
CIB1 Synergizes with EphrinA2 to Regulate Kaposi's Sarcoma-Associated Herpesvirus Macropinocytic Entry in Human Microvascular Dermal Endothelial Cells
KSHV envelope glycoproteins interact with cell surface heparan sulfate and integrins, and activate FAK, Src, PI3-K, c-Cbl, and Rho-GTPase signal molecules in human microvascular dermal endothelial (HMVEC-d) cells. c-Cbl mediates the translocation of virus bound α3β1 and αVβ3 integrins into lipid rafts (LRs), where KSHV interacts and activates EphrinA2 (EphA2). EphA2 associates with c-Cbl-myosin IIA and augmented KSHV-induced Src and PI3-K signals in LRs, leading to bleb formation and macropinocytosis of KSHV. To identify the factor(s) coordinating the EphA2-signal complex, the role of CIB1 (calcium and integrin binding protein-1) associated with integrin signaling was analyzed. CIB1 knockdown did not affect KSHV binding to HMVEC-d cells but significantly reduced its entry and gene expression. In contrast, CIB1 overexpression increased KSHV entry in 293 cells. Single virus particle infection and trafficking during HMVEC-d cell entry was examined by utilizing DiI (envelope) and BrdU (viral DNA) labeled virus. CIB1 was associated with KSHV in membrane blebs and in Rab5 positive macropinocytic vesicles. CIB1 knockdown abrogated virus induced blebs, macropinocytosis and virus association with the Rab5 macropinosome. Infection increased the association of CIB1 with LRs, and CIB1 was associated with EphA2 and KSHV entry associated signal molecules such as Src, PI3-K, and c-Cbl. CIB1 knockdown significantly reduced the infection induced EphA2, Src and Erk1/2 activation. Mass spectrometry revealed the simultaneous association of CIB1 and EphA2 with the actin cytoskeleton modulating myosin IIA and alpha-actinin 4 molecules, and CIB1 knockdown reduced EphA2's association with myosin IIA and alpha-actinin 4. Collectively, these studies revealed for the first time that CIB1 plays a role in virus entry and macropinocytosis, and suggested that KSHV utilizes CIB1 as one of the key molecule(s) to coordinate and sustain the EphA2 mediated signaling involved in its entry, and CIB1 is an attractive therapeutic target to block KSHV infection.
Published in the journal:
. PLoS Pathog 10(2): e32767. doi:10.1371/journal.ppat.1003941
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003941
Summary
KSHV envelope glycoproteins interact with cell surface heparan sulfate and integrins, and activate FAK, Src, PI3-K, c-Cbl, and Rho-GTPase signal molecules in human microvascular dermal endothelial (HMVEC-d) cells. c-Cbl mediates the translocation of virus bound α3β1 and αVβ3 integrins into lipid rafts (LRs), where KSHV interacts and activates EphrinA2 (EphA2). EphA2 associates with c-Cbl-myosin IIA and augmented KSHV-induced Src and PI3-K signals in LRs, leading to bleb formation and macropinocytosis of KSHV. To identify the factor(s) coordinating the EphA2-signal complex, the role of CIB1 (calcium and integrin binding protein-1) associated with integrin signaling was analyzed. CIB1 knockdown did not affect KSHV binding to HMVEC-d cells but significantly reduced its entry and gene expression. In contrast, CIB1 overexpression increased KSHV entry in 293 cells. Single virus particle infection and trafficking during HMVEC-d cell entry was examined by utilizing DiI (envelope) and BrdU (viral DNA) labeled virus. CIB1 was associated with KSHV in membrane blebs and in Rab5 positive macropinocytic vesicles. CIB1 knockdown abrogated virus induced blebs, macropinocytosis and virus association with the Rab5 macropinosome. Infection increased the association of CIB1 with LRs, and CIB1 was associated with EphA2 and KSHV entry associated signal molecules such as Src, PI3-K, and c-Cbl. CIB1 knockdown significantly reduced the infection induced EphA2, Src and Erk1/2 activation. Mass spectrometry revealed the simultaneous association of CIB1 and EphA2 with the actin cytoskeleton modulating myosin IIA and alpha-actinin 4 molecules, and CIB1 knockdown reduced EphA2's association with myosin IIA and alpha-actinin 4. Collectively, these studies revealed for the first time that CIB1 plays a role in virus entry and macropinocytosis, and suggested that KSHV utilizes CIB1 as one of the key molecule(s) to coordinate and sustain the EphA2 mediated signaling involved in its entry, and CIB1 is an attractive therapeutic target to block KSHV infection.
Introduction
Kaposi's sarcoma-associated herpes virus or human herpes virus 8 (HHV-8), a member of the lymphotrophic (γ2) herpesvirus subfamily, is etiologically linked to endothelial cell neoplasm Kaposi's sarcoma (KS), and B-cell neoplasms primary effusion lymphoma (PEL) or body cavity based B-cell lymphoma (BCBL), and multicentric Castleman's disease (MCD) [1], [2], [3]. KSHV infects a variety of target cells in vitro, including endothelial cells, B cells, monocytes, epithelial cells, and keratinocytes [4], and establishes default latent infection [3], [4].
Entry into the target cells is the most crucial step in the establishment of a successful infection for all viruses. Entry by KSHV is a complex multistep process involving a series of temporal interactions between multiple host cell surface molecules with multiple envelope glycoproteins of KSHV [4], [5], [6], [7]. KSHV binds to the human microvascular dermal endothelial (HMVEC-d) and human foreskin fibroblast (HFF) cells through an initial attachment to cell surface heparan sulfate (HS) molecules via a charge-based interaction between KSHV glycoproteins gB, gpK8.1A, ORF4, and gH which is blocked by the pretreatment of virus with soluble heparin [6], [8], [9], [10], [11]. Subsequent interaction of KSHV with multiple cell surface integrin receptors (α3β1, αVβ3, and αVβ5) induces the phosphorylation of host cell pre-existing focal adhesion kinase (FAK) signaling molecule and subsequent activation of Src, PI3-K, and Rho-GTPases (RhoA, Rac, and Cdc-42) effector signal molecules leading into actin polymerization dependent virus entry [12], [13], [14], [15], [16], [17]. Activation of diaphanous-2 by RhoA-GTPase induces microtubule acetylation, which facilitates the rapid transport of KSHV capsid towards the nucleus via dynein motor proteins [16]. Other downstream molecules activated by KSHV such as PKC-ζ, MEK, ERK1/2, and NFkB are essential for the initiation of viral and host gene expression early during infection [18], [19]. These studies revealed a novel paradigm that by interacting with integrins and a family of functionally related molecules at the cell surfaces early during infection, KSHV utilizes ligand mimicry as an opportunistic mechanism to subvert host signal molecules for its entry and successful infection. Further understanding of signal induction and host cell molecules modulated by KSHV are essential to develop designer drugs that can successfully block the entry and infection of KSHV.
KSHV exploits different entry pathways depending on the cell type. We have shown that KSHV productive entry in HFF cells occurs via clathrin mediated endocytosis [20] whereas macropinocytosis is utilized in HMVEC-d cells [15]. KSHV's interactions with receptors and clustering in HMVEC-d cells activated c-Cbl, an adaptor molecule, which plays a critical role in regulating KSHV induced membrane dynamicity by promoting actin-myosin rearrangement and macropinosome formation [21]. c-Cbl selectively monoubiquitinated and translocated KSHV bound α3β1 and αVβ3 integrins rapidly into the lipid raft (LR) region which resulted in the productive macropinocytic entry of virus, whereas non-LR (NLR) associated αVβ5 was polyubiquitinated leading to clathrin mediated entry that was targeted to lysosomes [22].
KSHV translocated into the LRs of HMVEC-d cells interacted with receptor tyrosine kinase EphA2 [23], and studies by us and others demonstrated that EphA2 is also one of the cellular entry receptor(s) for KSHV [24], [25]. We have demonstrated that KSHV interaction with HMVEC-d cell surface EphA2 results in the recruitment of the macropinosome associated c-Cbl-integrin-myosin IIA-complex in the LR regions as well as the amplification of Src, PI3K, and c-Cbl activation which resulted in the promotion of macropinocytic entry and infection [23]. In contrast, KSHV induced EphA2 coordinated integrin-associated signal assembly and amplification occurred in the NLR regions of HFF cells, leading into the c-Cbl modulated EphA2 polyubiquitination (K63 type) that regulated the clathrin mediated productive KSHV entry in HFF cells [26]. Taken together, these studies demonstrated that KSHV induced differential regulation of cellular signaling via EphA2 and c-Cbl is crucial for endocytosis during de novo infection.
Physiological macropinocytosis requires a subset of cell surface proteins and differential recruitment of the bulk of signal molecules to the plasma membrane in a temporal manner in response to a translocation of membrane lipid composition [27], [28]. Hence, identification of the specific regulator(s) promoting actin rich membrane protrusion formation during pathogen invasion has always been challenging [29]. Adaptor molecule c-Cbl and RTK EphA2 has been assigned to recruit multifunctional signal molecules that include several kinases, phosphatases, ubiquitin ligases, GTPases, cellular adaptors, and many other proteins to assemble a supra-molecular signalosome in non-viral systems [30]. Since c-Cbl and EphA2 are two important players in KSHV induced membrane blebbing, we explored the role of additional candidate signal molecule(s) that associates with LR regions of HMVEC-d cells to regulate macropinosome assembly and amplification of the integrin-EphA2 signal axis.
Calcium and integrin binding protein-1 (CIB1), a 22-kDa protein, was originally identified as a platelet specific integrin αIIb cytoplasmic tail binding partner and later observed to inhibit αIIbβ3 activation in megakaryocytes [31], [32]. Subsequent studies demonstrated that CIB1 is widely expressed in different human tissues [33] with a variety of binding partners including many kinases. CIB1 has been shown to interact with p21-activated kinase (PAK1), FAK, two polo-kinases (Plk) Fnk and Snk, DNA dependent protein kinses (DNA-PKcs), sphingosine kinase 1 (SK1), presinillin-2, Rac3, InsP3 receptor, and Pax3 [34], [35], [36], [37], [38], [39], [40], [41]. Studies have also shown that CIB1 is an enhancer of FAK, Erk1/2, and PAK1 kinase actions [42], [43]. Sequence analysis and the crystal structure of CIB1 revealed the presence of an N-terminal myristoylation sequence [44]. These observations corroborated the studies demonstrating stimulation dependent CIB1 translocation into detergent insoluble cytoskeletal fractions in platelets [33] and CIB1 directed translocation of sphingosine kinase1 (SK1) from the cytosol to plasma membranes of HeLa and HEK293T cells, thereby enhancing SK1 enzymatic activity [45]. During adhesion dependent signaling, when endothelial cells, fibroblasts and platelets stimulated with fibrinogen (Fg) were grown in fibronectin coated plates, the presence of CIB1 in filamentous actin membrane protrusions in an intact cytoskeleton dependent manner was observed [43], [46]. In vitro studies revealed that CIB1 functions as a regulator of platelet motility, spread and ploidy, megakaryocyte adhesion, and migration of endothelial cells [43], [47].
