
Genome-Wide Association Study in Asian Populations Identifies Variants in and Associated with Systemic Lupus Erythematosus
Systemic lupus erythematosus is a complex and potentially fatal autoimmune disease, characterized by autoantibody production and multi-organ damage. By a genome-wide association study (320 patients and 1,500 controls) and subsequent replication altogether involving a total of 3,300 Asian SLE patients from Hong Kong, Mainland China, and Thailand, as well as 4,200 ethnically and geographically matched controls, genetic variants in ETS1 and WDFY4 were found to be associated with SLE (ETS1: rs1128334, P = 2.33×10−11, OR = 1.29; WDFY4: rs7097397, P = 8.15×10−12, OR = 1.30). ETS1 encodes for a transcription factor known to be involved in a wide range of immune functions, including Th17 cell development and terminal differentiation of B lymphocytes. SNP rs1128334 is located in the 3′-UTR of ETS1, and allelic expression analysis from peripheral blood mononuclear cells showed significantly lower expression level from the risk allele. WDFY4 is a conserved protein with unknown function, but is predominantly expressed in primary and secondary immune tissues, and rs7097397 in WDFY4 changes an arginine residue to glutamine (R1816Q) in this protein. Our study also confirmed association of the HLA locus, STAT4, TNFSF4, BLK, BANK1, IRF5, and TNFAIP3 with SLE in Asians. These new genetic findings may help us to gain a better understanding of the disease and the functions of the genes involved.
Published in the journal:
. PLoS Genet 6(2): e32767. doi:10.1371/journal.pgen.1000841
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000841
Summary
Systemic lupus erythematosus is a complex and potentially fatal autoimmune disease, characterized by autoantibody production and multi-organ damage. By a genome-wide association study (320 patients and 1,500 controls) and subsequent replication altogether involving a total of 3,300 Asian SLE patients from Hong Kong, Mainland China, and Thailand, as well as 4,200 ethnically and geographically matched controls, genetic variants in ETS1 and WDFY4 were found to be associated with SLE (ETS1: rs1128334, P = 2.33×10−11, OR = 1.29; WDFY4: rs7097397, P = 8.15×10−12, OR = 1.30). ETS1 encodes for a transcription factor known to be involved in a wide range of immune functions, including Th17 cell development and terminal differentiation of B lymphocytes. SNP rs1128334 is located in the 3′-UTR of ETS1, and allelic expression analysis from peripheral blood mononuclear cells showed significantly lower expression level from the risk allele. WDFY4 is a conserved protein with unknown function, but is predominantly expressed in primary and secondary immune tissues, and rs7097397 in WDFY4 changes an arginine residue to glutamine (R1816Q) in this protein. Our study also confirmed association of the HLA locus, STAT4, TNFSF4, BLK, BANK1, IRF5, and TNFAIP3 with SLE in Asians. These new genetic findings may help us to gain a better understanding of the disease and the functions of the genes involved.
Introduction
Systemic lupus erythematosus (SLE) is a prototype autoimmune disease characterized by auto-antibody production and multi-organ damage. Genetic factors are known to play an important role in the disease, with the monozygotic twin concordance rate between 20–59, and the risk for siblings of affected individuals 30 times higher than that for the general population [1]–[3]. There are also population differences for the disease both in terms of genetic susceptibility and disease manifestations. African Americans, Hispanics and Asians all have higher disease prevalence than Caucasians; with Asians known to have more lupus nephritis than patients of European ancestry [4],[5].
Genome-Wide Association studies (GWAS) have dramatically changed the landscape of SLE genetics, with a pace of discovery the field has never seen before. In less than two years time, STAT4 [6], ITGAM [7]–[9], BLK [8], PXK and KIAA1542 [7], BANK1 [10] and TNFAIP3 [11],[12] and several other genes have been identified as associated with SLE [13]–[16]. More susceptibility loci were reported recently in two other GWAS on this disease [17],[18].
Despite varied disease prevalence and severity across different populations, it is noteworthy that most previous studies were conducted on patients of European ancestry with under-representation of other ethnicities. We previously examined some of the GWAS findings from populations of European origin [19]. In addition to confirming the association of STAT4 and BLK with SLE in our population, our data indicated differences between the Asian and Caucasian populations. For example, our study did not detect any significant disease association for PXK, a result that was later confirmed by an independent study on a Korean population [20]. Data from our study indicated that, although the risk alleles in ITGAM are rare in Asians (<2%), they are risk factors in our populations and are closely related to lupus nephritis in particular [21].
In this study, we first genotyped 320 SLE patients collected in Hong Kong by the Illumina 610-Quad Beadchip and analyzed the data against 1500 control individuals genotyped on the same platform. Selected SNPs were then replicated in four independent sample collections from Hong Kong, Shanghai and Anhui, (China), as well as Bangkok (Thailand). Genetic variants in and around two genes, ETS1 and WDFY4, were identified as associated with SLE with genome-wide significance. Functional characterization of the risk alleles also supported potential roles of these genetic variants in disease pathogenesis.
