
Cdk1 Targets Srs2 to Complete Synthesis-Dependent Strand Annealing and to Promote Recombinational Repair
Cdk1 kinase phosphorylates budding yeast Srs2, a member of UvrD protein family, displays both DNA translocation and DNA unwinding activities in vitro. Srs2 prevents homologous recombination by dismantling Rad51 filaments and is also required for double-strand break (DSB) repair. Here we examine the biological significance of Cdk1-dependent phosphorylation of Srs2, using mutants that constitutively express the phosphorylated or unphosphorylated protein isoforms. We found that Cdk1 targets Srs2 to repair DSB and, in particular, to complete synthesis-dependent strand annealing, likely controlling the disassembly of a D-loop intermediate. Cdk1-dependent phosphorylation controls turnover of Srs2 at the invading strand; and, in absence of this modification, the turnover of Rad51 is not affected. Further analysis of the recombination phenotypes of the srs2 phospho-mutants showed that Srs2 phosphorylation is not required for the removal of toxic Rad51 nucleofilaments, although it is essential for cell survival, when DNA breaks are channeled into homologous recombinational repair. Cdk1-targeted Srs2 displays a PCNA–independent role and appears to have an attenuated ability to inhibit recombination. Finally, the recombination defects of unphosphorylatable Srs2 are primarily due to unscheduled accumulation of the Srs2 protein in a sumoylated form. Thus, the Srs2 anti-recombination function in removing toxic Rad51 filaments is genetically separable from its role in promoting recombinational repair, which depends exclusively on Cdk1-dependent phosphorylation. We suggest that Cdk1 kinase counteracts unscheduled sumoylation of Srs2 and targets Srs2 to dismantle specific DNA structures, such as the D-loops, in a helicase-dependent manner during homologous recombinational repair.
Published in the journal:
. PLoS Genet 6(2): e32767. doi:10.1371/journal.pgen.1000858
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000858
Summary
Cdk1 kinase phosphorylates budding yeast Srs2, a member of UvrD protein family, displays both DNA translocation and DNA unwinding activities in vitro. Srs2 prevents homologous recombination by dismantling Rad51 filaments and is also required for double-strand break (DSB) repair. Here we examine the biological significance of Cdk1-dependent phosphorylation of Srs2, using mutants that constitutively express the phosphorylated or unphosphorylated protein isoforms. We found that Cdk1 targets Srs2 to repair DSB and, in particular, to complete synthesis-dependent strand annealing, likely controlling the disassembly of a D-loop intermediate. Cdk1-dependent phosphorylation controls turnover of Srs2 at the invading strand; and, in absence of this modification, the turnover of Rad51 is not affected. Further analysis of the recombination phenotypes of the srs2 phospho-mutants showed that Srs2 phosphorylation is not required for the removal of toxic Rad51 nucleofilaments, although it is essential for cell survival, when DNA breaks are channeled into homologous recombinational repair. Cdk1-targeted Srs2 displays a PCNA–independent role and appears to have an attenuated ability to inhibit recombination. Finally, the recombination defects of unphosphorylatable Srs2 are primarily due to unscheduled accumulation of the Srs2 protein in a sumoylated form. Thus, the Srs2 anti-recombination function in removing toxic Rad51 filaments is genetically separable from its role in promoting recombinational repair, which depends exclusively on Cdk1-dependent phosphorylation. We suggest that Cdk1 kinase counteracts unscheduled sumoylation of Srs2 and targets Srs2 to dismantle specific DNA structures, such as the D-loops, in a helicase-dependent manner during homologous recombinational repair.
Introduction
Homologous recombination (HR) is a fundamental DNA repair pathway and its deregulation is responsible for a variety of genomic rearrangements, including chromosome loss, DNA translocations and inversions, which are typical of the genetic alterations seen in tumor cells (reviewed in [1]). The mechanisms and proteins involved in HR have been well conserved throughout evolution and much of our knowledge on HR comes from studies conducted in the yeast Saccharomyces cerevisiae (reviewed in [2]–[5]). HR targets multiple DNA lesions, including single-stranded DNA (sDNA) breaks and DSBs, promoting their repair using a region of homology as a template. Diverse pathways can seal sDNA breaks, but the role of HR in DSB repair is essential. Different HR sub-pathways compete for DSB repair and some are less accurate than others [6],[7]. The position of DNA sequences involved in recombination and the extent of their homology influence the kinetics of DSB repair. Irreparable DNA breaks [8], or even those repaired slowly [9], appear to be sequestered to the nuclear periphery, through a mechanism resembling that used to tether telomeres at the nuclear membrane [10]. When a region of homology is found on both sides of a DSB, the preferred pathway of repair is gene conversion (GC). Among the initial steps in GC is the formation of Rad51 presynaptic nucleofilaments assisted by accessory factors. While Rad51 nucleation can occur directly at sDNA breaks, the ends of DSBs must be first processed to produce sDNA tails in order to recruit Rad51. Multiple factors with nuclease and/or helicase activities, including the Mre11/Rad50/Xrs2 complex, Sae2, Exo1, Dna2 and Sgs1 cooperate in 5′ to 3′ DSB resection (reviewed in [11]). Assembled Rad51 nucleofilaments invade and displace a duplex donor homologue DNA template leading to the formation of a D-loop structure. The D-loop is the site of DNA synthesis, which is promoted by extension of the 3′ invading strand. According to the canonical DSB repair model [12], the capture of the second end of the DSB generates a double Holliday junction (dHJ) whose resolution, by cutting or branch migration, influences the formation of crossover products associated with GC, that is, the extent of DNA exchanges associated with DSB repair. If the second DSB end is not captured, it can anneal to the invading strand evicted from the D-loop soon after DNA synthesis. In this process, called synthesis-dependent strand annealing (SDSA), GC is limited to DNA synthesized from donor strand and crossovers are prevented [4]. Another HR pathway, known as single strand annealing (SSA), is used when DSB repair occurs between direct repeats [4]. In this case resected homologous sequences anneal without DNA synthesis and DSB repair is associated with deletion of the sequence between the repeats. Notably, during SSA, a D-loop is not formed and Rad51 is not required. The formation of presynaptic Rad51 nucleofilaments is fundamental for HR commitment during GC. However, Rad51 could nucleate improperly on DNA or even be engaged into damaged filaments when other recombination factors are inactivated: in both cases HR is not proficient, rather it becomes toxic for other DNA transactions.