While in vitro binding assays evidenced that CIB1 is incapable of binding to any cellular integrins other than platelet specific αIIbβ3 integrin [32], CIB1 overexpressing CHO cell migration was inhibited upon Fn receptor function blocking anti-α5β1 antibody treatment [42]. A previous report also exhibited indirect association between CIB1 and integrins via associated signal component(s) such as FAK to facilitate downstream signaling cascade [42]. Upon Fn stimulation, CIB1 overexpressing cells exhibited increased migration in a Src, PI3-K, and Erk1/2 dependent manner [43]. Role(s) of cell surface integrins and membrane recruited Src and PI3-K kinases has been extensively studied during physiological as well as KSHV endocytosis processes [21], [48], [49], [50], [51], [52]. Although CIB1 has been implicated to promote integrin associated and immediate downstream kinase actions during cell migration [42], [43], whether the effector kinases are influenced by CIB1 to regulate endocytosis has not been studied before.
Owing to CIB1's expression in endothelial cells, localization in the actin membrane protrusions, and its ability to regulate directly or indirectly integrin associated and immediate downstream kinase actions, we hypothesized that CIB1 could be a potential candidate molecule recruited to the macropinosome complex during the KSHV endocytosis process. In this study conducted to test this hypothesis, we demonstrate that early during KSHV infection of HMVEC-d cells, CIB1 associates with membrane LRs, KSHV in induced membrane blebs, entry receptor EphA2, and bleb associated critical entry mediators such as Src, PI3-K, c-Cbl, and also with KSHV containing early macropinosomes. More importantly, knocking down CIB1 significantly inhibited KSHV entry and de novo infection due to a substantial reduction in EphA2 induced signal amplification. Our study also demonstrates that KSHV infection induced the simultaneous association of CIB1 and EphA2 with cytoskeletal binding partners such as myosin IIA and alpha-actinin4. These data strongly suggest the potential mechanism of how CIB1 facilitates KSHV macropinocytosis and demonstrates a new role for CIB1 as a modulator of pathological macropinocytosis and virus entry.
Materials and Methods
Cells and virus
Primary HMVEC-d cells (CC-2543; Clonetics, Walkersville, MD) were grown in endothelial cell basal medium 2 (EBM2; Clonetics). KSHV carrying BCBL-1 cells were grown in RPMI with 10% total bovine serum and 1% penicillin-streptomycin antibiotic solution [53]. Induction of KSHV lytic cycle in BCBL-1 cells by TPA (20 ng/ml), supernatant collection, and virus purification procedures were described previously [53]. DNA from purified KSHV was extracted and quantitated by real-time DNA-PCR using primers amplifying the KSHV ORF73 gene as described previously [54]. The same batch of purified KSHV was used for all sets of experiments.
Herpes simplex virus type-1 (HSV-1; KOS strain) virus stock (1.6×109 pfu/ml) that was propagated and quantified by plaque assay in vero cells was a generous gift from Dr. Karen Johnson (RFUMS) [55]. HSV-1 DNA copy number was quantified using real-time DNA-PCR with primers for HSV-1 envelope glycoprotein gB (forward primer, 5′ - TGTGTACATGTCCCCGTTTTACG-3′; reverse primer, 5′-GCGTAGAAGCCGTCAACCT-3′) [56].
DiI-labeled KSHV
The lipophilic carbocyanine dyes, DiI-1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate –[DiIC183] was used to label KSHV particles according to previously established methods [22], [57]. Briefly, 200 µl of 1 mg/ml of purified KSHV in TNE-30% sucrose buffer (TNE buffer: 0.01 M Tris-HCl, pH 7.4, 0.15 M NaCl, 0.05% EDTA) was incubated with 25 mM DiI in DMSO for 2 hours at RT with gentle mixing. To remove the unbound dye, a step 10%, 30%, and 55% w/v sucrose density gradient was used. The DiI labeled KSHV was layered on top of the 10% sucrose cushion and centrifuged at 55,000× g for 90 min at 4°C. The labeled virus was collected from the top of the 55% sucrose layer and passed through a 0.22 µm filter prior to use.
BrdU labeled KSHV
The thymidine analog 5-bromo-2-deoxyuridine (BrdU labeling reagent; Ref. 00103, Invitrogen) was filter sterilized. To metabolically label viral DNA during the synthetic phase of viral lytic replication, 1∶100 dilution of BrdU was added to the BCBL-1 cells at the day of TPA induction and on the second day of induction. BrdU labeled virus supernatant was collected on day 5 and purified as per procedures described previously [53].
Generation of HMVEC-d cells expressing CIB1 shRNA
A pool of lentivirus shRNA specific for human CIB1 and non-specific control shRNA were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). HMVEC-d cells were transduced with control lentivirus shRNA or CIB1 lentivirus shRNA according to the manufacturer's instructions and selected by puromycin hydrochloride (10 µg/ml).
Antibodies and reagents
Mouse monoclonal anti-KSHV gpK8.1A (4A4), LANA1, and rabbit anti-gB (UK-218) antibodies were raised in our laboratory [10], [14], [58], [59]. Rabbit anti-β3, phospho-EphA2, EphA2, Rab5, vimentin, annexin A2, phospho-Src, Src, myosin IIA, and mouse anti-phospho-Erk1/2 antibodies were from Cell Signaling Technology, Danvers, MA. Rabbit anti-CIB1 antibody was from Protein Tech, Chicago, IL. Mouse anti-CIB1 and Erk1/2, and rabbit anti-alpha actinin4 were from Millipore, Billerica, MA. Rabbit anti-caveolin-1, and mouse anti-β-actin and tubulin antibodies were from Sigma-Aldrich, St. Louis, MO. p85 PI3K were from BD Biosciences, San Diego, CA. Rabbit anti-transferrin and goat anti–flotillin-1 antibodies were from Abcam, Boston, MA. Mouse anti-Myc antibodies were from Santa Cruz. Anti-mouse and anti-rabbit antibodies linked to horseradish peroxidase were from KPL Inc., Gaithersburg, MD. 4′,6-diamidino-2-phenylindole (DAPI), rhodamine-conjugated dextran or transferrin, Alexa 488 conjugated LysoTracker, Alexa 488 conjugated phalloidin, Alexa 594 or 488 anti-rabbit, anti-mouse, and Alexa 633 anti-mouse secondary antibodies were from Molecular Probes, Invitrogen. Heparin was from Sigma-Aldrich, St. Louis, MO. EGTA-AM was from Chemicon International, Temecula, CA. Protein G Sepharose CL-4B was from Amersham Pharmacia Biotech, Piscataway, NJ. Purified recombinant baculovirus-expressed gBΔTM and ΔTMgpK8.1A proteins were generated in our laboratory [7], [10], [60].
Western blotting
Cells were lysed in RIPA buffer (15 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, 2 mM phenylmethylsulfonyl fluoride, and protease inhibitor mixture (Sigma)), sonicated, and centrifuged at 10,000 rpm at 4°C for 10 min. Protein concentrations were estimated by BCA protein assay reagent (Pierce, Rockford, IL). Equal concentrations of proteins were separated on SDS-PAGE, transferred to nitrocellulose and probed with the indicated specific primary antibodies followed by incubation with species-specific HRP-conjugated secondary antibody and chemiluminiscence based detection of immunoreactive protein bands (Pierce, Rockford, IL) according to the manufacturer's protocol. The bands were scanned and quantitated using the FluorChem FC2 and Alpha-Imager Systems (Alpha Innotech Corporation, San Leonardo, CA), with additional quantitation by ImageJ software.
Measurement of KSHV binding and entry by real-time DNA-PCR
For measuring binding and entry, HMVEC-d cells were either mock or KSHV infected (20 DNA copies per cell) at 4°C for 1 hour and 37°C for 2 hours, respectively. For binding experiments, cells were washed with HBSS to remove unbound virus and lysed immediately followed by DNA extraction using a DNeasy Kit (Qiagen, Valencia, CA). For entry experiments, cells were washed with HBSS, bound and non-internalized virus was removed by treatment with trypsin-EDTA for 5 min at 37°C [54], and DNA was extracted using the DNeasy Kit. For both binding and entry assays, extracted DNA was quantitated by amplification of the ORF73 gene by real-time DNA-PCR. The KSHV ORF73 gene cloned in the pGEM-T vector (Promega) was used as the external standard. A standard curve was generated and the relative copy numbers of viral DNA were calculated from the threshold cycle (Ct) value. A paired t test was used between control and shRNA transduced cells to obtain the P values for the percentage inhibition in entry.
Electron microscopy
HMVEC-d cells grown in 6 cm petri-dishes were washed three to four times with medium without serum. The cells were infected with purified KSHV (20 DNA copies/cell) at 37°C for 5 and 10 min in serum free medium. After each time point, cells were washed, fixed immediately with 2% paraformaldehyde and 2.5% gluteraldehyde in 0.1M Cacodylate buffer at RT by adding gently from the edge of the dish and kept for 20 to 30 minutes at RT. These were postfixed in 1% osmium tetroxide, dehydrated in a graded ethanol series, and embedded in 812 resin. Thin sections were made and visualized under a JEOL 100CXII transmission electron microscope.
Measurement of KSHV gene expression by real-time RT-PCR
To quantify KSHV gene expression, total RNA was extracted from mock or KSHV infected cells using an RNeasy Kit (Qiagen) according to the manufacturer's protocol. Isolated RNA was subjected to one step real time-RT-PCR using gene specific primers and Taqman probes (EZ RT-PCR core reagents, Applied Biosystems, Branchburg, NJ). The relative copy numbers of the transcripts were calculated from the standard curve, which was derived using the Ct values for different dilutions of in vitro-transcribed transcripts [54]. These values were normalized to each other using the values of GAPDH control reactions.
Measurement of HSV-1 gene expression by reverse transcription and q-RT-PCR
To quantify HSV-1 gene expression, total RNA was purified with RNeasy Kit (Qiagen) according to the manufacturer's instructions followed by a DNase digestion step (RQ-1 RNase free DNase product, Promega). Total RNA from HSV-1 infected shControl or shCIB1 transduced HMVEC-d cells was reverse transcribed with a high-capacity cDNA reverse transcription kit according to the suppliers (Applied Biosystems). Equal volumes of prepared cDNA were used for quantification of HSV-1 gene ICP0 and ICP4 transcripts with the Power SYBR Green PCR master mix (Applied biosystems) according to the manufacturer's protocol. The primers used were:
ICP0 - 5′ - AAGCTTGGATCCGAGCCCCGCCC-3′ (forward), and
5′-AAGCGGTGCATGCACGGGAAGGT-3′ (reverse);
ICP4 - 5′ - GACGTGCGCGTGGTGGTGCTGTACTCG-3′ (forward), and
5′ - GCGCACGGTGTTGACCACGATGAGCC-3′ (reverse) [61].
Samples were normalized relative to tubulin Ct. and primer pairs used for tubulin transcript detection were 5′-TCCAGATTGGCAATGCCTG-3′ (forward), and 5′-GGCCATCGGGCTGGAT-3′ (reverse). Relative changes in gene expression were analyzed using the 2−ΔΔCt method.
Immunofluorescence microscopy
HMVEC-d cells were grown in eight well chamber slides (Nalge Nunc International, Naperville, IL). Mock or KSHV infected cells were fixed with 4% paraformaldehyde for 15 min at room temperature followed by permeabilization (with 0.2% Triton X-100 for 5 min) and blocking with Image-iTFX signal enhancer (Invitrogen) for 15 min. The cells were then stained with specific primary antibodies and corresponding species-specific fluorescent dye conjugated secondary antibodies. For colocalization with dextran, transferrin, and LysoTracker, cells were incubated with the fluid-phase marker dextran Texas Red (40-kDa, 0.5 mg/ml; Invitrogen), or Alexa 594 transferrin (35 µg ml−1; Invitrogen), or Alexa 488 conjugated LysoTracker (1∶100, Invitrogen) at 37°C in the presence or absence of KSHV, followed by immunostaining with appropriate antibodies. Cells were imaged with a Nikon fluorescence microscope equipped with a Metamorph digital imaging system. Differential Interference Contrast (DIC) images were acquired with objectives equipped with DIC optics.
