
Genetic and Functional Dissection of and in Age-Related Macular Degeneration
A common haplotype on 10q26 influences the risk of age-related macular degeneration (AMD) and encompasses two genes, LOC387715 and HTRA1. Recent data have suggested that loss of LOC387715, mediated by an insertion/deletion (in/del) that destabilizes its message, is causally related with the disorder. Here we show that loss of LOC387715 is insufficient to explain AMD susceptibility, since a nonsense mutation (R38X) in this gene that leads to loss of its message resides in a protective haplotype. At the same time, the common disease haplotype tagged by the in/del and rs11200638 has an effect on the transcriptional upregulation of the adjacent gene, HTRA1. These data implicate increased HTRA1 expression in the pathogenesis of AMD and highlight the importance of exploring multiple functional consequences of alleles in haplotypes that confer susceptibility to complex traits.
Published in the journal:
. PLoS Genet 6(2): e32767. doi:10.1371/journal.pgen.1000836
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000836
Summary
A common haplotype on 10q26 influences the risk of age-related macular degeneration (AMD) and encompasses two genes, LOC387715 and HTRA1. Recent data have suggested that loss of LOC387715, mediated by an insertion/deletion (in/del) that destabilizes its message, is causally related with the disorder. Here we show that loss of LOC387715 is insufficient to explain AMD susceptibility, since a nonsense mutation (R38X) in this gene that leads to loss of its message resides in a protective haplotype. At the same time, the common disease haplotype tagged by the in/del and rs11200638 has an effect on the transcriptional upregulation of the adjacent gene, HTRA1. These data implicate increased HTRA1 expression in the pathogenesis of AMD and highlight the importance of exploring multiple functional consequences of alleles in haplotypes that confer susceptibility to complex traits.
Introduction
Genome-wide association studies (GWAS) have catalyzed significant progress towards elucidating the molecular basis of complex traits [1]. However, a substantial gap remains between association of a trait with a genomic segment and the identification of the causative allele(s). A locus for AMD, ARMS2 on 10q26, illustrates this challenge.
ARMS2 is one of several regions in the genome shown by GWAS to confer susceptibility to the disorder. However, in contrast to the other confirmed AMD susceptibility loci CFH (NM_000186) [2]–[8], C2/BF (NM_000063/NM_001710) and C3 (NM_000064) [9]–[11], which encompass a single gene; a region on 10q26 that contains three genes has been associated consistently with AMD susceptibility [12]–[15]. Recent data have refined this association to a haplotype that encompasses two genes, LOC387715 (NM_001099667), a primate-specific transcript with a proposed mitochondrial function [16] and HTRA1 (NM_002775), a multi-functional serine protease [17]–[18].
Initial reports describing the association between SNP rs11200638 in the 5′ end of HTRA1 and AMD focused on the possibility that the disease-associated allele of this SNP increased expression of HTRA1 [17]–[18]. However, the inability to verify this finding in heterologous expression systems led to the investigation of other alleles in the risk haplotype, and emphasis was placed on LOC387715 [16]. Consistent with this possibility, recent sequencing of the AMD-associated haplotype identified multiple SNPs, including an in/del, that are associated with an increased risk of AMD, and a decreased LOC387715 mRNA level with this AMD disease haplotype [19]. To delineate the causal genetic variations contained in the risk haplotype and to understand their functional roles in AMD susceptibility, we undertook genetic and functional investigations at the ARMS2 locus. We show that loss of LOC387715 is likely necessary but not sufficient to explain AMD susceptibility and that a common disease haplotype including the in/del and rs11200638 also has an effect on the transcriptional upregulation of the adjacent gene, HTRA1. These data, which implicate increased HTRA1 expression in the pathogenesis of AMD, suggest that the AMD risk conferred by this region is potentially driven by multiple variants, and highlight the importance of exploring multiple functional consequences of alleles in haplotypes that confer susceptibility to complex traits.
