
Structure of the Membrane Anchor of Pestivirus Glycoprotein E, a Long Tilted Amphipathic Helix
Erns is an essential virion glycoprotein with RNase activity that suppresses host cellular innate immune responses upon being partially secreted from the infected cells. Its unusual C-terminus plays multiple roles, as the amphiphilic helix acts as a membrane anchor, as a signal peptidase cleavage site, and as a retention/secretion signal. We analyzed the structure and membrane binding properties of this sequence to gain a better understanding of the underlying mechanisms. CD spectroscopy in different setups, as well as Monte Carlo and molecular dynamics simulations confirmed the helical folding and showed that the helix is accommodated in the amphiphilic region of the lipid bilayer with a slight tilt rather than lying parallel to the surface. This model was confirmed by NMR analyses that also identified a central stretch of 15 residues within the helix that is fully shielded from the aqueous layer, which is C-terminally followed by a putative hairpin structure. These findings explain the strong membrane binding of the protein and provide clues to establishing the Erns membrane contact, processing and secretion.
Published in the journal:
. PLoS Pathog 10(2): e32767. doi:10.1371/journal.ppat.1003973
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003973
Summary
Erns is an essential virion glycoprotein with RNase activity that suppresses host cellular innate immune responses upon being partially secreted from the infected cells. Its unusual C-terminus plays multiple roles, as the amphiphilic helix acts as a membrane anchor, as a signal peptidase cleavage site, and as a retention/secretion signal. We analyzed the structure and membrane binding properties of this sequence to gain a better understanding of the underlying mechanisms. CD spectroscopy in different setups, as well as Monte Carlo and molecular dynamics simulations confirmed the helical folding and showed that the helix is accommodated in the amphiphilic region of the lipid bilayer with a slight tilt rather than lying parallel to the surface. This model was confirmed by NMR analyses that also identified a central stretch of 15 residues within the helix that is fully shielded from the aqueous layer, which is C-terminally followed by a putative hairpin structure. These findings explain the strong membrane binding of the protein and provide clues to establishing the Erns membrane contact, processing and secretion.
Introduction
The genus Pestivirus belongs to the family Flaviviridae, together with the genera Hepacivirus, Flavivirus and Pegivirus. It represents a group of economically important animal viruses like classical swine fever virus (CSFV) and bovine viral diarrhea virus (BVDV) [1]. The economical damage caused by these viruses is not only due to the acute infection of the animals, but stems also from their ability to cause persistent infection of the fetus after infection of a pregnant animal [2], [3]. If an infection of the fetus by BVDV is established, a persistently infected calf is born. This calf often shows no signs of disease, but sheds huge amounts of infectious virus particles throughout its whole life, which leads to an efficient spreading of the virus.
The genome organisation and basic molecular features of pestiviruses are more similar to the hepacivirus HCV (human hepatitis C virus) than to the other members of the Flaviviridae. The positive sense single stranded RNA genome consists of about 12,300 nucleotides and contains one single open reading frame that codes for a single polyprotein precursor of ∼4000 amino acids [1]. This precursor is co - and posttranslationally processed by cellular and viral proteases to release the viral proteins. Compared to the HCV RNA, the pestivirus genome codes for two additional proteins: the non-structural protein Npro, and the structural protein Erns [1]. Both proteins interfere with the immune response of the infected animal and are important for establishing a persistent infection [4]. The two proteins exert two different functions. Npro is an autoprotease and leads to the degradation of IRF3 (interferon regulatory factor 3) in the infected cell via the proteasome [5], [6], [7], [8], [9], [10], and it also interferes with IRF7 dependent pathways [11]. This results in the deactivation of the innate immune response of the infected cell. In contrast, Erns is a viral glycoprotein that forms disulfide-linked homodimers and can be found together with the glycoproteins E1 and E2 on the surface of the enveloped virus particle [3], [12], [13]. Erns consists of 227 amino acids, has a molecular weight of 42–48 kDa, being heavily glycosylated except for its C-terminal region [3], [14], [15]. It is not only involved in the formation of infectious virus particles, but it is also secreted from the infected cells, and up to 50 ng/ml of the protein can be detected in the blood of infected animals [16]. In addition, Erns has an intrinsic RNase activity, which is very unusual for an RNA virus protein [17], [18], [19]. The structure of the RNase domain of Erns was recently determined, with the finding that it has a T2-RNase like fold [20] that confirmed data from sequence analysis studies [18]. T2-RNase represents a very old and unique RNase family whose members are broadly distributed in nature, but the functions of these RNases are unknown. The enzymatic activity of Erns is necessary for its activity as a virulence factor. It could be shown that the deactivation of the RNase activity by deletion of one amino acid in the active site of the protein caused attenuation of the virus in its natural host [21], [22].
Interestingly, attenuation was also observed in an RNase positive virus, in which Erns dimerization was prevented by mutation of the Cys residue that forms the intermolecular disulfide bond in the wt protein [23]. Moreover, an abrogation of the Erns RNase activity together with a deletion of the Npro coding sequence prevented the establishment of a persistent BVDV infection [4]. We and others postulated that the secreted version of Erns - and not the protein bound to the virus particle - represents the key player that interferes with the immune system. The secretion of Erns is controlled by the C-terminal end of the protein. In previous studies we have determined several parameters in the C-terminal region of Erns that are necessary for the retention/secretion of the protein [24], [25], [26]. The free C-terminus of Erns is formed upon cleavage of the Erns/E1 precursor protein by the cellular signal peptidase [27]. This cleavage site is a very unusual substrate for the signal peptidase, because the characteristic signal activating this peptidase is normally composed of a transmembrane helix followed by a so-called von Heijne sequence [28], [29]. The cleavage site between Erns and E1 contains such a von Heijne sequence, but the Erns C-terminus lacks a transmembrane helix and contains an amphipathic helix instead. Nevertheless, the C-terminus of Erns obviously has to fold in a certain conformation that is accepted as a substrate by the signal peptidase.
The C-terminus of Erns governs not only protein cleavage and secretion, but it is also important for the membrane binding of the protein. Sequence analysis predicts a helical fold for the C-terminus, which would bestow it with a marked amphipathic character [24], [25]. Fig. 1 shows the 2D flat projection of the 3D structure of the Erns anchor (Lys167 – Ala227) assuming a continuous α-helical conformation. The hydrophilic and hydrophobic faces are maintained throughout the entire length of the helix, which implies that it could bind flat onto the membrane surface. This amphipathic structure would explain the membrane anchoring, but not the action of the signal peptidase, nor the regulation of secretion. As an alternative arrangement, it has been recently suggested that the Erns membrane anchor might fold as a helical hairpin by forming a long ladder of salt bridges between its N-terminal and C-terminal helical segments, a so-called electrostatic ‘charge zipper’ [30]. The resulting amphiphilic hairpin would in principle have an appropriate length to span the lipid bilayer in a transmembrane alignment and could thus serve as a substrate for the signal peptidase.

To better understand the role of Erns and its mechanism of membrane anchoring and secretion, we have recently obtained some initial data on short peptide fragments from this region of the protein when bound to lipid bilayers [26], but the non-continuous nature of these sequences precluded any firm interpretation. Here, we show that the complete C-terminal anchor of Erns indeed adopts a continuous α-helical fold when bound to the membrane. In contrast to the simplistic pictures of a surface-bound helix or a transmembrane hairpin, however, our results show that the Erns C-terminus is slightly tilted with regard to the membrane surface, and a substantial stretch of residues is shielded from the aqueous phase by the hydrophobic environment, while the rest is located at the water/membrane interphase. This refined model represents the first example of a viral structural protein that is bound to a membrane via an amphipathic helix.
Results
Secondary structure of the Erns anchor
In a previous analysis we had examined three overlapping peptide fragments corresponding to the Erns anchor sequences of CSFV strain Alfort/Tübingen [26] and BVDV strain CP7 (data not shown). Their CD analysis revealed a strong tendency for the middle and the C-terminal part of the anchor sequence, represented by the two corresponding peptides, to fold as a helix. To verify these results for the entire Erns anchor (Lys167 – Ala227), we determined the secondary structure of this 61-residue domain in different environments by CD, as shown in Fig. 2. We first used a solution of 50% TFE in phosphate buffer (PB) pH 6.5 (Fig. 2 A, dashed line), which promotes intramolecular hydrogen bonds. This CD spectrum of the Erns anchor showed an overall helical fold, and the secondary structure deconvolution revealed a helix content of 80% for these data (Fig. 2 A, bar diagram). Thus, almost the complete Erns C-terminus is in principle able to adopt a helical conformation. To test whether this secondary structure is also present in pure phosphate buffer (PB), we measured the Erns anchor in PB pH 6.5 (Fig. 2 A, solid line) and pH 3 (Fig. 2 A, dotted line). However, the Erns anchor was only partially soluble at pH 6.5, leading to protein aggregation and turbidity in the aqueous sample. Therefore, the CD lineshape shows spectral artefacts caused by absorption flattening and differential scattering at wavelengths <215 nm, and the secondary structure analysis yielded only a very low degree of helix content. At pH 3, on the other hand, the protein is completely soluble in PB, and the secondary structure calculation revealed a helix content of about 50%.

To analyse the secondary structure of the Erns anchor in a membrane-like environment, we first used detergent micelles in low salt buffer. As the Erns anchor is positively charged we compared micelles of zwitterionic DPC and negatively charged SDS. Both systems supported a strong helical folding, and the secondary structure deconvolution revealed a helical fraction of over 60% (Fig. 2 A, bar diagram). We then used zwitterionic DMPC lipid vesicles, and a 1∶1 mixture of DMPC with anionic DMPG. Both systems induced a high degree of helical folding (∼70–80%) (Fig. 2 A, bar diagram), similar to 50% TFE. These results indicate that the Erns C-terminus has a helix content of up to 80% in a membrane (-mimicking) environment, and may therefore be present as a continuous amphipathic helix, as implicated in Fig. 1.