For confocal analysis, Olympus FV10i microscope was used for imaging and signal intensity line-scan analysis was performed on the enlarged regions of colocalization using the Fluoview1000 (Olympus) software.
Quantitative analysis of KSHV entry by immunofluorescence
Control or shRNA transduced cells were infected with BrdU labeled KSHV (20 DNA copies per cell) at 37°C for 30 min. Cells were washed with HBSS and treated with trypsin-EDTA for 5 min at 37°C, PFA-fixed, and Triton X-100 permeabilized according to the above mentioned protocol followed by a denaturation step with 4N hydrochloric acid (HCl) for 10 minutes at RT to expose incorporated BrdU residues [62]. Post-blocking with Image-iTFX signal enhancer (Invitrogen) for 15 min, cells were stained with mouse monoclonal anti-BrdU primary antibody followed by Alexa 488 conjugated secondary antibody staining and analyzed by fluorescence microscopy. At least five different fields each containing multiple cells were observed and analyzed as a proportion of DAPI-stained cells. A paired t test was used between control and shRNA transduced cells to obtain the P values for the percentage inhibition in entry.
Quantitative analysis of dextran uptake by flow cytometry
For quantitative analysis of dextran uptake, control or shRNA transduced HMVEC-d cells were incubated with FITC-dextran (40-kDa, 0.5 mg/ml; Invitrogen) and KSHV for 30 min at 37°C. Cells were harvested and fixed as described above and analyzed by flow cytometry [21]. To quantitate internalized KSHV and dextran, mean fluorescence intensity was determined by a Becton Dickinson LSRII flow cytometer and FlowJo software.
Immunoprecipitation
Cells were lysed in lysis buffer (25 mM Tris-HCl, pH: 7.5, 150 mM NaCl, 1% NP40, 2 mM EDTA, 10% Glycerol, and protease inhibitor mixture) and 200 µg of cell lysates prepared following the post-lysis steps as described above in the Western Blotting section. Lysates were incubated overnight with immunoprecipitating antibody at 4°C and the resulting immune complexes were captured by protein G-sepharose and analyzed by Western blots, using specific detection antibodies.
Lipid raft extraction and characterization
Lipid rafts were extracted by the non-detergent density gradient approach according to the manufacturer's protocol for the caveola/raft isolation kit (Sigma) [22], [23], [26], [63]. Briefly, uninfected and KSHV infected cells were lysed in mild 0.5 M sodium bicarbonate buffer [pH 11.0] (contains 500 mM sodium bicarbonate, 2 mM EDTA, 1 mM NaF, 1 mM orthovanadate, and sodium protease inhibitor cocktail) for 15 min at 4°C. Cell lysates were homogenized in a precooled Dounce homogenizer by 10 strokes and a subsequent sonication step for 10 seconds was done. OptiPrep reagent with different concentrations was used to prepare a discontinuous density gradient of five layers such as 35%, 30%, 25%, 20%, and 0% where two ml of 35% OptiPrep was placed as the bottom most gradient layer of the pre-cooled ultracentrifuge tube. Each OptiPrep gradient was layered over the other using a Pasteur pipette. The tubes were subjected to ultra-centrifugation at 45,000 rpm for 4 hr in a Beckman SWI 55 rotor. One ml of fractions were collected from the top of the ultra-centrifuge tube and pooled. Purity of the lipid-raft containing fractions was characterized by the presence of caveolin-1, and non-lipid rafts were confirmed by the presence of CD-71 by dot blot analysis.
Mass spectrometry
HMVEC-d cells were serum-starved for 8 hours and either mock or KSHV (20 DNA copies per cell) infected for 10 min. 200 µg of cell lysates were immunoprecipitated overnight at 4°C with mouse anti-CIB1 or rabbit anti-EphA2 antibodies. Immunoprecipitates were resolved using 4–20% gradient SDS-PAGE (Biorad, Hercules, CA) gel and were stained with Coomassie blue. The bands of interest specific for CIB1 and EphA2 immunoprecipitates were analyzed by mass spectrometry using an LC-ESI (electrospray ionization)-MS based approach at the Midwest Proteome Center, Rosalind Franklin University of Medicine and Sciences.
Plasmids and transfection
HEK293 cells were transiently transfected with Myc tagged CIB1 overexpressing plasmid (Addgene, Cambridge, MA). Transfection was performed using 5 µg of plasmid DNA, Lipofectamine 2000 (Invitrogen) and Opti-MEM (Invitrogen) according to the manufacturer's instructions. After 48 h, the cells were serum starved and either mock infected or infected with KSHV (30 DNA copies/cell) for 5 min. Cell lysates were prepared for use in immunoblotting, KSHV binding, and entry assays.
Results
Calcium and integrin binding protein 1 (CIB1) knockdown inhibits KSHV entry and de novo infection in HMVEC-d cells
To determine whether CIB1 plays a role during KSHV infection, CIB1 lentivirus encoding shRNA was used to knockdown CIB1 activity in HMVEC-d cells. CIB1 specific shRNA inhibited >90% of CIB1 protein expression as detected by Western blot with specific anti-CIB1 antibodies (Fig. 1A). The CIB1-shRNA pool did not have any off-target effects at the protein level on tubulin or KSHV entry receptor integrin β3 (Fig. 1A). In addition, we did not observe any reduction at the protein level of several entry associated signaling molecules in CIB1-shRNA transduced cells as compared to the effect of control shRNA-transduced cells. These results demonstrated that the pool of CIB1-shRNAs used here had minimal off-target effects.

To determine the stage of KSHV infection of HMVEC-d cells at which CIB1 plays a role, we first examined KSHV binding in control and CIB1-shRNA transduced cells. Almost similar levels of KSHV binding was detected in both CIB1-shRNA (∼10,000 bound DNA copies) and control shRNA (11,000 bound DNA copies) transduced cells (Fig. 1B). This demonstrated that CIB1 did not affect virus binding to its cell surface receptors. In contrast, when internalized viral DNA copies were measured by real-time DNA-PCR, compared to control shRNA, CIB1-shRNA significantly inhibited (∼70%) KSHV entry in HMVEC-d cells (Fig. 1C).
To track single virus particles by immunofluorescence assay, we infected HMVEC-d cells with DiI-labeled KSHV (viral envelope labeling; Fig. 1.D) or BrdU-labeled KSHV (viral DNA labeling; Figs. 1.E(i), and 1.E(ii)). As shown in Fig. 1D and 1E, we observed a consistent reduction in internalized KSHV in CIB1-shRNA transduced cells compared to control shRNA transduced HMVEC-d cells, which further suggested that CIB1 plays a role in KSHV entry. DiI or BrdU labeling procedures did not affect virus infectivity [22], [62].
To determine the effect of CIB1-shRNA on KSHV infection further, untransduced, control shRNA, and CIB1-shRNA transduced cells were infected with KSHV for 48 h and latent ORF73 gene expression was determined by real-time RT-PCR. In CIB1-shRNA transduced cells infected with KSHV, we observed >80% inhibition in ORF73 gene expression compared to control shRNA-KSHV cells (Fig. 2.A). The real-time RT-PCR results were also validated by the >85% reduction in the characteristic nuclear punctate LANA-1 (ORF73) staining after 48 h p.i. in CIB1 knockdown cells in contrast to control shRNA transduced cells (Figs. 2.B(i), and 2.B(ii)).

We further examined the effect of shCIB1 on another herpes virus family member, such as α-herpesviridae HSV-1 gene expression to determine whether the CIB1 action is specific to γ-herpesviridae KSHV infection of HMVEC-d cells. To test this, control shRNA, and CIB1-shRNA transduced HMVEC-d cells were infected with HSV-1 for 8 h and the expression of HSV-1 immediate early genes ICP0 and ICP4 expression was measured by quantitative real-time RT-PCR (Fig. 2.C). Both ICP0 and ICP4 transcript levels in CIB1-shRNA HMVEC-d cells were unchanged compared to sh-control HMVEC-d cells indicating that CIB1 is specifically involved in KSHV entry and primary infection. Taken together, these studies demonstrated that CIB1 plays a significant role in KSHV entry in the HMVEC-d cells.
CIB1 overexpression increases KSHV entry in 293 cells
Since CIB1-shRNA inhibited KSHV entry, we next determined whether CIB1 overexpression in 293 cells increased the entry of KSHV. Overexpressing CIB1 in natural target cells such as HMVEC-d is a better system to study the additive effect in KSHV entry. However, due to poor transfection efficiency in HMVEC-d cells, it was difficult to detect a sufficient increase in KSHV entry upon CIB1 overexpression by KSHV entry assays. Hence, we utilized the 293 cells only to reinforce the data with HMVEC-d cells. In addition, 293 cells have indeed been shown in several studies by us and others to be an accepted target to test and monitor the entry and infection of KSHV [9], [13], [14], [64], [65], [66]. C-terminal Myc-tagged CIB1 was transiently overexpressed in 293 cells using CIB1-Myc plasmid and CIB1 expression was checked by Western blot with anti-Myc antibodies with β-actin as a loading control (Fig. 2.D). When KSHV entry was assessed by internalized viral DNA copies, CIB1 overexpression increased virus entry by ∼60% compared to entry in control pcDNA transfected 293 cells (Fig. 2.E).
