the order of BS2>BS1>BS3. In addition, when 200 nM of rBosR was used, only a slight proportion (<10%) of ZM161 (BS3) was shifted (Fig. 7), and probe ZM161 could not be saturated even by 10,000 nM of BosR (data not shown). To more precisely assess the affinity of BosR for BS1 and BS2, we measured the amount of bound DNA as a function of BosR concentration in EMSA assays (Fig. 8A). The dissociation binding constants (Kd) for BS1 (ZM132) or BS2 (ZM127) were 210.2 or 36.6 nM, respectively. The relative affinities of these two DNA elements for BosR were also assessed by competition EMSA analysis (Fig. 8, B and C). Binding of labeled BS1 or BS2 was not inhibited by the non-specific competitor ZM126 (NS), but was inhibited by unlabeled (cold competitor) BS1 or BS2, respectively. Moreover, binding of labeled BS1 was inhibited approximately 90% by the addition of 200-fold unlabeled BS1, but was completely competed out by 50-fold unlabeled BS2 (Fig. 8B). The addition of 200-fold of unlabeled BS1 competed out only 15% of BS2 binding (Fig. 8C). These data indicate that BosR has a higher affinity for BS2 than for BS1.

Identification of a novel direct repeat sequence critical for BosR binding
In silico analysis indicated that BosR contained two potential CX2C Zn2+ binding motifs in its C-terminus (located at residues of 114–117 and 153–156). These types of Zn2+ structural sites are crucial for Fur dimerization and binding DNA as a homodimer [30], [53]. Accordingly, we found that BosR bound Zn tightly (Fig. 3D). Moreover, consistent with previous observations [28], our purified BosR appeared to exist principally as a dimer in solution (Fig. 3). Therefore, BosR may bind to the rpoS promoter as a homodimer, suggesting that the BosR binding sequence(s) may be a direct repeat (DR) sequence. In agreement with this assumption, close inspection of the three BosR BSs revealed a DR sequence (TAAATTAAAT) (Fig. 5C). Of note, this sequence also consists of two contiguous pentamer direct repeats (TAAAT). More specifically, BS1 contains one perfect DR at its 5′ sequence and one imperfect DR at its 3′ sequence; BS2 contains one perfect DR and one imperfect DR in the sequence upstream of the -24/-12 RpoN binding site; and BS3 contains one imperfect DR sequence at its 3′. Of note, in both BS1 and BS2, the DR1 and DR2 are located on opposite DNA strands.
We hypothesized that if the DR is essential for binding to BosR, mutational changes in the sequence should abolish or inhibit BosR binding. Along these lines, we initially synthesized two DNA fragments, ZM155 and ZM156, representing the 5′ or 3′ of BS1, respectively (Fig. 9A). Both ZM155 and ZM156 contain one DR sequence. After labeling these DNA fragments with digoxigenin, each DNA fragment was mixed with BosR and EMSAs were performed. As shown in Fig. 9B, BosR bound to both fragments. However, when the DR was mutated, the binding of BosR to either DNA fragment was abolished (Fig. 9B), strongly supporting the notion that the DR sequence is critical for BosR binding.

Using this same strategy, we also examined the two DRs in BS2. As shown in Fig. 10, although BosR still bound to probe ZM149 having sequences downstream of the -24/-12 site scrambled, BosR binding to BS2 was abolished when sequences upstream of the -24/-12 site were scrambled (ZM147), suggesting that the functional BosR binding sites are located in the sequence upstream of the -24/-12 site. Because sequences flanking the binding motif often play important roles in protein-DNA interactions, we synthesized another dsDNA (ZM212) to represent the 5′ of BS2, allowing added flanking sequences to the predicted DR sequences. EMSAs indicated that BosR still bound to probe (ZM213) with the DR1 mutated (Fig. 10B). When a mutation was introduced into DR2 (ZM214), BosR binding was dramatically reduced (Fig. 10B). Moreover, when both DR sequences were mutated, protein binding was completely abolished (Fig. 10B). These data suggest that DR1 and DR2 in BS2 are required for BosR binding.

Genome-wide distribution of the DR sequence essential for BosR binding
To identify other Bb genes potentially regulated by BosR, Katona et al. [28] performed a BlastN analysis based on the Per box consensus sequence. However, the DR identified in our current study is disparate from the Per box. To uncover additional BosR-regulated genes, we searched the Bb genome by using the Regulatory Sequence Analysis Tools (http://rsat.ulb.ac.be/rsat), and queried for genes containing a perfect DR sequence in putative promoter regions. Gene promoter regions were defined as sequences from −400 to +50 bp (relative to the ATG start codon). The results are shown in Table 2. A total of 60 Bb genes were found to harbor one or multiple perfect DR sequence in their promoter regions. Thirty-one genes were located on the main chromosome, and 29 genes were on linear or circular plasmids. Of the 31 chromosomal genes, 16 genes encode proteins with assigned functions and15 genes encode hypothetical proteins. More importantly, 13 of these genes were found to be regulated by BosR in our recent microarray analysis [34], further supporting that the DR sequence is important for BosR binding.