Results
The association of HLA, STAT4, BLK, BANK1, IRF5, TNFAIP3 with SLE
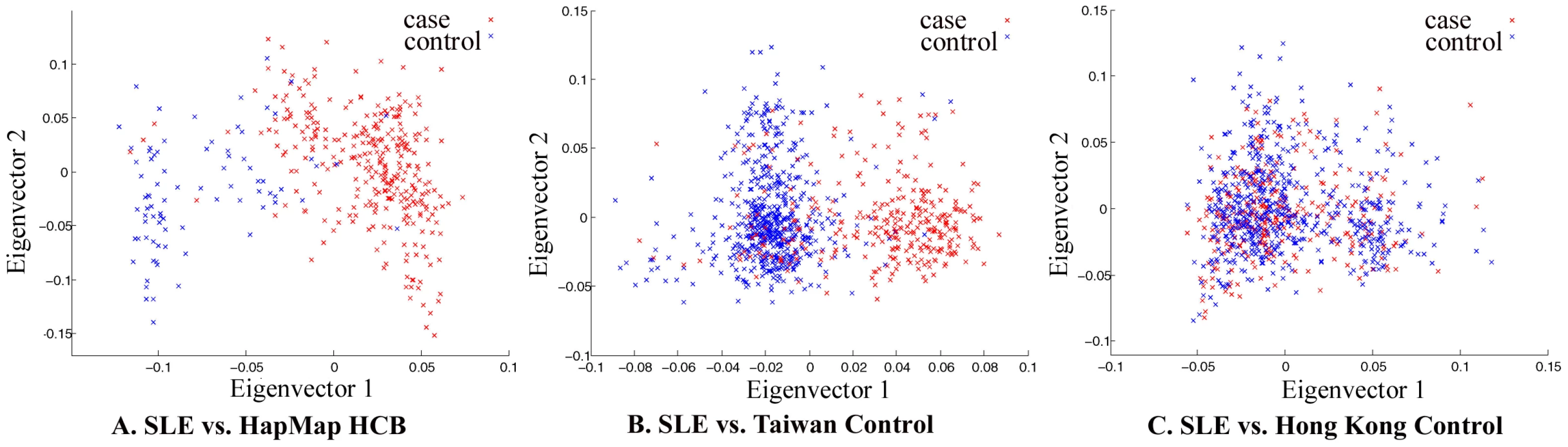
The whole-genome genotyping data was thoroughly examined by quality control measures and by population substructure analysis. Analysis of principal component using Eigenstrat [22] did reveal that the samples collected in Hong Kong clustered together, suggesting that confounding population substructure or admixture is not a major concern if Hong Kong controls were used in association analyses. It did indicate, however, that samples collected in Taiwan (obtained from deCODE Genetics) and Han Chinese in Beijing (HCB, available from HapMap) cluster very differently from Hong Kong samples, suggesting population substructure among Chinese living in different geographical regions and potential pitfalls in association studies when cases and controls are not well-matched (Figure 1).
Genome-wide association analysis confirmed significant association of some established susceptibility genes in our population, including SNPs in the HLA locus, STAT4, TNFSF4, BANK1, TNFAIP3, IRF5 and BLK (Table 1 and Figure 2). Similar to the Caucasian findings [12],[23], the risk allele for rs2230926 in TNFAIP3 is low in frequency but with a relatively large effect size in disease association. SNP rs9271366 located between HLA-DRB1 and HLA-DQA1 is the most significantly associated SNP in the whole genome in our study. Although the HLA locus has been consistently shown to be the locus conferring the largest effect size with SLE association, there is little overlap between previous GWAS findings from populations of European ancestry and our results. The most significant SNPs in the Caucasian data [7],[8] are either monomorphic in our population or are not associated with disease susceptibility based on our GWAS result, something worth further pursuit in future studies. Association of STAT4, BLK, IRF5, BANK1 and TNFSF4 with the disease has been reported in our population previously [19],[21],[24],[25].


To answer the question on whether there are still other genetic variants contributing to disease susceptibility, we reexamined the Q-Q plot comparing expected and observed P values by removing all the SNPs in the known susceptibility loci mentioned above. After removal of these SNPs, we still observed an excess of association signal (Figure 3), suggesting involvement of additional susceptibility loci for this disease. Since our GWAS involved a limited number of patients, and is therefore prone to false positive and false negative findings, we selected SNPs for replication based on both their significance in GWAS results as well as the function and expression pattern of the nearby genes. Selected SNPs were replicated first using Sequenom genotyping on limited number of additional samples (360 cases and 360 controls), and variants with significant association in the Sequenom data were then examined by TaqMan genotyping on a much expanded sample collection from four independent cohorts. SNPs in and around two genes, v-ets erythroblastosis virus E26 oncogene homolog 1 (avian) (ETS1) and the WDFY family member 4 (WDFY4) regions were chosen based both on initial GWAS data as well as their known function (in the case of ETS1) and expression pattern (in the case of WDFY4). Table 2 displays the SNPs in these two loci that showed disease association with a P value<0.01 from our GWAS data. Initial replication by Sequenom showed consistent results with the GWAS trend for these two genes and they were further tested in the remaining samples.