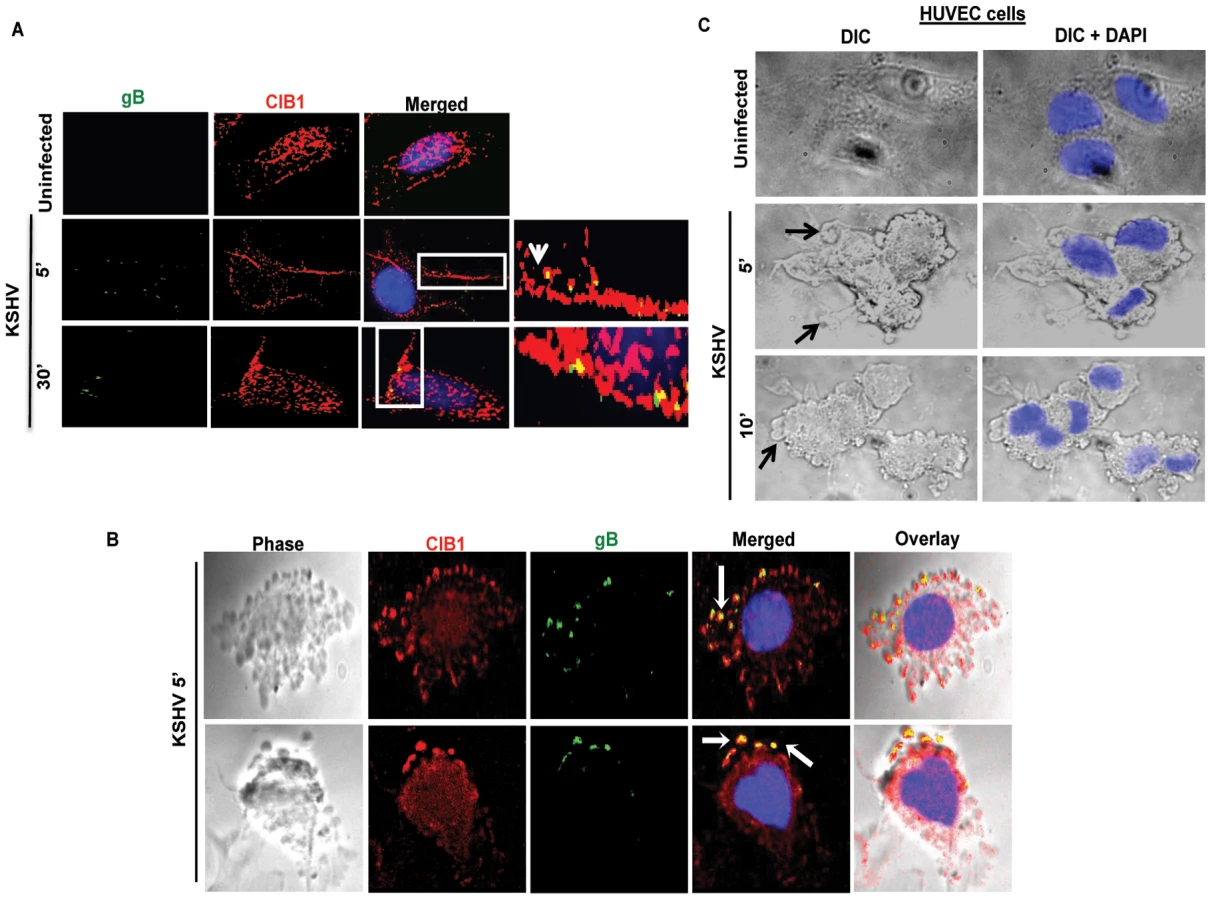
CIB1 colocalizes with KSHV and with KSHV induced membrane blebs in infected HMVEC-d and HUVEC cells
Several studies including ours have shown that KSHV enters the HMVEC-d, HUVEC, HFF, 293, THP-1 and B cells via endocytosis [9], [15], [20], [67]. Our earlier studies have demonstrated that KSHV exploits bleb mediated macropinocytosis to enter and establish infection in HMVEC-d cells [21]. Since we observed that CIB1-shRNA significantly inhibited KSHV infection in HMVEC-d cells, we tested whether CIB1 colocalized with KSHV in virus induced membrane bleb protrusions early during infection. In immunofluorescence studies, CIB1 colocalized with KSHV stained for viral envelope glycoprotein gB at 5 and 30 min p.i. (Fig. 3.A, white arrows). The DIC analysis also clearly demonstrated the CIB1-KSHV colocalization in cell membrane blebs at 10 min p.i. (Fig. 3.B, white arrows). DIC microscopic analysis also revealed bleb induction by KSHV in infected HUVEC cells (Fig. 3C, black arrows).
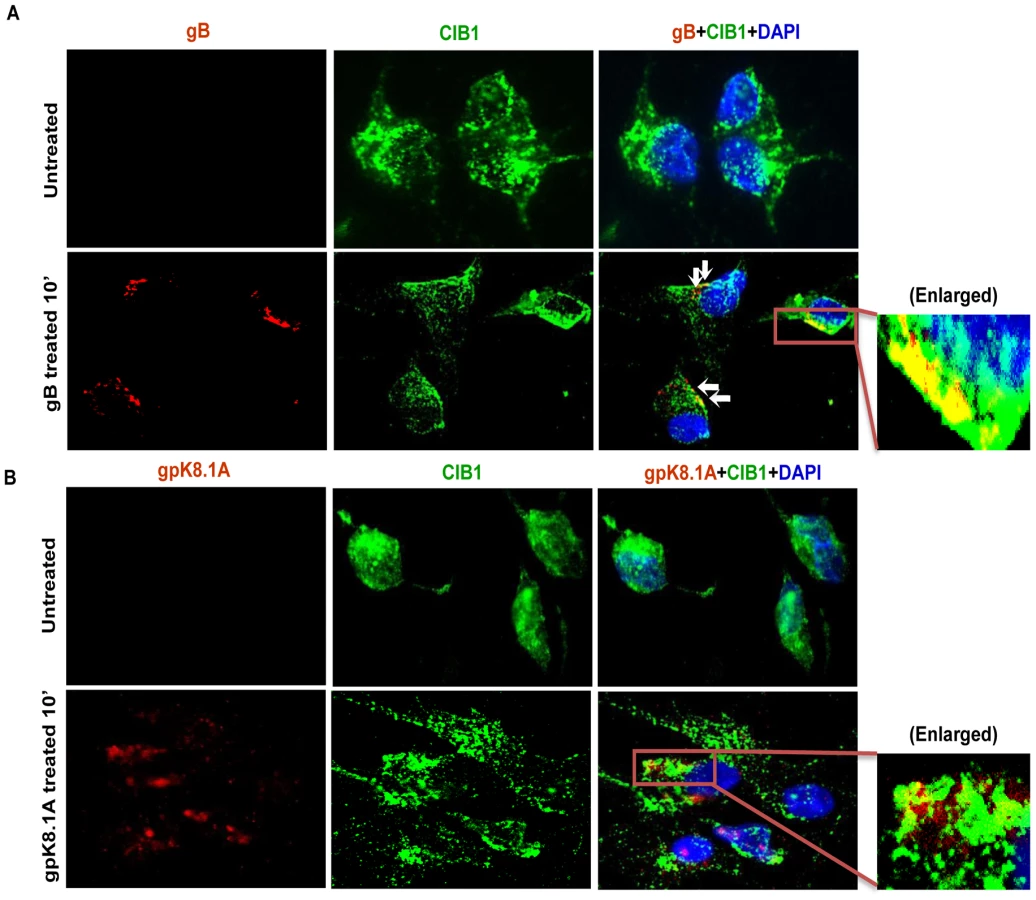
To determine whether purified recombinant KSHV envelope glycoproteins gB and gpK8.1A can show a similar effect, HMVEC-d cells were left untreated or incubated either with 3 µg of purified baculovirus-expressed soluble KSHV gBΔTM or ΔTMgpK8.1A proteins (without transmembrane and carboxy domains) [7], [10], [51], [60] at 4°C for 1 h, followed by 37°C for 10 min and analyzed by immunofluorescence. Addition of KSHV gBΔTM or ΔTMgpK8.1A proteins induced CIB1 rearrangement towards the cell periphery (Figs. 4.A and 4.B), and CIB1 colocalized substantially with gBΔTM (Fig. 4.A, white arrows) while colocalization with ΔTMgpK8.1A was less prominent (Fig. 4.B). This observation suggested that KSHV glycoprotein gB interacting with cell surface integrin molecules could be the major inducer for CIB1 engagement to the KSHV entry associated events.
In our earlier electron-microscopic studies of HMVEC-d cells, we observed the endocytic vesicles containing internalized KSHV particles [15]. Moreover, earlier morphological studies [21] as well as results shown in Fig. 3.B also identified characteristic bright phase macropinocytic membrane blebs associated with KSHV particles during de novo infection in HMVEC-d cells [21]. Consistent with these findings, when we conducted transmission electron microscopic studies of infected cells at 5 to 10 min p.i., we observed the association of enveloped KSHV particles with cell membrane protrusions and at various stages of wrapping and engulfment of KSHV by these membrane protrusions (Fig. 5, panel A-H, red arrows) resulting in KSHV in large endocytic vesicles (Fig. 5, panel I). Together, these evidences further validated that the induction of membrane protrusions is a critical event towards successful KSHV entry into endothelial cells.

Next, we examined the KSHV infected HMVEC-d and HUVEC endothelial cells for CIB1 localization in actin protrusions (phalloidin). We observed that compared to mock infected endothelial cells, as early as 5 min p.i., KSHV induced significant cell surface actin filament clusters at 5 min p.i. (Figs. 6.A and 6.B), which was consistent with our previous studies [15], [21]. CIB1 was prominently colocalized with actin in KSHV induced membrane blebs at 5 and 10 min p.i. in both HMVEC-d and HUVEC cells (Figs. 6.A and 6.B, white arrows). This suggested that CIB1 could be playing a role in KSHV infection induced actin polymerization, bleb induction and potentially in macropinocytosis.

CIB1 associates with KSHV productive trafficking in HMVEC-d cells
The colocalization of CIB1 with KSHV in membrane bleb protrusions prompted us to track KSHV and associated CIB1 during the immediate post-entry stage in infected HMVEC-d cells. We have shown earlier that during de novo KSHV infection in HMVEC-d cells, the majority of KSHV associated with blebs was sorted into Rab5 positive early endosomes leading to productive trafficking [23], whereas the few virus particles entering via clathrin-mediated endocytosis were directed to the non-infectious lysosomal degradative pathway [22]. We performed a triple colocalization assay for DiI-labeled KSHV (red), CIB1 (blue), and early endosomal or lysosomal compartment (green) to track internalized viruses associated with CIB1 and infected cell endosomes (Figs. 7.A and 7.B).

In uninfected HMVEC-d cells no appreciable colocalization of CIB1 was observed either in the early endosomal (Fig. 7.A, upper right most enlarged panel) or lysosomal compartments (Fig. 7.B, upper right most enlarged panel). After 10 min post-KSHV infection of HMVEC-d cells, minimal amounts of DiI-labeled KSHV did colocalize with LysoTracker (Fig. 7.A.(i), left most enlarged panel) whereas the majority of virus colocalized significantly with Rab5 positive early endosomes as shown by the yellow spots (Fig. 7.B.(i), left most enlarged panel). CIB1 (red) substantially colocalized with DiI-labeled KSHV (red) (Figs. 7.A.(iii), and 7.B.(iii), left most enlarged panels) as shown by the magenta spots at 10 min p.i., but not with LysoTracker (Fig. 7.A.(ii), left most enlarged panel). Hence, no triple colocalization (white spots) was observed between virus, CIB1, and lysosomal compartments (Fig. 7.A, right most enlarged panel). Line scan analysis (Fig. 7.A.(iv), bottom most panel) did not show any synchronous peak pattern for all three staining KSHV (red), CIB1 (blue), and LysoTracker (green), which suggested that CIB1 was not associated with non-productive KSHV trafficking. In contrast, CIB1 substantially colocalized with virus and Rab5 positive vesicles (Fig. 7.B.(ii), left most enlarged panel) at 10 min p.i., and prominent triple colocalization white spots were observed (Fig. 7.B, right most enlarged panel). Line scan analysis (Fig. 7.B.(iv), bottom most panel) also showed a synchronous colocalization intensity for all three stainings, which strongly indicated the role of CIB1 in KSHV productive trafficking.
CIB1 colocalizes with macropinocytic marker dextran early during KSHV infection of HMVEC-d cells
To explore whether CIB1-actin colocalization in KSHV induced membrane blebs (Fig. 6) and CIB1 association with KSHV containing Rab5 positive vesicles (Fig. 7) is biologically associated with the macropinocytosis of virus, colocalizations between CIB1 and the macropinocytosis marker dextran were examined. Compared to uninfected cells, at 5, 10, and 30 min p.i., CIB1 strongly colocalized with internalized dextran molecules (Fig. 8.A, white arrows). No significant colocalization between CIB1 and clathrin endocytosis marker transferrin was observed in uninfected and in KSHV infected HMVEC-d cells (Fig. 8.B), which also supported our observation of the absence of colocalization between CIB1 and KSHV in lysosomal compartments (Fig. 7.A). These results demonstrated that CIB1 plays a role selectively in KSHV macropinocytosis and productive trafficking.