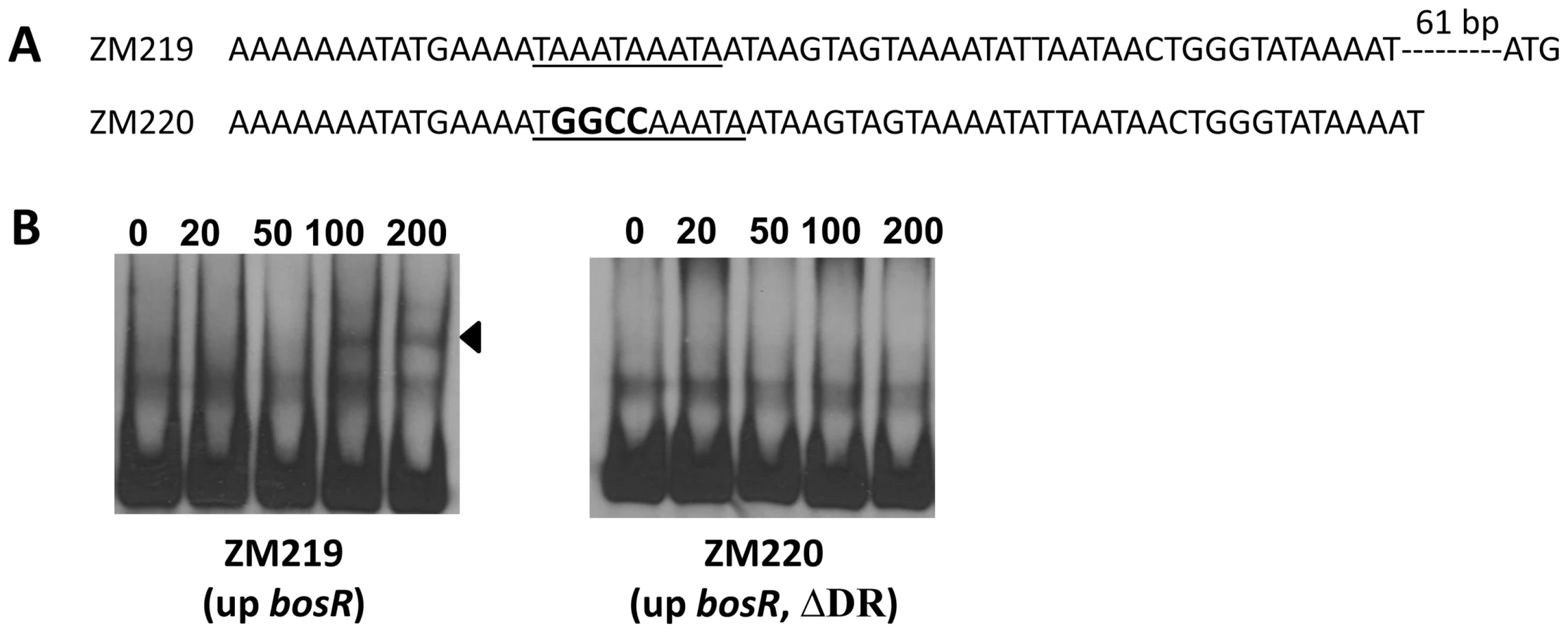
Previously, BosR was reported to bind to the Bb napA promoter (PnapA). Using footprinting assays, Boylan et al. [29] found that BosR protected a 50-bp region located at -222 to -173 (relative to the ATG start codon) in PnapA. Katona et al. [28], using EMSAs, reported that BosR also bound to a DNA fragment encompassing PnapA from −152 to +3. In addition, the latter researchers also reported that BosR bound to upstream regions of bosR. Interestingly, these two genes were not identified in our search (Table 2). However, when scrutinizing the upstream regions of bosR and napA, multiple imperfect DR sequences were detected (Fig. 11A, 12A). Therefore, EMSAs using synthesized dsDNA were employed to examine the roles of these imperfect DR sequences in BosR binding. Specifically, two dsDNA fragments, ZM215 and ZM217, were used to represent the BosR binding region in PnapA identified in previous studies [28]–[29]. As shown in Fig. 11B, BosR bound to both DNA fragments. When a mutation was introduced into the DR, binding of BosR to each probe was abolished. Similar data were also obtained for the probe ZM219 representing the bosR upstream region; BosR binding to the probe was abolished when the predicted DR was mutated (ZM220) (Fig. 12B). These data further substantiate the critical role of the DR in BosR binding.

BosR and RpoN bind to distinct sites in the rpoS promoter regions
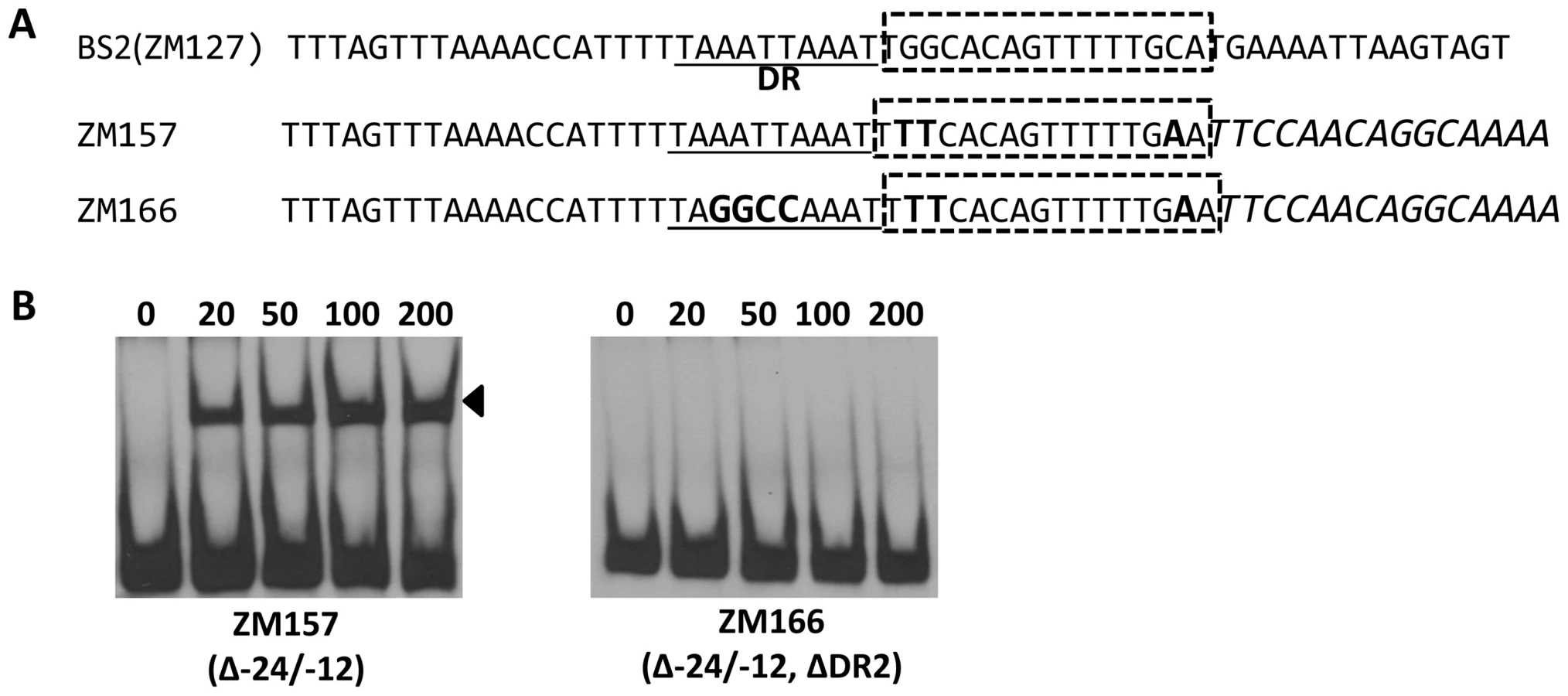
Bb RpoN activates rpoS directly through a canonical -24/-12 promoter, and mutation of G at -24 to T in the rpoS promoter resulted in a significant diminishment of in vitro RpoN binding and a dramatic decrease in rpoS expression [8], [11], [24]. Our analysis of BS2, the site exhibiting the highest affinity for BosR among the three BosR binding sites in the rpoS promoter, revealed that a perfect DR sequence is located just upstream and adjacent to the -24/-12 σ54 promoter (Fig. 13A). Given the importance of this locus for rpoS transcription, we further examined this site more closely using EMSA. As shown in Fig. 13B, when nucleotides in the -24/-12 site were mutated (ZM157, mutations of G-24T, G-25T, and C-12A), the binding of BosR was not altered. In contrast, when a mutation was introduced into the DR upstream of the -24/-12 site (ZM166, AATT replaced by GGCC), BosR binding was abolished (Fig. 13). These data strongly suggest that the key nucleotides for the binding of BosR and σ54 to the rpoS promoter are different.