Many in vivo studies suggest that Srs2, a member of UvrD family of DNA helicases conserved from bacteria to human, is involved in the removal of toxic Rad51 filaments from sDNA [13]–[17]. Further, the Srs2 protein disrupts presynaptic Rad51 filaments through its DNA translocase activity in vitro [18],[19]. This Srs2 anti-recombination activity requires a physical interaction with sumoylated PCNA, as it was evidenced in the absence of the post-replication repair (PRR) pathway, a context in which Srs2 prevents deadly the recombinational repair [20],[21]. Srs2 also exhibits 3′ to 5′ DNA helicase activity on duplex DNA [22]. Recent in vitro studies in yeast and plants suggest that Srs2 unwinds DNA structures mimicking a D-loop [23],[24]. Genetic evidence, indeed, suggests that Srs2 favors the SDSA pathway, since the loss of Srs2 results in an increase in crossover products [25]–[27]. Moreover, Srs2 is essential for DSB repair through either SSA or ectopic GC [25],[28],[29]; in SSA repair, Srs2 is required to mediate recovery from checkpoint-mediated arrest [29].
Since Srs2 affects HR in several ways, Srs2 functions in recombination are probably regulated. Previous studies demonstrated that Srs2 is a target of the cell cycle-dependent kinase (Cdk1) in vivo [30] and in vitro [31]. Cdk1 has been implicated in the DNA damage response and in DSB repair [32]; by monitoring repair of one HO-induced break, it was shown that Cdk1 is required both at the level of resection and at a step after Rad51-dependent strand invasion [33],[34]. It is known that Cdk1 triggers the resection of DSB ends by phosphorylating Sae2 [35], but other direct targets in DSB repair are unknown.
We found that srs2 mutants that are unable to undergo Cdk1-dependent phosphorylation can still remove toxic Rad51 nucleofilaments, but these srs2 mutants fail to promote homologous recombinational repair. Analysis on repair of a single HO-induced break through ectopic GC shows that the proper turnover of Srs2, at D-loop intermediates, is dependent on its modification by phosphorylation and this phosphorylation is essential for completion of the SDSA reaction that results in non-crossover products. Moreover, the phosphorylation-dependent role of Srs2 does not require an interaction with PCNA and does not affect the turnover of Rad51 at invading filaments. In the absence of Srs2 phosphorylation, the protein is sumoylated and this is the main cause of the recombinational repair defects seen in the nonphosphorylatable srs2 mutant. Thus, coordination of the sumoyaltion and phosphorylation modifications on Srs2 is essential during homologous recombinational repair.
Results
The C-terminus tail of Srs2 contains consensus sites for Cdk1-dependent phosphorylation and sumoylation
Saccharomyces cerevisiae Srs2 contains characteristic amino acid motifs important for ATP-binding and DNA-binding that are highly conserved among members of UvrD family [36]. All these motifs are located in the N-terminal domain of the Srs2 protein (grey box in Figure 1A) and are sufficient for the helicase activity [22], but not for translocase-dependent removal of Rad51 nucleofilaments, as tested in vitro [37],[38]. The C-terminal tail of Srs2 protein plays an important regulatory function, since it mediates protein-protein interactions, including interaction with Rad51 and PCNA [15], [21], [37]–[40]. Moreover, a cluster of five consensus sites for Cdk1 kinase is present in the C-terminal region of Srs2, while two additional sites are located in the helicase domain (Figure 1A; [39]). The last 138 amino acids (aa) of the Srs2 C-terminal tail are required for the interaction with PCNA [21] and also contain three consensus sites for sumoylation (Figure 1A).

We previously showed that changing the seven serine/threonine Cdk1 consensus sites to the unphosphorylatable residues alanine/valine abolished DNA damage-induced phosphorylation of Srs2, which can be monitored as an electrophoretic mobility shift on SDS-PAGE (Figure 1B; [39]). We then produced a new srs2 allele in which the same serine/threonine residues were changed to the negatively charged aspartic acid/glutamic acid residues, with the aim of producing a mutated version of Srs2 that mimics the constitutively phosphorylated protein isoform. As shown in Figure 1B, the levels of wt Srs2 and the two mutated Srs2 isoforms are similar, both in normal conditions and in response to DNA damage by methyl methanesulfonate (MMS)-treatment (data not shown). Henceforth, we will refer to the unphosphorylated and phosphorylated srs2 mutants, respectively, as srs2-7AV and srs2-7DE.
Srs2 phosphorylation promotes recombinational repair, but is not essential for the reversal of toxic Rad51 nucleofilaments accumulating at sDNA gaps
To investigate whether Cdk1-dependent phosphorylation of Srs2 is important for its roles in HR, we first evaluated cell survival of the two srs2 phospho-mutants following UV-light and zeocin treatments. Wild type (SRS2) and srs2Δ strains were used as controls. Previous studies have shown that the UV-sensitivity of srs2Δ strains is suppressed by mutations in RAD51, indicating that cell lethality is due to accumulation of toxic Rad51 nucleofilaments at gaps whose repair can occur in the absence of HR [16]. We found that srs2Δ and rad51Δ mutants are also sensitive to zeocin, a radiomimetic chemical that induces DSBs (Figure 2A and data not shown). Thus, zeocin-treatment induces DNA lesions whose repair is strictly HR-dependent and prevented in the absence of Srs2. As shown in Figure 2A, we found that both srs2-7AV and srs2-7DE mutants, as SRS2 strains, survive UV-light doses that kill srs2Δ mutants. Conversely, the srs2-7AV mutant, but not the srs2-7DE mutant, is sensitive to zeocin and, indeed, is even more sensitive than the srs2Δ strain. Previous reports showed that srs2Δ mutations are synthetically lethal with either sgs1Δ or rad27Δ mutations [13],[14],[41]. While the synthetic lethality of srs2Δ sgs1Δ double mutants is suppressed by rad51Δ [13], single rad27Δ mutants are themselves lethal in combination with rad51Δ [42]. Thus, the types of spontaneous DNA damage accumulating in sgs1Δ and rad27Δ mutants mirror those induced by UV and zeocin treatments: only in rad27Δ mutants and under zeocin treatment, HR is essential for DNA repair. We crossed the srs2-7AV and srs2-7DE phospho-mutants and srs2Δ as control with sgs1Δ or rad27Δ mutants. Heterozygous diploid mutants were sporulated and tetrad analysis was performed. As shown in Figure 2B, neither srs2Δ sgs1Δ nor srs2Δ rad27Δ double mutants form viable spores; the srs2-7AV mutation, but not the srs2-7DE mutation, is synthetically lethal with the rad27Δ mutation, while both srs2-7AV sgs1Δ mutants and srs2-7DE sgs1Δ mutant spores form colonies.
