
Deletion of the Huntingtin Polyglutamine Stretch Enhances Neuronal Autophagy and Longevity in Mice
Expansion of a stretch of polyglutamine in huntingtin (htt), the protein product of the IT15 gene, causes Huntington's disease (HD). Previous investigations into the role of the polyglutamine stretch (polyQ) in htt function have suggested that its length may modulate a normal htt function involved in regulating energy homeostasis. Here we show that expression of full-length htt lacking its polyglutamine stretch (ΔQ-htt) in a knockin mouse model for HD (Hdh140Q/ΔQ), reduces significantly neuropil mutant htt aggregates, ameliorates motor/behavioral deficits, and extends lifespan in comparison to the HD model mice (Hdh140Q/+). The rescue of HD model phenotypes is accompanied by the normalization of lipofuscin levels in the brain and an increase in the steady-state levels of the mammalian autophagy marker microtubule-associate protein 1 light chain 3-II (LC3-II). We also find that ΔQ-htt expression in vitro increases autophagosome synthesis and stimulates the Atg5-dependent clearance of truncated N-terminal htt aggregates. ΔQ-htt's effect on autophagy most likely represents a gain-of-function, as overexpression of full-length wild-type htt in vitro does not increase autophagosome synthesis. Moreover, HdhΔQ/ΔQ mice live significantly longer than wild-type mice, suggesting that autophagy upregulation may be beneficial both in diseases caused by toxic intracellular aggregate-prone proteins and also as a lifespan extender in normal mammals.
Published in the journal:
. PLoS Genet 6(2): e32767. doi:10.1371/journal.pgen.1000838
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000838
Summary
Expansion of a stretch of polyglutamine in huntingtin (htt), the protein product of the IT15 gene, causes Huntington's disease (HD). Previous investigations into the role of the polyglutamine stretch (polyQ) in htt function have suggested that its length may modulate a normal htt function involved in regulating energy homeostasis. Here we show that expression of full-length htt lacking its polyglutamine stretch (ΔQ-htt) in a knockin mouse model for HD (Hdh140Q/ΔQ), reduces significantly neuropil mutant htt aggregates, ameliorates motor/behavioral deficits, and extends lifespan in comparison to the HD model mice (Hdh140Q/+). The rescue of HD model phenotypes is accompanied by the normalization of lipofuscin levels in the brain and an increase in the steady-state levels of the mammalian autophagy marker microtubule-associate protein 1 light chain 3-II (LC3-II). We also find that ΔQ-htt expression in vitro increases autophagosome synthesis and stimulates the Atg5-dependent clearance of truncated N-terminal htt aggregates. ΔQ-htt's effect on autophagy most likely represents a gain-of-function, as overexpression of full-length wild-type htt in vitro does not increase autophagosome synthesis. Moreover, HdhΔQ/ΔQ mice live significantly longer than wild-type mice, suggesting that autophagy upregulation may be beneficial both in diseases caused by toxic intracellular aggregate-prone proteins and also as a lifespan extender in normal mammals.
Introduction
In vertebrates, the polyQ stretch within htt is located close to the protein's N-terminus, and separates a highly conserved 17 amino acid N-terminal domain (N1–17) that can act as a membrane association signal [1], from a proline-rich region that is implicated in protein-protein interactions [2]–[4]. Expansion of htt's polyQ stretch (>37Q) causes Huntington's disease (HD), a neurodegenerative disorder characterized by the appearance of cytoplasmic (neuropil) and nuclear aggregates of mutant htt, and selective cell death in the striatum and cortex [5]–[9]. Although the mechanism of pathogenesis is still unclear, HD is recognized as a toxic gain-of-function disease, where the expansion of the polyQ stretch within htt confers new deleterious functions on the protein. The extent to which the polyQ expansion affects normal htt function is also unclear, although there is accumulating evidence that loss of normal htt function likely contributes to HD pathogenesis [10]. The polyQ stretch is conserved in vertebrate htt, and its non-pathogenic size varies from 4Q in fish, to 37Q in humans [11]–[13]. However, the polyQ stretch is absent in Ciona and Drosophila htt, and present as only a short hydrophilic NHQQ stretch in sea urchin htt, suggesting that addition of a htt polyQ stretch may be a late evolutionary feature acquired sometime after protostome-deuterostome divergence [14].
In lymphoblastoid cell lines derived from HD patients, polyQ length (in both the normal and mutant htt alleles) affects energy status, with a longer polyQ stretch correlating with a reduced cellular ATP/ADP ratio [15]. Deletion of the normal short polyQ stretch (7Q) in mouse htt (ΔQ-htt) also results in elevated ATP levels in fibroblasts derived from embryonic and adult HdhΔQ/ΔQ mice [16]. In addition, adult HdhΔQ/ΔQ mice exhibit subtly enhanced performance on the rotarod, and altered behavior in the Barnes maze learning and memory test.
To assess ΔQ-htt function in the presence of expanded polyQ htt expression, we generated mice expressing both ΔQ-htt and 140Q-htt (Hdh140Q/ΔQ). We found that ΔQ-htt expression in the HD mouse model rescued behavioral/motor deficits, reduced the number of neuropil htt aggregates, normalized brain lipofuscin levels, and enhanced lifespan relative to the HD mouse model. Clearance of htt aggregates and the accumulation of lipofuscin are mediated by autophagy, a catabolic pathway that encompasses several distinct processes in mammalian cells [17]. Macroautophagy generally involves the non-selective turnover of bulk cytoplasmic contents, including organelles and aggregated protein, and is an essential pathway for the survival of organisms during nutrient deprivation [18]. Upregulation of autophagy reduces truncated mutant htt aggregation and toxicity in both in vitro and in vivo models [19]–[22], and recently, the acetylation of soluble full-length htt has also been reported to assist its recognition by the autophagic apparatus [23]. In HdhΔQ/+ and Hdh140Q/ΔQ mice, we observed enhanced microtubule-associated protein 1 light chain 3 (LC3, [24]) immunostaining, and increased levels of the LC3-II autophagic marker. Expression of ΔQ-htt, but not wild-type htt, induced the formation of autophagosomes in SK-N-SH neuroblastoma cells, and enhanced the clearance of truncated 74Q-htt aggregates in an autophagy-dependent process. Based on our observations, we hypothesize that deletion of the polyQ stretch within huntingtin enhances neuronal macroautophagy resulting in the more efficient clearance of neuropil mutant htt and phenotypic rescue in Hdh140Q/ΔQ mice. Moreover, we have observed that mice homozygous for ΔQ-htt expression live significantly longer than wild-type mice, an observation that is compatible with the view that enhancing constitutive autophagy may also be beneficial in normal ageing.