Results
Several studies have shown previously strong association of multiple single nucleotide polymorphisms (SNPs) in chromosome 10q26 encompassing PLEKHA1, LOC387715, and HTRA1with advanced AMD [12]–[24]. From our previous study [20], we identified a common disease haplotype TAT tagged by rs10490924, rs11200638 and rs2293870 that is significantly associated with a risk of AMD (P = 2.70×10−9), as well as a haplotype GGG that is modestly, yet significantly, associated with protection from AMD (P = 0.003). Next, to define the extent of the haplotype structures and capture all variants in the region, we undertook a re-sequencing effort of a region spanning ∼100 kb, starting in the 3′-UTR of PLEKA and ending ∼20kb downstream of the 3′ UTR of HTRA1 in six individuals with a homozygous risk haplotype and six individuals with a homozygous protective haplotype, followed by assessment of association of each discovered variant with AMD in 200 AMD cases and 200 normal controls (Table 1, Table S1, Table S2).

In agreement with recent work [19], we discovered that both risk and protective haplotypes span 21.5kb in the ARMS2 region, starting from upstream of LOC387715 to rs58077526, in intron 1 of HTRA1 (Figure 1, Table 1, Table S1). We confirmed recent data [19], according to which 22 SNPs tagged a risk and a protective haplotype including previously reported SNPs rs10490924, rs3750848, an in/del, rs3793917, and rs11200638 (Figure 1 and Table 1). Moreover, the reported 54 base pair insertion and 443 base pair deletion (in/del) at the 3′ end of LOC387715 resides exclusively on the disease risk haplotype that is strongly associated with a risk of AMD (P = 1.90×10−26) in a Utah case-control cohort (705 cases, 650 controls) (Table 2 and Table 3), as well as an independent replication cohort of northern European ancestry (442 cases, 434 controls ) (Table 2 and Table 3) and a third replication cohort of Han Chinese in China (138 cases, 591 controls ) (Table 2 and Table 3) [19].
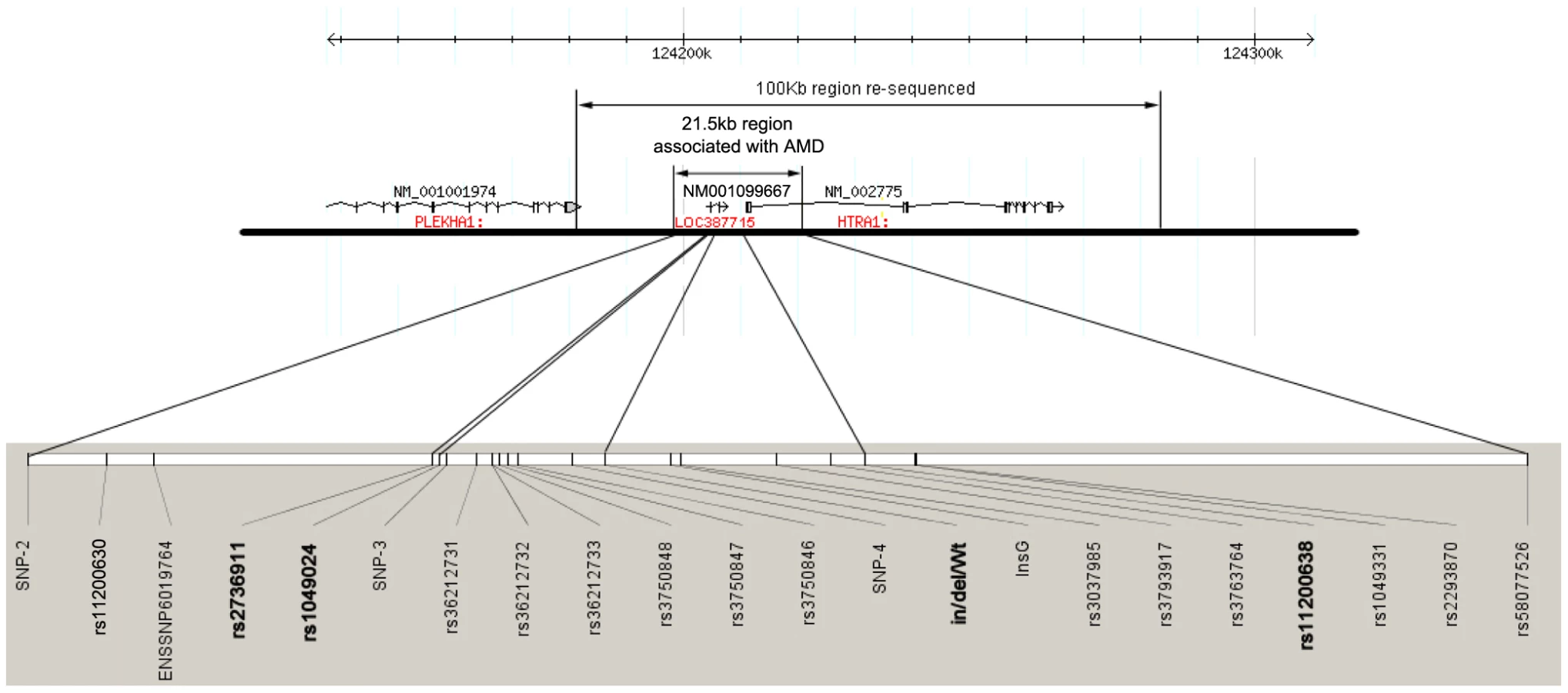


In contrast to the loss of function role of in/del in LOC387715, the T allele of SNP rs2736911, a non-synonymous coding SNP leading to a predicted premature stop (R38X) in LOC387715 is associated with a protective haplotype T-G-Wt-G (P = 4.00×10−4, Figure 2 and Table 2 and Table 4) defined by rs2736911, rs10490924, in/del/Wt and rs11200638. We replicated these findings twice and found that this haplotype is clearly not associated with AMD risk; rather it is (conservatively) neutral and possibly protective with regard to the disorder (P = 0.001 in the second replication cohort; P = 0.007 in third Han Chinese cohort, Table 4).