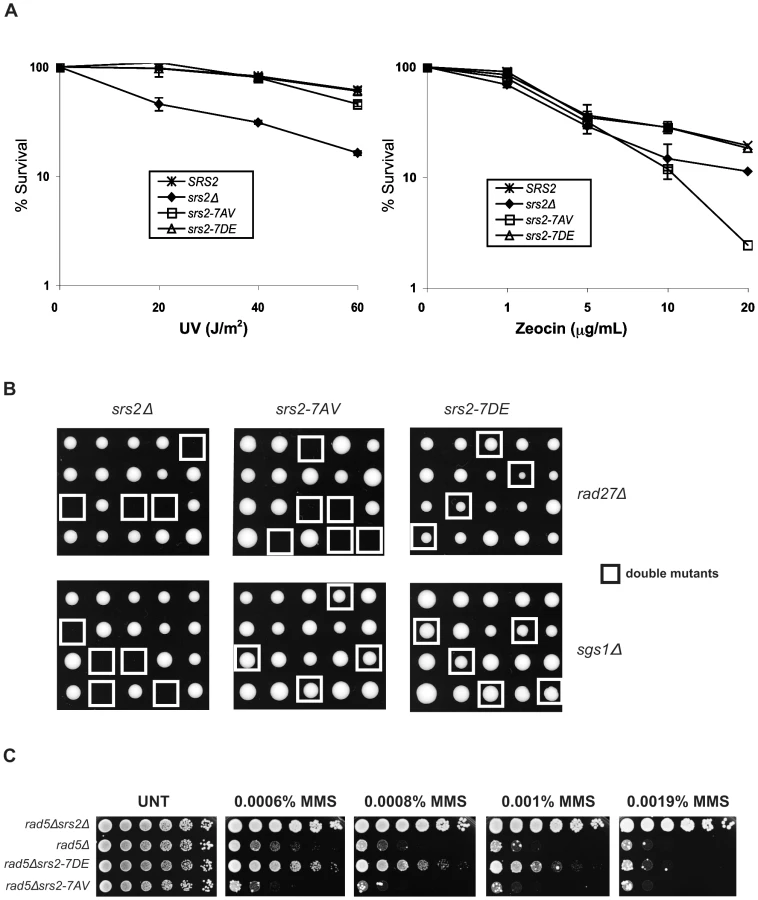
Hence, the phenotypes of srs2-7AV mutants suggest that Srs2 phosphorylation is dispensable for the reversal of toxic Rad51-dependent recombination intermediates induced at sDNA by UV or by the absence of Sgs1, but phosphorylation is required to promote recombinational repair in zeocin and in the absence of Rad27.
Previous data suggested that the Srs2 protein sensitizes postreplication repair (PRR) mutants, because it prevents HR [43],[44]. Accordingly, as show in Figure 2C, the sensitivity of rad5Δ mutants to MMS is alleviated by deleting SRS2. srs2 mutants encoding a protein that displays attenuated translocase activity also suppress the DNA damage sensitivity of PRR mutants, although they are not sensitive to DNA damaging agents by themselves [40]. Hence, we analyzed the srs2 phospho-mutants in a PRR mutant context, in which the importance of having an intact DNA translocation activity should be revealed. We constructed srs2 phospho-mutants in rad5Δ or rad18Δ backgrounds and then tested viability on medium containing MMS. We found that srs2-7AV mutation hypersensitizes rad5Δ and rad18Δ mutants to DNA damage, but, conversely, the srs2-7DE mutation partially suppresses the lethality of rad5Δ or rad18Δ mutation (Figure 2C and data not shown). Notably, srs2-7AV and srs2-7DE mutants are not sensitive to MMS, even at a higher MMS dose than those employed in Figure 2C (data not shown).
Thus, we conclude that, even in a PRR context, unphosphorylatable Srs2 can remove Rad51 at DNA gaps. On the other hand, the phosphorylated Srs2 protein isoform appears to be less proficient in the anti-recombinational role.
Srs2 phosphorylation is required for Rad51-dependent DSB repair
The observation that srs2-7AV mutants are sensitive to treatment with zeocin suggests that phosphorylation of Srs2 is important in DSB repair. To directly examine this, we tested the behavior of srs2 phospho-mutants in response to a single DSB created by a galactose-inducible HO endonuclease. Previous studies have shown that srs2Δ mutants can not survive a single HO-induced DSB when repair of this break occurs either by ectopic GC or by SSA [25],[28],[29]. While the GC pathway strictly depends on RAD51, SSA can occur in the absence of Rad51. There are important differences in the requirement for Srs2 in the two pathways: Srs2 is not required to complete DSB repair during SSA, but it is required for recovery from the DNA damage-induced cell cycle arrest [29]. RAD51 deletion rescues the checkpoint recovery defect in srs2Δ mutants [29]; thus, one hypothesis is that Rad51 accumulates on DNA contributing to the lethal checkpoint-induced arrest, since it can not be removed in absence of Srs2 [29],[45]. Conversely, during ectopic GC, srs2Δ mutants are unable to complete DSB repair, with a specific reduction in non-crossover products formation [25]. Since the region of DNA homology involved is limited in ectopic DSB repair, the formation of crossovers might be prevented because the formation of the dHJ intermediate is reduced [46]. Thus, the failure to carry out SDSA results in loss of non-crossover products and there is a marked reduction in DSB repair efficiency [25].
To analyze the requirement of Srs2 phosphorylation in the DSB repair response, we assayed cell survival of srs2 phospho-mutants in a SSA system in which DSB repair occurs between repeated sequences, one of which is located 25kb from the DSB and results in a chromosomal deletion [29] or in an ectopic GC system in which DSB repair occurs between chromosomes V and III [25]. In agreement with previous findings, the rate of cell survival of srs2Δ mutants is 2% in both the SSA and GC systems (Figure 3A and 3B). This high cell lethality in srs2Δ mutants correlates with inability to dephosphorylate the checkpoint kinase Rad53, which is activated in response to DSB induction (Figure 3A and 3B). Cell survival of srs2-7AV mutants is 25% in the GC system where they also fail to fully dephosphorylate Rad53 24 hours after DSB induction (Figure 3A and 3B). Survival of the srs2-7AV mutant is normal in the SSA system and survival of the srs2-7DE mutant is normal in both systems. Thus, Srs2 phosphorylation is necessary for cell survival when DSB repair proceeds through the Rad51-dependent GC pathway, but is dispensable in the SSA pathway, which does not require Rad51. As mentioned above, although SSA is Rad51-independent pathway, in absence of Srs2, Rad51 might improperly accumulate on DNA and interfere with checkpoint recovery [29],[45]. Since srs2-7AV survive DSB repair via SSA, this further strengthens the conclusion that Srs2 phosphorylation is not required for reversal of toxic Rad51-dependent intermediates.