To further validate the role of CIB1 in KSHV macropinocytosis, we performed a quantitative dextran uptake assay by flow cytometry at 30 min p.i.. Compared to KSHV infection in control shRNA transduced cells, CIB1 knockdown HMVEC-d cells showed a significant decrease in dextran uptake as measured by mean fluorescence intensity (Fig. 9.A). These results clearly demonstrated that CIB1 knockdown impaired KSHV induced macropinocytosis and suggested that CIB1 plays a role in macropinocytosis of KSHV.

CIB1 regulates KSHV internalization through macropinocytic bleb induction
Rab5 is a well-known marker for early endosomes as well as early-internalized macropinosomes [68], and our present studies in Fig. 7B demonstrated the triple colocalization of KSHV-Rab5-CIB1 as early as 10 min following de novo infection in HMVEC-d cells (Fig. 7.B). To further verify that CIB1 knockdown inhibits KSHV entry and hence productive trafficking, we determined the presence of KSHV in Rab5 positive vesicles. In control shRNA transduced cells, we observed internalized virus particles predominantly localizing in Rab5 positive vesicles (Fig. 9.B, upper enlarged panel, white arrows). In contrast, in CIB1-shRNA transduced cells, virions were not internalized or associated with Rab5 positive vesicles (Fig. 9.B); instead they were localized on the periphery of plasma membranes (Fig. 9.B, lower enlarged panel, white arrows). Together with the dextran uptake analysis, these results confirmed that CIB1 modulates KSHV entry via macropinocytosis in HMVEC-d cells.
We next determined the status of KSHV infection induced membrane blebbing events in CIB1 knockdown HMVEC-d cells. In IFA analyses, we observed that CIB1 knockdown resulted in a robust reduction (∼60%) of KSHV induced blebbing events compared to control shRNA treated cells (Figs. 10.A, and 10.B). Collectively, these results demonstrated that CIB1 plays a significant role in KSHV infection induced actin cytoskeletal rearrangement, bleb induction and macropinocytosis.

An increased level of CIB1 associates with lipid rafts (LRs) of HMVEC-d cells early during KSHV infection
Our studies have demonstrated that LRs play critical roles in KSHV entry into HMVEC-d cells as they act as a signal hub to facilitate clustering of entry receptors and signal molecules to promote actin remodeling, bleb induction, macropinocytosis and productive infection. From the preceding studies we theorized that CIB1 might be mediating its role in KSHV induced actin polymerization, blebbing and entry via its temporal association with LRs upon KSHV infection. To analyze CIB1 association with LRs of infected cells, LR and non-lipid raft regions (NLR) were fractionated from mock and KSHV infected HMVEC-d cells at 5, 10, and 30 min p.i.. LR and NLR fractions were initially characterized by dot blot analyses for caveolin-1 for LR purity and CD71 for purity of NLR fractions (data not shown) as described previously [22], and pooled fractions were subsequently examined by Western blots (Fig. 11.A). Although CIB1 was associated with LR fractions in uninfected and infected cells, upon KSHV infection, CIB1 significantly concentrated in infected-cell LRs as early as 5 min p.i. which was sustained during the observed time point of 30 min p.i. (Fig. 11.A, LR lanes 1–4). CIB1 was also associated with NLR at 5 min p.i., which was reduced by 30 min p.i. (Fig. 11.A, NLR, lanes 1–4). Detection of CIB1 in uninfected LRs may be due to a basal level of constitutive targeting of CIB1 to the LRs for cell adhesion. The KSHV receptor EphA2 level was also determined by Western blot from the same fractions. Consistent with our previous finding [23], EphA2 was observed strictly in uninfected HMVEC-d cell LR (Fig. 11.A, LR lanes 1–4) but not in NLR fractions (Fig. 11.A, NLR, lanes 1–4), which also remained unchanged with infection. Analysis of total cell lysates demonstrated that CIB1 expression during early KSHV infection remained unchanged (Fig. 11.B).

To confirm further our biochemical data, HMVEC-d cells were mock or KSHV infected and LR and CIB1 association was examined by IFA (Fig. 11.C). A scattered pattern of CIB1 with minimal colocalization with flotillin-1 was observed in uninfected cells (Fig. 11.C, top panels). In contrast, as early as 5 and 10 min p.i., clustered CIB1 colocalized significantly with LR marker flotillin-1 at the cell surfaces as well as in the cytosol (Fig. 11.C, second and third panels, white arrows). Taken together, temporal enrichment of CIB1 in infected cell LRs strongly supported the studies shown in Figs. 1–11 suggesting that CIB1 plays a role in KSHV macropinocytic entry.
CIB1 associates with KSHV entry receptor EphA2 early during infection of HMVEC-d cells
KSHV bound integrins αVβ3 and α3β1 translocate to the LRs leading into the interaction with EphA2, macropinocytosis and infection of HMVEC-d cells [23]. Since our results demonstrated that infection also induced the enhanced association of CIB1 with LRs of infected cells and CIB1 plays a role in macropinocytosis, we rationalized that CIB1 might interact with LR associated KSHV entry receptors especially EphA2 early during infection. To test this, uninfected and control and CIB1-shRNA transduced cells infected with KSHV were immunoprecipitated with anti-CIB1 and control IgG antibodies and Western blotted for EphA2 (Fig. 12.A, panel a). In uninfected cells, moderate association of EphA2 was observed and no EphA2 was detected in immunoprecipitates with control IgG antibodies (Fig. 12.A, panel a, lanes 1 and 2). In contrast, ∼2.4-fold more EphA2 was immunoprecipitated with CIB1 from infected cell than uninfected cell lysates (Fig. 12.A, panel a, lane 3). The specificity of this association was demonstrated by the significant 95% reduction by CIB1 knockdown (Fig. 12.A, panel a, lane 5). In addition, pretreatment of KSHV with heparin to block initial virus binding resulted in a significant reduction in CIB1-EphA2 association in infected cells and the observed residual CIB1-EphA2 association was comparable to the uninfected cells (Fig. 12.A, panel a, lane 4). These results together with the increased association of CIB1 in LRs of infected cells (Fig. 11), and the presence of EphA2 only in the LRs of HMVEC-d cells (Fig. 11) demonstrated that CIB1 associates with EphA2 during KSHV infection.

CIB1 associates with KSHV entry associated signal molecules early during infection of HMVEC-d cells
To determine the mechanism by which CIB1 plays a role in KSHV entry and infection, we next determined whether CIB1 associates simultaneously with components of the EphA2 assembled multi-molecular signal complex such as Src, PI3-K, and c-Cbl. Serum starved HMVEC-d cells were either mock or infected for 10 min, a time point of maximum bleb induction and virus internalization, immunoprecipitated with anti-CIB1 antibodies, and Western blotted for CIB1 (Fig. 12.A, panel e), and signal molecules such as Src, PI3K, and c-Cbl (Fig. 12.A). A robust increase in CIB1 associations with Src, PI3-K and c-Cbl about 1.5, 7.2, 3.8-fold, respectively, were observed in infected cells compared to the basal level association of these molecules with CIB1 in uninfected cells (Fig. 12.A, lanes 2 and 3 in panels b, c and d). Specificities of these associations were demonstrated by their significant reduction, ∼77%, 100%, and 95%, respectively, in CIB1 knockdown cells as well as by ∼50%, 72%, and 40%, respectively, in infection with heparin-treated KSHV (Fig. 12.A, lanes 4 and 5 in panels b, c and d). Total protein levels for EphA2, Src, PI3-K, c-Cbl, and CIB1 as determined by Western blotting using whole cell lysates (WCL) served as input controls and β-actin was used as loading control (Fig. 12.A, panels f–k).
Knockdown of CIB1 reduces the sustained activation of EphA2, Src and Erk1/2 early during KSHV infection of HMVEC-d cells
Simultaneous association of CIB1 with EphA2 and KSHV entry associated signal molecules prompted us to decipher the functional role of CIB1 during macropinosome associated signaling events. We first determined the activation of key macropinosome assembly regulator EphA2 in CIB1 shRNA transduced cells. In control shRNA transduced cells, KSHV induced ∼1.9, 2.8 and 1.9-fold EphA2 activation at 5, 10 and 30 min p.i., respectively, compared to basal levels in uninfected cells (Fig. 12.B, panel a, lanes 1–4). In contrast, in CIB1-shRNA transduced cells, EphA2 activation was significantly reduced and we observed only about 1.6, 0.77 and 0.73-fold activation at 5, 10 and 30 min p.i., respectively (Fig. 12.B, panel a, lanes 5–8). KSHV infection did not change the total EphA2 levels and CIB1-shRNA had no off-target effect on EphA2 at the protein level (Fig. 12.B, panel e). Tubulin was used as a loading control (Fig. 12B, panel h). These results demonstrated that CIB1 is essential for a robust and sustained activation of EphA2 during KSHV infection.
To confirm the specificity of CIB1's effect on EphA2 regulated signal amplification pathways, downstream entry associated signal molecules of EphA2 such as Src and post-entry KSHV gene expression associated signal molecule Erk1/2 were examined. In control shRNA transduced cells, compared to uninfected cells, KSHV induced ∼1.2, 1.7, and 3.3-fold Src activation at 5, 10 and 30 min p.i., respectively (Fig. 12.B, panel b, lanes 1–4). In contrast, in CIB1-shRNA transduced cells, Src activation was significantly reduced to about 0.9, 1.3, and 1.5-fold at 5, 10 and 30 min p.i., respectively (Fig. 12.B, panel b, lanes 5–8). Similarly, KSHV induced Erk1/2 activation was almost abolished by CIB1 knockdown. In control shRNA transduced cells, compared to uninfected cells, KSHV induced Erk1/2 activation was about 2.6, 3.5, and 2.5-fold at 5, 10 and 30 min p.i., respectively, whereas in CIB1-shRNA transduced cells, Erk1/2 activation was dramatically compromised with only 1, 1.2, and 1.3-fold activation at 5, 10 and 30 min p.i., respectively (Fig. 12.B, panel c, lanes 1–4). Total Src and Erk1/2 protein levels were unchanged by KSHV infection and by CIB1 knockdown (Fig. 6B, panels f and g). These results demonstrated that CIB1 plays roles in the sustained activation of EphA2 and its downstream target signal amplification of KSHV entry and gene expression associated Src and Erk1/2, respectively.
CIB1 and EphA2 associate with common actin modulating cytoskeletal molecules early during KSHV infection of HMVEC-d cells
Evidence presented thus far has demonstrated the association of CIB1 with known signal mediators of KSHV macropinocytosis such as EphA2, Src, PI3-K, and c-Cbl and thereby modulation of signal amplification leading to KSHV induced macropinosome formation. To identify the molecular partners required for CIB1 in coordinating EphA2 regulated membrane blebbing during KSHV infection; we used mass spectrometric (MS) analysis to identify common CIB1 and EphA2 interacting proteins (Table 1 and Table 2). HMVEC-d cells were mock or KSHV infected for 10 min and lysates were immunoprecipitated with anti-CIB1 or anti-EphA2 antibodies. Samples were separated by 4–20% gradient SDS-PAGE, followed by Coomassie blue staining and MS analysis (Fig. 13.A) which identified several novel CIB1 and EphA2 binding partners in the infected samples.