These findings present a paradox. Recent studies have shown the in/del to cause destabilization of LOC387715, suggesting that loss of function at that locus might confer risk to AMD. However, the introduction of the R38X mutation, which is also predicted to give rise to loss of LOC387715 message due to nonsense-mediated mRNA decay (NMD), is mildly protective. We considered two plausible alternatives; either that effect of R38X and the in/del have different effects on the transcript, or that loss of LOC387715 is insufficient to cause AMD. The first possibility is unlikely. Upon quantification of LOC387715 mRNA levels in seven placentas homozygous for the major disease haplotype, we found a 4.7-fold reduction in endogenous LOC387715 expression (Figure 3A), in agreement with recent published data [19]. However, we were also able to examine LOC387715 mRNA levels in five patients heterozygous for the R38X mutation, where we observed a 50% reduction (Figure 3A), suggesting that each of the R38X and in/del have the same mode of action.

Given these data, we considered the potential role of HTRA1, the only other locus encompassed by the major risk haplotype, in the pathogenesis of AMD. We have shown previously a three-fold increase expression of HTRA1 in the retinal pigmented epithelium (RPE) of patients with the risk haplotype [17], while others have also observed increased HTRA1 expression in AMD eyes in humans [25]. In addition, a functional SNP in the promoter region of HTRA1 is associated with increased HTRA1 expression in non-human primates with AMD-like phenotype [26]–[27]. We therefore expanded our analyses of mRNA levels in human placentas to determine the effect of a haplotype on endogenous expression. We found that, in addition to the observed decrease of LOC387715 message, the disease haplotype is also associated with a 2.7-fold increase in HTRA1 expression (Figure 3B). These data confirmed that the in/del or another component of the common risk haplotype affects LOC387715 mRNA stability. At the same time, it likely has an effect on HTRA1 expression. Although caution should be exercised when translating findings from one tissue to another, these data from placenta should serve as an indication for the effects of different haplotypes on the gene expression profiles in the eye.
To test this hypothesis further, we generated expression constructs that model the effect of the risk and protective haplotype on the HTRA1 promoter using a luciferase reporter assay on cultured human retinal pigment epithelial cells (Figure 4A and 4B). We observed a two-fold increase in luciferase expression in constructs that modeled the disease haplotype encompassing the in/del and the A allele of SNP rs11200638 (MT(L+in/del), Figure 4A and 4B). We detected no increase in luciferase expression from constructs that contained a short disease haplotype including the A risk allele of SNP rs11200638 but did not contain the in/del (MT(S), Figure 4A and 4B). These findings are consistent with Fritsche et al. and Kanda et al [16],[19], who reported an inability to verify that the A allele of SNP rs11200638 by itself alters transcriptional activity in heterologous cell systems. We did not observe increased luciferase expression either when we placed the in/del or A allele of SNP rs11200638 on a construct containing a protective haplotype, suggesting that the in/del or A allele of SNP rs11200638 by itself is insufficient to drive HTRA1 expression (WT(L+in/del), WT(L-A), Figure 4B).