Association of ETS1 with SLE
Making use of the remaining samples from Hong Kong not included in GWAS, and sample collections from Shanghai and Anhui, China, and Bangkok, Thailand, we went on to replicate the whole-genome findings on these two loci. SNPs in ETS1 listed in Table 2, rs12223943, rs7932088 and rs10893872, were examined in the expanded samples. SNPs rs10893872 and rs4937333 have absolute LD with each other, so only rs10893872 was chosen for replication. SNP rs1128334 was chosen in the place of rs6590330 for replication due to its high LD with rs6590330 (r2 = 0.97, Figure 4) and its relative position to the gene (3′-UTR) and predicted effect on microRNA binding. For SNPs rs12223943 and rs7932088, the association seen from whole-genome data was inconclusive (data not shown) in the replication stage and were not further pursued. We genotyped rs10893872 and rs1128334 in all the samples from the four cohorts and both were found to be highly associated with SLE (Table 3).


Independence test by logistic regression by Plink pointed to a major contribution from rs1128334. And indeed, the sequence around rs1128334 has high sequence conservation among different species (Figure 4). Haplotype analysis indicated that the TA haplotype formed by the two SNPs (rs10893872 and rs1128334) is the major risk haplotype, whilst the CG haplotype is the major protective haplotype (Table 4) with other haplotypes having low allele frequencies. Subphenotype analysis was also performed for these SNPs, and both SNPs were found to have larger effect sizes for patients with lupus nephritis in all four cohorts, although no statistical significance was reached in any cases. Analysis of other subphenotypes showed insignificant, or inconsistent results among different cohorts.

Allelic expression of ETS1 in PBMC
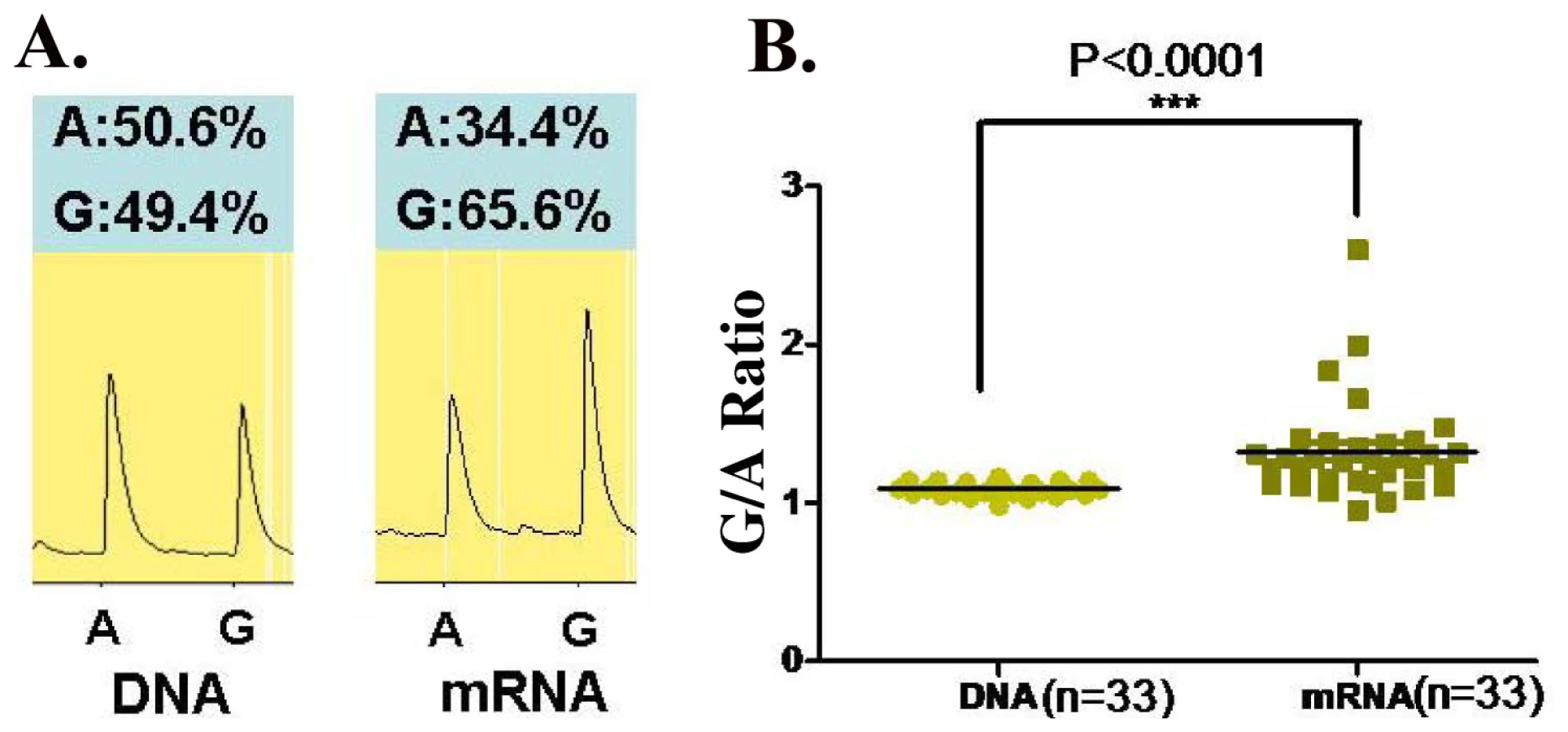
Since rs1128334 is located at the 3′-UTR region of the gene, it is predicted that it may have an effect on the expression level of ETS1. Therefore, we examined allelic expression of ETS1 gene for the two alleles of rs1128334, “A” and “G”, from healthy individuals heterozygous for this SNP (N = 33). This assay assesses directly whether the two alleles of the SNP correlate with different steady state mRNA levels. Pyrosequencing results from PBMC of healthy individuals heterozygous on the SNP showed a significantly higher expression from the “G” allele than from the risk “A” allele, with a P value<0.0001 (Figure 5).