Discussion
The essential role of the RpoN-RpoS pathway for virulence expression by the Lyme disease bacterium is now well documented [8]–[11], [13], [16]–[17], [24]–[25]. However, a number of the molecular details involved in the activation of this pathway have remained obscure. Whether Rrp2, as an EBP, is present as a dimer and assembles into a functional hexamer or heptamer when activated is unknown. It is also unclear how Rrp2 interacts with Eσ54 to modulate RpoN-dependent rpoS expression in the absence of demonstrable specific binding to the rpoS promoter or upstream region [8], [24], but there is precedence for this anomaly in another bacterial pathogen, Campylobacter jejuni (its EBP FlgR activates the σ54-dependent flagellar genes independent of DNA-binding) [54]. Moreover, the mammalian host factors that influence rpoS expression [12]–[13] have not been identified. Recently it was shown in Bb strain B31 that a mutation in bosR led to a loss of mouse infectivity, as well as a block in the expression of the virulence-associated factors OspC and DbpA [32], [34]–[35]. Given that both ospC and dbpA are under the control of RpoS [46]–[48], we proposed that BosR somehow was involved in the activation of the RpoN-RpoS regulatory pathway [34]. However, these data and the resultant hypothesis emanated from studies involving only one virulent strain (strain B31) of Bb. It has long been established that genetic regulators and control mechanisms can vary widely among strains of the same virulent species of pathogenic bacteria [36]–[38]. Hence, the question remained whether the novel RpoN-RpoS regulatory pathway and the important role of BosR were common to other virulent strains of Bb. The results of our study herein now confirm that the inactivation of bosR, which prevents activation of the RpoN-RpoS pathway by blocking the expression of rpoS, and, in turn, prevents expression of the virulence-associated genes ospC and dbpA, is not unique to a single virulent strain of Bb. BosR and its control over RpoN-RpoS activation thus appears to be an important global regulatory pathway essential for virulence expression by the Lyme disease spirochete.
Our findings also provide new insights into the function of BosR. BB0647 (BosR) was originally predicted to be a Fur homologue [7], [28]. Given the lack of precedence for interplay between a Fur homologue and an alternative sigma factor, such as RpoN or RpoS, in other bacteria, it was entirely unexpected that BosR would play a role in the induction of the RpoN-RpoS pathway [32], [34]. Furthermore, the nomenclature of “BosR”, as Borrelia oxidative stress regulator, was derived from the sequence similarity of BosR to the B. subtilis PerR, and an observation [29] that, in the heterologous E. coli, BosR activated the expression of Bb napA implicated in the oxidative stress response [55]. It was previously reported that NapA production was inhibited in a bosR mutant of Bb, and that the mutant displayed an in vitro growth defect [32]. Our former B31 mutant deficient in bosR exhibited no such growth defect, and the expression of napA was not significantly altered in the bosR mutant [34]; these same wild-type-like phenotypes were also observed in our current bosR mutant derived from strain 297 (Fig. 1B, Fig. S2B). Given these more recent findings, and the current paucity of compelling data that directly link BosR to an oxidative stress response in Bb, it is thus still premature to conclude that BosR is involved in modulating oxidative stress in Bb. Finally, despite the characterization of BosR as a Fur homologue, it also remains unclear whether BosR plays a role in regulating transition metal homeostasis in Bb.
Our EMSA data clearly indicated that BosR binds to the rpoS promoter, suggesting that BosR directly influences rpoS expression. Moreover, DNA footprinting assays revealed three BosR-protected regions in rpoS. The occupation of multiple, rather than one, binding sites might stabilize and secure BosR binding to the rpoS promoter region. BosR exhibited binding affinity for these three sites in the order of BS2>BS1>BS3. Among these three sites, BS2 was found juxtaposed to and partially overlapping with the -24/-12 RpoN binding site, whereas BS1 is located upstream of BS2. BS3 is located in sequences downstream of BS2, or more specifically, in the RpoS-encoding region. A previous study revealed that, when Bb was grown in vitro, a minimal PrpoS (starting from the -24/-12 RpoN binding site), which contains only one intact BosR BS (BS3), is able to express RpoS at the same level as PrpoS containing all three BosR BSs [24]. Furthermore, RpoS expressed from the minimal PrpoS restored mouse infectivity to the rpoS mutant [24]. These data imply that PrpoS with only BS3 is sufficient to drive rpoS transcription, suggesting that our data of BosR binding to BS3 may be physiologically relevant. However, BS1 and BS2 may be required to coordinate rpoS expression under different in vivo conditions of the two diverse niches of Bb's complex life cycle. Our unanticipated finding that BS3 for BosR is located within the RpoS-encoding region is rare but not unprecedented for transcriptional activators; binding sites for other bacterial regulatory proteins have been noted to occur in the coding regions of their target genes [56]–[57]. It is thus possible that BS3 located in the rpoS encoding region may somehow strengthen the binding of BosR to PrpoS, and then cooperate in opening the RpoN-RNAP closed complex. Or, BosR binding at BS3 might allow rpoS expression to be controlled more tightly, especially if rpoS transcription requires transient modulation.