We used Southern blotting with a probe that recognizes the MAT locus, to physically observe DSB repair in the GC system (Figure 3B). As mentioned above, in this system a DSB is induced at a MAT locus inserted into Chromosome V and is repaired using a unique uncleavable MAT-inc cassette on chromosome III (Figure 3B). Notably, crossovers that are associated with the GC event can be evaluated by restriction analysis, since crossovers give rise to chromosomal bands that differ in size from the parental chromosomes and the non-crossover GC products (Figure 3B). As shown in Figure 3C, DNA of SRS2 and srs2 mutants were analyzed by Southern blotting. DSB repair efficiency is about 30% in srs2Δ strain, in agreement with previous findings [25] and in srs2-7AV it is reduced to 70% compared to SRS2 or srs2-7DE (Figure 3C). Moreover, the percentage of crossovers associated with GC increases three-fold in srs2Δ and two-fold in srs2-7AV compared to SRS2 or srs2-7DE (Figure 3C). Similar to the srs2Δ mutants, the increase in crossovers is associated with a reduction in non-crossover repair efficiency in the srs2-7AV mutant (Figure 3C); thus, DSB repair defects in the absence of Srs2 phosphorylation likely indicate a specific failure to carry out repair via the SDSA pathway that results in non-crossover products.
Srs2 phosphorylation affects turnover of Srs2, but not turnover of Rad51, during strand invasion in DSB repair
Our analysis indicates that Srs2 phosphorylation is required for Rad51-dependent DSB repair. Although we found that Srs2 phosphorylation is not essential for the removal of toxic Rad51 nucleofilaments at DNA gaps or during DSB repair by SSA, it might be specifically required to remove Rad51-dependent recombination intermediates initiated at D-loop intermediate. To investigate this possibility, we analyzed Rad51 binding to DSBs by ChIP and Q-PCR in SRS2 and srs2 phospho-mutants. We used DNA primers that amplified the region of homology located on donor chromosome III. Using this strategy, proteins localizing either at broken or recipient chromosomes will be immunoprecipitated at the DSB when the invading strand is in duplex DNA, which most likely represents the D-loop. As shown in Figure 4A, Rad51 protein is undetectable at the donor MAT locus before HO induction, while it is loaded at the DSB with similar kinetics in SRS2 and all srs2 mutated strains. Thus, we conclude that Rad51-mediated strand invasion occurs with similar kinetics in SRS2 and srs2 mutants. We also conclude that Rad51 is removed from the DSB with similar kinetics in all contexts and strains analyzed. Thus, DSB repair defects in srs2Δ or srs2-7AV mutants are unrelated to an abnormal persistence of Rad51 after strand invasion.

Previous findings have indicated that Srs2 is loaded at DSBs [47]. We asked whether Srs2 phosphorylation could influence its ability to be recruited to DSBs in our GC system. Using the same ChIP strategy employed above, we found that Srs2 is sited at the invading strand with a three-fold enrichment (Figure 4B). The Srs2 and Srs2-7DE proteins are loaded and dislodged from DNA with kinetics resembling that of Rad51, but the Srs2-7AV protein accumulates only at later times and abnormally persists on DNA for at least 24 hours after DSB induction; notably, Rad51 has been displaced from DNA, when Srs2-7AV protein accumulates (Figure 4).
In summary, the data in Figure 4 suggest that Srs2 is loaded at the D-loop during GC and its proper recruitment is governed by Cdk1-dependent phosphorylation. However, the DSB repair defects in srs2-7AV or srs2Δ mutants appear not be related to inefficient metabolism of Rad51 nucleofilaments at donor DNA sequences.
Sumoylation of Srs2 is responsible for the recombination defects in srs2-7AV phosphomutants
In the course of our studies on Srs2 phosphorylation, we noticed that in response to massive DNA damage, such as treatment with 0.3% MMS, Srs2 accumulates as additional modified isoforms, which can be visualized as a ladder on SDS-PAGE analysis (Figure 5A). These Srs2 protein isoforms are recognized by SUMO-specific antibodies (Figure 5A). Preliminary characterization of Srs2 sumoylation indicates that none of the well-characterized SUMO ligases, including Siz1 and Siz2, are involved in this modification (Figure S1A). Three putative sumoylation sites have been mapped to the C-terminus tail of Srs2 (Figure 1A). Our data indicated that DNA damage-induced sumoylation of Srs2 was abolished in srs2-3KR mutants, in which the three lysine residues in the motifs identified as modified by SUMO were mutated to arginine (Figure 5A). Notably, the Srs2-3KR protein can be fully phosphorylated (Figure S1B). Intriguingly, while sumoylation of native Srs2 is induced at 0.3% MMS, the unphosphorylatable Srs2-7AV protein can be detected as SUMO-modified isoforms at ten-fold lower MMS doses (Figure 5A). Thus, while sumoylation and phosphorylation can occur independently, Srs2 accumulates in a sumoylated form in the absence of phosphorylation.

The biological relevance of Srs2 sumoylation is still obscure, as extensive studies of the phenotypes of the srs2-3KR mutant were inconclusive (D. Callahan and H. Klein, unpublished results). However, the finding that unphosphorylatable Srs2 is hyper-sumoylated prompted us to ask if the srs2-7AV mutant defects in recombinational repair might be related to Srs2 sumoylation. To test this, we mutagenized the sumoylation consensus sites in the srs2-7AV mutant to create the srs2-7AV3KR allele, which is simultaneously impaired for phosphorylation and sumoylation. We then tested the behavior of the srs2-7AV3KR mutant in the DSB repair GC system in which srs2-7AV mutant was highly sensitive (see Figure 3). We found that srs2-7AV3KR mutant survived DNA damage (Figure 5B); DSB repair is accomplished efficiently and a normal level of crossovers is seen in srs2-7AV3KR (Figure 5B). In addition, the srs2-7AV3KR mutant correctly reversed the checkpoint response after DSB induction and repair, as seen by Rad53 kinase dephosphorylation (data not shown). Furthermore, the srs2-3KR mutant, which is only impaired in sumoylation, can accomplish DSB repair (Figure 5B). To see if ablation of Srs2 sumoylation rescues the phosphorylation defects in recombinational repair in other contexts, we crossed the srs2-7AV3KR mutant with the rad27Δ mutant to generate rad27Δ srs2-7AV3KR double mutants. While the rad27Δ srs2-7AV double mutants never form viable spores (see Figure 2A), we found that rad27Δ srs2-7AV3KR double mutants developed into visible colonies (17/25 of total cases analyzed), although the double mutant grew very slowly (Figure 5C). This partial suppression highlights the importance of Srs2 protein modifications when it is likely that more than one lesion is formed.