Results
Rescue of Hdh140Q/+ motor and behavioral deficits in Hdh140Q/ΔQ mice
To evaluate the impact of expressing a version of wild-type htt lacking its short polyQ stretch on the motor and behavioral phenotypes exhibited by a mouse model for HD, HdhΔQ/+ mice were crossed with the CAG140 knock-in mouse expressing full-length htt with a chimeric human/mouse htt exon 1 containing an expanded stretch of 140 glutamines [25], (for a diagram of the knockin alleles used in this study, see Figure 1A). Hdh140Q/ΔQ, Hdh140Q/+, and wild-type control littermates were assessed using the accelerating rotarod, the Barnes maze, and an activity cage. Mice were tested on an accelerating rotating rod at 1, 5, and 19 months of age (Figure 1B). At one month of age, there were no significant differences between the wild-type controls, Hdh140Q/+ mice, and the Hdh140Q/ΔQ mice (n = 6 for each genotype at 1 and 5 months, n = 4 of each genotype at 19 months). A two-way repeated measures ANOVA showed no significant effect of genotype (F(2,6) = 0.87; P>0.05), although there was a significant trial day effect (F(4,6) = 13.00; P<0.001), indicating that all mice were learning to stay on the rod.

At five months of age, however, the Hdh140Q/+ mice performed poorly in comparison to both the wild-type control group and the Hdh140Q/ΔQ group (genotype effect; F(2,6) = 5.4; P<0.03). Interestingly, at five months, the Hdh140Q/ΔQ mice were indistinguishable from the wild-type controls. At 19 months of age, both the wild type and Hdh140Q/ΔQ mice still performed better than the Hdh140Q/+ mice and were indistinguishable from each other (genotype effect; F(2,4) = 6.5; P<0.04), although all mice were performing more poorly at 19 months relative to their performance at 5 months of age.
At five months of age, the mice were also tested on the Barnes maze, a measure of spatial learning and memory [26]. Wild-type mice produced better scores on the Barnes maze distance test than Hdh140Q/+ mice, but did not differ significantly from the Hdh140Q/ΔQ mice (n = 5 of each genotype) (Figure 1C). The distance score measures how effectively the mice are using spatial cues to locate the escape tunnel. A two way repeated measures ANOVA revealed a significant effect of genotype (F(2,4) = 5.96; P<0.02) and a significant effect of trial day (F(8,4) = 2.2; P<0.04). In addition, wild-type and Hdh140Q/ΔQ mice made fewer errors than Hdh140Q/+ mice before finding the Barnes maze target (Figure 1C). A two-way repeated measures ANOVA revealed a significant effect of genotype (F(2,4) = 25.28; P<0.001), and a significant effect of trial day [(F(8,4) = 3.33; P<0.003)].
At 6 and 20 months of age, the mice were also tested in an activity cage (n = 5 of each genotype) (Figure 1D). Previous analyses of Hdh140Q mice revealed that they exhibit a period of hyperactivity, followed by hypoactivity when tested at night in an activity cage [25]. Based on total horizontal activity, the Hdh140Q/+ mice were more hypoactive at night than the wild-type mice at 6 months, but the exploratory activity of the Hdh140Q/ΔQ mice did not differ significantly from wild-type controls (one-way ANOVA F(2,12) = 6.63; P<0.02; post-hoc analysis wild-type versus Hdh140Q/+, P<0.02). At 20 months of age, one-way ANOVA revealed an overall difference in activity levels as well (F(2,14) = 6.78; P<0.02). Bonferroni post-hoc analysis showed the Hdh140Q/+ mice to be significantly hypoactive when compared to the Hdh140Q/ΔQ mice (P<0.01).
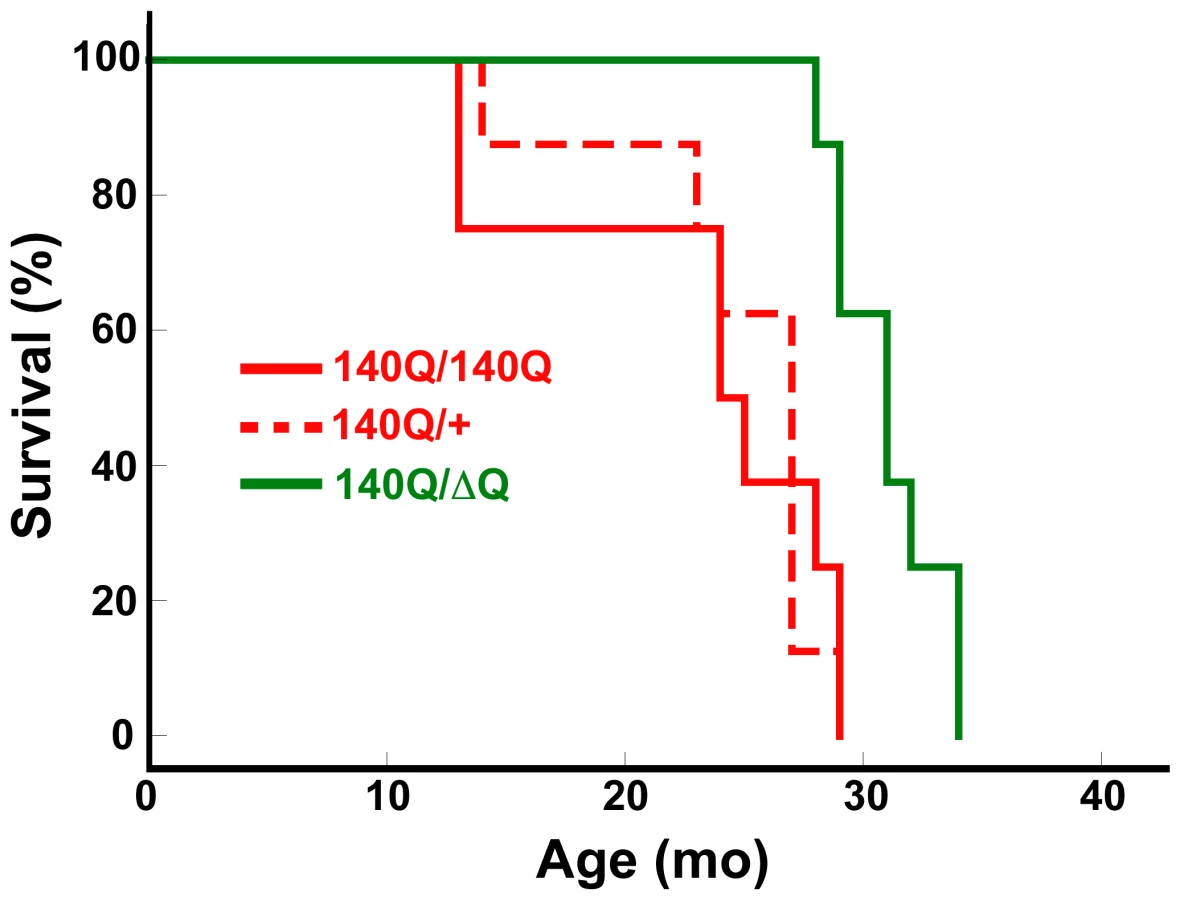
Hdh140Q/ΔQ mice also exhibited a significant increase in their lifespan (median age of 31+/−0.8 months) in comparison to either Hdh140Q/+ or Hdh140Q/140Q mice (median ages of 24+/−2.3 and 27+/−1.7 months, respectively, Hdh140Q/+ versus Hdh140Q/ΔQ log-rank test, χ2 = 11.7, P<0.002; Hdh140Q/140Q versus Hdh140Q/ΔQ log-rank test, χ2 = 9.9, P<0.003 for n = 8 females of each genotype) (Figure 2). However, we could not detect any significant difference in the lifespan of the Hdh140Q/+ and Hdh140Q/140Q mice (log-rank test, χ2 = 0.03, P = 0.958).