A major finding of this study is the identification of a novel DNA binding sequence for BosR. As a Fur homologue, BosR dimers were reported [28]–[29] to bind in vitro to the Bb napA promoter, the upstream regions of bosR and bb0646, and DNA containing a Fur box (GATAATGATAATCATTATC) or Per box (TTATAAT-ATTATAA). In general, the Fur box is interpreted as two 9-bp inverted repeats (GATAATGAT), or two heptamer inverted repeats (TGATAAT), or three hexamer repeats (GATAAT), whereas the Per box is recognized as two inverted repeat (TTATAAT) [53], [58]. Despite the facts that the rpoS promoter contains neither a Fur (or Per) box, nor has significant similarity with the Bb napA promoter or the upstream sequences of bosR and bb0646, BosR binds to the rpoS promoter. More importantly, we identified a DR sequence (TAAATTAAAT) that is critical for BosR binding. This assertion is strongly supported by several lines of evidence. First, the DR sequence is present in all three BosR BSs. Second, the DR sequence was identified in the promoter regions of 13 genes already known to be influenced by BosR. Third, imperfect DR sequences are present in previously-established BosR-binding DNA fragments, such as PnapA and a bosR upstream region. Finally, mutations in the DR severely reduced or completely abolished DNA binding by BosR. Of note, the DR sequence is markedly different from the direct or inverted repeats present in Fur or Per boxes. Thus, BosR appears to be able to recognize different DNA sequences, including the Fur box consensus, the Per box consensus, and the rpoS promoter element (containing the DR sequence). Such promiscuous DNA recognition activity has been observed previously for the Bradyrhizobium japonicum Fur protein [59]; in addition to binding to the Fur box consensus, B. japonicum Fur also binds in vitro to the irr promoter (with similar affinity), but, the irr promoter does not contain a Fur box. Rather, it contains three essential direct repeat sequences of TGCATC that differ markedly from the direct repeats (GATAAT) or inverted repeats (GATAATGAT) in the Fur box [59]. The mechanistic details of this anomaly remain unknown. One possibility for BosR is that its binding properties in vitro may depend largely on DNA conformation. However, when analyzing the conformation of the dsDNA (including DNA containing mutated DRs) used in our EMSAs by PREDICTOR (http://www.farwer.staff.shef.ac.uk/PREDICTOR), which is a program calculating the three-dimensional atomic structure of dsDNA, no obvious differences in DNA conformation were revealed. Moreover, although BosR is predicted to share a similar three dimensional structure with Fur and the PerR protein (Fig. S3), BosR may harbor some subtle, unique structural feature(s) (undetected by protein modeling) that confer its DNA binding traits. Alternatively, under different in vivo conditions (tick vector or mammalian hosts), BosR may display alternative structural conformations that differentially regulate gene expression. As such, differing conformations may prompt BosR to bind to different DNA sequences (or with varying affinities). It is perhaps noteworthy that, after decades of intensive work, there is still much controversy over the molecular mechanisms and biochemistry of how Fur operates as a transcriptional repressor [60]. Thus, it is not surprising that there is yet much to learn regarding the molecular mechanism(s) that allow BosR to act as a regulator. Nonetheless, our finding that BosR, as a Fur or PerR homolog, recognizes disparate DNA sequences not only hints that the well-established model for Fur (or PerR)-DNA interaction may warrant further refinements, but also suggests that BosR employs mechanisms different from Fur or PerR to regulate gene expression. In addition, the recognition that BosR depends on a novel direct repeat for its binding to the rpoS promoter serves as a strong foundation for further mechanistic studies.
Several aspects of our study engender a number of provocative possibilities surrounding the function of BosR as a transcriptional enhancer for rpoS. First, our mutagenesis experiments revealed that different key nucleotides are required for BosR or RpoN binding to the rpoS promoter, implying that BosR and RpoN may bind to different faces of the DNA helix (comprising the rpoS promoter). Second, that BS2 is immediately adjacent to the -24/-12 RpoN binding site tempts speculation that BosR and RpoN (and possibly Rrp2) may interact with one another at the -24/-12 site to initiate rpoS transcription. However, results from E. coli-based two-hybrid assays thus far have failed to show interactions between BosR and RpoN (or Rrp2) (unpublished data). These results, however, are not unexpected, because there is no precedence for interactions between Fur/PerR proteins and RpoN or an EBP (e.g. Rrp2) in any bacterial system. Despite that, it is not impossible that BosR may transiently interact with RpoN or Rrp2 in vivo. It also remains possible that BosR may act as a critical molecule to recruit Rrp2 and/or RpoN to the rpoS promoter and the Eσ54-CC. The binding of BosR dimers to one face of the rpoS promoter at multiple sites may lead to DNA bending or other conformational changes that may facilitate the binding of Eσ54 to the -24/-12 site on the other strand of the promoter.
BosR, Fur, and PerR share structural similarity (Fig. S3) and all three are zinc - (or other metal-) containing regulatory proteins. However, there are other key features of BosR that markedly distinguish it from its putative Fur or PerR homologs. With Fur, metal-dependent DNA binding acts primarily as a repressor to avoid cellular iron toxicity [30], [53], [58], [60]. Its positive regulatory role is often indirect, via the Fur-regulated anti-sense regulatory small RNA, RhyB [30], [60]. In the case of PerR, transcriptional activation of its target genes (involved in protecting the bacterial cell against oxidative stress) occurs when the metal-bound PerR dissociates from the promoter [51], [60]. Of the three regulators, BosR is the only one that works in concert with an alternative sigma factor (RpoN) and an EBP (Rrp2), and the only one that appears to activate gene transcription directly by DNA binding. However, at this time we cannot rule out the less likely possibility that BosR may prevent the binding of a repressor that blocks rpoS transcription. Nonetheless, from the metal (zinc) content of rBosR, it is tempting to speculate that BosR's DNA-binding activity may be metal-dependent. Although zinc was found in rBosR, the question remains whether zinc is the physiologically relevant metal that confers normal activity to native BosR. It is thus not out of the realm of possibility that other metal(s) may be physiological relevant during Bb's existence in ticks or mammalian hosts. Further studies are warranted to investigate these possibilities, although many of the potential experimental approaches have substantial obstacles.