Association of WDFY4 with SLE
Two of the SNPs in WDFY4 that showed the most significant association with the disease in our GWAS, SNPs rs10857650 and rs877819, were selected for further replication. In addition, three nonsynonymous SNPs in this gene not genotype by the Illumina 610-Quad Beadchip, rs2170132 (Ser1528Pro), rs7097397 (Arg1816Gln) and rs2292584 (Pro3118Leu), were also selected to test for disease association, aiming at identifying functional variants in this gene. SNPs rs2170132 and rs2292584 did not show significant difference between the cases and the controls in the Hong Kong cohort (Table 5) and Thai samples (data not shown) and were not further tested in other cohorts. Genotyping results on rs10857650 using TaqMan showed significant discordance with results from the Illumina Beadchip, and was thus removed from further analysis.

SNP rs7097397 and rs877819 were confirmed to have significant association with the disease (Table 3). Conditional logistic regression test indicate that the nonsynonymous SNP coded by rs7097397 is probably the functional variant, with P = 1.01×10−5 when controlling the effect of rs877819. Independent contribution from rs877819 is questionable, with a P value of 0.088 considering the effect of rs7097397 in the same test. The two SNPs have intermediate LD (r2 = 0.44, Figure 6A). The genetic result is consistent with the fact that the arginine residue at 1816 in WDFY4 protein is well conserved among orthologs in different mammals (Figure 6B). Preliminary analysis on subphenotype stratification suggests that this nonsynonymous change may be more closely involved in male and early onset patients in a case only analysis (onset age 12 and below vs. above, OR = 1.96, P = 0.0021; male vs. female: OR = 1.52, P = 0.023).

Association of the SNPs in the two genes with disease risk was also corrected by logistic regression using age and sex as covariates, and the associations found in this study all remain highly significant after all the corrections.
Discussion
Several GWAS on SLE have been conducted on populations of European ancestry [7],[8], but populations of Asian or African ancestry were seriously underrepresented. Only during the process of submission of our current work, a GWAS study on Chinese populations was reported [18]. Considering the population differences in both disease prevalence and clinical manifestations, GWAS on non-Caucasian populations may have novel findings and help to elucidate the differences between populations.
An interesting analysis result from our GWAS data is the difference between Hong Kong samples and samples collected in Taiwan and Beijing, shown by principal component analysis (Figure 1). It suggests population substructure for Chinese living in different regions, which may cause spurious findings in association studies when cases and controls are not well matched. With most of the genetic variants of relatively larger effect sizes already being identified, GWAS becomes more susceptible to effects from mismatches between cases and controls in dealing with SNPs of smaller effect sizes. Our analysis echoed two very recent reports delineating population substructures in Chinese populations living in different geographical regions [26],[27].
Ets-1 is a member of the ETS family of transcription factors that share a unique Ets DNA binding domain. They control a wide variety of cellular processes including cell proliferation and differentiation [28]. Ets-1-deficient mice develop lupus-like disease characterized by high titers of IgM and IgG autoantibodies, immune complex-mediated glomerulonephritis, and local activation of complement [29]. Ets-1 is also involved in many cellular abnormalities that are known to participate in SLE pathogenesis as illustrated in Figure 7.
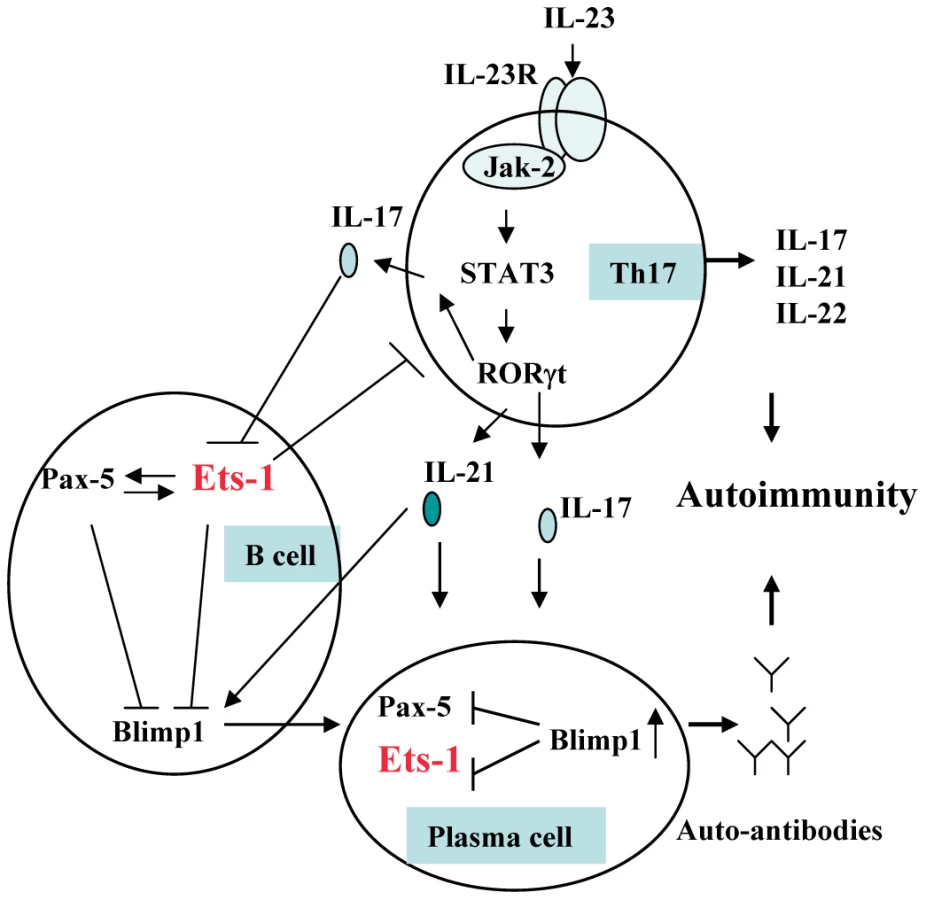
Ets-1 is a negative regulator of terminal differentiation of B cells and plays critical roles in maintaining B cell identity [30]–[32]. Ets-1-deficient B cells were present in normal numbers but have a large proportion of IgM plasma cells [33]. Ets-1 blocks the function of B-lymphocyte-induced maturation protein 1 (Blimp-1), an essential transcription factor for plasma cells [34]. The number and frequency of plasma cells were known to correlate with disease activity and the titer of anti-dsDNA antibodies in SLE [35],[36].