Because of the potentially ambiguous nature of in vitro luciferase assays, we repeated these experiments in an in vivo system, whereby the constructs containing the disease haplotype including the in/del and the protective haplotype described earlier (Figure 4A) were electroporated into the RPE of wild-type adult mice. Luciferase activity was assayed four days after transfection. Consistent with the in vitro data, we observed a significant increase in normalized luciferase activity in a construct bearing the disease haplotype tagged by the in/del and A allele of SNP rs11200638 (P = 0.032) (Figure 4C), whereas no increased luciferase activity was observed in a construct with the in/del on a protective haplotype (P = 0.180).
Discussion
The AMD-associated region on 10q26 poses an interesting problem. Multiple lines of evidence support the causality of both LOC387715 and HTRA1 in conferring risk to AMD. At the same time, emerging data suggests that mutations in either gene alone might be insufficient to confer such risk. The association signal is best explained by a common disease haplotype including an in/del in the 3′ UTR of LOC387715 and the A allele of SNP rs11200638. Although there is evidence that AMD patients exhibit loss of LOC387715 message, the fact that haploinsufficiency in LOC387715 alone (through the R38X mutation) does not confer risk to AMD, suggests that either that transcript is unrelated to the disorder, or that additional events within the risk haplotype must occur. Although we cannot formally reject the hypothesis that loss of LOC387715 is irrelevant to the disease, the spatiotemporal expression pattern of this gene and its exclusive emergence with the evolution of the macula in non-human primates, provide partial evidence for its role in AMD pathogenesis [14],[16],[19],[26]. At the same time, the evidence for the involvement of HTRA1 upregulation is likewise compelling, including i) the upregulation of the transcript in the major risk haplotype, and ii) the observed increased HTRA1 expression in AMD eyes.
One can outline reasonable hypotheses to explain each aspect of the discrepant data. Alternatively, a single hypothesis of dual causality might explain all the observations. Specifically, we speculate that concomitant downregulation of LOC387715 and upregulation of HTRA1 can explain the disease. This theoretical binary model is consistent with the fact that i) the common AMD-associated haplotype affects both transcripts and ii) the protective haplotype containing the R38X LOC387715 allele is associated with normal HTRA1 expression (Figure 3B). Moreover, we suggest that if changes in either gene alone were sufficient to confer AMD susceptibility, one might have expected to discover rare alleles that recapitulate the effect of the in/del (such as rare loss of function mutations in LOC387715 or activating mutations in HTRA1), neither of which have been discovered to date by us or by other groups. The primate-specific nature of LOC387715 renders this binary model intractable in model organisms. Nonetheless, this model may be tested in the monkey using genetic manipulations.
Materials and Methods
Patients
This study was approved by the Institutional Review Boards of the University of Utah, University of California San Diego, Johns Hopkins University, and Sichuan Provincial People's Hospital. Subjects gave informed consent prior to participation. Participants underwent a standard examination, which included visual acuity measurements, dilated slit lamp biomicroscopy, and stereoscopic color fundus photography. Grading was carried out with the classification established by AREDS [28]. Diagnosis of advanced AMD was based on the presence of GA or CNV (equivalent to AREDS category 4 or 5). Control subjects were >60 years of age, with no signs of AMD, defined as no drusen or RPE abnormalities in the Utah collection, and controls in the Hopkins cohort and the Chinese cohort were defined as being >60 years old, having fewer than 5 small drusen (<63 um), and no RPE abnormalities. Patient characteristics of the case-control series are listed in Table 5.