Taken together, the data in Figure 5 indicate that Srs2 is sumoylated in vivo. Sumoylation of Srs2 is not required for DSB repair, but the recombinational repair defects in unphosphorylatable srs2-7AV mutants are largely related to the unscheduled sumoylation of the protein.
Cdk1-targeted role of Srs2 is exerted independently of its interaction with PCNA
The sumoylation consensus sites are located in the last 138 residues of the C-terminus tail of Srs2 (Figure 1A), which also mediates the interaction with PCNA [21]. Hence, we asked if this tail is important for the Cdk1-dependent role of Srs2. As shown in Figure 6, we found that the srs2−ΔC138 mutant is viable after induction of a HO-mediated DSB and also when combined with a rad27Δ. Conversely, unphosphorylatable srs2-7AV mutants lacking the PCNA-interaction domain (srs2-7AVΔC138) are lethal in both contexts. These data suggest that Cdk1 targets Srs2 to promote recombinational repair independently of the interaction with PCNA and sumoylation. Moreover, elimination of sumoylation sites, but not deletion of the Srs2 tail containing the same sites, suppresses the recombination defects in the srs2-7AV mutant.

Discussion
Increasing evidence suggests that cell cycle-dependent kinase Cdk1 is required for the DNA damage response [32], so changing the general view that in the presence of DNA lesions Cdk1 has to be inhibited to allow sufficient time for DNA repair. It rather suggests that Cdk1 is actively involved in DNA repair. Cdk1 phosphorylates Sae2, which is part of the DSB resection machinery, and is needed to promote HR [35]. Sae2 is the only Cdk1 target that functions in DSB repair identified thus far, although Cdk1 is required at later steps during HR [33]. Cdk1 also phosphorylates the budding yeast Srs2 protein [30],[31] a member of the evolutionarily conserved family of UvrD proteins, which displays both DNA unwinding [22] and sDNA translocation activities in vitro [18],[19]. The DNA translocase activity of Srs2 is essential to disrupt toxic Rad51 nucleofilaments and prevent unwanted recombination events, while the helicase activity of Srs2, both in yeast and plants, is thought to be required to dismantle DNA structures mimicking a D-loop [23],[24].
One key finding in the present study is that Cdk1-dependent phosphorylation of Srs2 is required to complete the SDSA pathway, thus Srs2 is a novel target for Cdk1 in DSB repair. Srs2 phosphorylation controls the quality of DSB repair by preventing crossover outcome. Since we found that Srs2 phosphorylation is not required for the removal of toxic Rad51 filaments, we suggest that Cdk1 targets Srs2 helicase to dismantle D-loop structures in order to favor non-crossover products.
Srs2 phosphorylation promotes recombinational repair, but is dispensable for reversal of toxic Rad51 nucleofilaments at gaps
Recombination can be both prevented and stimulated in srs2 mutants, suggesting a dual role for Srs2 in HR. The finding that Srs2 is a DNA translocase that antagonizes the formation of unscheduled Rad51 filaments explains certain srs2 phenotypes in HR that are suppressed by ablating RAD51; these include the synthetic lethality with sgs1 mutants or high sensitivity to UV-light [13],[16]. Nevertheless, srs2 mutants are defective in Rad51-dependent DSB repair [25],[28] or lethal when combined with rad27Δ mutants [14],[41]. These are contexts in which HR is essential to restore DNA lesions and the activity of Srs2 is required to promote homologous recombinational repair.
In this study we analyzed the recombination phenotypes of two srs2 mutants that mimicked either the constitutive unphosphorylated (srs2-7AV) or Cdk1-dependent phosphorylated (srs2-7DE) protein isoforms. We found that srs2-7AV unphosphorylatable mutants display only a subset of srs2Δ phenotypes and, in particular, they do not display those phenotypes that are suppressed by RAD51 deletion. In fact, srs2-7AV mutants are not UV-sensitive or synthetically lethal with sgs1Δ, but are non-viable when combined with rad27 mutants or treated with the DSB-inducing drug zeocin. Thus, functions of Srs2 in preventing unscheduled recombination or in allowing efficient recombinational repair are genetically separable. The phosphorylation of Srs2 is dispensable for the removal of toxic Rad51 nucleofilaments assembled at gaps, while it is essential to promote recombinational repair.
Srs2 phosphorylation is required during DSB repair to complete the SDSA pathway
In accordance with the finding that Srs2 phosphorylation is essential to promote recombination, we found that it is also required for Rad51-mediated DSB repair. In particular, we have been able to show that Srs2 phosphorylation is necessary to complete SDSA in DSB repair. ChIP data on Rad51 are consistent with the idea that strand invasion is not affected and that Rad51 protein does not persist on the D-loops in srs2Δ or srs2-7AV mutants, although we cannot rule out that presynaptic filament assembly may somehow be affected in the absence of Srs2 or its phosphorylation. ChIP analysis conducted on Srs2 suggests that the protein is found at DSBs upon strand invasion, thus it is likely loaded at D-loops. Taken together, these data are consistent with a role of phosphorylated Srs2 in SDSA pathway, but another helicase/translocase may be implicated in removing Rad51 at the D-loops. We favour the idea that Cdk1 targets Srs2 to dismantle the D-loop intermediate in SDSA (Figure 7) perhaps after DNA synthesis has extended the invading strand. Srs2 helicase activity might be stimulated by binding to the D-loop structure and/or by interaction with other recombination factors. ChIP data conducted on unphosphorylatable Srs2-7AV at the invading strand suggest that the mutated protein accumulates at later times and is not rapidly dislodged from DNA as the wild-type protein. The fact that unphosphorylatable Srs2 appears glued at the D-loops is evocative of a protein working very inefficiently and whose turnover is largely prevented. It is likely that the unscheduled accumulation of the protein on the DNA might contribute to impaired cell viability and, consistent with this idea, the lethal phenotype of srs2-7AV mutant in response to DSBs is dominant (Figure S2).