The common proteins pulled down with anti-CIB1 or anti-EphA2 antibodies were grouped according to functional categories (Table 1). However, the lists of all identified proteins are not reported and are beyond the scope of present study. Categories that match closely to the functional relevance in context to the study are reported in the table. Interestingly, cytoskeletal motility and membrane ruffling mediators such as myosin IIA, alpha actinin-4, annexin A2, and vimentin were detected in both EphA2 and CIB1 immunoprecipitates. We validated the MS data by co-IP analysis and all of these molecules did show temporal association with EphA2 and CIB1 early during KSHV infection of HMVEC-d cells (Fig. 13.B). Simultaneous association of these molecules with EphA2 and CIB1 were observed as early as 5 min p.i. which decreased by 30 min p.i. (Fig. 13.B). The total protein levels of these molecules remained unchanged with KSHV infection (Fig. 13.B). These results strongly suggested that besides its role in the sustained activation of EphA2, Src and Erk1/2, CIB1's regulation of KSHV macropinocytosis may possibly be mediated through the interactions with these actin-modulating molecules.
Among the various molecules identified by MS, alpha actinin and myosin family members are known to regulate the turnover dynamics of sub-membranous actin cortex homeostasis and the cellular blebbing process [69], [70]. Our earlier studies have demonstrated that myosin IIA is an EphA2 downstream critical cytoskeletal effector facilitating KSHV induced membrane blebbing and bleb retraction [21], [23]. Therefore, we further examined whether CIB1 association facilitates the interaction of EphA2 with cytoskeletal cross linker alpha actinin-4, and motor protein myosin IIA, respectively. To address this question, we immunoprecipitated EphA2 from untransduced and CIB1-shRNA transduced HMVEC-d cells at 10 min p.i. and subsequently Western blotted for alpha actinin-4 and myosin IIA (Fig. 13.C). KSHV infection increased the EphA2 association with alpha actinin-4 and myosin IIA by a significant 1.4 and 1.6-fold, respectively, compared to uninfected HMVEC-d cells (Fig. 13.C, top two lanes, lanes 1–3). However, CIB1-shRNA transduction significantly reduced EphA2 association with alpha actinin-4 and myosin IIA by 100% and 40%, respectively, compared to KSHV infected control cells (Fig. 13.C, top two lanes, lane 4).
The cytoskeletal proteins studied here are functionally known to be involved in cell migration. Although these proteins are abundant, in our co-IP studies they demonstrated KSHV infection dependent temporal association with CIB1 and EphA2 simultaneously which suggested that it is a specific directed event. Moreover, when infection with heparin-treated KSHV was used as specificity control and such associations were reduced significantly (Fig. 13.C, top two lanes, lane 5). Hence, these results suggested that simultaneous association of cytoskeletal proteins with EphA2 and CIB1 is a specific event induced during KSHV infection. Total protein levels of these molecules remained unchanged with KSHV infection and CIB1-shRNA transduction (Fig. 13.C).
We also observed a significant reduction in KSHV induced EphA2 association with PI3-K and c-Cbl upon CIB1-shRNA transduction (data not shown). Reduction in EphA2 association with these molecules could be due to the reduced EphA2 activity with CIB1-shRNA transduction as observed (Fig. 12.B).
Taken together, these results demonstrated that that simultaneous association of cytoskeletal proteins with EphA2 and CIB1 is specific, and CIB1 synergizes with EphA2 to regulate signal assembly and possibly also cytoskeletal cross talk to aid in KSHV macropinocytosis.
Discussion
Our comprehensive biochemical and morphological studies presented here identified several novel roles for CIB1 such as: (i) CIB1 is a player in KSHV macropinocytosis in HMVEC-d cells; (ii) CIB1 is involved in KSHV productive trafficking in HMVEC-d cells; (iii) CIB1 acts as a signal amplifier facilitating EphA2 initiated signal amplification, and (iv) CIB1 also mimics an adaptor function in aiding EphA2 to facilitate cytoskeletal cross linker alpha actinin4 and motor protein myosin IIA association possibly to generate the mechanical driving force for actin modulation and macropinosome formation during KSHV entry.
Macropinocytosis, an actin-driven endocytic process, is not directly associated with a specific cellular cargo and lacks any single characteristic molecular marker [27], [28]. Actin cytoskeletal rearrangement is a critical process at the very early stages of macropinocytosis and several viruses induce varieties of critical multi-stepped signaling cascades to exploit actin filament dynamics to facilitate entry into the host cell [29], [71], [72]. The molecular aspects of macropinosomes show variations depending on the virus and according to their cellular tropism [73], [74], [75], [76]. Since the physiological macropinocytic process is not a completely understood event, the variations in molecular engagement during virus induced macropinosome formation are also difficult to delineate. KSHV macropinocytosis in HMVEC-d cells is a complex event. Our earlier studies have shown that the KSHV induced membrane blebbing process initiates by virus triggered EphA2 receptor activation in the LR, association with selective LR translocated integrin receptors, further clustering of LR associated diverse molecular players including kinases such as Src and PI3-K, GTPase family member RhoA, adaptor protein c-Cbl, actin binding motor protein myosin IIA, and subsequent amplification of these signal cascades results in KSHV productive trafficking into the Rab5 positive early macropinosome [23]. While it was known that KSHV induced E3 ubiquitin ligase c-Cbl mediates monoubiquitination of bleb associated integrin receptors, actin cytoskeleton and myosin IIA to promote actomyosin dependent KSHV macropinocytosis, the complete molecular identity of the KSHV induced macropinosome at different stages of macropinocytosis remained largely unknown [21], [22]. Thus, the present study identifying simultaneous association of CIB1 with KSHV entry receptor EphA2, associated Src, PI3-K, and c-Cbl signal molecules along with cytoskeletal myosin IIA and alpha actinin-4 to facilitate KSHV induced bleb associated signal amplification, and regulating actin modulation provides additional knowledge in understanding the mechanism of KSHV induced macropinosome formation in HMVEC-d cells, a natural in vivo target cell.
Our functional studies with CIB1-shRNA transduced HMVEC-d cells and CIB1 overexpressing 293 cells cumulatively provided strong evidence for the necessity of CIB1 in KSHV entry (Fig. 14). CIB1 belongs to the CIB family and shares high structural homologies with other members such as CIB2, CIB3, and CIB4 by 59%, 62%, and 64%, respectively [44]. We did not study other CIB family members as all of them are very recently identified, lacks reagents and relevance to study the function. Currently, only chromosomal localization has been assigned for CIB3 [44] and there is no published functional data on CIB3. CIB4 was named based on the automated computational analysis of the human genome [44]. Finally, we also ruled out the possibility of any compensatory mechanism by CIB2 as its mRNA is expressed mostly in the developing central nervous system and developing and adult skeletal muscles [77], [78], [79].

Earlier studies by us and others have demonstrated that different KSHV glycoproteins interact with multiple cell surface integrin receptors such as α3β1, αVβ3, and αVβ5 as well as EphA2 on HMVEC-d cells, followed by receptor clustering and subsequent activation or membrane localization of downstream signal molecules [4], [5], [14]. Our present studies demonstrate the engagement of CIB1 with KSHV entry associated molecules drives the signal amplification events. Since CIB1 is so far not reported to be regulated by phosphorylation, it was not possible to determine which KSHV glycoproteins are involved in CIB1 induction by a direct read out such as phosphorylation after incubating target cells with individual envelope glycoproteins. Nevertheless, results shown in Fig. 4 suggested that KSHV glycoprotein gB, that has been shown to interact with cell surface integrins [10], [51], is probably involved in the induction of CIB1 recruitment to the KSHV induced signal complex.
Biochemical and morphological evidences from the current study demonstrated enrichment of CIB1 in infected HMVEC-d cell membrane LR fractions (Fig. 11) and translocation of a pool of CIB1 to the membrane actin protrusions early during de novo KSHV infection (Fig. 6). In our fractionation studies, we clearly demonstrated that in HMVEC-d cells, KSHV induced CIB1 translocation from membrane NLR to the LR fraction as early as 5 min p.i. that was sustained till 30 min p.i., which was comparable to another previous study observing complete translocation of CIB1 into the Triton X-100 detergent insoluble fraction in agonist-activated platelets [33]. CIB1 co-IPed with FAK after KSHV infection (data not shown) which could be also a reason for the minimal level of CIB1 association with NLR as FAK doesn't transcolate to LR after KSHV infection [22], [23]. Since KSHV internalization is a rapid process, CIB1 and KSHV association at 30 min p.i. also indicates the downstream role of CIB1 regulated potential effector(s) in post-entry macropinosome trafficking stages, which needs further investigation.
Strong colocalization of CIB1 with LR marker flotillin-1 at the cell periphery at 5 min post-KSHV infection and increased colocalization both at the cell periphery and in the cytosol at 10 min p.i. confirmed the biochemical data for KSHV induced temporal association of CIB1 with LRs. Interestingly, the CIB1 staining pattern was also coherent with flotillin-1 as both the molecules were more clustered with infection, compared to a diffused pattern in the uninfected HMVEC-d cells (Fig. 11.C). The CIB1 staining pattern in uninfected HMVEC-d cells (Fig. 11.C) is consistent with the earlier reports determining cellular distribution of CIB1 in a different cellular system [38]. A KSHV induced clustered cytosolic CIB1 staining pattern could be anticipated due to (i) CIB1 interaction with cytosolic downstream effector proteins, and/or (ii) CIB1 localization in a subcellular organelle membrane raft. This could be advantageous for KSHV macropinocytosis for several reasons such as: (a) CIB1 could be translocating some of the cytosolic effector kinases to the LRs similar to the fact that CIB1 mediated PDK1 and SK1 translocation upon fibronectin treatment in HeLa and HEK 293 cells, respectively [45], [80]; (b) CIB1 might interact with organelle associated proteins as shown during CIB1 and PS2 reticular co-staining in ER upon co-transfection into HeLa cells [38]. Collectively, clustered cellular redistribution of CIB1 early during KSHV infection might predict a beneficial role for the immediate KSHV post-entry stages such as macropinosome trafficking, immune evasion, and nuclear delivery of viral DNA.
Our co-immunoprecipitation studies provided evidence for KSHV induced distinct simultaneous association of CIB1 with EphA2 receptor and signal molecules such as Src, PI3-K, and c-Cbl (Fig. 12.A). Although Src association with CIB1 upon fibrinogen stimulation in platelets was shown before [43], KSHV infection induced detectable interactions of EphA2, PI3-K, and c-Cbl in CIB1 immunoprecipitates is an interestingly novel observation in HMVEC-d cells. Early during entry, KSHV utilizes a series of HMVEC-d cell pre-existing kinases that possess SH2 and SH3 binding sites, which create subsequent binding sites for immediate downstream molecules leading into the activation and maintenance of signal cascades [4], [5], [23]. However, CIB1 structure lacks any anti-parallel beta sheets required to form traditional SH2 (a central anti-parallel beta sheet surrounded by two alpha helices) and SH3 domains (five anti parallel beta strands packed perpendicularly to two perpendicular beta sheets) [81], [82]. Available crystal structural information characterized CIB1 as an EF hand (basic helix-loop-loop helix) family protein, or more specifically a compact alpha helical protein with four EF hands [44]. Based on our biochemical co-IP and fractionation data, we propose that KSHV induced plasma membrane LR translocation of CIB1 enables its interaction directly or indirectly with EphA2 assembled kinase member(s), thereby recruiting CIB1 as a component of KSHV induced bleb associated signal complex. Further detailed studies are essential to determine the domains of CIB1 mediating these interactions.