Reduction of neuropil htt aggregates in Hdh140Q/ΔQ mice
To determine if the rescue of behavioral phenotypes in the Hdh140Q/ΔQ mice correlated with a change in the number and distribution of htt aggregates, we examined Hdh140Q/+, Hdh140Q/ΔQ, and HdhΔQ/+ (control) brains (n = 4 of each genotype) using an antibody recognizing aggregated mutant htt in inclusions (MW8 [27]) (Figure 3A). At 4 months of age, we were unable to detect htt aggregates in either Hdh140Q/+ or Hdhı4˜Q˜ΔQ mice. Starting at 6 months of age, however, we observed a small, but similar number of nuclear aggregates in the striatum of both genotypes. In contrast, there was a significant reduction in the number of striatal neuropil aggregates observed at 6 months of age in the Hdh140Q/ΔQ brain in comparison to the Hdh140Q/+ brain, P<0.001 (Figure 3B). At 1 year and 2 years of age, the aggregate load increases dramatically in the Hdh140Q/+ brain, with the number of striatal neuropil aggregates growing more quickly with age than the number of nuclear aggregates (Figure 3B). The significant reduction in the number of striatal neuropil aggregates that was observed at 6 months of age in the Hdh140Q/ΔQ striatum was also observed in the striatum of Hdh140Q/ΔQ mice at 1 year and 2 years of age, P<0.001 and P<0.05, respectively. In the cortex, a similar marked decrease in Hdh140Q/ΔQ neuropil aggregates was observed at 6 months, 1 year, and 2 years of age (P<0.001–P<0.005) (Figure S1). In both striatum and cortex, nuclear aggregates were also reduced significantly at 1 year of age, but the magnitude of the decrease was less than that observed for the neuropil aggregates.

Lipofuscin deposits are reduced in Hdh140Q/ΔQ mice
Increased lipofuscin has been observed in the HD brain and in the R6/2 transgenic mouse model for HD [28]–[30]. Accumulating in the lysosomes of neurons and other post-mitotic cells, lipofuscin is a yellowish-brown autofluorescent aging pigment that is composed of oxidized lipid and aldehyde cross-linked protein [31]. Lipofuscin is believed to be the byproduct of the incomplete autophagic catabolism of cellular organelles, such as mitochondria that are rich in iron. Iron and peroxide-catalyzed oxidation of incompletely digested lipid and protein results in the slow accumulation of lipofuscin in autolysosomes at a rate that correlates with metabolic activity and age of the organism [32]. In HD, oxidative stress may enhance the formation of lipofuscin, resulting in the appearance of large perinuclear lipofuscin deposits in neurons. In aged cells with high levels of lipofuscin, autophagy is diminished [33],[34], and in C. elegans, lower levels of lipofuscin in age-matched worms correlated with greater motility, suggesting that lipofuscin accumulation reflects biological versus chronological age [35].
We compared the extent of lipofuscin accumulation in the striatum and cortex of wild-type, HdhΔQ/+, Hdh140Q/+, and Hdh140Q/ΔQ mice at 4 months, 6 months, 1 year, and 2 years of age (n = 4 mice of each genotype) (Figure 4A and 4B). Consistent with prior observations in the R6/2 HD transgenic mouse model and in postmortem HD brain tissue, we observed a significant increase in lipofuscin (measured as the pixel area of deposits in confocal images) in the striatum and cortex of Hdh140Q/+ mice as they aged in comparison to wild-type mice, P<0.05 to P<0.001 (Figure 4B). Lipofuscin accumulation was greater in the striatum, relative to the cortex in the Hdh140Q/+ brain. In both the Hdh140Q/ΔQ cortex and striatum, however, neuronal lipofuscin accumulation was similar to that observed in wild type controls at all ages examined.

Altered autophagy in HdhΔQ/+ and Hdh140Q/ΔQ mice
To determine if clearance of the neuropil htt aggregates and the reduction in lipofuscin in the Hdh140Q/ΔQ brain may be related to altered autophagy, we performed immunohistochemical analyses and western blot analyses of cellular fractions obtained from wild-type, HdhΔQ/+, Hdh140Q/+, and Hdh140Q/ΔQ whole brains and dissected brain regions, respectively, using an antibody to LC3. LC3 is encoded by the mammalian homolog of the yeast Atg8 gene, and is widely used as a marker for autophagy in mammalian cells because it associates tightly with autophagic membranes beginning at vesicle nucleation, and ending with its turnover in autolysosomes [24]. Western blotting with antibodies recognizing the N-terminus of LC3 detects two species with apparent molecular weights of 18 kD (LC3-I) and 16 kD (LC3-II). LC3 is processed proteolytically at its C terminus to form cytosolic LC3-I, which is conjugated to phosphatidylethanolamine on autophagosome membranes to form LC3-II. LC3-II associates specifically with autophagosome and autolysosome membranes, and LC3 vesicle numbers or levels of LC3-II correlate with autophagosome numbers [24],[36].
LC3 immunostaining was enhanced in the striatum of Hdh140Q/ΔQ mice beginning at 6 months of age in comparison to age-matched wild-type, HdhΔQ/+, and Hdh140Q/+ mice (n = 4 of each genotype, Figure S2). At 1 year of age, the Hdh140Q/ΔQ striatum continued to exhibit enhanced LC3 immunostaining, and at 2 years of age, elevated LC3 immunostaining was now detected in both the HdhΔQ/+ and Hdh140Q/ΔQ striatum (Figure 5A). In contrast, LC3 immunostaining in the Hdh140Q/+ striatum at 1 year and 2 years of age was not increased substantially in comparison to age-matched wild-type controls. Moreover, co-localization of LC3 immunostaining with neuropil htt aggregates was observed in the Hdh140Q/ΔQ striatum at 1 and 2 years of age, but was difficult to detect in the Hdh140Q/+ striatum (Figure 5A).

To confirm that the enhanced LC3 immunostaining observed in the HdhΔQ/+ and Hdh140Q/ΔQ striatum was due to an increase in LC3-II levels, dissected striata from 2 year old wild-type, HdhΔQ/+, Hdh140Q/+, and Hdh140Q/ΔQ mice (n = 4 of each genotype) were homogenized and separated into supernatant (NP40-soluble) and pellet (NP40-insoluble) fractions, and then analyzed by western blotting with an antibody that recognizes both LC3-I and LC3-II (Figure 5B and 5C). In the soluble protein fractions, an increase in LC3-II was observed in the Hdh140Q/ΔQ striatum. Interestingly, LC3-II was also enriched in the striatal pellet fractions from both HdhΔQ/+ and Hdh140Q/ΔQ mice. In contrast, LC3-II was present at only low levels in the wild-type and Hdh140Q/+ pellet fractions. A corresponding western blot analysis of LC3 levels in total (unfractionated ) protein extracts from 2 year old mice revealed an increase in LC3-II in both the HdhΔQ/+ and Hdh140Q/ΔQ samples (Figure S3B). We note that we also observed an enrichment of both the autophagy protein beclin 1 and lysosome-associated membrane protein type 1 (Lamp1) levels in the 800×g low-speed P1 fractions from HdhΔQ/+ and Hdh140Q/ΔQ striatal extracts prepared by lysis in the absence of detergent (Figure S3A). Overall levels of beclin 1 and Lamp1 in total brain extract, however, were similar in all genotypes (Figure S3B). Lamp1 is a marker for late endososomes, amphisomomes (formed after autophgagosome-late-endosome fusion), dense autolysosomes and lysosomes that are enriched in the 800×g P1 fraction [37], and these observations, together with our findings related to the alterations in beclin 1 and LC3-II fractionation, suggest that the subcellular distribution of several components of the autophagy pathway are altered by ΔQ-htt expression.