Our data indicate that the proportion of srs2-7AV cells that do not survive DSB repair via GC is higher than the one, which fails to repair DNA lesion (Figure 3). This suggests that a fraction of srs2-7AV cells might die because of checkpoint-mediated arrest, as in srs2Δ mutants [25]. However, Srs2 phosphorylation is not required for recovery during DSB repair by SSA, that is, when Srs2 is probably engaged to remove toxic Rad51-depedent DNA structures, rather than working at the D-loop intermediate [45]. Thus, the checkpoint recovery defect in srs2-7AV mutants might have different causes during DSB repair by GC or SSA; as described below, perhaps some aspects of recovery defect in srs2 mutants in GC could be explained considering that Cdk1-dependent phosphorylation was no longer required for Srs2 recombination activity, if sumoylation is also prevented.
Cdk1 activity prevents unscheduled sumoylation of Srs2
We found that Srs2 sumoylation can be detected in vivo in response to heavy DNA damage. Protein modification is prevented ablating three lysine residues located in the extreme C-terminus tail of Srs2. Sumoylation and Cdk1-dependent phosphorylation modifications of Srs2 are independent events, but when phosphorylation fails, sumoylated Srs2 accumulates. There is a functional relationship between these two DNA damage induced modifications, since ablation of sumoylation residues largely rescues the recombinational repair phenotypes of srs2-7AV mutants.
What may be the mechanism for the toxicity of sumoylation in the absence of phosphorylation? Sumoylation of Srs2 alone appears unnecessary for many of its recombination functions (D. Callahan and H.Klein, unpublished results); here we show that it is not essential in DSB repair (see Figure 5B and Figure 6). While the biological significance of Srs2 sumoylation waits to be elucidated, we speculate that it might be important for degradation of Srs2 protein. Srs2 can interact physically with Slx5 [39], that in complex with Slx8, has been implicated in degradation of sumoylated proteins bound to irreparable DNA breaks at the nuclear periphery [8],[9]. Our data suggest that Cdk1-dependent phosphorylation of Srs2 counteracts its sumoylation, which takes over only in response to massive DNA damage. Thus, in a possible scenario, unphosphorylated and sumoylated Srs2 is trapped at DSB and becomes channeled via the Slx5/Slx8 pathway to the nuclear periphery (Figure 7).
Since this emergency nuclear periphery pathway intervenes to degrade proteins in response to irreparable DSBs, it might normally act on phosphorylated and sumoylated Srs2 and, therefore, Srs2-7AV cannot be eliminated. Conversely, after successful DSB repair, phosphorylated Srs2 could be recycled by other routes, and independent of sumoylation. Intriguingly, the unscheduled Srs2-dependent sequestration of DSBs to the periphery might explain the checkpoint recovery defects in srs2-7AV and perhaps also that of srs2Δ, if we imagine that another unregulated DNA helicase takes over in the absence of Srs2. Our studies did not show any obvious alterations in Srs2 protein levels in srs2 phospho-mutants and/or SUMO-mutants (M.Saponaro and G.Liberi, unpublished results), but local protein degradation events at damaged DNA could be relevant.
Elimination of sumoylation compensates for the absence of phosphorylation of Srs2 in DSB repair, but paradoxically this rescue requires the last 138 residues of Srs2 that are not normally necessary for DSB repair. Hence, this suppression might require interaction with other factors. Preventing sumoylation in the unphosphorylatable Srs2 rescues recombination defects that ensue after a single DSB, but the importance of these Srs2 modifications become evident when many breaks occur, as in the rad27Δ mutants.
Roles of Srs2 modifications in replication-induced DNA damage
We found that Srs2 phosphorylation is essential for recombinational repair of spontaneous damage occurring during S-phase in rad27Δ mutants. Similar to the response to DSBs, sumoylation of Srs2 is a main cause of death in srs2-7AV phospho-mutants. It is more difficult to predict the kind of damage which requires phosphorylated Srs2 in rad27Δ mutants. Rad27 is required for Okazaki DNA fragment processing [48] and in its absence, Srs2 might dismantle DNA and/or RNA structures that block HR. In any case, based on our conclusion that Srs2 phosphorylation is not essential for the processing of toxic Rad51 filaments, we think it more probable that the helicase activity, rather than translocase activity, is crucial for the survival in rad27Δ mutants. This proposed role of phosphorylated Srs2 in replication might seem at odds with the role suggested for Srs2 in preventing recombinational repair during S-phase through recruitment by sumoylated PCNA [20],[21]. However, in PRR mutants, Srs2 is proposed to be recruited by PCNA to disrupt Rad51 filaments at DNA gaps, while in the absence of Rad27, we are considering that Srs2 acts in a PCNA-independent and phosphorylation-dependent role as a helicase, rather than as a translocase. Importantly, srs2-7DE mutants slightly suppress the MMS sensitivity of PRR mutants, suggesting that the phosphorylated Srs2 is less efficient as a DNA translocase than the non-phosphorylated isoform. This is unmasked in PRR mutants, where it is likely that many sDNA breaks occur. Srs2 phosphorylation might modulate its interaction with PCNA, a hypothesis that will be interesting to test in the future.
Concluding remarks
Our data indicate that Srs2 is a new target of Cdk1 kinase in DSB repair, acting at the level of strand invasion, rather than during DNA end resection. Srs2 phosphorylation is required to limit the extent of DNA exchanges during DSB repair with a function that is genetically separable from its role in processing toxic Rad51 filaments. We suggest that Cdk1-mediated phosphorylation might control, throughout the interaction with PCNA and/or other factors, the ability of Srs2 to function as a translocase or a helicase that inhibits or allows HR depending on the context. Furthermore, our data unravel a novel aspect of Cdk1-dependent regulation in counteracting untimely sumoylation events, which might become toxic for recombination if not properly scheduled.