Genotyping
SNPs were genotyped by SNaPshot on an ABI 3100XL analyzer (ABI, Foster City, CA, USA) according to the manufacturer's instructions. PCR and SNaPshot primers are listed in Table S2. All SNPs had a genotyping success rate >98% and accuracy >99% as judged by random re-sequencing of 20% of samples in all three case-control series.
Re-sequencing of haplotypes
To discover all variations in the AMD susceptibility locus at 10q26, we undertook a re-sequencing of 100kb in the region including genes PLEKHA1, LOC387715 and HTRA1. We chose DNA samples from six individuals with a homozygous risk haplotype and six individuals with a homozygous protective haplotype for re-sequenbcing analysis. We designed PCR primer pair for amplification of 1kb amplicon; each amplicon had a 50bp overlap with next amplicons. The PCR amplicons were directly purified using a Qiaquick kit (Qiagen, Valencia, CA, USA) and sequenced using forward and reverse primers using the BigDye Terminator v3.1 Cycle Sequencing Kit (ABI, Foster City, CA, USA) according to the manufacturer's instructions. Then the sequencing results were annotated using the NCBI genomic DNA sequence information (http://www.ncbi.nlm.nih.gov, build 36).
Statistical analysis
All SNP genotyping results were screened for deviations from Hardy-Weinberg equilibrium with no SNP showing significant deviation (p>0.05). The chi-squared test for allelic trend for an additive model or dominant allele model over alleles was performed with PEPI version 4.0 [29]. Odds ratios and 95% confidence intervals were calculated by conditional logistic regression with SPSS version 13.0. Linkage disequilibrium (LD) structure was examined with Haploview (version 4.0) [30]. Default settings were used, creating 95% confidence bounds on D' to define pair-wise SNPs in strong LD [31]. Haploview was also used for allelic association tests.
Endogenous expression levels of HTRA1 and LOC387715
Total RNA was extracted from human placentas, and the first strand cDNA was generated by reverse transcript using reverse transcript kit (Invitrogen, Carlsbad, CA, USA). Real time PCR was performed for HTRA1 mRNA qualification using ABI human HTRA1 probe real time PCR kit (ABI, Foster city, CA, USA). RT-PCR was performed to qualify LOC387715 mRNA levels using primers 5′-atggcagctggcttggcc-3′ and 5′-ttgctgcagtgtggatgatag-3′ with ex taq polymerase (TaKaRa Bio USA, Mountain View, CA USA). GAPDH was used as the internal control. Human placenta tissues were harvested and genotyped. We measured RNA levels in seven individuals with a homozygous disease haplotype (C-T-in/del-A, Figure 3A and 3B), five individuals with a homozygous protective haplotype (C-G-Wt-G, Figure 3A and 3B), and five individuals with a homozygous non-risk haplotype with respect to in/del/Wt and a heterozygous CT allele at rs2736911 (C/T-G-Wt-G, Figure 3A and 3B). Significance was examined using SPSS's independent sample t-test.
Heterologous Luciferase assays
A 4423 bp DNA fragment containing the −1 to −4423 position from the HTRA1 translation start site including either the risk haplotype (MT(L+in/del), Figure 4A) or protective haplotype (WT(L-G), Figure 4A) was PCR amplified from genomic DNA of an individual with a homozygote risk haplotype or an individual with a protective haplotype using the following primers: forward: cgacgcgtcggatgcagccaatcttctcctaac; reverse: agatctcgagcccggcgactctggcggcggcggcggtg). A DNA fragment containing −1 to 2100bp from the HTRA1 translation site including either the risk haplotype (MT(S), Figure 4A) or protective hapotype (WT(S), Figure 4A) was amplified from genomic DNA of an individual with a homozygous risk haplotype or protective haplotype using the primers, cggggtaccaactcctgggctcaaaggat and ccgctcgagtccgcgcctggccggggtccctcag. Constructs were subcloned into the Mlu I-Xho I site of the pGL3-basic vector (Promega, Madison, WI, USA). All constructs were verified by restriction enzyme digestion and complete bidirectional DNA sequencing. The wild type haplotype constructs carrying in/del (WT(L+in/del), Figure 4A)was generated by subcloning of a fragment containing in/del cutting from MT(L+in/del) using Mlu I-Nhe I. The wild type haplotype constructs carrying A allele at rs11200638 (WT (L-A), Figure 4A) was generated using site-directed mutagenesis kit (Agilent Technologies, La Jolla, CA). A positive control plasmid (pGL3-control Vector) containing an SV40 enhancer and promoter driving luciferase reporter was obtained from Promega (Madison, WI, USA).