It was proposed recently, that htt's association with the ER via its N1–17 domain allows it function as a sensor of ER stress, and to potentially regulate autophagy [1],[38]. In previous work, we found no obvious difference in the nuclear/cytoplasmic localization of ΔQ-htt in comparison to wild-type htt in early passage wild-type and HdhΔQ/ΔQ primary mouse embryonic fibroblasts (PMEFs) [16]. To analyze further the subcellular localization of wild-type - and ΔQ-htt together with markers for the ER (calnexin), and to assess a marker for autophagy (LC3), we performed immunocytochemistry on passage 5 (P5) cultures of wild-type and HdhΔQ/ΔQ primary mouse embryonic fibroblasts (PMEFs) (Figure S4). P5 cultures of wild-type fibroblasts are actively dividing, while P5 cultures of HdhΔQ/ΔQ fibroblasts are, in contrast, undergoing replicative senescence [16]. Wild-type - and ΔQ-htt were detected in both the cytoplasm and nucleus, and perinuclear localization of wild-type - and ΔQ-htt with the ER marker, calnexin was also detected in both Hdh+/+ and HdhΔQ/ΔQ PMEFs (Figure S4A, S4B). However, nuclear localization of htt appeared to be increased in those cells with a more senescent morphology (i.e. more flattened/spread appearance). Perinuclear LC3 immunoreactivity was also enhanced in the HdhΔQ/ΔQ PMEFs with a senescent morphology (Figure S4C), suggesting the possibility for increased autophagy in those HdhΔQ/ΔQ PMEFs undergoing replicative senescence.
Expression of ΔQ-htt enhances autophagosome synthesis in vitro
An alteration to autophagy resulting in the increased steady-state levels of LC3-II can be attributed to either enhanced autophagic flux, or to a block in a later step within the pathway that would interfere with the turnover of LC3-II in the autolysosome [39]. To determine if ΔQ-htt can enhance autophagosome synthesis, we transfected SK-N-SH neuroblastoma cells with full-length wild-type (7Q-htt) or ΔQ-htt cDNA expression constructs (diagrams in Figure S5), and monitored the levels of LC3-II 24 h post-transfection by western blotting (Figure 6A). The levels of LC3-II were increased significantly in the ΔQ-htt transfected cells in comparison to either control vector - or 7Q-htt-transfected cells. To monitor autophagy by an alternative method, we also transfected HeLa cells with an EGFP-LC3 expression construct, together with pCDNA3.1 (vector control), 7Q-htt or ΔQ-htt in a 1∶3 ratio (Figure S6). The proportion of EGFP-positive cells with >10 EGFP-LC3 vesicles was assessed and expressed as an odds ratio with 95% confidence limits. ΔQ-htt transfection, but not 7Q-htt transfection, increased the proportion of cells with EGFP-LC3 vesicles. To measure autophagosome synthesis, the cDNA constructs were also transfected in the presence or absence of the antibiotic bafilomycin A1, a vacuolar H+ ATPase inhibitor that suppresses turnover of LC3-II in autolysosomes [40]–[42]. Thus, measuring the levels of LC3-II in the presence of bafilomycin A1 measures LC3-II formation, as the antibiotic blocks LC3-II degradation. The levels of LC3-II were increased significantly in the bafilomycin A1-treated and ΔQ-htt-transfected cells in comparison to the bafilomycin A1 - treated cells alone or the bafilomycin A1-treated and 7Q-htt-transfected cells, suggesting that ΔQ - but not 7Q-htt expression results in increased autophagosome synthesis (Figure 6B). To confirm that an increase in LC3-II formation resulting from ΔQ-htt expression is enhancing autophagic activity that can remove another autophagy substrate, 7Q - or ΔQ-htt constructs were transfected into Atg5+/+ (autophagy-competent) and Atg5−/− (autophagy-deficient) mouse embryonic fibroblasts [43], together with an EGFP-tagged 74Q-htt exon 1 construct (EGFP-HDQ74) expressing an N-terminal fragment of mutant htt that forms aggregates readily in vitro [44]. Aggregate formation in EGFP-positive cells 48 h post-transfection was assessed by calculating odds ratios with 95% confidence limits [44]–[46] (Figure 6C). The proportion of cells with EGFP-HDQ74 aggregates was significantly reduced in Atg5+/+ cells transfected with ΔQ-htt, but not in Atg5−/− cells. Interestingly, 7Q-htt overexpression also reduced aggregate load in both Atg5+/+ and in Atg5−/− cells. These data suggest that while ΔQ-htt can induce autophagic clearance of mutant htt aggregates, 7Q-htt overexpression may induce a reduction in aggregate numbers or formation via an autophagy-independent mechanism in our in vitro system. Taken altogether, these data support the hypothesis that ΔQ-htt expression can stimulate autophagosome formation and the Atg5-dependent clearance of htt aggregates.

Importantly, we saw no difference in autophagy in 7Q-htt-overexpressing cells versus empty vector transfected cells (Figure 6A and 6B), or when comparing huntingtin knockout (Hdhex4/5/Hdhex4/5 [47]) mouse embryonic stem cells (Hdh−/−) which were either transfected with empty vector of with wild-type full-length 17Q-Htt (Figure 6D), suggesting that the ability of htt to induce autophagy is a specific consequence of the loss of its polyQ tract.
ΔQ-htt expression stimulates autophagy via an mTOR–independent pathway
A central regulator of metabolism and autophagy in both invertebrates and vertebrates is TOR (Target of Rapamycin) kinase, and inhibition of TOR kinase activity by rapamycin and its analogs has been used successfully to stimulate autophagic clearance of mutant htt aggregates in both Drosophila and mouse models for HD [48]. To determine if the activity of mammalian TOR (mTOR) is inhibited by ΔQ-htt expression, we examined the phosphorylation status of mTOR in the striatum of two year old wild-type, HdhΔQ/+, Hdh140Q/+, Hdh140Q/140Q, and Hdh140Q/ΔQ mice, and also the phosphorylation status of downstream targets of mTOR in our in vitro system (Figure 7). Phospho-mTOR (p-mTOR) levels correlate positively with mTOR kinase activity and inversely with mTOR inhibition and the activation of macroautophagy [48], although autophagy can also be regulated by mTOR-independent pathways. We observed no difference in p-mTOR levels in the supernatant fractions of all genotypes examined (Figure 7A). However, we did detect an enrichment of p-mTOR in the striatal pellet fractions from the Hdh140Q/+ and Hdh140Q/140Q brains. This association of p-mTOR with the pellet fraction likely represents p-mTOR association with htt aggregates, as was observed previously both in vitro, and in a transgenic HD mouse model [48].