Ets-1 is also a negative regulator of Th17 cell differentiation, and naïve CD4+ T cells deficient in Ets-1 undergo greatly enhanced differentiation into Th17 cells when cultured in vitro under Th17-skewing conditions [37]. Th17 cells with specificity for self-antigens are known to be highly pathogenic and lead to the development of inflammation and severe autoimmunity [38]. Higher plasma IL-17, IL-23 and higher number of Th17 cells in SLE patients were reported and correlated positively with SLE disease index (SLEDAI) [39]–[41]. IL-17 and IL-21 produced by Th17 cells may also induce B cell terminal differentiation [42]–[44].
In this study, we found that in the PBMC of healthy individuals, expression of ETS1 from the risk “A” allele is reduced comparing to that from the “G” allele. The expression level of ETS1 may be tightly regulated. It was shown that resting T cells express high levels of ETS1 mRNA and protein, which decreased to very low levels upon T cell activation [45]. Lower expression of ETS1 for the risk allele carriers may play a role in disease pathogenesis through increased differentiation and activity of both plasma cells and Th17 cells.
It is likely that the association SNPs identified in this study may affect the response of ETS1 gene to other upstream signals. SNP rs1128334 locates in the 3′-UTR of ETS1, and rs10893872 is in absolute LD with rs4937333, another SNP that is also located in the 3′-UTR of the gene and both SNPs are on putative microRNA (miRNA) binding sites. In a recent study by Du et al, the expression level of a microRNA, miR-326, was found to be related to disease severity in patients with multiple sclerosis and mice with experimental autoimmune encephalomyelitis. ETS1 was shown to be the major target of miR-326, through downregulation of which miR-326 promoted the generation of Th17 cells both in vitro and in vivo [46]. Another microRNA, miRNA-146a, was also found to be involved in SLE pathogenesis [47].
WDFY family member 4 (WDFY4, NCBI GeneID: 57705) codes for a huge protein (3184 amino acid residues) with unknown function. Its closest paralog is WD repeat and FYVE domain containing 3 (WDFY3, NCBI GeneID: 23001). Similar to WDFY3, WDFY4 does contain WD40 domains and a BEACH (Beige and chediak-kigashi) domain, although FYVE zinc finger domain is truncated. Its sequence is well conserved among various species. For example, there is an 84% sequence similarity between the human protein and its orthologs from Bos Taurus and Canis lupus familiaris. And there is a 42% identity between the human protein and protein XP_701288.3 in Danio Rerio (zebra fish). Although protein XP_701288.3 is annotated as WDFY3 in NCBI, it has a higher sequence similarity with human WDFY4 than with WDFY3.
WD40 domain is found in a number of eukaryotic proteins that cover a wide variety of functions including adaptor/regulatory modules in signal transduction, pre-mRNA processing and cytoskeleton assembly, while the BEACH domains are implicated in membrane trafficking. While very little is known about the potential function of this well conserved protein, an interesting phenomenon is that the gene is predominantly expressed in immune tissues such as lymph node, spleen, thymus and tonsil (UniGene Hs.287379, http://www.ncbi.nlm.nih.gov/UniGene/ESTProfileViewer.cgi?uglist=Hs.287379), unlike WDFY3, which is expressed in a wide variety of tissues and organs.
After submission of our work, Han et al reported their GWAS work on SLE on Chinese populations [18] and reported genome-wide significant association signals on ETS1 (rs6590330, downstream of the gene) and WDFY4 (rs1913517, in the intron of both WDFY4 and LRRC18, leucine rich repeat containing 18). Independent identification of these two genes in SLE susceptibility underlined the validity of the findings. Identifying susceptibility genes provided a good start in our effort to elucidate the disease mechanisms and the functions of the genes involved, although many questions remain to be answered. Characterization of the functions of the genes in autoimmunity may eventually help us to translate genetic findings into clinical and therapeutic applications.