Materials and Methods
Strains and plasmids
Genotypes of the strains used in this study are listed in Table S1. Deletion strains were obtained by the one-step PCR method and multiple mutant strains were derived from meiotic segregants of appropriate crosses. The srs2-7DE phospho-mutant was constructed by a site-directed mutagenesis strategy already described to construct the srs2-7AV mutant [39]. Mutations in SRS2 were introduced at the seven consensus sites for the Cdk1 kinase (T604D, S698E, S879E, S893E, S938E, S950E and S965E). Construction of srs2-3KR strain, containing mutations K1081R, K1089R and K1142R at SUMO-consensus sites, will be described in detail elsewhere. srs2-7AV3KR mutant was constructed as follow: a NAT selection cassette was integrated downstream of the srs2-3KR mutated gene. DNA primers were designed to amplify a DNA region containing both the 3KR mutations and the NAT cassette. This DNA region was then used to replace, by transformation, the C-terminus of the srs2-7AV. A similar PCR-mediated strategy was used to delete the C-terminus-PCNA interaction domain in both wild type and srs2-7AV mutants. SRS2 and srs2 mutants were also cloned into the low copy-number Ycplac22 vector by gap-repair procedure or using PCR-based strategies described above and were used in all HO-based experiments. As tested by Western blotting, protein levels are similar when Srs2 or its mutated versions were expressed from SRS2 chromosomal locus or from the Ycplac22 centromeric plasmid.
UV and drug treatments
Log-phase cells were spread on YPD plates, irradiated with UV light (254 nm) and incubated in the dark; cell survival was compared to that of untreated controls. Log-phase cultures were incubated with different doses of zeocin (Invitrogen) for 1 hour and cell survival was calculated by comparing the plating efficiency with untreated cells. The UV and zeocin curves are the average of three independent experiments. Spot assays were performed by evaluating the growth of serially diluted cultures on synthetic complete medium containing adenine at a final concentration of 0.7 g/liter, with or without MMS (SIGMA).
DSB repair efficiency, crossover frequency, and viability measurements
Relative frequencies of survival of cultures plated on glucose and galactose using the SSA and GC DSB HO-inducible systems were calculated as previously described [25],[29]. Product formation and analysis of crossover formation were assessed by Southern blotting analyses as described in [25]. The results shown are the average of three to five independent experiments.
ChIP experiments and quantitative PCR
Asynchronous cultures were grown overnight at 28°C in YEP media containing 2% raffinose. When cultures reached mid log-phase, 15 µg/ml nocodazole was added to synchronize the cells in G2/M. Expression of the HO endonuclease was induced by addition of 2% galactose for the indicated times. ChIP analysis was carried out as previously described [49]. Samples were incubated with 1% Formaldehyde for 20 min with the anti-Rad51 ChIP and 45 minutes with the anti-Srs2 ChIP. Immunoprecipitation was carried out with clarified extracts using anti-Rad51 (kindly gift of Patrick Sung, Yale University, New Heaven) or anti-Srs2 (Santa Cruz Biotechnology) antibodies overnight at 4°C. Levels of immunoprecipitated DNAs were measured by quantitative real-time PCR using the SYBR Green technique (SYBR Green PCR Master Mix, Applied Biosystems) and run in an Applied Biosystems 7500 Fast Real-Time PCR System. Sequences of the DNA primers are listed in the Table S2. Dissociation stage curves were checked to test primer specificity. The results were analyzed with the 2-DCT method as previously described [50]. For the ChIP anti-Rad51 experiments the relative enrichment was determined by the fold increase of ChIPed DNA relative to that prior to DSB induction; for the ChIP anti-Srs2 the absolute 2-DCT variation after the subtraction of the 2-DCT of the ChIP carried out in parallel in a srs2Δ mutant is shown. The total amount of DNA is normalized respect to an unrelated locus near ARS305.
Protein analysis and detection of the sumoylated forms
Proteins were extracted using a TCA protocol [30]. Western blotting analysis was performed as previously described [30] with anti-Rad53 [51] and anti-Srs2 (Santa Cruz Biotechnology) antibodies. Immuno-precipitation of Srs2 was carried out with an anti-Srs2 antibody or without antibody as a control on proteins extracted by the TCA protocol using a 50 ml culture of cells at a density of 107 cells/ml. After a series of washings with JS buffer (50 mM Hepes pH 7,5, 150 mM NaCl, 1,5 mM MgCl2, 1% glycerol, 5 mM EGTA, 1% Triton X-100), proteins were resuspended in a suitable volume of Laemmli buffer and separated on a 10% acrylamide SDS-PAGE gel. The blots were probed with a rabbit anti-SUMO antibody (kindly gift of Xiaolan Zhao, Memorial Sloan-Kettering Cancer Center, New York) and a HRP-labeled secondary antibody (Amersham). Blots were then stripped with the commercial solution Restore Western Blot Stripping Buffer (Thermo Scientific) and probed with the anti-Srs2 antibody (Santa Cruz Biotechnology).
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. Keen-KimD
NooraieF
RaoPN
2008 Cytogenetic biomarkers for human cancer. Front Biosci 13 5928 5949
2. PaquesF
HaberJE
1999 Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63 349 404
3. KroghBO
SymingtonLS
2004 Recombination proteins in yeast. Annu Rev Genet 38 233 271
4. San FilippoJ
SungP
KleinH
2008 Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77 229 257
5. LiuY
WestSC
2004 Happy Hollidays: 40th anniversary of the Holliday junction. Nat Rev Mol Cell Biol 5 937 944
6. JainS
SugawaraN
LydeardJ
VazeM
Tanguy Le GacN
2009 A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev 23 291 303
7. AgmonN
PurS
LiefshitzB
KupiecM
2009 Analysis of repair mechanism choice during homologous recombination. Nucleic Acids Res 37 5081 5092
8. NagaiS
DubranaK
Tsai-PflugfelderM
DavidsonMB
RobertsTM
2008 Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 322 597 602
9. OzaP
JaspersenSL
MieleA
DekkerJ
PetersonCL
2009 Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev 23 912 927
10. ConradMN
LeeCY
WilkersonJL
DresserME
2007 MPS3 mediates meiotic bouquet formation in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 104 8863 8868
11. MimitouEP
SymingtonLS
2009 Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci 34 264 272
12. SzostakJW
Orr-WeaverTL
RothsteinRJ
StahlFW