To confirm our in vivo analyses, SK-N-SH cells were transfected with either 7Q - or ΔQ-htt expression constructs and the phosphorylation status of two targets of mTOR kinase activity were assessed 24 h post-transfection (Figure 7B). The levels of phospho-S6 kinase and phospho-S6 ribosomal protein were not significantly different in the cells transfected with 7Q - or ΔQ-htt, supporting the hypothesis that ΔQ-htt's upregulation of autophagy is not mediated by a reduction in mTOR kinase activity.
ΔQ-htt expression extends lifespan in the mouse
Our observations suggest that expression of a version of htt lacking its normal stretch of polyQ can enhance autophagic clearance of neuropil mutant htt inclusions. During normal aging, misfolded and aggregated proteins accumulate due to an apparent decline in the function of lysosomal degradation pathways [49]. In Caenhorhabditis elegans, autophagy is an essential component in the mechanism that extends lifespan upon dietary restriction. Knockdown of essential autophagy genes, for example, shortens lifespan in C. elegans, and suppresses lifespan extension induced by dietary restriction, reduced mitochondrial function, and alterations in insulin/IGF-1 or TOR signaling [50]. Moreover, enhancing basal levels of autophagy in the nervous system of Drosophila by Atg8a overexpression increases both longevity and resistance to oxidative stress [51]. However, the ability of autophagy upregulation to extend mammalian lifespan has not previously been tested. To determine if ΔQ-htt expression has an effect on longevity in the absence of 140Q-htt expression, we assessed the lifespan of HdhΔQ/ΔQ mice in comparison to wild-type mice (n = 15 mice of each genotype). While the wild-type controls lived to 28+/−1.3 months (median age +/ − s.e.m.), the HdhΔQ/ΔQ mice lived to a median age of 33+/−1.1 months, representing an 18% extension of lifespan (log-rank test, c2 = 9.6, P<0.005) (Figure 8). The oldest HdhΔQ/ΔQ mouse survived for approximately 3.5 years in our colony, compared to 3 years for the oldest wild-type mouse.

Discussion
We provide evidence that expression of a version of mouse htt that lacks its short 7Q polyglutamine domain can stimulate the formation of autophagosomes in vitro and enhance the clearance of htt neuropil aggregates, ameliorate behavioral/motor phenotypes, and extend lifespan in a mouse model for HD. When expressed in homozygosity, ΔQ-htt can also significantly extend lifespan in the mouse. Recently, it was proposed that htt may participate directly in autophagy because of its structural similarity with mTOR, and also because it co-localizes partially with autophagosomes [38]. These results, and our data, do not address directly whether or not htt has a normal function involved in autophagy. However, our results do suggest that deletion of htt's short 7Q stretch can enhance basal autophagy, and are compatible with a hypothesis suggesting that htt's polyQ stretch may modulate a normal function for htt in this process. Expansion of htt's polyQ stretch beyond the pathogenic threshold may, in contrast, suppress such a function, and account for our observation that we did not observe autophagy induction in the Hdh140Q/+ brain. In this scenario, ΔQ-htt's gain-of-function in autophagy would be dominant to a potential loss-of-function in autophagy caused by the expansion of the polyQ stretch.
The rescue of motor and behavioral phenotypes in the Hdh140Q/ΔQ mice starting at 5–6 months of age correlates with a reduction in neuropil htt aggregates, and a normalization of lipofuscin levels. Neuropil aggregates are an early phenotypic feature of HD, and our results are compatible with a recent study demonstrating that clearance of cytoplasmic htt aggregates by the expression of an intrabody specific for aggregated htt can rescue motor and behavioral deficits in a transgenic mouse model for HD [52]. The reduced number of nuclear inclusions observed in the Hdh140Q/ΔQ brain at one year of age may be due to htt's ability to shuttle between the nucleus and cytoplasm [1]. The stimulation of autophagy in other HD mouse models, for example, can also reduce htt nuclear aggregate number [48]. The normalization of lipofuscin deposits in the Hdh140Q/ΔQ brain may be the consequence of reduced oxidative stress that is secondary to the reduction in mutant htt aggregate load. Although we had previously detected increased lipofuscin in the HdhΔQ/ΔQ brain [16], we observed similar levels of lipofuscin in the HdhΔQ/+ and wild-type brain. As lipofuscin is an end-product of autophagy, the increased lipofuscin accumulating in the HdhΔQ/ΔQ brain may represent the increased turnover of mitochondria due to the expression of two HdhΔQ alleles. In this case, increased lipofuscin accumulation could represent increased autophagic activity instead of increased oxidative stress.
The mitochondrial-lysosomal axis theory of postmitotic cellular ageing posits that during ageing, autophagic capacity decreases, mitochondrial turnover declines, and damaged mitochondria and protein aggregates accumulate [53]. Old or damaged mitochondria will produce less ATP and more superoxide radicals leading to increased oxidative stress. This causes a positive feedback loop resulting in further damage. There is accumulating evidence that enhanced autophagy correlates with increased longevity in C. elegans and Drosophila. In C. elegans, autophagy is an essential component in the mechanism that extends lifespan upon dietary restriction [50]. Knockdown of essential autophagy genes, for example, shortens lifespan in C. elegans, and suppresses lifespan extension induced by dietary restriction, reduced mitochondrial function, and alterations in insulin/IGF-1 or TOR signaling. In a complementary experiment, enhancing basal levels of autophagy in the nervous system of Drosophila by Atg8a overexpression increases both longevity and resistance to oxidative stress [51].
The 18% increase in HdhΔQ/ΔQ lifespan relative to wild-type controls is comparable to that observed with mouse mutations in insulin signaling pathways that result in increased longevity. Mice heterozygous for a knock-out of the insulin-like growth factor 1 receptor gene (Igf1r) exhibit a 26% increase in mean lifespan [54], while mice expressing a mutant insulin receptor gene in adipose tissue exhibit an 18% increase in lifespan [55]. Mice lacking expression of the insulin receptor substrate 1 gene (Irs1−/−) also live 18% longer than wild-type mice [56], and brain-specific knock-out of one Irs2 allele in the mouse results in an 18% extension of lifespan [57]. Our findings do not permit us to determine if increased lifespan in the HdhΔQ/ΔQ mice is the consequence of neuronal or global expression of ΔQ-htt. Neuronal-specific overexpression of Atg8a in Drosophila is sufficient to extend lifespan [51], but analogous experiments in mice have not yet been performed.
The effect of ΔQ-htt expression on HdhΔQ/ΔQ lifespan appears, at first, to be incompatible with the premature replicative senescence phenotype that we observed in HdhΔQ/ΔQ PMEFs cultured in vitro [16]. However, the detection of both increased senescence-associated (SA)-β-galactosidase staining [16],[58], and LC3 immunoreactivity (Figure S4C) in senescent PMEFs supports the data of Narita and colleagues suggesting that upregulating autophagy may facilitate the mitotic senescence transition in vitro [59]. In this scenario, ΔQ-htt expression may have opposite effects on cellular senescence and mammalian lifespan. A similar, apparently contradictory, response to mammalian SIRT1 expression (a homolog of the yeast Sir2 factor involved in extending replicative lifespan) has been described in PMEFs where absence of SIRT1 expression increases replicative lifespan [60]. Thus, in these examples, replicative lifespan in vitro may not always correlate positively with organismal lifespan.