Materials and Methods
Ethics statement
This study was conducted according to the principles expressed in the Declaration of Helsinki. The Hong Kong study was approved by the Institutional Review Board of the University of Hong Kong and Hospital Authority, Hong Kong West Cluster, New Territory West Cluster, and Hong Kong East Cluster. The study on Shanghai, Anhui and Thai samples was approved by the Institutional Review Board of Renji Hospital, Research Ethics Committee of Anhui Medical University and the Ethics Committee of the Faculty of Medicine, Chulalongkorn University, respectively. All patients provided written informed consent for the collection of samples and subsequent analysis.
Subjects
1073 SLE samples collected in Hong Kong were from four hospitals in Hong Kong Island and the New Territories: Queen Mary Hospital, Tuen Mun Hospital, Queen Elizabeth Hospital and Pamela Youde Nethersole Eastern Hospital. The patients were all of self-reported Chinese ethnicity living in Hong Kong. The average onset age was 28 years old and the ratio of female to male patients was 9∶1. About half of the patients had renal involvement, and about 70% tested positive for anti-dsDNA antibodies. The SLE samples collected in Shanghai were patients attending Renji Hospital of Jiaotong University Medical School, a tertiary referral hospital covering Shanghai and the surrounding areas. There is an 8∶1 female to male patient ratio, and about 52% of patients have lupus nephritis. 951 SLE patients collected in Anhui were all self-reported Chinese ethnicity living in Anhui province, central China. They were recruited from the Departments of Rheumatology at Anhui Provincial Hospital and the First Affiliated Hospital of Anhui Medical University, both located in Hefei, Anhui province, about 450 km from Shanghai. The average onset age was 31 years old and the ratio of female to male patients was 17∶1. 314 Thai patients with SLE (female∶male ratio = 14∶1) attending King Chulalongkorn Memorial Hospital, a tertiary referral center in Bangkok were also recruited in this study. Medical records were reviewed to confirm that all subjects met the revised criteria of the American College of Rheumatology for SLE diagnoses [48].
Controls used in the GWAS stage were from both healthy individuals and from other studies conducted in the same institution genotyped with the same platform. For the replication stage, Hong Kong controls were healthy blood donors kindly contributed by the Hong Kong Red Cross and were all of self-reported Chinese ethnicity living in Hong Kong. Controls from Shanghai and Anhui were selected from a pool of healthy blood donors recruited from Renji Hospital (Shanghai) and Hefei City (Anhui), respectively, with an effort to match for the age and sex of corresponding SLE patients. Thai controls were recruited from unrelated voluntary healthy donors from the same ethnic background and geographic area as the Thai SLE patients.
Genome-wide association study
320 (27 males, 293 females) SLE patients were genotyped by Illumina 610-Quad Human Beadchip with a total number of SNPs reaching 620,901. 24 individuals were removed due to low call rate or hidden first degree relationship. A total number of 104,395 SNPs were also removed from the initial analysis on the ground of low genotyping call rate (<90%, 30,133 SNPs) and/or low minor allele frequency (MAF) (<0.005, 102,970 SNPs). 2,285 SNPs were also removed due to violation of Hardy-Weinberg equilibrium in controls (P values<0.0001). After quality control measures, 314 case (27 males, 287 females) and 1484 controls (840 males and 644 females) were analyzed on a total of 514,221 SNPs. The call rate for the remaining SNPs reached 0.999 with a genome-wide inflation factor of 1.03, which is an indication of good match between the cases and controls. To overcome any potential effect from the heterogeneity in the controls, three independent comparisons were initially conducted by separating the controls into three different subsets and only SNPs reaching significance in all subsets in association analysis were selected for further replication.
The SNPs were analyzed for association with the disease by means of comparison of the minor allele frequency in patients and controls (basic allelic test) as well as other tests using Plink [49]. Linkage disequilibrium (LD) patterns were analyzed and displayed by HaploView [50]. Association of the SNPs with disease risk was also corrected by logistic regression using age and sex as covariates. Average odds ratios (OR) and P values jointly analyzed from four sample collections were obtained by Cochran-Mantel-Haenszel (CMH) test of disease association conditional on SNP frequency differences among different populations. Test of independent contributions of a SNP controlling for the effect of other SNPs in the same locus was done by conditional logistic regression as well as haplotype analyses. Subphenotype stratification was performed by comparing cases with and without a given subphenotype.
Genotyping in replication stage
SNPs rs1128334, rs10893872, rs7097397, rs10857650, and rs877819 were genotyped by TaqMan SNP genotyping method using assay-on-demand probes and primers (Applied Biosystems, Foster City, CA94404, USA). Some of the initial screening was also done using Sequenom MassARRAY iPLEX Gold system. Genotyping accuracy was confirmed by direct sequencing of PCR products for some randomly chosen samples. Genotyping concordance between Illumina Human 610-Quad Beadchip and TaqMan SNP genotyping method was also examined on selected samples and probes: rs877819 has a concordance rate of 99.64% (1 out of 277 differed by the two platforms); rs10893872 has a concordance rate of 99.16% (1 out of 119 samples differed). SNP rs1128334 and rs7097397 were examined by direct sequencing of selected samples and showed complete consistence between TaqMan and sequencing (50 samples each). The results of rs10857650 were discarded due to low concordance between the Illumina Beadchip data and the TaqMan results.