Our findings also reveal a new aspect of bacterial σ54-dependent gene activation and expands our understanding of transcriptional regulation by alternative sigma factors in general. Traditionally, for all known bacterial σ54-dependent genes, transcriptional activation requires only the cognate activator EBP (ATPase) [3]–[4]. For some σ54-dependent promoters, maximal induction relies on one or several auxiliary factors. For example, IHF, as a DNA-bending protein, can promote the interaction between Eσ54 and an EBP via DNA looping. In this case, however, IHF acts only as an architectural element to facilitate formation of the loop. It is the EBP, rather than the IHF protein, that modulates σ54-dependent gene expression [61]. In E. coli and Salmonella typhimurium, in the presence of arginine, the arginine repressor ArgR can induce the expression of the σ54-dependent astCADBE operon [62]–[63]. Nonetheless, ArgR plays only an accessory, rather than essential, role in the expression of astCADBE. In the absence of ArgR, genes are still expressed, although at more moderate levels. In response to flavonoids, the Azorhizobium caulinodans transcriptional activator NodD induces the transcription of NifA-RpoN-controlled NodA operon at an early stage [64]. In mature nitrogen-fixing nodules, the nodA gene is still transcribed in the nodD mutant in response to nitrogen-oxygen availability. In addition to the putative EBP (Rrp2), BosR is directly involved in the transcriptional activation of σ54-dependent rpoS in Bb. Unlike ArgR, NodD, and other accessory factors involved in maximizing the induction of σ54-dependent genes, BosR is essential for rpoS transcription in Bb. To our knowledge, this is the only demonstration, in any bacterial σ54-dependent transcriptional system, that transcription of a σ54-dependent gene requires an additional transcriptional activator. Bioinformatics indicate that homologues of BosR and σ54 are not only conserved in other Borrelia species (such as B. garinii and B. afzelii), but may be encoded in numerous other bacterial species including Bordetella, Burkholderia, Shewanella, Campylobacter, Clostridium, Bacillus, Listeria, and others. Given this wide distribution of BosR homologs, our study may have broader significance in understanding the regulatory control over RpoN - or RpoS-dependent genes in other pathogenic bacteria.
Materials and Methods
Strains and culture conditions
Strains and plasmid used in this study are listed in Table 3. Infectious Bb strain 297 [65] was used as the WT strain throughout this study. Bb was routinely cultured at 37°C and 5% CO2 in either BSK-II medium [66] or BSK-H medium (Sigma) supplemented with 6% rabbit serum (Pel-Freeze). When appropriate, supplements were added to media at following concentrations: kanamycin, 160 µg/ml; streptomycin, 150 µg/ml. Spirochetes were enumerated by dark-field microscopy.

Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animal procedures were approved by Institutional Animal Care and Use Committee at UT Southwestern Medical Center (Animal Protocol Number 0165-07-14-1).
Construction of bosR mutants and complement
The bosR mutant OY08 was created by allelic exchange in Bb 297 using a suicide vector pOY24 [34]. The mutation in bosR was cis-complemented by transforming a suicide vector, pOY83 [34], into the bosR mutant, generating OY33. Transformation of Bb was performed as previously described [67]. Plasmid contents of all Bb strains were determined by PCR using specific primers.
Generation of lac-inducible gene expression constructs
To artificially control BosR or RpoS expression in Bb, gene expression constructs were generated using a newly-developed lac-based inducible expression system [45]. First the PflaB-BblacI-PpQE30 cassette from pJSB252 [45] was ligated into pJD54 digested with BglII and BamHI, which generated pOY99.2. Then rpoS or bosR was amplified from Bb 297 and cloned into pOY99.2 digested with NdeI and BglII, generating pOY110 or pOY112, respectively. In these constructs, rpoS or bosR transcription was directly controlled by the IPTG-inducible T5 promoter in pQE30 (PpQE30).
Bb infection of mice
The infectivity of Bb clones was assessed using the murine needle-challenge model of Lyme borreliosis [43]–[44]. C3H/HeN mice (Charles River Laboratories) were infected via intradermal injection with various concentrations of Bb. At 4 weeks post inoculation, mice were sacrificed and skin, heart, and joint tissues were collected and cultured in BSK supplemented with 1× Borrelia antibiotic mixture (Sigma). The outgrowth of spirochetes in these cultures was assessed using darkfield microscopy.
Recombinant BosR expression and purification
Recombinant BosR was produced in E. coli using the Champion pET SUMO protein expression system (Invitrogen). Briefly, bosR was amplified using primers ZM69F and ZM69R, and ligated into pET SUMO vector through TA cloning such that the resultant construct pOY73 encoded a fusion protein with a His6 –SUMO tag at its N terminus. Constructs were confirmed using PCR amplification, restriction digestion, and sequence analysis. The resulting construct, pOY73, was then transformed into E. coli strain BL21-DE3. After induction with 1 mM IPTG (Sigma), recombinant His6 –SUMO-tagged BosR was purified using a Ni-NTA spin column under native conditions according to the manufacturer's instruction (Qiagen). The N-terminal His6 –SUMO tag was removed via cleavage with SUMO protease (Invitrogen) at 30°C for 4 h in the buffer A containing 20 mM Tris, 20 mM NaCl, 100 mM L-arginine, pH 7.5. The protease digestion mixture was concentrated and buffer exchanged with buffer A using an Amicon ultracentrifuge filter device (Millipore) with a 10,000 molecular weight exclusion limit. The concentrated protein was applied to a HiLoad 16/60 Superdex 200 prep grade column and purified on an Äkta fast performance liquid chromatography system (GE Healthcare) using buffer A. Subsequent to elution, peak fractions were analyzed by SDS-PAGE and Western Blot. At this stage, the protein was pure to apparent homogeneity and predominantly present as dimer. Fractions containing pure BosR with a homogeneity >95% were pooled. Protein concentration was determined using the BCA protein assay kit (Thermo Scientific). Rat polyclonal antibody against BosR, Ab-BosR, was generated as previously described [34].
Metal content analysis
Metal contents in protein or buffer solutions (as references) were measured using inductively coupled plasma atomic emission spectrometry (ICP-AES), by the Research Analytical Laboratory, University of Minnesota. Proteins were demetallated by dialyzing samples with 10 mM ETDA for 24 h, as described previously [49]. Three independent tests were performed, and average metal concentrations with standard deviations were presented.