Orientation of the Erns amphipathic helix relative to the membrane surface
To examine the orientation of the helical sequence relative to the membrane surface we used oriented CD (OCD). In macroscopically oriented membrane samples it is possible to estimate the tilt angle of a membrane bound helix from the intensity of the negative band at 208 nm in the OCD spectrum, which is polarized parallel to the helix axis [31], [32], [33]. If the helix adopts an alignment parallel to the membrane surface, as expected for Erns, the minimum at 208 nm has a stronger intensity than the minimum around 223 nm. In contrast, a transmembrane helix that lies perpendicularly to the membrane surface shows no negative band at 208 nm, or even some positive ellipticity (Supporting Information Fig. S1). The OCD spectra of the Erns anchor, recorded in oriented DMPC or DMPC/DMPG (1∶1) (Fig. 2 B), show a pronounced minimum at 208 nm with nearly the same intensity as the one at 223 nm. A transmembrane alignment of the Erns anchor, as had been speculated for the Erns/E1 precursor [30], can thus be excluded for the mature processed Erns under these conditions. However, because the intensity of the 208 nm minimum is less than the intensity of the 223 nm band, the helix does not seem to be aligned completely parallel to the membrane surface. Without taking any minor secondary structure elements into account (e.g. disordered regions, which have a minimum at 198 nm), the Erns anchor appears to be slightly tilted within the membrane.
Orientation of the Erns anchor is independent of concentration
Many amphipathic helices, especially antimicrobial peptides, are known to interact with each other in a concentration dependent manner, which often results in changes in their membrane alignment. At low concentrations these peptides exhibit a mostly parallel orientation with regard to the membrane surface, but at higher concentrations the self-assembly of these molecules leads to a re-alignment and the formation of a transmembrane pore [33], [34], [35], [36], [37], [38]. In the case of Erns, the possibility of self-assembly via an intermolecular charge zipper had been suggested [30], or helix-helix interactions via a GxxxG motif might be conceivable. To test for a putative concentration-dependent change in the secondary structure (e.g. aggregation or oligomerization) or in the orientation of the helical segment of Erns, we compared several samples with different lipid/protein ratios. The CD spectra of all four tested concentrations (P/L ratios) in the DMPC/DMPG 1∶1 lipid mixture were essentially identical (Fig. 2C). Neither did the OCD spectra at the same ratios of 1∶20, 1∶50 and 1∶100, as displayed in Fig. 2D, show any change in the alignment of the helical segment. (The OCD sample at 1∶200 did not yield a reliable spectrum due to technical problems resulting from the very low protein concentration.) In summary, these CD and OCD data demonstrate that the Erns anchor does not show a concentration-dependent change in its global secondary structure nor in its orientation in the membrane. Most importantly, the data recorded here give no indication at all of a transmembrane alignment at any concentration.
Secondary structure of the far C-terminal region of Erns
The observed peripheral location of the C-terminal part of the Erns membrane anchor is highly intriguing, because this architecture represents a so far unknown type of substrate for the cellular signal peptidase, which usually requires a transmembrane helix upstream of the cleavage site. To investigate the structure of the C-terminal fragment in more detail, we expressed a truncated construct named ErnsΔN, which represents the far C-terminal 34 amino acids of the anchor sequence (Arg194 – Ala227). To analyse the secondary structure of this C-terminal Erns anchor fragment (Fig. 2 E), we used 50% TFE (dashed line), DPC micelles (straight line), and DMPC vesicles (not shown). Unfortunately, the CD spectrum of ErnsΔN in DMPC vesicles showed scattering artefacts which lead to an error-prone CD spectrum and prevented an exact analysis. To allow comparison with the subsequent NMR analysis, we examined the C-terminal region also in small lipid bicelles with DHPC/DMPC (4∶1) (dotted line). The corresponding CD spectra of ErnsΔN showed a high percentage of helical folding (∼70–90%) in all three systems (Fig. 2 E, bar diagram). This means that the Erns anchor can fold as a long amphipathic helix not only in detergent micelles and lipid vesicle suspensions, but also in bicelles.
The OCD spectrum of the C-terminal fragment in Fig. 2F shows a less intensive band at 208 nm than the complete Erns anchor at a P/L ratio of 1∶50. This means that either the N-terminal region of the Erns anchor is less tilted in the membrane than the C-terminal region, or the N-terminal elongation pulls the C-terminal region into a more parallel orientation. Importantly, the band at 208 nm still has a distinct negative signal amplitude, which excludes the possibility that this region of the Erns anchor could be inserted into the membrane in a transmembrane alignment.
Detailed NMR structure analysis of the far C-terminal region of the Erns anchor
Further information on the orientation of the Erns anchor in the membrane, the positions of individual residues, and the water accessibility of individual NH groups was obtained from liquid-state NMR spectroscopy. Although the full-length Erns anchor yielded good quality 1H15N-HSQC spectra in DHPC/DMPC bicelles, as well as in DPC and SDS micelles, the corresponding 3D-15N-HSQC-NOESY and TOCSY spectra suffered from line broadening that impeded sequential assignment (Supporting Information Figure S2). The stretch Trp203-Gly212 could be tentatively assigned based on characteristic proton shifts, but the assignment could only be safely confirmed later by comparison with the spectra of ErnsΔN. Therefore, the detailed NMR analysis was done for the truncated ErnsΔN corresponding to the 34 C-terminal residues of Erns. Backbone assignment (1H, 15N, 13Cα, 13Cβ) was achieved for all residues, except for the first two in DHPC/DMPC (4∶1) bicelles.
We sought to determine whether parts of ErnsΔN are protected from the solvent, by titration with the paramagnetic agent Gd-DOTA and by dissolution of a lyophilized sample of ErnsΔN/DHPC/DMPC in D2O. Supporting Information Figure S3 shows that residue protection factors of ErnsΔN with Gd DOTA at 0.5 mM are uniform along the entire sequence. D2O exchange revealed no stably protected residues either (data not shown), given that already a single exchange event leads to a disappearance of the signal. Similarly, titration with paramagnetic agents shows a protection only when the residues are deeply inserted in the hydrophobic interior of the bicelle [39]. We therefore determined also the water accessibility of the NH groups with the more sensitive CLEANEX-PM pulse sequence [40], which measures proton exchange rates. The CLEANEX pulse sequence applies a water-selective excitation pulse prior to a chemical exchange sequence in which magnetization transfer from water protons to 15N-bound exchangeable protons occurs, followed by a 1H15N-HSQC-type experiment. The chemical exchange efficiency depends on the accessibility of the 15N-bound proton, and thus reports on its location in the bicelle or its stable participation in a hydrogen bond. Here, a single encounter with a water molecule may already lead to a measurable magnetization, meaning that for partially solvent-accessible amide protons this method is more sensitive than the other two, because the protein exchange time is limited to 100 ms.
The assigned 1H15N-HSQC of ErnsΔN is shown in Fig. 3A. Judging from the nuclear Overhauser effect (NOE) and chemical shift anisotropy (CSI) patterns (Fig. 4A, middle part), the helix extends from Leu215 or Glu216, corresponding to 50% helical content. These findings support the CD data above, that showed a high degree of helical folding in the same membrane-mimetic environment (data above). The remaining residues in the C-terminus show some HN-HN contacts as well as NOEs between Trp222, Phe223 and Tyr226 (Fig. 4B), which suggests a loop-like structure, but the quality of the spectra did not yield enough NOEs for a full 3D structure determination. Nonetheless, the HN-HN contacts together with the loop-like contacts in the C-terminus (Fig. 4A, upper part) and the water accessibility measurements argue in favour of a dynamic loop-like conformation of the far C-terminus rather than a firmly folded α-helical structure.


A comparison of the 15N-HSQC and the CLEANEX spectra (Fig. 3B) revealed that only 13 amide protons are engaged in exchange (i.e. they give cross-peaks in the assigned CLEANEX spectrum). We may thus conclude that the remaining 20 amide groups are protected from exchange and are therefore assume a stable secondary structure within a more hydrophobic environment characterized by a low dielectric constant. A comparison with the water-HN NOEs in a 1H15N-NOESY experiment confirmed our analysis. To obtain the water accessibility of each NH group, we calculated the normalized proton exchange rate by dividing the intensity of each CLEANEX peak by the intensity of the corresponding 1H-15N HSQC peak. Fig. 3C shows that a long stretch of 15 amino acids in the middle of ErnsΔN does not have any water contact at all and should therefore be protected within the bicelle. This stretch comprises not only hydrophobic amino acids, but also contains two Thr, one Arg, two Lys and one Gln. Most remarkably, the side chains of Gln207 and Trp203, which are positioned on the edge of the hydrophobic face of the helix, are not in contact with water either, thus again supporting the model of an amphiphilic helix that is embedded in and protected by the membrane. In contrast, the N-terminal region of ErnsΔN up to Leu200 shows a high normalized proton exchange rate that is characteristic of a water-exposed surface-location. Surprisingly, most of the C-terminal residues, including the very last amino acid Ala227, are again shielded from water.
15N-NMR relaxation analysis (Fig. 4A, lower part) suggests that the residues comprising the helical part are fairly rigid. The T1, T2 and hetNOE values show a tendency to gradually change from residue 201 towards the N-terminus and from residue 215 or 216 towards the C-terminus, indicating increasing flexibility towards the ends. Besides the immediate N-terminus up to residue 196 (in accordance with the lack of data for the first residues), the last two residues of the C-terminus are highly flexible, which is evident from their high T2 values and low to negative hetNOEs. Together with the fact that these two and the preceding residues at the utmost C-terminal end are protected from contact with water and show an interconnecting network of NOEs, a picture emerges where the C-terminus forms a stable but semi-flexible structure within the bicelle, whereas the N-terminus is unstructured and exposed on the surface of the bicelle.