Although CIB1 was co-immunoprecipitated with most of the previously established EphA2 associated signal molecules at 10 min post - KSHV infection, we were unable to co-IP CIB1 with EphA2 associated β1 and β3 integrins despite using multiple detergents and buffer conditions (data not shown). Similarly, an earlier study was also unable to co-IP endogenous CIB1 with αIIbβ3 integrin since such hydrophobic interactions tend to fall apart by the use of any detergents [33] although such interactions were conveniently observed by other methods such as yeast two-hybrid screening and isothermal calorimetric assay [32]. Based on our co-IP study, we conclude that CIB1 might not associate directly with KSHV entry receptor integrins in HMVEC-d cells and co-IP studies are not suitable to detect possible indirect associations through protein-protein hydrophobic interactions.
Structurally, CIB1 has 4 EF hands capable of sensing divalent cations (Ca2+/Mg2+) through EF III and IV [44]. Like other EF hand family members, whether Ca2+ binding on the C-terminal of CIB1 could potentiate it as a Ca2+-dependent myristoyl-switch protein regulating CIB1 membrane targeting is largely unknown [44]. However, CIB1's interactions with its known binding partner(s) are reported to be both Ca2+ dependent [45] as well as independent [38]. A recent report demonstrated that KSHV could induce Ca2+ through plasma membrane L-type Ca2+ channels in an Src dependent manner in human umbilical vein endothelial cells (HUVEC) [83]. Further studies are essential to determine whether calcium plays a role in KSHV infection induced CIB1's associations with EphA2 and associated signal molecules.
Selective modulation of cellular signaling is a critical event for KSHV endocytosis regulation in HMVEC-d cells [23], [26], and the current study reveals the unique role of CIB1 as a signal amplifier during KSHV induced signaling events. Although CIB1 was known to promote actions of several cellular kinases via a yet unidentified mechanism [42], [43], the role of CIB1 in facilitating kinase action during endocytosis was not shown before. Inhibition of sustained EphA2 activity upon CIB1 shRNA transduction, and subsequent inhibition of downstream Src and Erk1/2, strongly advocated for a crucial role for CIB1 during KSHV macropinocytosis (Fig. 12). Our functional studies analyzing KSHV induced dextran uptake, virus and macropinosome association, and membrane blebbing clearly have established an important role for CIB1 during KSHV entry into HMVEC-d cells.
Through an LC-MS/MS proteomic approach we have identified and validated simultaneous association of alpha actinin-4 and myosin IIA with EphA2 and CIB1 as early as 5 min post-KSHV infection in HMVEC-d cells (Fig. 13). EphA2 signaling was well known for actin crosstalk promoting cytoskeletal assembly or disassembly under different conditions depending on cell type [84], [85]. CIB1 previously was identified to translocate onto the platelet cytoskeleton simultaneously with αIIbβ3 integrin in an agonist dependent manner [33] and cellular CIB1 was also reported to interact with IQ motifs in myo1c regulatory domain possibly linking actin cytoskeleton to cellular membranes [86]. However, CIB1 and EphA2 coordinated signaling during agonist induced cytoskeletal rearrangement under physiological or pathological conditions was not studied before. In CIB1-shRNA transduced cells, reduction of EphA2 association with alpha actinin-4 and myosin IIA demonstrated that CIB1 mimics function of an adaptor molecule to regulate EphA2 and cytoskeletal cross talk. Although alpha actinin and myosin family members were known to regulate actin homeostasis and cellular blebbing processes [69], [70], the involvement of CIB1 in influencing blebbing events is an exciting observation. Further studies are necessary to decipher the role of the KSHV triggered EphA2-alpha actinin 4-myosin IIA triad in KSHV entry and infection.
Overall, our studies identify CIB1 as a host factor regulating a virus primary infection as well as endocytosis for the first time. This study also provides evidences that CIB1 can act as a signal amplifier thereby integrating signal crosstalk to facilitate macropinocytosis by supporting KSHV induced actin modulation events. Thus CIB1 can serve as a potential target to intervene in KSHV infection.
Zdroje
1. ChangY, CesarmanE, PessinMS, LeeF, CulpepperJ, et al. (1994) Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266 : 1865–1869.
2. CesarmanE, ChangY, MoorePS, SaidJW, KnowlesDM (1995) Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332 : 1186–1191.
3. Ganem D (2007) Kaposi's sarcoma-associated herpesvirus. Fields virology 5th ed., vol. 2: p. 2875–2888.
4. ChandranB Early events in Kaposi's sarcoma-associated herpesvirus infection of target cells. J Virol 84 : 2188–2199.
5. ChakrabortyS, VeettilMV, ChandranB Kaposi's Sarcoma Associated Herpesvirus Entry into Target Cells. Front Microbiol 3 : 6.
6. AkulaSM, PramodNP, WangFZ, ChandranB (2001) Human herpesvirus 8 envelope-associated glycoprotein B interacts with heparan sulfate-like moieties. Virology 284 : 235–249.
7. WangFZ, AkulaSM, PramodNP, ZengL, ChandranB (2001) Human herpesvirus 8 envelope glycoprotein K8.1A interaction with the target cells involves heparan sulfate. J Virol 75 : 7517–7527.
8. BirkmannA, MahrK, EnsserA, YagubogluS, TitgemeyerF, et al. (2001) Cell surface heparan sulfate is a receptor for human herpesvirus 8 and interacts with envelope glycoprotein K8.1. J Virol 75 : 11583–11593.
9. AkulaSM, WangFZ, VieiraJ, ChandranB (2001) Human herpesvirus 8 interaction with target cells involves heparan sulfate. Virology 282 : 245–255.
10. WangFZ, AkulaSM, Sharma-WaliaN, ZengL, ChandranB (2003) Human herpesvirus 8 envelope glycoprotein B mediates cell adhesion via its RGD sequence. J Virol 77 : 3131–3147.
11. HahnA, BirkmannA, WiesE, DorerD, MahrK, et al. (2009) Kaposi's sarcoma-associated herpesvirus gH/gL: glycoprotein export and interaction with cellular receptors. J Virol 83 : 396–407.
12. NaranattPP, AkulaSM, ZienCA, KrishnanHH, ChandranB (2003) Kaposi's sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: implications for infectivity. J Virol 77 : 1524–1539.
13. VeettilMV, Sharma-WaliaN, SadagopanS, RaghuH, SivakumarR, et al. (2006) RhoA-GTPase facilitates entry of Kaposi's sarcoma-associated herpesvirus into adherent target cells in a Src-dependent manner. J Virol 80 : 11432–11446.
14. VeettilMV, SadagopanS, Sharma-WaliaN, WangFZ, RaghuH, et al. (2008) Kaposi's sarcoma-associated herpesvirus forms a multimolecular complex of integrins (alphaVbeta5, alphaVbeta3, and alpha3beta1) and CD98-xCT during infection of human dermal microvascular endothelial cells, and CD98-xCT is essential for the postentry stage of infection. J Virol 82 : 12126–12144.
15. RaghuH, Sharma-WaliaN, VeettilMV, SadagopanS, ChandranB (2009) Kaposi's sarcoma-associated herpesvirus utilizes an actin polymerization-dependent macropinocytic pathway to enter human dermal microvascular endothelial and human umbilical vein endothelial cells. J Virol 83 : 4895–4911.
16. NaranattPP, KrishnanHH, SmithMS, ChandranB (2005) Kaposi's sarcoma-associated herpesvirus modulates microtubule dynamics via RhoA-GTP-diaphanous 2 signaling and utilizes the dynein motors to deliver its DNA to the nucleus. J Virol 79 : 1191–1206.
17. KrishnanHH, Sharma-WaliaN, StreblowDN, NaranattPP, ChandranB (2006) Focal adhesion kinase is critical for entry of Kaposi's sarcoma-associated herpesvirus into target cells. J Virol 80 : 1167–1180.
18. SadagopanS, Sharma-WaliaN, VeettilMV, RaghuH, SivakumarR, et al. (2007) Kaposi's sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J Virol 81 : 3949–3968.
19. Sharma-WaliaN, KrishnanHH, NaranattPP, ZengL, SmithMS, et al. (2005) ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J Virol 79 : 10308–10329.
20. AkulaSM, NaranattPP, WaliaNS, WangFZ, FegleyB, et al. (2003) Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) infection of human fibroblast cells occurs through endocytosis. J Virol 77 : 7978–7990.
21. Valiya VeettilM, SadagopanS, KerurN, ChakrabortyS, ChandranB Interaction of c-Cbl with myosin IIA regulates Bleb associated macropinocytosis of Kaposi's sarcoma-associated herpesvirus. PLoS Pathog 6: e1001238.
22. ChakrabortyS, ValiyaVeettilM, SadagopanS, PaudelN, ChandranB c-Cbl-mediated selective virus-receptor translocations into lipid rafts regulate productive Kaposi's sarcoma-associated herpesvirus infection in endothelial cells. J Virol 85 : 12410–12430.
23. ChakrabortyS, VeettilMV, BotteroV, ChandranB (2012) Kaposi's sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc Natl Acad Sci U S A 109: E1163–1172.
24. HahnAS, KaufmannJK, WiesE, NaschbergerE, Panteleev-IvlevJ, et al. (2012) The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi's sarcoma-associated herpesvirus. Nat Med 18 : 961–966.
25. BoshoffC (2012) Ephrin receptor: a door to KSHV infection. Nat Med 18 : 861–863.
26. DuttaD, ChakrabortyS, BandyopadhyayC, Valiya VeettilM, AnsariMA, et al. (2013) EphrinA2 regulates clathrin mediated KSHV endocytosis in fibroblast cells by coordinating integrin-associated signaling and c-Cbl directed polyubiquitination. PLoS Pathog 9: e1003510.
27. KerrMC, TeasdaleRD (2009) Defining macropinocytosis. Traffic 10 : 364–371.
28. JonesAT (2007) Macropinocytosis: searching for an endocytic identity and role in the uptake of cell penetrating peptides. J Cell Mol Med 11 : 670–684.
29. MarshM, HeleniusA (2006) Virus entry: open sesame. Cell 124 : 729–740.
30. DikicI, SzymkiewiczI, SoubeyranP (2003) Cbl signaling networks in the regulation of cell function. Cell Mol Life Sci 60 : 1805–1827.
31. YuanW, LeisnerTM, McFaddenAW, WangZ, LarsonMK, et al. (2006) CIB1 is an endogenous inhibitor of agonist-induced integrin alphaIIbbeta3 activation. J Cell Biol 172 : 169–175.
32. NaikUP, PatelPM, PariseLV (1997) Identification of a novel calcium-binding protein that interacts with the integrin alphaIIb cytoplasmic domain. J Biol Chem 272 : 4651–4654.
33. ShockDD, NaikUP, BrittainJE, AlahariSK, SondekJ, et al. (1999) Calcium-dependent properties of CIB binding to the integrin alphaIIb cytoplasmic domain and translocation to the platelet cytoskeleton. Biochem J 342 Pt 3 : 729–735.
34. LeisnerTM, LiuM, JafferZM, ChernoffJ, PariseLV (2005) Essential role of CIB1 in regulating PAK1 activation and cell migration. J Cell Biol 170 : 465–476.