Human RPE cells were split into 24-well plates and cotransfected 24 hours later with 1ng of the transfection control Renilla luciferase plasmid pTK-RL (Promega, Madison, WI) and 200ng of one of the following plasmids: pGL3-basic, pGL3-control, WT(L-A), WT(L-G), WT(L+in/del), MT(L+in/del), WT(S) and MT(S). Transfections (n = 6, three preps for each construct and 2 transfections for each preps) were done using the Fugene-6 protocol according to manufacturer's specifications (Roche Applied Science, Mannheim, Germany). Forty-eight hours after transfection, cells were washed with PBS twice and luciferase activities measured with the Dual-Luciferase Assay Kit (Promega, Madison, WI). Fold induction was derived relative to normalized reporter activity.
In vivo transfection by electroporation
In vivo transfection was done by subretinal injection of reporter plasmids followed by electroporation as described [32]. Briefly, C57BL/6 mice (4–5 weeks old) were given a subretinal injection of 1µl of PBS containing 0.5 ug of pGL3 containing one of the firefly luciferase constructs described above and 0.25 ng of Renilla luciferase plasmid ( pRL-CMV, Promega, Madison, WI) for normalization. After injection, two steel electrodes separated by about 3.0 mm were placed on the posterior sclera and an ECM830 electroporator (BTX, San Diego, CA, USA) was used to deliver eight 50 ms electric pulses separated by 100 ms with voltage set at 30V. The eyes were enucleated 65–68h after electroporation and the cornea and lens were removed. Posterior eyecups were minced and homogenized in 100 µl of Reporter Lysis Buffer (Promega, Madison, WI). Firefly and Renilla luciferase activities were measured using 40µl of lysate and a Dual-Luciferase Reporter Assay System (Promega, Madison, WI). The firefly luciferase value was divided by the Renilla luciferase value to give the normalized luciferase activity. The normalized luciferase activity of each test plasmid injection group was divided by the normalized luciferase activity of empty-PGL3 and pRL-CMV injection group to give the relative luciferase activity ratio. Statistical analysis was done using ANOVA and Bonferroni/Dunn test.
Supporting Information
Zdroje
1. ManolioTA
BrooksLD
CollinsFS
2008 A HapMap harvest of insights into the genetics of common disease. J Clin Invest 118 1590 1605
2. ZareparsiS
BranhamKE
LiM
ShahS
KleinRJ
2005 Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration. Am J Hum Genet 77 149 153
3. KleinRJ
ZeissC
ChewEY
TsaiJY
SacklerRS
2005 Complement factor H polymorphism in age-related macular degeneration. Science 308 385 389
4. HainesJL
HauserMA
SchmidtS
ScottWK
OlsonLM
2005 Complement factor H variant increases the risk of age-related macular degeneration. Science 308 419 421
5. HagemanGS
AndersonDH
JohnsonLV
HancoxLS
TaiberAJ
2005 A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A 102 7227 7232
6. EdwardsAO
RitterR3rd
AbelKJ
ManningA
PanhuysenC
2005 Complement factor H polymorphism and age-related macular degeneration. Science 308 421 424
7. LiM
Atmaca-SonmezP
OthmanM
BranhamKE
KhannaR
2006 CFH haplotypes without the Y402H coding variant show strong association with susceptibility to age-related macular degeneration. Nat Genet 38 1049 1054
8. MallerJ
GeorgeS
PurcellS
FagernessJ
AltshulerD
2006 Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet 38 1055 1059
9. GoldB
MerriamJE
ZernantJ
HancoxLS
TaiberAJ
2006 Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet 38 458 462
10. YatesJR
SeppT
MatharuBK
KhanJC
ThurlbyDA
2007 Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med 357 553 561
11. MallerJB
FagernessJA
ReynoldsRC
NealeBM
DalyMJ