Structural influence of the C-terminus
Earlier cell culture studies had reported that the C-terminus of Erns (BVDV Strain CP7) has a major influence on the retention/secretion of the protein. The deletion of the last five C-terminal amino acids (FGAYA) led to a dramatic increase in the amount of secreted Erns [24]. A recently conducted cell culture experiment of the Erns protein from CSFV strain Alford/Tübingen showed that even the loss of the four C-terminal residues (GAYA) has a major impact on protein secretion. In these experiments the protein secretion rate increased from below 10% (wt protein) to 35% (truncated version) (unpublished data). Since the major part of the membrane binding of the Erns anchor is supposedly contributed by the central region of the amphipathic helix, we wondered whether the C-terminus may have any influence on the conformation of the membrane anchor, thereby modulating its membrane association. To answer this question, we expressed and analysed yet another, C-terminally trunctated protein ErnsΔNΔC, a variant of ErnsΔN lacking the last 6 amino acids (WFGAYA). The assignment of ErnsΔN could be easily transferred to the 1H-15N HSQC spectrum of this shortened version (Fig. 5A). The corresponding CLEANEX spectrum (Fig. 5B) and the calculated normalized proton exchange rates (Fig. 5C) indicated that in ErnsΔNΔC only 8 amino acids located in the middle of the sequence were shielded from water. In addition, two further amino acids, Thr201 on the N-terminal side and Lys214 on the C-terminal side of the shielded sequence showed no water contact.
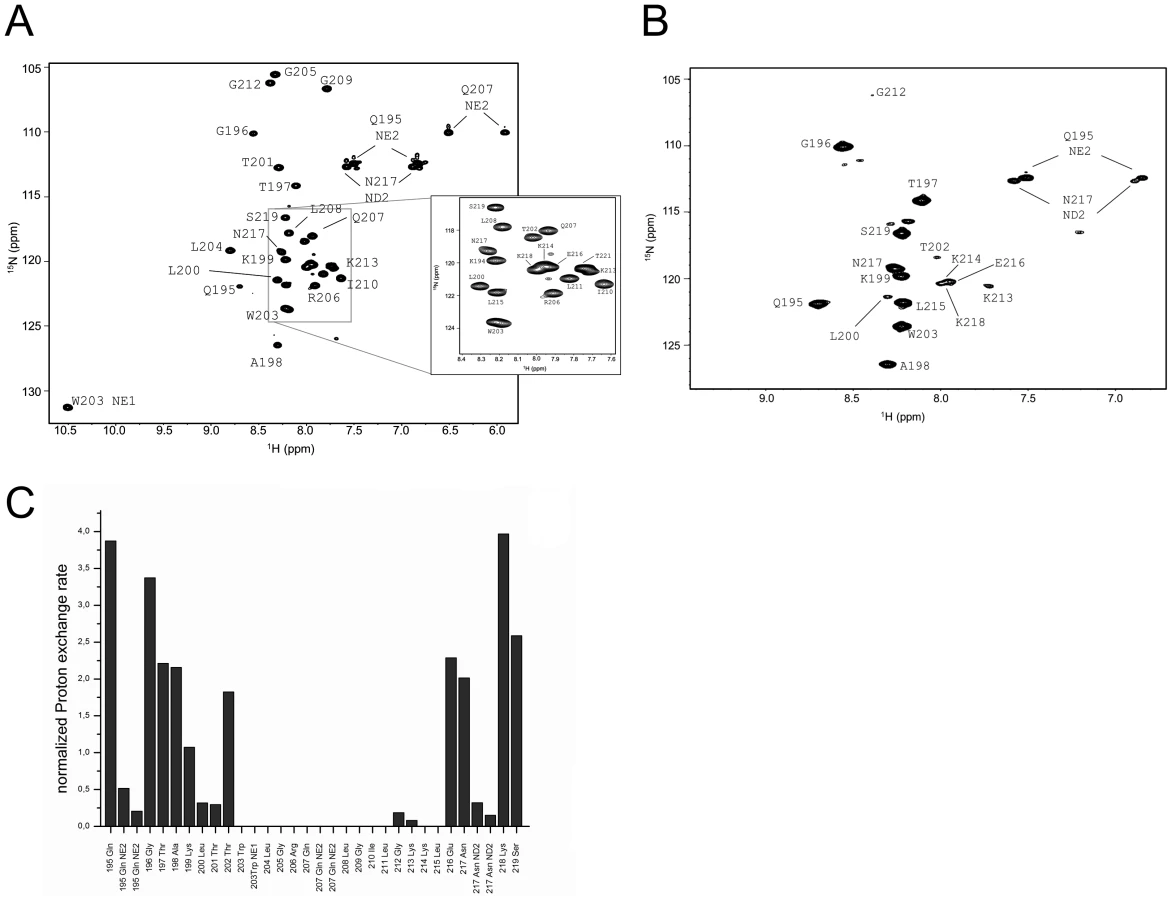
Comparison of the normalized proton exchange rates of ErnsΔN and ErnsΔNΔC in Fig. 6A reveals several differences resulting from the deletion of the 6 C-terminal amino acids. For the N-terminal amino acids, ErnsΔN and ErnsΔNΔC yielded nearly the same values (except for the peptide bond NH group of Gln195), hence the conformation and water-exposed location of these residues should be equivalent in both proteins. However, residues Thr201, Gly212 and Lys213, which in the case of ErnsΔN are embedded in the bicelle, now seem to be located somewhat closer to the lipid/water interphase in ErnsΔNΔC, as seen by the small but significant change in the proton exchange rate. Especially the peptide bond of Trp203 shows a high proton exchange rate in ErnsΔNΔC and should therefore in the truncated fragment be located at the membrane surface where it is exposed to water. Interestingly, the NH side group of Trp203 did not show a change in its water accessibility upon truncation, but is still located in a hydrophobic environment. Most notably, the water accessibility of the C-terminal amino acids is increased in ErnsΔNΔC. The peptide NH group of the four C-terminal amino acids Glu216, Asn217, Lys218 and Ser219 all exhibit an enhanced proton exchange rate once the last six amino acids are deleted. Moreover, the Thr202 NH group in ErnsΔNΔC shows a proton exchange rate that is comparable to the values determined for the aforementioned four amino acids. Interestingly, the side chain NH of Asn217 showed nearly the same low proton exchange rate in both proteins. This indicates that only the peptide bond has changed its position in ErnsΔNΔC whereas the side chain is still protected from water.

The results of the water accessibility experiments with ErnsΔN and ErnsΔNΔC are summarized in Fig. 6B. It is obvious that the deletion of the last six amino acids results in a change in water accessibility of upstream sequences and increases the number of NH bonds that are engaged in fast exchange. This occurs especially in the formerly water shielded central region of the Erns protein. Interestingly enough, the flanking amino acids Thr201 and Lys214 did not gain direct access to water, but the accessibility of the peptide NH groups increased at both sides of the formerly shielded domain upon C-terminal truncation. In both deletion constructs, there remains a continuous stretch of 8 amino acids showing no water accessibility, including the side chain of Gln207. This finding of a discontinuity in the protection pattern at residues 216–219 does not support the picture of a perfectly straight amphipathic helix in the C-terminal region.
Structural model of the C-terminal region
To elucidate the degree of helicity of membrane-bound Erns by an independent method, we performed all-atom Monte Carlo (MC) simulations with an all-atom intramolecular force field [41] that was recently used to describe the reversible folding of various proteins [42], [43]. To speed up the simulation, we employed an implicit membrane model with three layers providing discrete dielectric environments [44]. ErnsΔN (Fig. 7B) as well as the entire Erns anchor (Fig. 7A) were simulated at temperatures ranging from 220K to 400K, starting from completely helical conformations that were placed in the proximity of the membrane surface. The region from Thr172 to Lys220 remained helical, while the N - and C-terminal regions showed partial unfolding with increasing temperature (see Supporting Information Fig. S4A). A similar picture emerged for ErnsΔN, as illustrated in Fig. 7B, which remained helical from Leu200 to Lys214. The last six C-terminal residues are unfolded for both peptides (Supporting Information Fig. S4A/C). Both peptides remained bound to the membrane surface in all simulations, with the hydrophobic residues facing inward. Moreover, the helix was slightly tilted (Supporting Information Fig. S5).
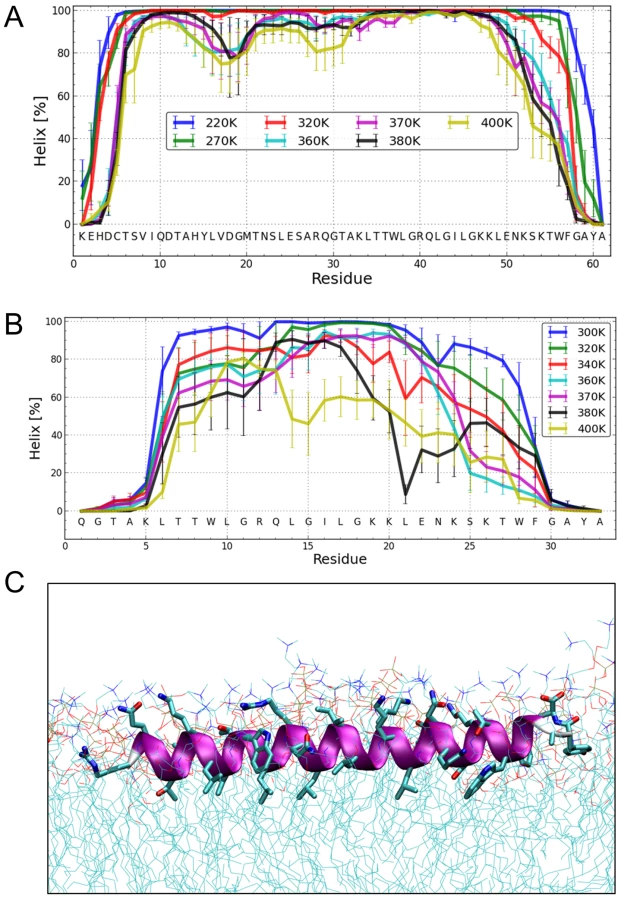
The Monte Carlo simulations can give a good prediction of the local secondary structure of ErnsΔN, but the implicit membrane model is not suitable to gain an impression of the depth of insertion or of the exact tilt angle of the protein within the membrane. We therefore conducted all-atom molecular dynamics (MD) simulations in an explicit lipid bilayer composed of 512 DMPC molecules, in order to obtain further information on the membrane insertion of ErnsΔN (Fig. 7C). The resulting MD model shows a pronounced helical conformation of ErnsΔN and a slightly tilted orientation in the membrane with an angle of 15° relative to the membrane surface. The peptide maintained a stable helical conformation throughout all simulations, but showed a kink around residue 202 when pulled into the membrane. Besides using the conventional MD approach of allowing the peptide to approach the membrane from the aqueous phase, the simulation in Fig. 7C was started from a position deep within the membrane. This should be a more relevant physiological starting position, since at least part of the structure should be located within the membrane when the C-terminus is generated by signal peptidase cleavage of the E/E1 precursor. The peptide maintained a stable helical conformation throughout the simulation, while the membrane initially bent to compensate for the peptide translocation when the protein was pulled into the membrane core. However, after 35 ns of free MD, the peptide was back to its original position near the membrane surface and assumed its stable orientation there.