Allele-specific transcription quantification
Thirty-three healthy individuals heterozygous for rs1128334 were chosen to assess the relative ETS1 mRNA levels from the two alleles, “A” and “G”, by pyrosequencing [51],[52]. In the meantime, DNA detection ratio was used as a control for amplification efficiency. Briefly, total RNA was extracted from peripheral blood mononuclear cell (PBMC) from each individual. RNA samples were then treated with DNAase to eliminate genomic DNA contamination before being reverse-transcribed into cDNA using oligo-dT primer. cDNA was then amplified by PCR using transcript-specific primers, together with DNA from the same individuals. The cDNA and DNA PCR products were purified using the Qiaquick PCR purification kit, and then subjected to allele quantitative pyrosequencing. The sequencing primer was designed using Pyrosequencing Assay Design Software v.1.0. Reactions were performed on a Biotage PSQ96MA machine, and allele quantification was analyzed using PSQMA 2.1 software. The average G/A cDNA expression ratio of each individual was normalized by the G/A DNA ratio from the same sample. Paired student' t test was used to compare the normalized expression level from the “A” and “G” alleles.
Zdroje
1. SestakAL
ShaverTS
MoserKL
NeasBR
HarleyJB
1999 Familial aggregation of lupus and autoimmunity in an unusual multiplex pedigree. J Rheumatol 26 1495 1499
2. Ramos-NiembroF
Alarcon-SegoviaD
1978 Familial aspects of mixed connective tissue disease (MCTD). I. Occurrence of systemic lupus erythematosus in another member in two families and aggregation of MCTD in another family. J Rheumatol 5 433 440
3. ArnettFC
ShulmanLE
1976 Studies in familial systemic lupus erythematosus. Medicine (Baltimore) 55 313 322
4. MokCC
LauCS
2003 Lupus in Hong Kong Chinese. Lupus 12 717 722
5. WongSN
TseKC
LeeTL
LeeKW
ChimS
2006 Lupus nephritis in Chinese children–a territory-wide cohort study in Hong Kong. Pediatr Nephrol 21 1104 1112
6. RemmersEF
PlengeRM
LeeAT
GrahamRR
HomG
2007 STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 357 977 986
7. HarleyJB
Alarcon-RiquelmeME
CriswellLA
JacobCO
KimberlyRP
2008 Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 40 204 210
8. HomG
GrahamRR
ModrekB
TaylorKE
OrtmannW
2008 Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med 358 900 909
9. NathSK
HanS
Kim-HowardX
KellyJA
ViswanathanP
2008 A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet 40 152 154
10. KozyrevSV
AbelsonAK
WojcikJ
ZaghloolA
Linga ReddyMV
2008 Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 40 211 216
11. GrahamRR
CotsapasC
DaviesL
HackettR
LessardCJ
2008 Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet
12. MusoneSL
TaylorKE
LuTT
NitithamJ
FerreiraRC
2008 Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet
13. WebbR
MerrillJT
KellyJA
SestakA
KaufmanKM
2009 A polymorphism within IL21R confers risk for systemic lupus erythematosus. Arthritis Rheum 60 2402 2407
14. JacobCO
ZhuJ
ArmstrongDL
YanM
HanJ
2009 Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A 106 6256 6261
15. LiuK
LiQZ
Delgado-VegaAM
AbelsonAK
SanchezE
2009 Kallikrein genes are associated with lupus and glomerular basement membrane-specific antibody-induced nephritis in mice and humans. J Clin Invest 119 911 923
16. EdbergJC
WuJ
LangefeldCD
BrownEE
MarionMC
2008 Genetic variation in the CRP promoter: association with systemic lupus erythematosus. Hum Mol Genet 17 1147 1155
17. GatevaV
SandlingJK
HomG
TaylorKE
ChungSA
2009 A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet 41 1228 1233
18. HanJW
ZhengHF
CuiY
SunLD
YeDQ
2009 Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet 41 1234 1237
19. YangW
NgP
ZhaoM
HirankarnN
LauCS
2009 Population differences in SLE susceptibility genes: STAT4 and BLK, but not PXK, are associated with systemic lupus erythematosus in Hong Kong Chinese. Genes Immun 10 219 226
20. KimI
KimYJ
KimK
KangC
ChoiCB
2009 Genetic studies of systemic lupus erythematosus in Asia: where are we now? Genes Immun
21. YangW
ZhaoM
HirankarnN
LauCS
MokCC
2009 ITGAM is associated with disease susceptibility and renal nephritis of systemic lupus erythematosus in Hong Kong Chinese and Thai. Hum Mol Genet 18 2063 2070
22. PriceAL
PattersonNJ
PlengeRM
WeinblattME
ShadickNA
2006 Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38 904 909
23. Cunninghame GrahamDS
GrahamRR
MankuH
WongAK
WhittakerJC
2008 Polymorphism at the TNF superfamily gene TNFSF4 confers susceptibility to systemic lupus erythematosus. Nat Genet 40 83 89
24. ChangYK