2007 Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet 39 1200 1201
12. FisherSA
AbecasisGR
YasharBM
ZareparsiS
SwaroopA
2005 Meta-analysis of genome scans of age-related macular degeneration. Hum Mol Genet 14 2257 2264
13. JakobsdottirJ
ConleyYP
WeeksDE
MahTS
FerrellRE
2005 Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet 77 389 407
14. RiveraA
FisherSA
FritscheLG
KeilhauerCN
LichtnerP
2005 Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet 14 3227 3236
15. SchmidtS
HauserMA
ScottWK
PostelEA
AgarwalA
2006 Cigarette smoking strongly modifies the association of LOC387715 and age-related macular degeneration. Am J Hum Genet 78 852 864
16. KandaA
ChenW
OthmanM
BranhamKE
BrooksM
2007 A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degeneration. Proc Natl Acad Sci U S A 104 16227 16232
17. YangZ
CampNJ
SunH
TongZ
GibbsD
2006 A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science 314 992 993
18. DewanA
LiuM
HartmanS
ZhangSS
LiuDT
2006 HTRA1 promoter polymorphism in wet age-related macular degeneration. Science 314 989 992
19. FritscheLG
LoenhardtT
JanssenA
FisherSA
RiveraA
2008 Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet 40 892 896
20. GibbsD
YangZ
ConstantineR
MaX
CampNJ
2008 Further mapping of 10q26 supports strong association of HTRA1 polymorphisms with age-related macular degeneration. Vision Res 48 685 689
21. JakobsdottirJ
ConleyYP
WeeksDE
FerrellRE
GorinMB
2008 C2 and CFB genes in age-related maculopathy and joint action with CFH and LOC387715 genes. PLoS ONE 3 e2199 doi:10.1371.journal.pone.0002199
22. DeangelisMM
JiF
AdamsS
MorrisonMA
HarringAJ
2008 Alleles in the HtrA serine peptidase 1 gene alter the risk of neovascular age-related macular degeneration. Ophthalmology 115 1209 1215 e1207
23. HughesAE
OrrN
PattersonC
EsfandiaryH
HoggR
2007 Neovascular age-related macular degeneration risk based on CFH, LOC387715/HTRA1, and smoking. PLoS Med 4 e355 doi:10.1371/journal.pmed.0040355
24. SeddonJM
FrancisPJ
GeorgeS
SchultzDW
RosnerB
2007 Association of CFH Y402H and LOC387715 A69S with progression of age-related macular degeneration. JAMA 297 1793 1800
25. ChanCC
ShenD
ZhouM
RossRJ
DingX
2007 Human HtrA1 in the archived eyes with age-related macular degeneration. Trans Am Ophthalmol Soc 105 92 97; discussion 97–98
26. FrancisPJ
AppukuttanB
SimmonsE
LandauerN
StoddardJ
2008 Rhesus monkeys and humans share common susceptibility genes for age-related macular disease. Hum Mol Genet 17 2673 2680
27. SinghKK
KrawczakM
DawsonWW
SchmidtkeJ
2009 Association of HTRA1 and ARMS2 gene variation with drusen formation in rhesus macaques. Exp Eye Res 88 479 482
28. 1999 The Age-Related Eye Disease Study (AREDS): design implications. AREDS report no. 1. Control Clin Trials 20 573 600
29. AbramsonJH
GahlingerPM
2001 PEPI, ver. 4.0: Computer Programs for Epidemiologists Salt Lake City Sagebrush Press
30. BarrettJC
FryB
MallerJ
DalyMJ
2005 Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21 263 265
31. GabrielSB
SchaffnerSF
NguyenH
MooreJM
RoyJ
2002 The structure of haplotype blocks in the human genome. Science 296 2225 2229
32. KachiS
OshimaY
EsumiN
KachiM
RogersB
2005 Nonviral ocular gene transfer. Gene Ther 12 843 851
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 2
Nejčtenější v tomto čísle
- Genome-Wide Association Study in Asian Populations Identifies Variants in and Associated with Systemic Lupus Erythematosus
- Nucleoporins and Transcription: New Connections, New Questions
- Nuclear Pore Proteins Nup153 and Megator Define Transcriptionally Active Regions in the Genome
- The Genetic Interpretation of Area under the ROC Curve in Genomic Profiling