SDS-PAGE and immunoblot analysis
SDS-PAGE and immunoblot analysis were carried out as previously described [34]. Briefly, purified protein samples or a volume of whole cell lysate equivalent to 4×107 spirochetes were loaded per lane on a 12.5% acrylamide gel. Resolved proteins were either stained with Coomassie brilliant blue or transferred to nitrocellulose membrane for immunoblot analysis. BosR was detected using the anti-BosR rat polyclonal antibody, Ab-BosR. Rrp2, RpoS, OspC, and DbpA were detected using anti-Rrp2 monoclonal antibody 5B8-100-A1, anti-RpoS monoclonal antibody 6A7-101, anti-OspC monoclonal antibody 1B2-105A, or anti-DbpA monoclonal antibody 6B3, respectively. To confirm equal loading of bacteria in each lane, immunoblotting for the flagellar core protein (FlaB) was performed using a chicken IgY anti-FlaB antibody. Immunoblots were developed colorimetrically using 4-chloro-1-napthol as the substrate or by chemiluminescence using ECL Plus Western Blotting Detection system (Amersham Biosciences).
Electrophoretic mobility shift assay (EMSA)
Primers used in EMSA are listed in Table S1. PCR-amplified or synthesized DNA probes were end-labeled with digoxigenin using recombinant terminal transferase (Roche Applied Science). The labeled probe (30 fmol) was mixed with various amounts of purified BosR in 20 µl of the gel shift binding buffer containing 20 mM Hepes (pH 7.5), 50 µg/ml poly[d(A-T)], 5% (w/v) glycerol, 1 mM DTT, 100 µg/ml BSA, 1 mM MgCl2, and 50 mM KCl. After being incubated at 37°C for 30 min, the samples were analyzed by 5% non-denaturing polyacrylamide gel electrophoresis at 80V for 1–3 h. Then DNA was transferred onto a positively charged Nylon membrane (Roche Applied Science, USA) by electroblotting. The digoxigenin-labeled probes were subsequently detected by an enzyme immunoassay using an antibody (anti-digoxigenin-AP, Fab fragments) and the chemiluminescent substrate disodium 3-(4-methoxyspiro {l,2-dioxetane-3,2′-(5′-chloro)tricyclo[3.3.1.13,7]decan}-4-yl) phenyl phosphate (CSPD) (Roche Applied Science, USA).
DNase I footprinting assays
For DNase I footprinting, DNA probe (PrpoS) containing the rpoS promoter was PCR-amplified using primers 88F and 88R. The resultant DNA was labeled with T4 polynucleotide kinase (NEB) and γ-32P-ATP (PerkinElmer). A 50-µl reaction containing the radiolabeled probe (300 fmol) and various amounts of BosR was incubated in the gel shift binding buffer at 37°C for 30 min. Then incubated DNA was digested with 0.01 unit of DNase I (Invitrogen) at room temperature for 2 min. The reaction was terminated by adding 100 µl of DNase I stop solution containing 200 mM Tris-HCl (pH 7.5), 50 mM EDTA, 2% SDS, 200 µg/ml of proteinase K, and 250 µg/ml of glycogen, followed by phenol/chloroform extraction. Then DNA was precipitated with 20 µl of 3M ammonium acetate, and 600 µl of 100% ethanol at −20°C overnight. The precipitated DNA was washed with 70% ethanol and air-dried. The pellet was resuspended in 10 µl of formamide dye (90% formamide, 1× TBE, and 0.02% bromophenol blue/xylene cyanol) and analyzed in a 6% polyacrylamide/7 M urea gel at 75 W for 2 h. The gel was transferred onto a chromatography paper (Fisher), dried, and exposed in a PhosphorImager screen. The signals were detected by Typhoon 9200 PhosphorImager (GE Healthcare).
RNA isolation and RT-PCR
Spirochetes were grown in BSK at 37°C under 5% CO2, and harvested when bacterial growth reached a density of 5×107 cells per ml. Total RNA was isolated using Trizol (Invitrogen) according to the instructions. After genomic DNA was digested using RNase-free DNase I (GenHunter Corporation), RNA was further purified using RNeasy Mini Kit (Qiagen). cDNA was generated from 1 µg of RNA using the SuperScript III Platinum Two-step qRT-PCR kit according to the manufacturer's protocol (Invitrogen).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. GruberTM
GrossCA
2003
Multiple sigma subunits and the partitioning of bacterial transcription space.
Annu Rev Microbiol
57
441
466
2. GhoshT
BoseD
ZhangX
2010
Mechanisms for activating bacterial RNA polymerase.
FEMS Microbiol Rev
34
611
627
3. RappasM
BoseD
ZhangX
2007
Bacterial enhancer-binding proteins: unlocking sigma54-dependent gene transcription.
Curr Opin Struct Biol
17
110
116
4. WigneshwerarajS
BoseD
BurrowsPC
JolyN
SchumacherJ
2008
Modus operandi of the bacterial RNA polymerase containing the sigma54 promoter-specificity factor.
Mol Microbiol
68
538
546
5. BurgdorferW
BarbourAG
HayesSF
BenachJL
GrunwaldtE
1982
Lyme disease-a tick-borne spirochetosis?
Science
216
1317
1319
6. SteereAC
GrodzickiRL
KornblattAN
CraftJE
BarbourAG
1983
The spirochetal etiology of Lyme disease.
N Engl J Med
308
733
740
7. FraserCM
CasjensS
HuangWM
SuttonGG
ClaytonR
1997
Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi.
Nature
390
580
586
8. BurtnickMN
DowneyJS
BrettPJ
BoylanJA
FryeJG
2007
Insights into the complex regulation of rpoS in Borrelia burgdorferi.
Mol Microbiol
65
277
293
9. HubnerA
YangX
NolenDM
PopovaTG
CabelloFC
2001
Expression of Borrelia burgdorferi OspC and DbpA is controlled by a RpoN-RpoS regulatory pathway.
Proc Natl Acad Sci U S A
98
12724
12729
10. OuyangZ
BlevinsJS
NorgardMV
2008
Transcriptional interplay among the regulators Rrp2, RpoN and RpoS in Borrelia burgdorferi.