YangW
ZhaoM
MokCC
ChanTM
2009 Association of BANK1 and TNFSF4 with systemic lupus erythematosus in Hong Kong Chinese. Genes Immun 10 414 420
25. SiuHO
YangW
LauCS
ChanTM
WongRW
2008 Association of a haplotype of IRF5 gene with systemic lupus erythematosus in Chinese. J Rheumatol 35 360 362
26. ChenJ
ZhengH
BeiJX
SunL
JiaWH
2009 Genetic Structure of the Han Chinese Population Revealed by Genome-wide SNP Variation. Am J Hum Genet
27. XuS
YinX
LiS
JinW
LouH
2009 Genomic Dissection of Population Substructure of Han Chinese and Its Implication in Association Studies. Am J Hum Genet
28. DittmerJ
2003 The biology of the Ets1 proto-oncogene. Mol Cancer 2 29
29. WangD
JohnSA
ClementsJL
PercyDH
BartonKP
2005 Ets-1 deficiency leads to altered B cell differentiation, hyperresponsiveness to TLR9 and autoimmune disease. Int Immunol 17 1179 1191
30. MaierH
ColbertJ
FitzsimmonsD
ClarkDR
HagmanJ
2003 Activation of the early B-cell-specific mb-1 (Ig-alpha) gene by Pax-5 is dependent on an unmethylated Ets binding site. Mol Cell Biol 23 1946 1960
31. GarvieCW
HagmanJ
WolbergerC
2001 Structural studies of Ets-1/Pax5 complex formation on DNA. Mol Cell 8 1267 1276
32. PufallMA
GravesBJ
2002 Ets-1 flips for new partner Pax-5. Structure 10 11 14
33. BoriesJC
WillerfordDM
GrevinD
DavidsonL
CamusA
1995 Increased T-cell apoptosis and terminal B-cell differentiation induced by inactivation of the Ets-1 proto-oncogene. Nature 377 635 638
34. JohnSA
ClementsJL
RussellLM
Garrett-SinhaLA
2008 Ets-1 regulates plasma cell differentiation by interfering with the activity of the transcription factor Blimp-1. J Biol Chem 283 951 962
35. JacobiAM
OdendahlM
ReiterK
BrunsA
BurmesterGR
2003 Correlation between circulating CD27high plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum 48 1332 1342
36. GrammerAC
LipskyPE
2003 B cell abnormalities in systemic lupus erythematosus. Arthritis Res Ther 5 Suppl 4 S22 27
37. MoisanJ
GrenninglohR
BettelliE
OukkaM
HoIC
2007 Ets-1 is a negative regulator of Th17 differentiation. J Exp Med 204 2825 2835
38. BettelliE
KornT
KuchrooVK
2007 Th17: the third member of the effector T cell trilogy. Curr Opin Immunol 19 652 657
39. WongCK
HoCY
LiEK
LamCW
2000 Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2 cytokine (IL-4) concentrations in patients with systemic lupus erythematosus. Lupus 9 589 593
40. WongCK
LitLC
TamLS
LiEK
WongPT
2008 Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in auto-immunity. Clin Immunol 127 385 393
41. YangJ
ChuY
YangX
GaoD
ZhuL
2009 Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum 60 1472 1483
42. HsuHC
YangP
WangJ
WuQ
MyersR
2008 Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol 9 166 175
43. DoreauA
BelotA
BastidJ
RicheB
Trescol-BiemontMC
2009 Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol 10 778 785
44. EttingerR
SimsGP
RobbinsR
WithersD
FischerRT
2007 IL-21 and BAFF/BLyS synergize in stimulating plasma cell differentiation from a unique population of human splenic memory B cells. J Immunol 178 2872 2882
45. BhatNK
ThompsonCB
LindstenT
JuneCH
FujiwaraS
1990 Reciprocal expression of human ETS1 and ETS2 genes during T-cell activation: regulatory role for the protooncogene ETS1. Proc Natl Acad Sci U S A 87 3723 3727
46. DuC
LiuC
KangJ
ZhaoG
YeZ
2009 MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol 10 1252 1259
47. TangY
LuoX
CuiH
NiX
YuanM
2009 MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum 60 1065 1075
48. HochbergMC
1997 Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40 1725
49. PurcellS
NealeB
Todd-BrownK
ThomasL
FerreiraMA
2007 PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81 559 575
50. BarrettJC
FryB
MallerJ
DalyMJ
2005 Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21 263 265
51. AhmadianA
GharizadehB
GustafssonAC
SterkyF
NyrenP
2000 Single-nucleotide polymorphism analysis by pyrosequencing. Anal Biochem 280 103 110
52. SunKH
TangSJ
ChenCY
LeeTP
FengCK
2005 Monoclonal ribosomal P autoantibody inhibits the expression and release of IL-12, TNF-alpha and iNOS in activated RAW macrophage cell line. J Autoimmun 24 135 143
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 2
Nejčtenější v tomto čísle
- Genome-Wide Association Study in Asian Populations Identifies Variants in and Associated with Systemic Lupus Erythematosus
- Nucleoporins and Transcription: New Connections, New Questions
- Nuclear Pore Proteins Nup153 and Megator Define Transcriptionally Active Regions in the Genome
- The Genetic Interpretation of Area under the ROC Curve in Genomic Profiling