35. NaikMU, PhamNT, BeebeK, DaiW, NaikUP (2011) Calcium-dependent inhibition of polo-like kinase 3 activity by CIB1 in breast cancer cells. Int J Cancer 128 : 587–596.
36. KauselmannG, WeilerM, WulffP, JessbergerS, KonietzkoU, et al. (1999) The polo-like protein kinases Fnk and Snk associate with a Ca(2+) - and integrin-binding protein and are regulated dynamically with synaptic plasticity. EMBO J 18 : 5528–5539.
37. WhiteC, YangJ, MonteiroMJ, FoskettJK (2006) CIB1, a ubiquitously expressed Ca2+-binding protein ligand of the InsP3 receptor Ca2+ release channel. J Biol Chem 281 : 20825–20833.
38. StablerSM, OstrowskiLL, JanickiSM, MonteiroMJ (1999) A myristoylated calcium-binding protein that preferentially interacts with the Alzheimer's disease presenilin 2 protein. J Cell Biol 145 : 1277–1292.
39. HollenbachAD, McPhersonCJ, LagutinaI, GrosveldG (2002) The EF-hand calcium-binding protein calmyrin inhibits the transcriptional and DNA-binding activity of Pax3. Biochim Biophys Acta 1574 : 321–328.
40. HaatajaL, KaartinenV, GroffenJ, HeisterkampN (2002) The small GTPase Rac3 interacts with the integrin-binding protein CIB and promotes integrin alpha(IIb)beta(3)-mediated adhesion and spreading. J Biol Chem 277 : 8321–8328.
41. WuX, LieberMR (1997) Interaction between DNA-dependent protein kinase and a novel protein, KIP. Mutat Res 385 : 13–20.
42. NaikMU, NaikUP (2003) Calcium-and integrin-binding protein regulates focal adhesion kinase activity during platelet spreading on immobilized fibrinogen. Blood 102 : 3629–3636.
43. NaikMU, NaikUP (2011) Contra-regulation of calcium - and integrin-binding protein 1-induced cell migration on fibronectin by PAK1 and MAP kinase signaling. J Cell Biochem 112 : 3289–3299.
44. GentryHR, SingerAU, BettsL, YangC, FerraraJD, et al. (2005) Structural and biochemical characterization of CIB1 delineates a new family of EF-hand-containing proteins. J Biol Chem 280 : 8407–8415.
45. JarmanKE, MorettiPA, ZebolJR, PitsonSM (2010) Translocation of sphingosine kinase 1 to the plasma membrane is mediated by calcium - and integrin-binding protein 1. J Biol Chem 285 : 483–492.
46. NaikUP, NaikMU (2003) Association of CIB with GPIIb/IIIa during outside-in signaling is required for platelet spreading on fibrinogen. Blood 102 : 1355–1362.
47. KostyakJC, NaikMU, NaikUP (2012) Calcium - and integrin-binding protein 1 regulates megakaryocyte ploidy, adhesion, and migration. Blood 119 : 838–846.
48. KakigiA, OkadaT, TakedaT, TakedaS, TaguchiD, et al. (2010) Endocytosis of MPO in marginal cells is regulated by PKC, protein phosphatase, ERK and PI3-K signaling cascades, but not by PKA and MEK signaling cascades. ORL J Otorhinolaryngol Relat Spec 72 : 188–195.
49. MettlenM, PlatekA, Van Der SmissenP, CarpentierS, AmyereM, et al. (2006) Src triggers circular ruffling and macropinocytosis at the apical surface of polarized MDCK cells. Traffic 7 : 589–603.
50. YorkRD, MolliverDC, GrewalSS, StenbergPE, McCleskeyEW, et al. (2000) Role of phosphoinositide 3-kinase and endocytosis in nerve growth factor-induced extracellular signal-regulated kinase activation via Ras and Rap1. Mol Cell Biol 20 : 8069–8083.
51. Sharma-WaliaN, NaranattPP, KrishnanHH, ZengL, ChandranB (2004) Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-rho GTPase signal pathways and cytoskeletal rearrangements. J Virol 78 : 4207–4223.
52. DonepudiM, ReshMD (2008) c-Src trafficking and co-localization with the EGF receptor promotes EGF ligand-independent EGF receptor activation and signaling. Cell Signal 20 : 1359–1367.
53. AkulaSM, PramodNP, WangFZ, ChandranB (2002) Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108 : 407–419.
54. KrishnanHH, NaranattPP, SmithMS, ZengL, BloomerC, et al. (2004) Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J Virol 78 : 3601–3620.
55. JohnsonKE, ChikotiL, ChandranB (2013) Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virol 87 : 5005–5018.
56. VargheseS, RabkinSD, NielsenGP, MacGarveyU, LiuR, et al. (2007) Systemic therapy of spontaneous prostate cancer in transgenic mice with oncolytic herpes simplex viruses. Cancer Res 67 : 9371–9379.
57. Le BlancI, LuyetPP, PonsV, FergusonC, EmansN, et al. (2005) Endosome-to-cytosol transport of viral nucleocapsids. Nat Cell Biol 7 : 653–664.
58. ZhuFX, LiX, ZhouF, GaoSJ, YuanY (2006) Functional characterization of Kaposi's sarcoma-associated herpesvirus ORF45 by bacterial artificial chromosome-based mutagenesis. J Virol 80 : 12187–12196.
59. SadagopanS, Sharma-WaliaN, VeettilMV, BotteroV, LevineR, et al. (2009) Kaposi's sarcoma-associated herpesvirus upregulates angiogenin during infection of human dermal microvascular endothelial cells, which induces 45S rRNA synthesis, antiapoptosis, cell proliferation, migration, and angiogenesis. J Virol 83 : 3342–3364.
60. ZhuL, PuriV, ChandranB (1999) Characterization of human herpesvirus-8 K8.1A/B glycoproteins by monoclonal antibodies. Virology 262 : 237–249.
61. KalamvokiM, RoizmanB (2010) Interwoven roles of cyclin D3 and cdk4 recruited by ICP0 and ICP4 in the expression of herpes simplex virus genes. J Virol 84 : 9709–9717.
62. RosenkeK, FortunatoEA (2004) Bromodeoxyuridine-labeled viral particles as a tool for visualization of the immediate-early events of human cytomegalovirus infection. J Virol 78 : 7818–7822.
63. SongKS, LiS, OkamotoT, QuilliamLA, SargiacomoM, et al. (1996) Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J Biol Chem 271 : 9690–9697.
64. PanH, XieJ, YeF, GaoSJ (2006) Modulation of Kaposi's sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J Virol 80 : 5371–5382.
65. FordPW, HamdenKE, WhitmanAG, McCubreyJA, AkulaSM (2004) Vascular endothelial growth factor augments human herpesvirus-8 (HHV-8/KSHV) infection. Cancer Biol Ther 3 : 876–881.
66. George PaulA, Sharma-WaliaN, KerurN, WhiteC, ChandranB Piracy of prostaglandin E2/EP receptor-mediated signaling by Kaposi's sarcoma-associated herpes virus (HHV-8) for latency gene expression: strategy of a successful pathogen. Cancer Res 70 : 3697–3708.
67. RappoccioloG, HenslerHR, JaisM, ReinhartTA, PeguA, et al. (2008) Human herpesvirus 8 infects and replicates in primary cultures of activated B lymphocytes through DC-SIGN. J Virol 82 : 4793–4806.
68. LanzettiL, PalamidessiA, ArecesL, ScitaG, Di FiorePP (2004) Rab5 is a signalling GTPase involved in actin remodelling by receptor tyrosine kinases. Nature 429 : 309–314.
69. TinevezJY, SchulzeU, SalbreuxG, RoenschJ, JoannyJF, et al. (2009) Role of cortical tension in bleb growth. Proc Natl Acad Sci U S A 106 : 18581–18586.
70. FritzscheM, LewalleA, DukeT, KruseK, CharrasG (2013) Analysis of turnover dynamics of the submembranous actin cortex. Mol Biol Cell 24 : 757–767.
71. MercerJ, HeleniusA (2012) Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr Opin Microbiol 15 : 490–499.
72. MercerJ, SchelhaasM, HeleniusA (2010) Virus entry by endocytosis. Annu Rev Biochem 79 : 803–833.
73. GobeilLA, LodgeR, TremblayMJ (2013) Macropinocytosis-like HIV-1 internalization in macrophages is CCR5 dependent and leads to efficient but delayed degradation in endosomal compartments. J Virol 87 : 735–745.
74. KriegerSE, KimC, ZhangL, MarjomakiV, BergelsonJM (2013) Echovirus 1 entry into polarized caco-2 cells depends on dynamin, cholesterol, and cellular factors associated with macropinocytosis. J Virol 87 : 8884–8895.
75. CoyneCB, ShenL, TurnerJR, BergelsonJM (2007) Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2 : 181–192.
76. MercerJ, HeleniusA (2008) Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320 : 531–535.
77. BlazejczykM, SobczakA, DebowskaK, WisniewskaMB, KirilenkoA, et al. (2009) Biochemical characterization and expression analysis of a novel EF-hand Ca2+ binding protein calmyrin2 (Cib2) in brain indicates its function in NMDA receptor mediated Ca2+ signaling. Arch Biochem Biophys 487 : 66–78.
78. HagerM, BigottiMG, MeszarosR, CarmignacV, HolmbergJ, et al. (2008) Cib2 binds integrin alpha7Bbeta1D and is reduced in laminin alpha2 chain-deficient muscular dystrophy. J Biol Chem 283 : 24760–24769.
79. DashDP, SilvestriG, HughesAE (2006) Fine mapping of the keratoconus with cataract locus on chromosome 15q and candidate gene analysis. Mol Vis 12 : 499–505.
80. BinZ (2007) Regulation of cell survival by CIB1, a new modulator of phosphoinositide-dependent protein kinase-1 (PDK1). FASEB 6: LB12.
81. NguyenJT, TurckCW, CohenFE, ZuckermannRN, LimWA (1998) Exploiting the basis of proline recognition by SH3 and WW domains: design of N-substituted inhibitors. Science 282 : 2088–2092.
82. WaksmanG, KominosD, RobertsonSC, PantN, BaltimoreD, et al. (1992) Crystal structure of the phosphotyrosine recognition domain SH2 of v-src complexed with tyrosine-phosphorylated peptides. Nature 358 : 646–653.
83. YeFC, ZhouFC, NithiananthamS, ChandranB, YuXL, et al. (2013) Kaposi's sarcoma-associated herpesvirus induces rapid release of angiopoietin-2 from endothelial cells. J Virol 87 : 6326–6335.
84. CarterN, NakamotoT, HiraiH, HunterT (2002) EphrinA1-induced cytoskeletal re-organization requires FAK and p130(cas). Nat Cell Biol 4 : 565–573.
85. SalaitaK, GrovesJT (2010) Roles of the cytoskeleton in regulating EphA2 signals. Commun Integr Biol 3 : 454–457.
86. TangN, LinT, YangJ, FoskettJK, OstapEM (2007) CIB1 and CaBP1 bind to the myo1c regulatory domain. J Muscle Res Cell Motil 28 : 285–291.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 2
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Suppression of RNA Silencing by a Plant DNA Virus Satellite Requires a Host Calmodulin-Like Protein to Repress Expression
- Reversible Silencing of Cytomegalovirus Genomes by Type I Interferon Governs Virus Latency
- Identification of Host-Targeted Small Molecules That Restrict Intracellular Growth
- Implication of PMLIV in Both Intrinsic and Innate Immunity