Upregulation of autophagy has the potential to be a therapeutic strategy for Huntington's disease and related disorders. Although rapamycin and its analogs have proven to be very useful in stimulating increased clearance of N-terminal truncated mutant htt aggregates in various animal models for HD via a “pulsitile” upregulation of autophagy, our data suggest that tonic long-term autophagy upregulation via ΔQ-htt expression is not associated with overt side-effects. This genetic method for autophagy upregulation is apparently mTOR independent based on our inability to detect a significant decrease in soluble p-mTOR levels in vivo, and alterations in the phosphorylation status of downstream mTOR kinase targets in vitro. In this regard, we have identified a series of novel compounds that influence autophagy in an mTOR-independent fashion [45],[61]. Although further work is required to elucidate the pathway responsible for ΔQ-htt's affect on autophagy, our findings support the view that the development of both genetic and small molecule-based therapeutic strategies aimed at stimulating the autophagic clearance of aggregated protein may be of use in both the treatment of neurodegenerative disease, and in lifespan extension.
Methods
Generation of mice
Hdh+/+, HdhΔQ/+, Hdh140Q/+, and Hdh140Q/ΔQ mice were obtained from heterozygous intercrosses between HdhΔQ/+ and Hdh140Q/+ mice that were maintained in a mixed 129/Sv and C57BL6 background. HdhΔQ/ΔQ and Hdh140Q/140Q mice were obtained from HdhΔQ/+ and Hdh140Q/+ intercrosses, respectively. All protocols for animal use were approved by the Institutional Animal Care and Use Committee of the University of Virginia, and were in accordance with NIH guidelines. For routine genotyping, PCR was used to confirm the presence of the different Hdh alleles: ΔQ allele; ΔQ-for = 5′-GACGGGCCCAAGATGG-3′ and ΔQ-rev = 5′-GGCGGTGGAAACGACTT-3′ amplify a 226 bp product from only the ΔQ allele, while Epi-for = 5′-GCGTAGTGCCAGTAGGCTCCAAG-3′and Epi-rev = 5′-CTGAAACGACTTGAGCGACTCGAAAG-3′ flank the site of the FLAG epitope in the ΔQ allele and amplify either a 112 bp product from the wild-type allele or a 136 bp product from the ΔQ allele. 140Q allele; 140-for = 5′-CTGCACCGACCGTGAGTCC-3′and 140-rev = 5′-GAAGGCACTGGAGTCGTGAC-3′ flank a small intron-1 deletion created during the generation of the 140Q allele. A wild-type allele (or ΔQ allele) will generate a 235 bp product, while the 140Q allele will generate a 150 bp product. To verify that the mean CAG repeat length in the Hdh140Q allele was similar in the Hdh140Q/+ and Hdh140Q/ΔQ mice that were used for our analyses, the CAG repeat was amplified using CAG-1 = 5′-CTTCGAGTCCCTCAAGTCCTTC-3′and CAG-2 = 5′-GGTGGCGGCTGTTGCTGCTG-3′ (data not shown). These oligonucleotides are specific for the human sequence surrounding the CAG repeat in the Hdh140Q allele and will generate a ∼450 bp product using the Expand High Fidelity PCR system (Roche Molecular Diagnostics).
Motor and behavioral analyses
The accelerating rotarod test was performed at 1, 5, and 19 months of age (n = 6 mice of each genotype at 1 and 5 months of age, and n = 4 mice of each genotype at 19 months of age; all mice in the same cohort) as described [16]. At each time-point, there were 3 separate testing sessions of 5 days (3 trials per day) to control for environmental factors. The Barnes maze testing was performed at 5 months of age according to methods described previously (n = 5 mice of each genotype) [16]. There were 3 separate testing sessions of 9 days to control for environmental factors. Activity testing was performed at 6 and 20 months of age (separate cohorts) according to methods described previously, except that tests were performed between the hours of 7 pm - 6 am, as open field activity is dependent upon the resting state of the mouse, with more activity anticipated during nocturnal hours [25]. There were 3 separate testing sessions with a mix of genotypes in each session.
Immunohistochemical and immunocytochemical analyses
Rapidly frozen mouse brains were sectioned at 14 µm using a cryostat (Bright Instrument Co.). Sections were washed briefly in PBS, fixed for 10 min in 4% paraformaldehyde in 0.1M phosphate buffer pH 7.4 or in 4% paraformaldehyde for 10 min, followed by rinse in PBS and a second fixation step in 100% methanol for 15 min on ice (both conditions yielded identical results, data not shown). Sections were washed in PBS before blocking with 5% donkey serum, 0.1% Triton ×100, in PBS for 1 h at RT, and then incubated o/n at 4°C with primary antibody diluted in 5% donkey serum, 0.1% Triton ×100 in PBS. Primary antibodies used were: rabbit polyclonal LC3 (1∶100, Novus Biologicals), and mouse monoclonal MW8 (1∶70, Developmental Studies Hybridoma Bank). Following the primary antibody incubation, sections were washed in PBS three times and incubated with secondary antibody (donkey anti-mouse, rabbit or guinea pig-Cy3 or –FITC, Jackson Immunologicals) together with the fluorescent DNA stain To-Pro-3 iodide (Invitrogen) for 1 h at RT. Sections were then washed with PBS before treatment to suppress lipofuscin autofluorescence by incubating sections sequentially in 75% ethanol for 5 min, lipofuscin eliminator reagent (Chemicon/Millipore) for 5 min, and 5 min in 75% ethanol. Sections were then mounted with Vectashield (Vector Laboratory), and examined using a Nikon C1-confocal microscope.
For immunocytochemical analyses, PMEFs were seeded at a concentration of 1×104 cells/ml onto 4-well chamber slides (Nunc). Two days following plating, the cells were washed briefly two times with PBS, fixed in 4% paraformaldehyde in 0.1 M phosphate buffer pH 7.4 for 10 min at RT followed with a 10 min incubation in cooled 100% methanol on ice, and then washed three times for 5 min at RT in PBS. The cells were blocked in PBS containing 5% donkey serum, 0.1% Triton X-100, and donkey anti-mouse IgG FAB (1∶400) for 1 h at RT. The cells were then washed three times in PBS (10 min each wash at RT), and then blocked again in PBS containing 5% donkey serum, 0.1% Triton X-100 for 1.5 h at RT. The cells were incubated with primary antibody diluted in blocking solution for 2 h at RT, and then washed three times for 5 min at RT in PBS. The cells were then incubated with secondary antibody diluted in blocking solution, and then washed again three times for 5 min each at RT in PBS. Slides were then immersed in 70% ethanol for 5 min and then treated with 1 drop of autoflorescence eliminator reagent (Millipore/Chemicon) for an additional 5 min. Slides were coverslipped in aqueous mounting medium and imaged using an Olympus BX51 microscope equipped with an Olympus MagnaFire CCD camera. Primary antibodies used were: goat anti-Calnexin (C-20), Santa Cruz Biotechnology, 1∶200; mouse anti-LC3 (5F10), Nanotools, 1∶100; mouse anti-FLAG M2 (F3165), Sigma, 1∶100; and mouse anti-htt MAB2166, Millipore, 1∶100.