1983 The double-strand-break repair model for recombination. Cell 33 25 35
13. GangloffS
SoustelleC
FabreF
2000 Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat Genet 25 192 194
14. KleinHL
2001 Mutations in recombinational repair and in checkpoint control genes suppress the lethal combination of srs2Delta with other DNA repair genes in Saccharomyces cerevisiae. Genetics 157 557 565
15. BurgessRC
LisbyM
AltmannovaV
KrejciL
SungP
2009 Localization of recombination proteins and Srs2 reveals anti-recombinase function in vivo. J Cell Biol 185 969 981
16. AboussekhraA
ChanetR
AdjiriA
FabreF
1992 Semidominant suppressors of Srs2 helicase mutations of Saccharomyces cerevisiae map in the RAD51 gene, whose sequence predicts a protein with similarities to procaryotic RecA proteins. Mol Cell Biol 12 3224 3234
17. MalikPS
SymingtonLS
2008 Rad51 gain-of-function mutants that exhibit high affinity DNA binding cause DNA damage sensitivity in the absence of Srs2. Nucleic Acids Res 36 6504 6510
18. VeauteX
JeussetJ
SoustelleC
KowalczykowskiSC
Le CamE
2003 The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423 309 312
19. KrejciL
Van KomenS
LiY
VillemainJ
ReddyMS
2003 DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423 305 309
20. PapouliE
ChenS
DaviesAA
HuttnerD
KrejciL
2005 Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell 19 123 133
21. PfanderB
MoldovanGL
SacherM
HoegeC
JentschS
2005 SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 436 428 433
22. RongL
KleinHL
1993 Purification and characterization of the SRS2 DNA helicase of the yeast Saccharomyces cerevisiae. J Biol Chem 268 1252 1259
23. DupaigneP
Le BretonC
FabreF
GangloffS
Le CamE
2008 The Srs2 helicase activity is stimulated by Rad51 filaments on dsDNA: implications for crossover incidence during mitotic recombination. Mol Cell 29 243 254
24. BlanckS
KobbeD
HartungF
FenglerK
FockeM
2009 A SRS2 homolog from Arabidopsis thaliana disrupts recombinogenic DNA intermediates and facilitates single strand annealing. Nucleic Acids Res
25. IraG
MalkovaA
LiberiG
FoianiM
HaberJE
2003 Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115 401 411
26. RobertT
DervinsD
FabreF
GangloffS
2006 Mrc1 and Srs2 are major actors in the regulation of spontaneous crossover. Embo J 25 2837 2846
27. Welz-VoegeleC
Jinks-RobertsonS
2008 Sequence divergence impedes crossover more than noncrossover events during mitotic gap repair in yeast. Genetics 179 1251 1262
28. AylonY
LiefshitzB
Bitan-BaninG
KupiecM
2003 Molecular dissection of mitotic recombination in the yeast Saccharomyces cerevisiae. Mol Cell Biol 23 1403 1417
29. VazeMB
PellicioliA
LeeSE
IraG
LiberiG
2002 Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell 10 373 385
30. LiberiG
ChioloI
PellicioliA
LopesM
PlevaniP
2000 Srs2 DNA helicase is involved in checkpoint response and its regulation requires a functional Mec1-dependent pathway and Cdk1 activity. Embo J 19 5027 5038
31. UbersaxJA
WoodburyEL
QuangPN
ParazM
BlethrowJD
2003 Targets of the cyclin-dependent kinase Cdk1. Nature 425 859 864
32. WohlboldL
FisherRP
2009 Behind the wheel and under the hood: functions of cyclin-dependent kinases in response to DNA damage. DNA Repair (Amst) 8 1018 1024
33. IraG
PellicioliA
BalijjaA
WangX
FioraniS
2004 DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431 1011 1017
34. AylonY
LiefshitzB
KupiecM
2004 The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. Embo J 23 4868 4875
35. HuertasP
Cortes-LedesmaF
SartoriAA
AguileraA
JacksonSP
2008 CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455 689 692
36. LeeJY
YangW
2006 UvrD helicase unwinds DNA one base pair at a time by a two-part power stroke. Cell 127 1349 1360
37. AntonyE
TomkoEJ
XiaoQ
KrejciL
LohmanTM
2009 Srs2 disassembles Rad51 filaments by a protein-protein interaction triggering ATP turnover and dissociation of Rad51 from DNA. Mol Cell 35 105 115
38. ColavitoS
Macris-KissM
SeongC
GleesonO
GreeneEC
2009 Functional significance of the Rad51-Srs2 complex in Rad51 presynaptic filament disruption. Nucleic Acids Res
39. ChioloI
CarotenutoW
MaffiolettiG
PetriniJH
FoianiM
2005 Srs2 and Sgs1 DNA helicases associate with Mre11 in different subcomplexes following checkpoint activation and CDK1-mediated Srs2 phosphorylation. Mol Cell Biol 25 5738 5751
40. Le BretonC
DupaigneP
RobertT
Le CamE
GangloffS
2008 Srs2 removes deadly recombination intermediates independently of its interaction with SUMO-modified PCNA. Nucleic Acids Res 36 4964 4974
41. DebrauwereH
LoeilletS
LinW
LopesJ
NicolasA
2001 Links between replication and recombination in Saccharomyces cerevisiae: a hypersensitive requirement for homologous recombination in the absence of Rad27 activity. Proc Natl Acad Sci U S A 98 8263 8269
42. SymingtonLS
1998 Homologous recombination is required for the viability of rad27 mutants. Nucleic Acids Res 26 5589 5595
43. AboussekhraA
ChanetR
ZgagaZ
Cassier-ChauvatC
HeudeM
1989 RADH, a gene of Saccharomyces cerevisiae encoding a putative DNA helicase involved in DNA repair. Characteristics of radH mutants and sequence of the gene. Nucleic Acids Res 17 7211 7219
44. PalladinoF
KleinHL
1992 Analysis of mitotic and meiotic defects in Saccharomyces cerevisiae SRS2 DNA helicase mutants. Genetics 132 23 37
45. HarrisonJC
HaberJE
2006 Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet 40 209 235
46. PradoF
AguileraA
2003 Control of cross-over by single-strand DNA resection. Trends Genet 19 428 431
47. CarterSD
VigasovaD
ChenJ
ChovanecM
AstromSU
2009 Nej1 recruits the Srs2 helicase to DNA double-strand breaks and supports repair by a single-strand annealing-like mechanism. Proc Natl Acad Sci U S A
48. LiuY
KaoHI
BambaraRA
2004 Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem 73 589 615
49. LuccaC
VanoliF
Cotta-RamusinoC
PellicioliA
LiberiG
2004 Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene 23 1206 1213
50. LivakKJ
SchmittgenTD
2001 Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 402 408
51. FioraniS
MimunG
CalecaL
PicciniD
PellicioliA
2008 Characterization of the activation domain of the Rad53 checkpoint kinase. Cell Cycle 7 493 499
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 2
Nejčtenější v tomto čísle
- Genome-Wide Association Study in Asian Populations Identifies Variants in and Associated with Systemic Lupus Erythematosus
- Nucleoporins and Transcription: New Connections, New Questions
- Nuclear Pore Proteins Nup153 and Megator Define Transcriptionally Active Regions in the Genome
- The Genetic Interpretation of Area under the ROC Curve in Genomic Profiling