Discussion
Erns represents one of the four known pestiviral structure proteins. The protein is crucial for the formation of infectious viral particles and plays a major role also during the infection of cells. But aside from this elementary function, Erns represents also a virulence factor of pestiviruses. In this context it is important that Erns has an intrinsic RNase activity. RNases are only very rarely found in RNA viruses [45], and to our knowledge the pestivirus Erns is the only viral structural protein displaying such enzymatic activity. This RNase activity is crucial for the virulence of the virus. It could be shown that pestiviruses containing an RNase negative Erns protein are viable and able to replicate to nearly wild type virus titers, but are attenuated in their natural hosts [21], [22]. Most importantly, the RNase activity is a major factor for establishing a persistent infection upon transplacental infection of the fetus in a pregnant host animal [4].
Erns is not only concentrated within the cell at the site of virus budding, but it is also secreted to some extent from an infected cell and is thus found in the blood of infected animals [15], [16]. According to the working hypotheses put forward so far, the virulence factor activity of the Erns RNase is supposedly linked directly to its secretion from infected cells. Accordingly, an analysis of Erns membrane anchoring and the mechanisms underlying its partial secretion are of major importance for understanding its activity. It had been proposed earlier that Erns should be bound to the host membranes and to the virion via interaction with the E2 protein that contains a typical transmembrane region [46]. Regardless of the question whether an Erns/E2 heterodimer is formed in all pestiviruses, the results presented here and data published before [24], [25], [26] clearly show that the Erns C-terminus represents an intrinsic membrane anchor per se. The amphiphilic region serves to attach the protein to lipid bilayers and enables its intracellular retention in the absence of any other viral protein. Accordingly, the hypothesis of indirect membrane anchoring of Erns achieved via interaction with E2 - as put forward by Lazar et al. [46] - is refuted by the data available now.
The structure of the Erns N-terminal RNase domain was successfully determined by X-ray crystallography, yielding important clues to its enzymatic functions [20]. Unfortunately, the full-length Erns protein containing its C-terminal membrane anchor region could not be crystallized, so that structural data on this functionally important domain are missing. We have therefore investigated the structure of the C-terminal domain using several complementary methods. First, we could demonstrate in cell culture experiments that this domain does indeed serve as the membrane anchor [24], [25]. However, in those experiments the membrane anchor did not bind to membranes as tightly as a typical transmembrane helix. Sequence prediction and model building showed that the Erns membrane anchor could be structured as a long amphipathic helix. Mutations interfering with the proposed amphipathic character of the C-terminal region affected the membrane anchoring as well as the secretion of the protein [25], [26].
Peripheral membrane binding via amphipathic helices is quite common for both cellular and viral proteins. Cellular amphipathic helices are typically located at the N-terminus and consist of only 3–4 helical turns. Such proteins are found in the cytoplasm or the ER and have a broad range of important biological functions. For example, such structures are used in the cell for measuring membrane curvature [47], for generating membrane vesicles [48], [49], or for establishing protein-protein interactions [50]. The amphipathic helices are generally oriented parallel to the membrane surface and act, in the case of proteins involved in the budding of membrane vesicles, as a membrane anchor of these proteins. The local curvature of the membrane involved in vesicle budding is usually not induced by the amphipathic helix itself, but e.g. by the bent form of the proteins [49] or by protein-protein crowding [51].
The considerable length of the Erns amphipathic helix (up to about 50 amino acids in TFE) is more reminiscent of amphipathic helices found in another group of proteins, such as cytolytic peptides. These peptides usually consist of an amphipathic helix composed of 23–31 amino acids [52]. Melittin, a toxic component of bee venom, for example, binds to cellular membranes and forms a membrane pore by oligomerization, which leads to the damage of the target cell. Similarly, the antimicrobial peptide PGLa is a straight amphiphilic α-helix, which binds flat to the membrane surface, whereupon it can start to tilt and eventually assemble transiently as an oligomeric transmembrane pore [34], [36], [38]. The melittin and PGLa helices can be slightly tilted into the membrane like the Erns anchor [35], [37], [53], but the Erns anchor most likely lacks the ability of a concentration depending structural re-orientation according to our experiments.
Several viral proteins are known to contain short amphipathic helices, such as the Brome-Mosaic-Virus protein 1a [54], or the NS3, NS4A, NS-4B and NS-5A proteins of hepatitis C virus [55], [56]. Also for pestiviruses membrane binding of NS5A was demonstrated to occur via an amphipathic helix similar to the closely related hepatitis C virus [57]. But all these systems represent non-structural proteins, and the amphipathic helix is only used for membrane attachment and oligomerization to build up the replication complexes on the cytoplasmic membrane surface [58]. To our knowledge Erns is the only structural viral protein that is anchored via an amphipathic helix. This amphipathic helix is unusually long and located in the C-terminal region. It combines several functions, as it is not only important for the membrane binding of the protein, but also for the control of its secretion/retention, its intracellular location [26], and for cleavage of the glycoprotein Erns/E1precursor by the cellular signal peptidase [27]. All these different demands have to be supported by the specific conformation and particular sequence of the Erns C-terminal region.
Our analyses revealed that the Erns anchor is predominantly helical when bound to the membrane, but that the stability of the helix varies considerably over the sequence, with 15 residues representing a core helix that can be extended towards both sides. Monte Carlo (MC) simulations support the experimental data, as they yielded a similar extent of helicity and could identify the stretch Leu200 – Lys214 as the most stable helical region, in perfect agreement with CD and NMR data. Both the OCD analyses and the simulations revealed a slight tilt of the helix with respect to the membrane surface (Fig. 2F and Supporting Information Fig. S5, respectively).
Since the MC analyses do not provide information on the location of the anchor in the membrane, molecular dynamics (MD) simulations were conducted. The MD analyses also revealed a helical conformation with a slight tilt. The helix was, embedded just below the lipid headgroup region of the membrane. Unlike the other data, the helix in the MD simulations extended virtually over the complete length of the peptide. Thus, the helix content of this simulated structure is significantly higher than what was observed experimentally in the CD, NMR and MC analyses, which had shown unwinding of the N - and C-terminal residues. This discrepancy is most likely due to an overestimation of the H-bond energy in the force field.
As a general consensus of the CD, OCD, NMR, and MC simulation data, and in part also MD simulation results, at least part of the helix should be inserted more deeply in the membrane than the surrounding amino acids. This conclusion is in agreement with the data obtained by the NMR CLEANEX experiments showing that the central region of the amphipathic helix does not exchange the NH protons with water protons. Likewise, the side chain protons of Trp203 and Gln207 are protected and thus seem to be located in the hydrophobic interior of the bicelle. Nonetheless, the immersion within the bicelle is either not very deep or not perfectly stable, because all residues experience the influence of the paramagnetic agent gadolinium (Supporting Information Fig. S3). This ion induces strong relaxation that leads to a disappearance of the signals within a minimum radius of 5 Å [59]. The fact that the NMR signals of the full-length Erns anchor show dynamic exchange most probably with the free unstructured protein also indicates that the interaction with the membrane-mimicking bicelles is only of moderate stability.
Interestingly, the N-terminal end of ErnsΔN is located on the bicelle surface or protrudes into the solvent, given its high water accessibility, while the amino acids of the far C-terminal end are shielded from water including the last C-terminal residue Ala227. Together with the observed NOE pattern, this result suggests that the C-terminal end of the sequence is at least temporarily located within the membrane, being quite flexible with regard to its secondary structure, as most clearly seen for Ala227, the very last C-terminal residue of the protein. Ala227 does not show any water contact, which argues for a membrane immersed location, but the relaxation and Het-NOE measurements revealed a flexible conformation. Any particular stable C-terminal conformation, such as a putative turn close to the water/lipid interphase was not reproduced in the MD simulations, possibly due to an overestimation of the helical hydrogen-bonding. However, the MC simulation revealed a significant probability for a turn-like structure at the C-terminus (Supporting Information Fig. S4D), and the same pattern was also revealed by MC simulations of the full-length Erns anchor (Supporting Information Fig. S4B).
This model is supported by the pronounced influence that the native Erns C-terminus has on the central helix, as seen from the increased water accessibility in the truncated construct ErnsΔNΔC. This effect was not restricted to the C-terminal region of the immersed part, but was also seen for residues located upstream at the N-terminal end of the inserted helix. Thus, the C-terminal end of the Erns anchor has an effect on the immersion of the whole inserted segment. On the other hand, the N-terminal region of the protein did not show such an interesting effect, as all amide groups had strong water contacts.