Quantification of htt aggregates and EGFP-LC3 vesicles
Tissue sections
Sections were imaged with a 60× objective using a Nikon C1 confocal microscope, and the numbers of protein aggregates were counted manually (blind to genotype) from 14 µm coronal and sagittal sections through the striatum. Neuropil and nuclear inclusions were counted separately. A z-stack was performed using the confocal microscope to confirm the presence of MW8-positive aggregates in both the cytoplasm and the nucleus (data not shown). Htt aggregates in the cortex from the same sections used to obtain the striatal htt aggregates counts were also quantified.
Cell culture analyses
The percentage of EGFP-positive cells with EGFP-HDQ74 aggregates was determined as previously described [44]–[46]. Analysis and acquisition of images were done with a Nikon Eclipse E600 fluorescence microscope (plan-apo 60×/1.4 oil immersion lens). Quantification of cells with EGFP-LC3 vesicles was performed as described previously [45].
Lipofuscin imaging and quantification
Lipofuscin accumulation analyses were performed using 14 µm fresh frozen brain sections. Sections were fixed for 15 min on ice in 100% methanol, washed in PBS, and then incubated with To-Pro-3 iodide (1∶10,000 dilution in PBS) for 1 h at RT. Sections were then washed in PBS and mounted using Vectashield. Confocal images were acquired in the green and red channels (lipofuscin has a broad autofluorescent emission spectrum from 500 nm to 650 nm). Yellow pixel areas corresponding to the lipofuscin deposits were quantified using ImagePro 4.5 (Media Cybernetics) software from 8 images of the ventral striatum or parietal cortex obtained from each brain (n = 4 brains of each genotype for each age analyzed).
Tissue fractionation
Dissected striata from an individual brain were homogenized on ice in 500 µl 50 mM Tris-HCl pH 8.5, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 0.5% NP-40 supplemented with 5 mM NaF, 1 mM Na3VO4, and a protease inhibitor mixture (Complete –EDTA tablets, Roche). The tissue homogenate (total or unfractionated sample) was then centrifuged for 10 min at 4°C at 16,100×g to obtain crude cytoplasmic (supernatant) and nuclear pellet fractions. The pellet was suspended by dounce homogenization in 100 µl homogenization buffer, and incubated with 0.2 mg/ml final concentration of DNAse I for 60 min on ice. The suspension was then centrifuged for 10 min at 4°C at 16,100×g to obtain the final pellet fraction. The pellet was resuspended in 100 µl homogenization buffer, and the protein concentration in the supernatant and pellet samples was determined using the BCA assay (Pierce). Typically, there was a 20-fold excess of protein recovered from the supernatant fraction relative to the pellet fraction. 30 µg of each fraction was analyzed by western blotting. Although some htt N-terminal fragments can be solubilized by SDS-PAGE sample buffer extraction of the pellet fraction, the majority of htt material in the pellet consists of aggregates (Figure S7). For the generation of a P1 low-speed pellet fraction (Figure S3A), striatal tissue from each brain was dounce-homogenized on ice in 500 µl 15 mM Tris-HCl pH 7.6, 0.25 M sucrose, 1 mM MgCl2, 2.5 mM EDTA, 1 mM EGTA, 1 mM DTT, 5 mM NaF, 1 mM Na3VO4, and a protease inhibitor mixture (Complete –EDTA tablets, Roche), and then centrifuged for 10 min at 4°C at 800×g to obtain a crude cytoplasmic supernatant fraction and a P1 pellet fraction containing nuclei and dense secondary lysosomes.
Plasmids
The EGFP-HDQ74 construct expressing a truncated htt exon 1 fragment with 74Q fused to an EGFP reporter was described previously [44]. The EGFP-LC3 expression plasmid was a gift from T. Yoshimori, while the full-length 17Q-Htt construct was a gift from M. R. Hayden (described in [62]). The full-length 7Q-htt and ΔQ-htt expression constructs (Figure S5) were assembled from a genomic fragment containing mouse exon 1 with a portion of the flanking intron 1, a portion of the full-length mouse htt cDNA extending from exon 2 through exon 67 including a synthetic 3′splice acceptor site, and a poly(A) addition sequence from the bovine growth hormone gene. The mouse exon 1 fragments contained either wild-type sequences encoding the 7Q stretch, or sequences derived from our HdhΔQ targeting construct lacking the polyQ stretch. A 3×FLAG epitope tag was also inserted at the htt N-terminus between amino acids 1 and 2. A phosphoglycerol kinase (pgk) gene promoter was used to drive expression of the 7Q - and ΔQ-htt constructs in the transfected cells.
Cell culture and transfection
SK-N-SH cells, wild-type Atg5 (Atg5+/+), and Atg5-deficient (Atg5−/−), HeLa cells, wild-type P5, and HdhΔQ/ΔQ P5 mouse embryonic fibroblasts (MEFs) were maintained in DMEM (D6546, Sigma) supplemented with 10% FBS, 100 U/ml penicillin/streptomycin and 2 mM L-glutamine (Sigma) in a 37°C, 5% CO2 humidified incubator. Hdhex4/5/Hdhex4/5 knock-out (Hdh−/−) mouse ES cells were cultured on 0.1% gelatine coated tissue culture flasks in DMEM (D6546, Sigma) supplemented with 15% FBS, 1× L-Glutamine, 1× penicillin/streptomycin, 1× essential amino acids, 3.5 ml (per 500 ml media) 2-mercaptoethanol (Sigma) and 1000 U/ml ESGRO (ESG1107 with LIF, Chemicon/Millipore), and incubated in a 37°C, 5% CO2 humidified incubator. Cells were transfected with DNA constructs for 4 h using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol, and either processed for western blotting analysis 24 h post-transfection by harvesting the cells and lysing the cell pellet on ice for 30 min in SDS-PAGE sample buffer (62.5 mM Tris-HCl pH 6.8, 2% SDS, 5% β-mercaptoethanol, 10% glycerol, 0.01% bromophenol blue) or fixed with 4% paraformaldehyde (Sigma) 48 h post-transfection and mounted with ProLong Gold antifade reagent containing 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) for aggregation analysis.