Microbiology
154
2641
2658
11. SmithAH
BlevinsJS
BachlaniGN
YangXF
NorgardMV
2007
Evidence that RpoS (sigmaS) in Borrelia burgdorferi is controlled directly by RpoN (sigma54/sigmaN).
J Bacteriol
189
2139
2144
12. BrooksCS
HeftyPS
JolliffSE
AkinsDR
2003
Global analysis of Borrelia burgdorferi genes regulated by mammalian host-specific signals.
Infect Immun
71
3371
3383
13. CaimanoMJ
IyerR
EggersCH
GonzalezC
MortonEA
2007
Analysis of the RpoS regulon in Borrelia burgdorferi in response to mammalian host signals provides insight into RpoS function during the enzootic cycle.
Mol Microbiol
65
1193
1217
14. SchwanTG
PiesmanJ
GoldeWT
DolanMC
RosaPA
1995
Induction of an outer surface protein on Borrelia burgdorferi during tick feeding.
Proc Natl Acad Sci U S A
92
2909
2913
15. YangX
GoldbergMS
PopovaTG
SchoelerGB
WikelSK
2000
Interdependence of environmental factors influencing reciprocal patterns of gene expression in virulent Borrelia burgdorferi.
Mol Microbiol
37
1470
1479
16. FisherMA
GrimmD
HenionAK
EliasAF
StewartPE
2005
Borrelia burgdorferi sigma54 is required for mammalian infection and vector transmission but not for tick colonization.
Proc Natl Acad Sci U S A
102
5162
5167
17. YangXF
AlaniSM
NorgardMV
2003
The response regulator Rrp2 is essential for the expression of major membrane lipoproteins in Borrelia burgdorferi.
Proc Natl Acad Sci U S A
100
11001
11006
18. XuH
CaimanoMJ
LinT
HeM
RadolfJD
2010
Role of acetyl-phosphate in activation of the Rrp2-RpoN-RpoS pathway in Borrelia burgdorferi.
PLoS Pathog
6
9
e1001104
19. GrimmD
TillyK
ByramR
StewartPE
KrumJG
2004
Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals.
Proc Natl Acad Sci U S A
101
3142
3147
20. PalU
YangX
ChenM
BockenstedtLK
AndersonJF
2004
OspC facilitates Borrelia burgdorferi invasion of Ixodes scapularis salivary glands.
J Clin Invest
113
220
230
21. BlevinsJS
HagmanKE
NorgardMV
2008
Assessment of decorin-binding protein A to the infectivity of Borrelia burgdorferi in the murine models of needle and tick infection.
BMC Microbiol
8
82
22. ShiY
XuQ
McShanK
LiangFT
2008
Both decorin-binding proteins A and B are critical for overall virulence of Borrelia burgdorferi.
Infect Immun
76
1239
1246
23. WeeningEH
ParveenN
TrzeciakowskiJP
LeongJM
HookM
2008
Borrelia burgdorferi lacking DbpBA exhibits an early survival defect during experimental infection.
Infect Immun
76
5694
5705
24. BlevinsJS
XuH
HeM
NorgardMV
ReitzerL
2009
Rrp2, a sigma54-dependent transcriptional activator of Borrelia burgdorferi, activates rpoS in an enhancer-independent manner.
J Bacteriol
191
2902
2905
25. LybeckerMC
SamuelsDS
2007
Temperature-induced regulation of RpoS by a small RNA in Borrelia burgdorferi.
Mol Microbiol
64
1075
1089
26. LybeckerMC
AbelCA
FeigAL
SamuelsDS
2010
Identification and function of the RNA chaperone Hfq in the Lyme disease spirochete Borrelia burgdorferi.
Mol Microbiol
78
622
635
27. OuyangZ
HeM
OmanT
YangXF
NorgardMV
2009
A manganese transporter, BB0219 (BmtA), is required for virulence by the Lyme disease spirochete, Borrelia burgdorferi.
Proc Natl Acad Sci U S A
106
3449
3454
28. KatonaLI
TokarzR
KuhlowCJ
BenachJ
BenachJL
2004
The fur homologue in Borrelia burgdorferi.
J Bacteriol
186
6443
6456
29. BoylanJA
PoseyJE
GherardiniFC
2003
Borrelia oxidative stress response regulator, BosR: a distinctive Zn-dependent transcriptional activator.
Proc Natl Acad Sci U S A
100
11684
11689
30. CarpenterBM
WhitmireJM
MerrellDS
2009
This is not your mother's repressor: the complex role of fur in pathogenesis.
Infect Immun
77
2590
2601
31. PoseyJE
GherardiniFC
2000
Lack of a role for iron in the Lyme disease pathogen.
Science
288
1651
1653
32. HydeJA
ShawDK
SmithRIii
TrzeciakowskiJP
SkareJT
2009
The BosR regulatory protein of Borrelia burgdorferi interfaces with the RpoS regulatory pathway and modulates both the oxidative stress response and pathogenic properties of the Lyme disease spirochete.
Mol Microbiol
74
1344
1355
33. HydeJA
ShawDK
SmithR3rd
TrzeciakowskiJP
SkareJT
2010
Characterization of a conditional bosR mutant in Borrelia burgdorferi.
Infect Immun
78
265
274
34. OuyangZ
KumarM
KariuT
HaqS
GoldbergM
2009
BosR (BB0647) governs virulence expression in Borrelia burgdorferi.
Mol Microbiol
74
1331
1343
35. SamuelsDS
RadolfJD
2009
Who is the BosR around here anyway?
Mol Microbiol
74
1295
1299
36. DmitrievAV
McDowellEJ
ChausseeMS
2008
Inter - and intraserotypic variation in the Streptococcus pyogenes Rgg regulon.
FEMS Microbiol Lett
284
43
51
37. LuoF
LizanoS
BessenDE
2008
Heterogeneity in the polarity of Nra regulatory effects on streptococcal pilus gene transcription and virulence.
Infect Immun
76
2490
2497
38. RibardoDA
McIverKS
2006
Defining the Mga regulon: Comparative transcriptome analysis reveals both direct and indirect regulation by Mga in the group A streptococcus.
Mol Microbiol
62
491
508
39. OjaimiC
MulayV
LiverisD
IyerR
SchwartzI
2005
Comparative transcriptional profiling of Borrelia burgdorferi clinical isolates differing in capacities for hematogenous dissemination.