The structural model of the Erns C-terminus presented here does not answer all open questions about the biological mode of action of the Erns anchor, because it does not allow any conclusions about the mechanism governing the equilibrium of Erns retention and secretion. Nevertheless, it gives a first hint on how the interaction of the protein with the membrane occurs. Although the Erns protein lacks a transmembrane domain or a GPI anchor, which are usually responsible for tight membrane association, its unusually long amphipathic helix confers strong membrane binding. This anchor is not only attached to the membrane via the hydrophobic face of the amphiphilic helix, but because of the tilt part of the helix including its far C-terminal end is inserted into the membrane. This may facilitate the arrangement of lipids around the inserted peptide and may lower the free energy of the system to stabilize the membrane/protein interaction. It thus appears feasible that the Erns anchor could bind more strongly to membranes than a typical amphipathic helix on the bilayer surface, thereby providing the firm membrane association that is crucial for a viral surface protein. Nevertheless, the membrane affinity of this anchor is considerably lower than that conferred by a transmembrane helix [24], which seems to be an important prerequisite for the observed secretion of Erns. Erns represents the first membrane protein for which anchoring via an amphipathic helix is described, and this unusual type of membrane attachment can be hypothesized to be of functional importance. The structure adopted by this anchor in a membrane could contribute to the known equilibrium between retention and secretion by adjusting the binding force at an appropriate level. This hypothesis is in agreement with the observation that C-terminally truncated Erns is more efficiently secreted [24], [26], because the observed increased water accessibility of the central part of this truncated helix suggests that this region becomes less deeply immersed and consequently has a decreased binding affinity. Whether this proposed decrease of binding (alone) is responsible for increased secretion of the mutant protein cannot be decided yet. Alternatively, a different orientation of the protein in the membrane could interfere with the interaction between Erns and other proteins important for its membrane association. Further investigation is necessary to answer the question whether other mutations enhancing secretion [25], [26] also alter the immersion and binding affinity of the helix. In a rather speculative working hypothesis, one might propose that either a slight truncation of the C-terminus or its unfolding as a result of (changing the) interaction with a partner molecule could destabilize lipid binding of the anchor and lead to secretion. So far, nobody has achieved a detailed analysis of the primary structure of the secreted protein, and any data concerning putative interaction partners of the anchor sequence are missing, so that both possibilities have to be investigated in future work. For the time being, the present data support a model that provides strong membrane binding, which, however, can be modulated by changes affecting the far C-terminal end or the overall structure of the anchor.
The structure model presented here displays the monomeric form of the protein but in vivo, a considerable amount of Erns is found as a homodimer covalently linked via a disulfide bond between the Cys residues at position 171 of the protein [23], [60], [61], [62]. Nevertheless, we had no indication of dimer formation when analyzing the full-length anchor containing Cys171. The mass spectra of the purified protein did not reveal the presence of the dimeric form and the NMR analyses proved furthermore that every nucleus was found in only one defined surrounding. This observation implies that only one conformation - whether dimer or monomer - was present in the samples. For a dimer, this result could only be obtained in the case of ideal symmetry. Moreover, we conducted NMR analyses on the Erns anchor both with and without the addition of DTT and were not able to identify any differences between these spectra (not shown). Most importantly, the NMR analyses leading to the structural model were conducted with a C-terminal part of the anchor lacking Cys171.Fraom biochemical analyses of the Erns protein it is known that the ability to form dimers is massively weakened when the disulfide linkage is prevented by mutation of Cys171 [23]. Taken together, these results strongly support the notion that the Erns anchor or fragments thereof analyzed in our experiments were monomers. Erns dimerization is important in vivo, however, and the dimeric form is found in virions, infected cells, and also in the supernatant of infected cells. Knocking out Erns dimer formation by mutation of Cys171 doesn't interfere with virus viability but leads to virus attenuation in the natural host [23], so dimer formation plays a role for Erns function. Even though we do not have any data on the structure of a membrane anchored Erns dimer, it is tempting to speculate on the structure of such molecule. We can conclude from the data on a peptide corresponding to the N-terminal part of the CSFV Alfort/Tübingen Erns membrane anchor [26], as well as from the crystal structure of residues 1 to 165 of the BVDV NCP7 Erns [20] that Cys171 is located in a rather flexible region of the protein, linking the enzymatically active N-terminal domain to the C-terminal membrane anchor. Hence, contact of the two monomers in this region should not be sterically hindered. In plane binding of the membrane anchor helices of both monomers parallel to the bilayer surface according to our structural model would also not interfere with protein/protein interaction between the regions containing Cys171. The angle between the two anchor helices in a two dimensional projection should be flexible, since there is no reason to postulate any particular fixed arrangement. Furthermore, as pointed out above, we have no indication that two or more anchor molecules could engage in a parallel or antiparallel alignment of the two amphipathic helices of an Erns dimer. Whether the two regions around Cys171 of the two monomers form a parallel stem–like structure or whether they cross each other cannot be answered yet and also the membrane topology of this contact region is unclear at the moment. Further experimental work would be necessary to get an idea of the structure of the Erns membrane anchor in a dimeric state. One important step towards elucidation of this point would be to identify residues that are part of the dimerization interface in the region around Cys171.
The model established here for the Erns C-terminus describes only the folding of the fully processed protein. However, Erns is not translated as a single protein but rather as a part of the pestivirus polyprotein that is posttranslationally cleaved into the different viral proteins. The N-terminus of Erns is generated upon cleavage by the cellular signal peptidase [27], which is also responsible for several other steps of pestivirus polyprotein processing [15]. The signal peptidase usually cleaves after a so called von Heijne sequence that is preceded by a transmembrane helix [28], [29]. It has long been puzzling why and how the C-terminal end of Erns can be generated by signal peptidase, given that this sequence lacks a transmembrane element [27]. The structural model for the Erns C-terminus presented here cannot explain this conundrum, as we found no indication for a transmembrane orientation of the C-terminal segment. Therefore, the structure of the uncleaved Erns C-terminus should differ from the final form of the processed C-terminus to allow the cleavage by the cellular signal peptidase. It seems likely that the Erns C-terminus adopts an alternative transient structure during processing, while being tethered to the membrane via the transmembrane region of the E1 protein that follows downstream in the polyprotein. This sequence of events could explain why the cleavage between Erns and E1 is delayed compared to other polyprotein processing steps [15].
Recently, a new structural motif named “charge zipper” was identified in the membrane binding domains of various proteins [30]. The contact between two amphipathic helices is stabilized through electrostatic interactions between charged amino acids on the two apposed segments, such that a long ladder of salt bridges is formed between them, either leading to an intramolecular hairpin, or resulting in intermolecular oligomerization. Such a charge zipper motif was also identified in the C-terminal membrane anchor of Erns. Folding of this region into a helical hairpin would allow a transmembrane insertion of the anchor, since the charged amino acids would be shielded within the structure, while hydrophobic residues would be present on the outer face and could be easily inserted into the membrane. The resulting structure could provide a (transient) transmembrane helical hairpin structure upstream of the Erns/E1 processing site, which might explain its cleavage by the signal peptidase. After cleavage, a structural rearrangement of the released Erns C-terminus could occur, resulting in the identified structure of the mature Erns C-terminus that allows membrane anchoring of Erns and control of secretion. This charge zipper hypothesis is highly interesting but has to be analyzed in detail with further experiments.
The Erns membrane anchor thus combines several very different functions in a rather short stretch of the sequence. These functions are important for the life cycle of pestiviruses. They rely on a specific structure that is probably established through different transition states, and which promotes membrane binding and signal peptidase processing. The final structure of the mature Erns C-terminus presented here as a model describes for the first time a new way of anchoring a viral surface protein via an unusually long amphipathic helix to a membrane. This arrangement represents a new perception of protein/membrane interaction that might also be relevant for other peripheral membrane proteins. Importantly, the structural model of the Erns C-terminus reveals not only a new possible fold of an amphipathic membrane anchor, but it also provides new insight into the biochemical requirements of the Erns protein and paves the way towards a better understanding of the virulence function of this fascinating system.
Materials and Methods
Construction of plasmids
Plasmid pd29G was used as a starting point for all expression constructs. It consists of the plasmid pETZ2-1 (kindly provided by Gunter Stier [63]), which codes for a Z2 domain to enhance the water solubility of the fusion protein, a C-terminal His-TAG for protein purification and a N-terminal TEV protease cleavage site to allow removal of the complete TAG from the desired expression product. The insertion in pd29G consists of a cDNA coding for part of the viral polyprotein from BVDV strain CP7 Erns (Lys167 - Ala227) after four cloning derived amino acids (GAMA). The cDNA sequence was optimized for the bacterial expression of the protein. Plasmid pd29G-1, containing the sequence information for the protein ErnsΔN (Arg194 - Ala227), was generated by a QuikChange (QC) PCR using ‘CCTGTATTTTCAGGGCCGTCAGGGGACCGC’ as forward and ‘GCGGTCCCCTGACGGCCCTGAAAATACAGG’ as reverse primer. QC PCR was conducted according to the protocol by Strategene (Heidelberg, Germany). The expression plasmid of ErnsΔNΔC (Arg194 – Thr221; plasmid pd29G-1-2) was established from pd29G-1 via QuikChange mutagenesis with primers ‘GAAAACAAAAGCAAAACCTAATAACTCGAGCACCACCACC’ and ‘GGTGGTGGTGCTCGAGTTATTAGGTTTTGCTTTTGTTTTC’. The established constructs were all verified by nucleotide sequencing with the BigDye terminator cycle sequencing Kit (PE Applied Biosystems, Weiterstadt, Germany). Sequence analysis and alignments were done with MultiAlin [64]. Cloning was done using standard procedures [65].
Expression and purification of proteins
The expression of proteins was done with E.coli strain BL21(DE3) in standard LB-Medium for the CD measurement, or in minimal medium containing 15N (15NH4Cl, ISOTEC, Sigma-Aldrich, USA) and 13C (13C6 D-Glucose, Cambridge Isotope Laboratories, Inc, USA) for the NMR analysis. 1 l of medium was inoculated with an overnight culture until the OD600 of the mixture was between 0.05 and 0.1. The bacterial growth at 37°C and 220 rpm was observed until an OD600 of 0.8 was reached. At this point protein expression was induced by addition of IPTG (final concentration 0.5 mM). For expression in minimal medium, 2.5 ml 20% 13C6-Glucose per liter of culture was added to the medium. The bacteria were incubated at 20°C and 220 rpm and harvested after 3 h. After centrifugation for 10 min at 5000 g and 4°C the bacteria were resuspended in 15 ml Lysisbuffer [50 mM NaH2PO4 pH 8.0, 300 mM NaCl, 30 mM Imidazol, Lysozym, 6% TritonX-100, 1 tab. Roche Complete Protease inhibitor without EDTA (Roche, Mannheim, Germany)] per liter of minimal medium. After 10 min incubation at room temperature the bacteria were lysed by three freeze-thaw cycles performed with liquid nitrogen and warm water and sonification for 6×30 sec on ice (Branson Sonifier B15, level 7, cycle 80%). The insoluble debris was removed by centrifugation (Beckman JA17 rotor, 30 min, 31000 g) at 4°C.
The purification was started with a 5 ml Ni-NTA column (Protino Ni-NTA Columns, Macherey-Nagel, Germany) on an FPLC system (LKB GradiFrac, Pharmacia Biotech, Freiburg) with a flow rate of 3 ml/min. The UV absorbance at 280 nm was measured with a connected absorbance recorder (LKB Optical Unit, LKB REC102, Pharmacia Biotech, Freiburg) to identify protein containing fractions. A step gradient of 50 mM and 100 mM imidazole was used to prevent unspecific protein binding, and elution was accomplished with 300 mM imidazole. Afterwards, the protein containing fractions were pooled and ultrafiltrated (Amicon Ultra-15, Millipore). The retentate was diluted in TEV-Buffer (Invitrogen, USA) and again ultrafiltrated until the NaCl concentration was less than 5 mM. The protein concentration was determined by measuring the absorbance at 280 nm to calculate the amount of AcTEV-Protease (Invitrogen, USA) that was needed to cleave off the N-terminal Z2-TAG. It was assumed that 10 U AcTEV-Protease could cleave 2.16 mg substrate. To prevent oxidation, the solution was overlayed with CO2 and incubated for several days at room temperature. Each day a 1 µl sample of the solution was collected and analyzed to check the cleavage process. After cleavage was completed, the cleavage products were separated with a reverse phase HPLC (BT9200 Titan HPLC pump, Eppendorf, Hamburg) using a C4-Reprosil 300 column (Dr. Maisch GmbH, Ammerbuch-Entringen, Germany) and a gradient from 20% to 60% acetonitrile with 0.05% trifluoroacetic acid. The absorbance at 280 nm was recorded (BT9520 IN UV/Vis detector, LKB REC101, Pharmacia Biotech, Freiburg) and the eluent was collected in 2 ml fractions (Foxy Jr., ISCO) and analyzed. Positive fractions were pooled and lyophilized for several days. Afterwards, the molecular mass of the protein was measured using a MALDI-MS (Ultraflex I, Bruker) to determine the achieved isotope labeling, the purity and the absence of oxidation products.
Circular dichroism (CD) spectroscopy
Phosphate buffer (PB), 2,2,2-trifluoroethanol (TFE) and sodium dodecyl sulfate (SDS) were obtained from VWR (Darmstadt, Germany). The detergent n-dodecylphosphocholine (DPC) and the phospholipids DMPC 1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (DMPC), 1,2-dimyristoyl-sn-glycero-3-phosphatidylglycerol (DMPG) and 1,2-dihexanoyl-sn-glycero-3-phosphatidylcholine (DHPC) used for vesicle and bicelle preparation were purchased from Avanti Polar Lipids (Alabaster, AL, USA). A weighed amount of lyophilized Erns protein was dissolved in deionized water for preparing a 50 µM stock solution. SDS and DPC were used in a concentration of about 10 mM in 10 mM PB pH 6.5 with a protein/detergent ratio of 1∶600. The lipid powders of DMPC and DMPG were dissolved in 50∶50 chloroform/methanol (v/v) to get lipid stock solutions of ∼7 mM. Aliquots of these stock solutions were mixed in a glass vial and thoroughly vortexed to obtain the DMPC/DMPG mixture (1∶1 molar ratio). Subsequently, the organic solvent was removed under a gentle stream of nitrogen, followed by overnight incubation under vacuum. The DMPC or DMPC/DMPG lipid film that had formed in the vial was dispersed by the addition of 200 µl PB and homogenized by vigorous vortexing for 7×1 min and by 7 freeze-thaw cycles. Afterwards, small unilamellar vesicles were formed by sonication of the multilamellar vesicles for 4 min in a strong ultrasonic bath (UTR 200, Hielscher, Germany). The sonication procedure was repeated 3 times (with intermittent cooling of the water in the ultrasonic bath to room temperature with ice, to avoid overheating the samples). To prepare bicelles, a weighed amount of DHPC was first dissolved in 10 mM PB by sonification. An aliquot of this solution was used to dissolve a weighed amount of DMPC. Afterwards, the bicelle dispersion was homogenized by vortexing and freeze-thaw cycles as described above. Due to the significant technical challenges of examining bicelles in optical spectroscopy, which are caused by much stronger light dispersion compared to sonicated small unilamellar vesicles, the protein concentration of the sample was calculated by the UV-VIS absorption of a stock solution, from which the corresponding dilution factor could be determined.
To prepare the samples for CD analysis, an aliquot of the Erns protein stock solution was added to PB, to a 50∶50 mixture of TFE/PB (v/v), to SDS or DPC micelles, or to the corresponding lipid dispersions. The final protein concentration in PB, TFE/PB and in micellar environment was 15 µM. In the lipid vesicle samples the protein concentration was adjusted in the range from 13–27 µM and the lipid concentration between 0.7–1.8 mM, resulting in peptide-to-lipid (P/L) ratios of ∼1∶20, 1∶50, 1∶100 and 1∶200. A 20 µM protein and 2 mM (total) lipid concentration and a P/L ratio of 1∶100 was set up in the bicelle samples by dissolving the lyophilized ErnsΔN protein directly in the bicelle dispersion.
CD spectra were recorded on a J-815 spectropolarimeter (Jasco, Groß-Umstadt, Germany) in rectangular quartz glass cells of 1-mm path length (Suprasil; Hellma, Müllheim, Germany) between 260 and 185 nm at 0.1-nm intervals. The temperature was set to 25°C for the peptide solutions in PB, the 50% TFE mixture, and the micellar solutions, and at 30°C for the vesicle or bicelle suspensions (i.e., well above the lipid phase transition temperature of 23°C for DMPC and DMPG) using a water thermostat-regulated cell holder. Three repeat scans at a scan rate of 10 nm/min, 8 s response time, and 1 nm bandwidth were averaged for each sample and for the baseline of the respective peptide-free sample. After subtracting the baseline spectra from the sample spectra, CD data were processed with the adaptive smoothing method in the Jasco Spectra Analysis software. To calculate the mean residue ellipticities required for quantitative secondary structure estimation, the concentration of the peptide stock solutions was determined from the UV absorbance of the respective peptide at 280 nm. For better comparison of the spectra of the different samples, the calculated mean residue ellipticity (MRE) is shown in the graphs.
Secondary structure analyses were performed using the CDSSTR program [66], [67] with the implemented singular value decomposition (SVD) algorithm; by the CONTIN-LL [68], [69] program, which is based on the ridge regression algorithm; and by the SELCON-3 [70], [71] program, which incorporates the self-consistent method together with the SVD algorithm to assign protein secondary structure. The three algorithms are provided by the DICHROWEB online server [72], [73]. The quality of the fit between experimental and back-calculated spectrum according to the secondary structure fractions was assessed from the normalized root mean square deviation (NRMSD), with a value <0.1 considered as a good fit [72].
Oriented circular dichroism (OCD) spectroscopy
Oriented protein-lipid samples for OCD measurements were prepared by depositing the proteolipid vesicles (with DMPC and DMPC/DMPG, as described above) on a planar quartz glass substrate. Each sample was generated by spotting a 60–80 µl aliquot of the vesicle sample onto a 20 mm diameter quartz glass plate (SUPRASIL, Hellma, Jena) as a ∼12 mm central circular spot, and dried under a gentle stream of air. Afterwards, the sample was rehydrated for 15 h at 30°C and 97% relative humidity in an OCD sample cell using a reservoir of saturated K2SO4 solution. The in-house built OCD cell can be integrated in a J-810 spectropolarimeter as an accessory, and further details on the OCD sample preparation and measurements have been described [33], [74]. The thin oriented bilayers formed during hydration of the sample minimize the possibility of undesired spectral artifacts caused by linear dichroism or absorption flattening. The OCD spectra were recorded as an average of 8 scans with a 45° rotation of the cell after each scan to further reduce spectral artifacts due to linear dichroism arising from imperfections in the sample, strain in the quartz glass windows, or imperfect alignment of the window. For OCD measurements the same data acquisition parameters were used as in the conventional CD experiments described above. Background spectra of pure lipid bilayers (without protein) were subtracted from all OCD spectra. In order to compare the different OCD spectra in a better way all spectra were normalized to match their ellipticity around the minimum at ∼220 nm.
NMR spectroscopy
The NMR analysis was carried out in a lipid bicelle system with a total lipid concentration of 200 mM in PBS (50 mM KH2PO4 pH 6.8, 50 mM NaCl), using DHPC and DMPC (Avanti Polar Lipids, Alabaster, AL, USA) at a ratio of 4∶1. For sample preparation, the weighed amount of DH/MPC was dissolved in 450 µl PBS by vortexing and sonification until the solution was clear. Afterwards, the required amount of DH/MPC was added and the mixture was again sonified. Thereafter, 0.5 µmol of the lyophilized protein and 50 µl D2O were added. After another round of sonification the insoluble material was removed by centrifugation for 20 sec. at 320 rcf to get a clear solution. The protein/lipid ratio obtained by this procedure was 1∶222.
NMR experiments were performed on a Bruker Avance I 600 MHz spectrometer with a broadband triple resonance probe, or on a Bruker Avance III 600 MHz spectrometer with a cryo probe and a Z-gradient.
The 15N-HSQC and CLEANEX experiments [40], and titration with Gd 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (Gd-DOTA, Sigma-Aldrich) were typically acquired with 2 - 4 scans and a total of 128 increments in the indirect dimension, between 23 and 50°C. Titration with Gd-DOTA was performed on 0.5 mM sample of ErnsΔN in either DHPC/DMPC or DPC micelles. A CLEANEX spin-lock field of 4.8 kHz was applied for a mixing time of 100 ms. 3D 15N-HSQC NOESY and 15N-HSQC-TOCSY experiments were performed at 23°C with a mixing time of 120 ms (NOESY) and 60 ms (TOCSY). 200–250 increments in the 1H dimension and 55 increments in the 15N dimension were acquired. A 3D 13C-HMQC NOESY was acquired in 200 mM deuterated DPC (Avanti Polar Lipids, Alabaster, AL, USA). 1H 15N 13C triple resonance experiments (HNCACB, HNCA, CBCA(CO)NH) at 23°C [75] were used to assign the 1HN, 13C and 15N resonances. 15N longitudinal (R1) and transversal (R2) relaxation as well as the heteronuclear NOE (het-NOE) were measured at 27°C. Relaxation delays varied between 10.8 and 3466.8 ms for R1, and 14.4 to 259.2 ms for R2 [76]. One duplicate point was included to test for instabilities. The heteronuclear NOE was determined as the signal intensity ratio of 1HN/N crosspeaks with and without 1H saturation. All experiments were recorded in an interleaved manner [77]. The water signal was suppressed with a combination of the water-flip-back and the WATERGATE scheme in all cases.