Infect Immun
73
6791
6802
40. WangG
van DamAP
SchwartzI
DankertJ
1999
Molecular typing of Borrelia burgdorferi sensu lato: taxonomic, epidemiological, and clinical implications.
Clin Microbiol Rev
12
633
653
41. Labandeira-ReyM
SkareJT
2001
Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1.
Infect Immun
69
446
455
42. PurserJE
NorrisSJ
2000
Correlation between plasmid content and infectivity in Borrelia burgdorferi.
Proc Natl Acad Sci U S A
97
13865
13870
43. AkinsDR
BourellKW
CaimanoMJ
NorgardMV
RadolfJD
1998
A new animal model for studying Lyme disease spirochetes in a mammalian host-adapted state.
J Clin Invest
101
2240
2250
44. BartholdSW
de SouzaMS
JanotkaJL
SmithAL
PersingDH
1993
Chronic Lyme borreliosis in the laboratory mouse.
Am J Pathol
143
959
971
45. BlevinsJS
RevelAT
SmithAH
BachlaniGN
NorgardMV
2007
Adaptation of a luciferase gene reporter and lac expression system to Borrelia burgdorferi.
Appl Environ Microbiol
73
1501
1513
46. EggersCH
CaimanoMJ
RadolfJD
2004
Analysis of promoter elements involved in the transcriptional initiation of RpoS-dependent Borrelia burgdorferi genes.
J Bacteriol
186
7390
7402
47. OuyangZ
HaqS
NorgardMV
2010
Analysis of the dbpBA upstream regulatory region controlled by RpoS in Borrelia burgdorferi.
J Bacteriol
192
1965
1974
48. YangXF
LybeckerMC
PalU
AlaniSM
BlevinsJ
2005
Analysis of the ospC regulatory element controlled by the RpoN-RpoS regulatory pathway in Borrelia burgdorferi.
J Bacteriol
187
4822
4829
49. DesrosiersDC
SunYC
ZaidiAA
EggersCH
CoxDL
2007
The general transition metal (Tro) and Zn2+ (Znu) transporters in Treponema pallidum: analysis of metal specificities and expression profiles.
Mol Microbiol
65
137
152
50. AlthausEW
OuttenCE
OlsonKE
CaoH
O'HalloranTV
1999
The ferric uptake regulation (Fur) repressor is a zinc metalloprotein.
Biochemistry
38
6559
6569
51. TraoreDA
El GhazouaniA
IlangoS
DupuyJ
JacquametL
2006
Crystal structure of the apo-PerR-Zn protein from Bacillus subtilis.
Mol Microbiol
61
1211
1219
52. SheikhMA
TaylorGL
2009
Crystal structure of the Vibrio cholerae ferric uptake regulator (Fur) reveals insights into metal co-ordination.
Mol Microbiol
72
1208
1220
53. EscolarL
Perez-MartinJ
de LorenzoV
1999
Opening the iron box: transcriptional metalloregulation by the Fur protein.
J Bacteriol
181
6223
6229
54. JoslinSN
HendrixsonDR
2008
Analysis of the Campylobacter jejuni FlgR response regulator suggests integration of diverse mechanisms to activate an NtrC-like protein.
J Bacteriol
190
2422
2433
55. LiX
PalU
RamamoorthiN
LiuX
DesrosiersDC
2007
The Lyme disease agent Borrelia burgdorferi requires BB0690, a Dps homologue, to persist within ticks.
Mol Microbiol
63
694
710
56. MitraR
DasHK
DixitA
2005
Identification of a positive transcription regulatory element within the coding region of the nifLA operon in Azotobacter vinelandii.
Appl Environ Microbiol
71
3716
3724
57. ShimadaT
IshihamaA
BusbySJ
GraingerDC
2008
The Escherichia coli RutR transcription factor binds at targets within genes as well as intergenic regions.
Nucleic Acids Res
36
3950
3955
58. BaichooN
HelmannJD
2002
Recognition of DNA by Fur: a reinterpretation of the Fur box consensus sequence.
J Bacteriol
184
5826
5832
59. FriedmanYE
O'BrianMR
2003
A novel DNA-binding site for the ferric uptake regulator (Fur) protein from Bradyrhizobium japonicum.
J Biol Chem
278
38395
38401
60. LeeJW
HelmannJD
2007
Functional specialization within the Fur family of metalloregulators.
Biometals
20
485
499
61. GoosenN
van de PutteP
1995
The regulation of transcription initiation by integration host factor.
Mol Microbiol
16
1
7
62. KiupakisAK
ReitzerL
2002
ArgR-independent induction and ArgR-dependent superinduction of the astCADBE operon in Escherichia coli.
J Bacteriol
184
2940
2950
63. LuCD
AbdelalAT
1999
Role of ArgR in activation of the ast operon, encoding enzymes of the arginine succinyltransferase pathway in Salmonella typhimurium.
J Bacteriol
181
1934
1938
64. GaoM
D'HaezeW
De RyckeR
HolstersM
2001
Dual control of the nodA operon of Azorhizobium caulinodans ORS571 by a nod box and a NifA-sigma54-type promoter.
Mol Genet Genomics
265
1050
1059
65. HughesCA
KodnerCB
JohnsonRC
1992
DNA analysis of Borrelia burgdorferi NCH-1, the first northcentral U.S. human Lyme disease isolate.
J Clin Microbiol
30
698
703
66. PollackRJ
TelfordSR3rd
SpielmanA
1993
Standardization of medium for culturing Lyme disease spirochetes.
J Clin Microbiol
31
1251
1255
67. SamuelsDS
1995
Electrotransformation of the spirochete Borrelia burgdorferi.
Methods Mol Biol
47
253
259
68. RevelAT
BlevinsJS
AlmazanC
NeilL
KocanKM
2005
bptA (bbe16) is essential for the persistence of the Lyme disease spirochete, Borrelia burgdorferi, in its natural tick vector.
Proc Natl Acad Sci U S A
102
6972
6977
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 2
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Genetic Mapping Identifies Novel Highly Protective Antigens for an Apicomplexan Parasite
- Type I Interferon Signaling Regulates Ly6C Monocytes and Neutrophils during Acute Viral Pneumonia in Mice
- Infections in Cells: Transcriptomic Characterization of a Novel Host-Symbiont Interaction
- The ESCRT-0 Component HRS is Required for HIV-1 Vpu-Mediated BST-2/Tetherin Down-Regulation