Spectra were processed using the nmrPipe software package [78] and analyzed with NMRView [79]. The normalized proton exchange rate was calculated by the intensity of the peak in the CLEANEX spectra divided by the intensity of the correlated peak in the 15N-HSQC spectra
Monte Carlo simulations
Using the Monte Carlo simulation package SIMONA [80] we performed a total of 1.2×109 steps, each of which changed a single dihedral, for both Erns systems in the all-atom AMBER99SB-ILDN force field [41], in combination with an accurate implicit description of the solvent and membrane interactions [44]. Starting from an ideal all-helical conformation in the proximity of the outermost membrane layer, all simulations showed a quick attachment to the membrane surface. Depending on the simulation temperature (Erns - 220K, 270K, 320K, 360K, 370K, 380K, 400K; ErnsΔN - 300K, 320K, 340K, 360K, 370K, 380K, 400K), the secondary structure varies over the sequence. We determined the standard deviation for the per-residue secondary structure information from five distinct populations per temperature, using the secondary structure assignment package DSSP [81]. For each population we averaged the fraction of secondary structure elements over every 10,000th step, neglecting the initial 8×105 steps to allow for equilibration.
Molecular dynamics simulations of ErnsΔN
MD simulations were conducted using the molecular simulation package GROMACS 4.5.5 [82]. The AMBER99SB-ILDN force field [41] was used for the peptide, together with the SLIPID force field for the DMPC bilayer. ErnsΔN (Arg194 – Ala227) was constructed as an ideal helix with an acetylated N-terminus using the program xleap from the AmberTools package [83]. The peptide membrane complex was formed during an unrestrained membrane binding simulation of 10 ns length, by placing the peptide molecule parallel to a pre-equilibrated lipid bilayer at a distance of 1.8 nm above the lipid headgroups, at an elevated temperature of 480 K to speed up insertion. During the binding simulation, hydrogen bonds within the peptide were restrained to prevent unfolding. After cooling down, the system was equilibrated at 303 K with position restraints of 1000 kJ/(mol nm2) on the peptide for 500 ps. After this, an unrestrained MD simulation of 500 ns length was conducted using a Nose-Hover thermostat [84] and Parrinello-Rahman barostat [85], with semiisotropic pressure coupling. A time step of 2 fs was used together with the LINCS algorithm [86] to constrain bonds involving hydrogen atoms. Long range electrostatics were treated via PME combined with a 1.4 nm direct space cutoff for vdW and Coulomb interactions.
For another set of pulling simulations, the GROMACS pull code was used. The peptide was pulled into the membrane with a force constant of 10000 kJ/(mol nm2) and a pull rate of 0.2 pm/ps along the membrane normal for 10 ns. Then, the system with the peptide in the center of the bilayer was equilibrated for 500 ps using position restraints of 1000 kJ/(mol nm2) on the peptide. After that, another unrestrained MD simulation of 75 ns length was conducted.
Supporting Information
Zdroje
1. Lindenbach BD, Thiel HJ, Rice CM (2007) Flaviviridae: The Viruses and Their Replication. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia, New York: Lippincott - Raven Publishers. pp. 1101–1152.
2. MoennigV, PlagemannPG (1992) The pestiviruses. Advances in virus research 41 : 53–98.
3. Thiel HJ, Plagemann PGW, Moennig V (1996) Pestiviruses. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. Philadelphia, New York: Lippincott - Raven Publishers. pp. 1059–1073.
4. MeyersG, EgeA, FetzerC, von FreyburgM, ElbersK, et al. (2007) Bovine viral diarrhea virus: prevention of persistent fetal infection by a combination of two mutations affecting Erns RNase and Npro protease. Journal of virology 81 : 3327–3338.
5. SeagoJ, HiltonL, ReidE, DoceulV, JeyatheesanJ, et al. (2007) The Npro product of classical swine fever virus and bovine viral diarrhea virus uses a conserved mechanism to target interferon regulatory factor-3. The Journal of general virology 88 : 3002–3006.
6. RuggliN, SummerfieldA, FiebachAR, Guzylack-PiriouL, BauhoferO, et al. (2009) Classical swine fever virus can remain virulent after specific elimination of the interferon regulatory factor 3-degrading function of Npro. Journal of virology 83 : 817–829.
7. La RoccaSA, HerbertRJ, CrookeH, DrewTW, WilemanTE, et al. (2005) Loss of interferon regulatory factor 3 in cells infected with classical swine fever virus involves the N-terminal protease, Npro. Journal of virology 79 : 7239–7247.
8. HiltonL, MoganeradjK, ZhangG, ChenYH, RandallRE, et al. (2006) The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. Journal of virology 80 : 11723–11732.
9. ChenZ, RijnbrandR, JangraRK, DevarajSG, QuL, et al. (2007) Ubiquitination and proteasomal degradation of interferon regulatory factor-3 induced by Npro from a cytopathic bovine viral diarrhea virus. Virology 366 : 277–292.
10. BauhoferO, SummerfieldA, SakodaY, TratschinJD, HofmannMA, et al. (2007) Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. Journal of virology 81 : 3087–3096.
11. FiebachAR, Guzylack-PiriouL, PythonS, SummerfieldA, RuggliN (2011) Classical swine fever virus N(pro) limits type I interferon induction in plasmacytoid dendritic cells by interacting with interferon regulatory factor 7. Journal of virology 85 : 8002–8011.
12. WeilandE, StarkR, HaasB, RumenapfT, MeyersG, et al. (1990) Pestivirus glycoprotein which induces neutralizing antibodies forms part of a disulfide-linked heterodimer. Journal of virology 64 : 3563–3569.
13. WeilandF, WeilandE, UngerG, SaalmullerA, ThielHJ (1999) Localization of pestiviral envelope proteins E(rns) and E2 at the cell surface and on isolated particles. The Journal of general virology 80 (Pt 5) 1157–1165.
14. HulstMM, MoormannRJ (2001) Erns protein of pestiviruses. Methods in enzymology 342 : 431–440.
15. RümenapfT, UngerG, StraussJH, ThielHJ (1993) Processing of the envelope glycoproteins of pestiviruses. Journal of virology 67 : 3288–3294.
16. MagkourasI, MatzenerP, RumenapfT, PeterhansE, SchweizerM (2008) RNase-dependent inhibition of extracellular, but not intracellular, dsRNA-induced interferon synthesis by Erns of pestiviruses. The Journal of general virology 89 : 2501–2506.
17. HulstMM, HimesG, NewbiginE, MoormannRJ (1994) Glycoprotein E2 of classical swine fever virus: expression in insect cells and identification as a ribonuclease. Virology 200 : 558–565.
18. SchneiderR, UngerG, StarkR, Schneider-ScherzerE, ThielHJ (1993) Identification of a structural glycoprotein of an RNA virus as a ribonuclease. Science 261 : 1169–1171.
19. WindischJM, SchneiderR, StarkR, WeilandE, MeyersG, et al. (1996) RNase of classical swine fever virus: biochemical characterization and inhibition by virus-neutralizing monoclonal antibodies. Journal of virology 70 : 352–358.
20. KreyT, BontemsF, VonrheinC, VaneyMC, BricogneG, et al. (2012) Crystal structure of the pestivirus envelope glycoprotein E(rns) and mechanistic analysis of its ribonuclease activity. Structure 20 : 862–873.
21. MeyerC, Von FreyburgM, ElbersK, MeyersG (2002) Recovery of virulent and RNase-negative attenuated type 2 bovine viral diarrhea viruses from infectious cDNA clones. Journal of virology 76 : 8494–8503.
22. MeyersG, SaalmullerA, ButtnerM (1999) Mutations abrogating the RNase activity in glycoprotein E(rns) of the pestivirus classical swine fever virus lead to virus attenuation. Journal of virology 73 : 10224–10235.
23. TewsBA, SchurmannEM, MeyersG (2009) Mutation of cysteine 171 of pestivirus E(rns) RNase prevents homodimer formation and leads to attenuation of classical swine fever virus. Journal of virology 83 : 4823–4834.
24. FetzerC, TewsBA, MeyersG (2005) The carboxy-terminal sequence of the pestivirus glycoprotein E(rns) represents an unusual type of membrane anchor. Journal of virology 79 : 11901–11913.
25. TewsBA, MeyersG (2007) The pestivirus glycoprotein Erns is anchored in plane in the membrane via an amphipathic helix. The Journal of biological chemistry 282 : 32730–32741.
26. BurrackS, AberleD, BurckJ, UlrichAS, MeyersG (2012) A new type of intracellular retention signal identified in a pestivirus structural glycoprotein. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 26 : 3292–3305.
27. BintintanI, MeyersG (2010) A new type of signal peptidase cleavage site identified in an RNA virus polyprotein. The Journal of biological chemistry 285 : 8572–8584.
28. NilssonI, JohnsonAE, von HeijneG (2002) Cleavage of a tail-anchored protein by signal peptidase. FEBS letters 516 : 106–108.
29. NilssonI, WhitleyP, von HeijneG (1994) The COOH-terminal ends of internal signal and signal-anchor sequences are positioned differently in the ER translocase. The Journal of cell biology 126 : 1127–1132.
30. WaltherTH, GottseligC, GrageSL, WolfM, VargiuAV, et al. (2013) Folding and self-assembly of the TatA translocation pore based on a charge zipper mechanism. Cell 152 : 316–326.
31. WuY, HuangHW, OlahGA (1990) Method of oriented circular dichroism. Biophysical journal 57 : 797–806.
32. OlahGA, HuangHW (1988) Circular dichroism of oriented alpha-helices. II. Electric field oriented polypeptides. The Journal of Chemical Physics 89 : 6956–6962.
33. BürckJ, RothS, WadhwaniP, AfoninS, KanithasenN, et al. (2008) Conformation and membrane orientation of amphiphilic helical peptides by oriented circular dichroism. Biophysical journal 95 : 3872–3881.
34. AfoninS, GrageSL, IeronimoM, WadhwaniP, UlrichAS (2008) Temperature-dependent transmembrane insertion of the amphiphilic peptide PGLa in lipid bilayers observed by solid state 19F NMR spectroscopy. Journal of the American Chemical Society 130 : 16512–16514.
35. GlaserRW, SachseC, DurrUH, WadhwaniP, AfoninS, et al. (2005) Concentration-dependent realignment of the antimicrobial peptide PGLa in lipid membranes observed by solid-state 19F-NMR. Biophysical journal 88 : 3392–3397.
36. GrageSL, AfoninS, UlrichAS (2010) Dynamic transitions of membrane-active peptides. Methods in molecular biology 618 : 183–207.
37. StrandbergE, TiltakD, EhniS, WadhwaniP, UlrichAS (2012) Lipid shape is a key factor for membrane interactions of amphipathic helical peptides. Biochimica et biophysica acta 1818 : 1764–1776.
38. StrandbergE, ZerweckJ, WadhwaniP, UlrichAS (2013) Synergistic insertion of antimicrobial magainin-family peptides in membranes depends on the lipid spontaneous curvature. Biophysical journal 104: L9–11.
39. HiltyC, WiderG, FernandezC, WuthrichK (2004) Membrane protein-lipid interactions in mixed micelles studied by NMR spectroscopy with the use of paramagnetic reagents. Chembiochem : a European journal of chemical biology 5 : 467–473.
40. HwangTL, van ZijlPC, MoriS (1998) Accurate quantitation of water-amide proton exchange rates using the phase-modulated CLEAN chemical EXchange (CLEANEX-PM) approach with a Fast-HSQC (FHSQC) detection scheme. Journal of biomolecular NMR 11 : 221–226.
41. Lindorff-LarsenK, PianaS, PalmoK, MaragakisP, KlepeisJL, et al. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78 : 1950–1958.
42. Lindorff-LarsenK, PianaS, DrorRO, ShawDE (2011) How fast-folding proteins fold. Science 334 : 517–520.
43. ShawDE, MaragakisP, Lindorff-LarsenK, PianaS, DrorRO, et al. (2010) Atomic-level characterization of the structural dynamics of proteins. Science 330 : 341–346.
44. SJS CBMWWi (2013) SLIM: An Improved Generalized Born Implicit Membrane Model. Journal of Chemical Theory and Computation
45. Meyers G, Rümenapf T, Ziebuhr J (2011) Viral RNase Involvement in Strategies of Infection. In: Nicholson AW, editor. Ribonucleases: Springer Berlin Heidelberg. pp. 135–165.
46. LazarC, ZitzmannN, DwekRA, Branza-NichitaN (2003) The pestivirus E(rns) glycoprotein interacts with E2 in both infected cells and mature virions. Virology 314 : 696–705.
47. CuiH, LymanE, VothGA (2011) Mechanism of membrane curvature sensing by amphipathic helix containing proteins. Biophysical journal 100 : 1271–1279.
48. BhatiaVK, MadsenKL, BolingerPY, KundingA, HedegardP, et al. (2009) Amphipathic motifs in BAR domains are essential for membrane curvature sensing. The EMBO journal 28 : 3303–3314.
49. PrinzWA, HinshawJE (2009) Membrane-bending proteins. Critical reviews in biochemistry and molecular biology 44 : 278–291.
50. PednekarD, WangY, FedotovaTV, WojcikiewiczRJ (2011) Clustered hydrophobic amino acids in amphipathic helices mediate erlin1/2 complex assembly. Biochemical and biophysical research communications 415 : 135–140.
51. StachowiakJC, SchmidEM, RyanCJ, AnnHS, SasakiDY, et al. (2012) Membrane bending by protein-protein crowding. Nature cell biology 14 : 944–949.
52. RaghuramanH, ChattopadhyayA (2007) Melittin: a membrane-active peptide with diverse functions. Bioscience reports 27 : 189–223.
53. BernecheS, NinaM, RouxB (1998) Molecular dynamics simulation of melittin in a dimyristoylphosphatidylcholine bilayer membrane. Biophysical journal 75 : 1603–1618.
54. LiuL, WestlerWM, den BoonJA, WangX, DiazA, et al. (2009) An amphipathic alpha-helix controls multiple roles of brome mosaic virus protein 1a in RNA replication complex assembly and function. PLoS pathogens 5: e1000351.
55. GouttenoireJ, MontserretR, KennelA, PeninF, MoradpourD (2009) An amphipathic alpha-helix at the C terminus of hepatitis C virus nonstructural protein 4B mediates membrane association. Journal of virology 83 : 11378–11384.
56. PeninF, BrassV, AppelN, RamboarinaS, MontserretR, et al. (2004) Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. The Journal of biological chemistry 279 : 40835–40843.
57. MoradpourD, BrassV, PeninF (2005) Function follows form: the structure of the N-terminal domain of HCV NS5A. Hepatology 42 : 732–735.
58. GouttenoireJ, RoingeardP, PeninF, MoradpourD (2010) Amphipathic alpha-helix AH2 is a major determinant for the oligomerization of hepatitis C virus nonstructural protein 4B. Journal of virology 84 : 12529–12537.
59. GO (2010) Protein NMR using paramagnetic ions. Annu Rev Biophys 39 : 387–405.
60. ThielHJ, StarkR, WeilandE, RumenapfT, MeyersG (1991) Hog cholera virus: molecular composition of virions from a pestivirus. Journal of virology 65 : 4705–4712.
61. van GennipHG, HesselinkAT, MoormannRJ, HulstMM (2005) Dimerization of glycoprotein E(rns) of classical swine fever virus is not essential for viral replication and infection. Archives of virology 150 : 2271–2286.
62. LangedijkJP, van VeelenPA, SchaaperWM, de RuAH, MeloenRH, et al. (2002) A structural model of pestivirus E(rns) based on disulfide bond connectivity and homology modeling reveals an extremely rare vicinal disulfide. Journal of virology 76 : 10383–10392.
63. BogomolovasJ, SimonB, SattlerM, StierG (2009) Screening of fusion partners for high yield expression and purification of bioactive viscotoxins. Protein expression and purification 64 : 16–23.
64. CorpetF (1988) Multiple sequence alignment with hierarchical clustering. Nucleic acids research 16 : 10881–10890.
65. Green MR, Sambrook J (2012) Molecular Cloning: A Laboratory Manual (Fourth Edition): Cold Spring Harbor Laboratory Press.
66. SreeramaN, VenyaminovSY, WoodyRW (2000) Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis. Analytical biochemistry 287 : 243–251.
67. JohnsonWC (1999) Analyzing protein circular dichroism spectra for accurate secondary structures. Proteins 35 : 307–312.
68. van StokkumIH, SpoelderHJ, BloemendalM, van GrondelleR, GroenFC (1990) Estimation of protein secondary structure and error analysis from circular dichroism spectra. Analytical biochemistry 191 : 110–118.
69. ProvencherSW, GlocknerJ (1981) Estimation of globular protein secondary structure from circular dichroism. Biochemistry 20 : 33–37.
70. SreeramaN, WoodyRW (1993) A self-consistent method for the analysis of protein secondary structure from circular dichroism. Analytical biochemistry 209 : 32–44.
71. SreeramaN, VenyaminovSY, WoodyRW (1999) Estimation of the number of alpha-helical and beta-strand segments in proteins using circular dichroism spectroscopy. Protein science : a publication of the Protein Society 8 : 370–380.
72. WhitmoreL, WallaceBA (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic acids research 32: W668–673.
73. LobleyA, WhitmoreL, WallaceBA (2002) DICHROWEB: an interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics 18 : 211–212.
74. WindischD, HoffmannS, AfoninS, VollmerS, BenamiraS, et al. (2010) Structural role of the conserved cysteines in the dimerization of the viral transmembrane oncoprotein E5. Biophysical journal 99 : 1764–1772.
75. BlombergN, SattlerM, NilgesM (1999) 1H, 15N, and 13C resonance assignment of the PH domain from C. elegans UNC-89. Journal of biomolecular NMR 15 : 269–270.
76. FarrowNA, MuhandiramR, SingerAU, PascalSM, KayCM, et al. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33 : 5984–6003.
77. KayLE, TorchiaDA, BaxA (1989) Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 28 : 8972–8979.
78. DelaglioF, GrzesiekS, VuisterGW, ZhuG, PfeiferJ, et al. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. Journal of biomolecular NMR 6 : 277–293.
79. JohnsonBA (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol 278 : 313–352.
80. StrunkT, WolfM, BriegM, KleninK, BiewerA, et al. (2012) SIMONA 1.0: an efficient and versatile framework for stochastic simulations of molecular and nanoscale systems. Journal of computational chemistry 33 : 2602–2613.
81. KabschW, SanderC (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 : 2577–2637.
82. BerendsenHvdS, D; van DrunenR (1995) GROMACS: A message-passing parallel molecular dynamics implementation. Computer Physics Communications 91 : 43–56.
83. Case DA, Darden TA, Cheatham, III TE, Simmerling CL, Wang J, et al.. (2012) AMBER 12. University of California, San Francisco.
84. NoséS (1984) A molecular dynamics method for simulations in the canonical ensemble. Molecular Physics 52 : 255–268.
85. ParrinelloM, RahmanA (1981) Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics 52 : 7182–7190.
86. HessB, BekkerH, BerendsenHJC, FraaijeJGEM (1997) LINCS: A linear constraint solver for molecular simulations. Journal of computational chemistry 18 : 1463–1472.
87. de Jongh HH, GoormaghtighE, Killian JA (1994) Analysis of circular dichroism spectra of oriented protein-lipid complexes: toward a general application. Biochemistry 33 (48) 14521–14528.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 2
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Suppression of RNA Silencing by a Plant DNA Virus Satellite Requires a Host Calmodulin-Like Protein to Repress Expression
- Reversible Silencing of Cytomegalovirus Genomes by Type I Interferon Governs Virus Latency
- Identification of Host-Targeted Small Molecules That Restrict Intracellular Growth
- Implication of PMLIV in Both Intrinsic and Innate Immunity