Western blotting
Samples (30 µg unless otherwise noted) were fractionated on SDS-PAGE, and then transferred electrophoretically onto 0.45 µm PVDF membranes (Invitrogen). Membranes were processed for western blotting using standard procedures. Antibody dilutions used were: rabbit polyclonal beclin 1 (H300; 1∶100, Santa Cruz Biotechnology; 1∶250), LC3 (1∶2,000 to 1∶5,000), guinea pig polyclonal p62/SQSTM1 (American Research Products; 1∶1,000), MW8 (1∶1,000), ubiquitin (DakoCytomation; 1∶1000), 1C2 (Chemicon/Millipore; 1∶5,000), anti-htt MAB2166 (Chemicon/Millipore; 1∶5000), β-actin (MP Biomedicals; 1∶50,000), and the following antibodies from Cell Signaling Technology used at 1∶1,000 dilution: rabbit monoclonal anti-Lamp1 (3243), rabbit anti-p70 S6 kinase (9202), rabbit anti-phospho-p70 S6 kinase (Thr389) (9205), rabbit anti-S6 ribosomal protein (2217), and rabbit anti-phospho-S6 ribosomal protein (Ser235/236) (2211). Blots were incubated 5 min in chemiluminescence reagent (SuperSignal West Dura, Pierce or an ECL detection kit, G.E. Healthcare) prior to film exposure. For densitometry, films in the linear exposure range were scanned on a flatbed scanner, and analyzed using the Image J program (Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, MD, USA, http://rsb.info.nih.gov/ij/, 1997–2005). Levels of protein in each sample were normalized to actin, and the levels in the wild-type samples, with the exception of the mTOR/p-mTOR blots which were normalized to the band intensity of an abundant high-molecular weight protein visible on the blots after staining with Ponceau S.
Statistical analyses
For behavioral tests, data was analyzed using the SigmaStat program (Systat Software). One-way ANOVA, two-way repeated measures ANOVA (with Bonferroni or Holm-Sidak post-hoc tests), and unpaired Student t-tests were used to analyze data. Significance was accepted at P<0.05. Mantel-Cox log rank tests on the Kaplan-Meier survival data were performed using SPSS 16.0.1 (SPSS Inc.). For quantification of htt aggregate number, lipofuscin deposit area, and levels of autophagy markers in subcellular fractions, Student t-tests were used. For analysis of LC3-II, S6K, and S6P levels in the in vitro cell culture experiments, a factorial ANOVA test using STATVIEW v4.53 (Abacus Concepts) was performed on the densitometric data, where the control condition was set to 100%. Error bars denote s.e.m. Pooled estimates for the changes in EGFP-HDQ74 aggregate formation resulting from perturbations assessed in multiple experiments, and the quantification of EGFP-LC3-positive vesicle numbers, were calculated as odds ratios with 95% confidence intervals. Odds ratios and P values were determined by unconditional logistical regression analysis, using the general log-linear analysis option of SPSS 9 software (SPSS Inc.), as previously described [42], [44]–[46],[61]. Experiments were performed in triplicate at least twice. ***, P< 0.001; **, P<0.01; *, P<0.05.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. AtwalRS
XiaJ
PinchevD
TaylorJ
EpandRM
2007 Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum Mol Genet 16 2600 2615
2. FaberPW
BarnesGT
SrinidhiJ
ChenJ
GusellaJF
1998 Huntingtin interacts with a family of WW domain proteins. Hum Mol Genet 7 1463 1474
3. GaoYG
YanXZ
SongAX
ChangYG
GaoXC
2006 Structural Insights into the specific binding of huntingtin proline-rich region with the SH3 and WW domains. Structure 14 1755 1765
4. LiuYF
DethRC
DevysD
1997 SH3 domain-dependent association of huntingtin with epidermal growth factor receptor signaling complexes. J Biol Chem 272 8121 8124
5. The Huntington's Disease Collaborative Research Group 1993 A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72 971 983
6. DiFigliaM
SappE
ChaseKO
DaviesSW
BatesGP
1997 Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277 1990 1993
7. MyersRH
VonsattelJP
StevensTJ
CupplesLA
RichardsonEP
1988 Clinical and neuropathologic assessment of severity in Huntington's disease. Neurology 38 341 347
8. de la MonteSM
VonsattelJP
RichardsonEPJr
1988 Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington's disease. J Neuropathol Exp Neurol 47 516 525
9. VonsattelJP
MyersRH
StevensTJ
FerranteRJ
BirdED
1985 Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44 559 577
10. CattaneoE
ZuccatoC
TartariM
2005 Normal huntingtin function: an alternative approach to Huntington's disease. Nature Rev Neurosci 6 919 930
11. BaxendaleS
AbdullaS
ElgarG
BuckD
BerksM
1995 Comparative sequence analysis of the human and pufferfish Huntington's disease genes. Nature Genet 10 67 76
12. HarjesP
WankerEE
2003 The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci 28 425 433
13. KarlovichCA
JohnRM
RamirezL
StainierDY
MyersRM
1998 Characterization of the Huntington's disease (HD) gene homologue in the zebrafish Danio rerio. Gene 217 117 125
14. TartariM
GissiC
Lo SardoV
ZuccatoC
PicardiE
2008 Phylogenetic comparison of huntingtin homologues reveals the appearance of a primitive polyQ in sea urchin. Mol Biol Evol 25 330 338
15. SeongIS
IvanovaE
LeeJM
ChooYS
FossaleE
2005 HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum Mol Genet 14 2871 2880
16. ClaboughEB
ZeitlinSO
2006 Deletion of the triplet repeat encoding polyglutamine within the mouse Huntington's disease gene results in subtle behavioral/motor phenotypes in vivo and elevated levels of ATP with cellular senescence in vitro. Hum Mol Genet 15 607 623
17. SarkarS
RubinszteinDC
2008 Huntington's disease: degradation of mutant huntingtin by autophagy. FEBS J 275 4263 4270
18. MizushimaN
2007 Autophagy: process and function. Genes Dev 21 2861 2873
19. FilimonenkoM
StuffersS
RaiborgC
YamamotoA
MalerodL
2007 Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179 485 500
20. YamamotoA
CremonaML
RothmanJE
2006 Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol 172 719 731
21. RubinszteinDC
GestwickiJE
MurphyLO
KlionskyDJ
2007 Potential therapeutic applications of autophagy. Nat Rev Drug Discov 6 304 312
22. SarkarS
RavikumarB
FlotoRA
RubinszteinDC
2009 Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 16 46 56
23. JeongH
ThenF
MeliaTJJr
MazzulliJR
CuiL
2009 Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 137 60 72
24. KabeyaY
MizushimaN
UenoT
YamamotoA
KirisakoT
2000 LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19 5720 5728
25. MenalledLB
SisonJD
DragatsisI
ZeitlinS
ChesseletMF
2003 Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol 465 11 26
26. BarnesCA
1979 Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol 93 74 104
27. KoJ
OuS
PattersonPH
2001 New anti-huntingtin monoclonal antibodies: implications for huntingtin conformation and its binding proteins. Brain Res Bull 56 319 329
28. GoebelHH
HeipertzR
ScholzW
IqbalK
Tellez-NagelI
1978 Juvenile Huntington chorea: clinical, ultrastructural, and biochemical studies. Neurology 28 23 31
29. Tellez-NagelI
JohnsonAB
TerryRD
1974 Studies on brain biopsies of patients with Huntington's chorea. J Neuropathol Exp Neurol 33 308 332
30. DaviesSW
TurmaineM
CozensBA
RazaAS
MahalA
1999 From neuronal inclusions to neurodegeneration: neuropathological investigation of a transgenic mouse model of Huntington's disease. Philos Trans R Soc Lond B Biol Sci 354 981 989
31. UchidaK
2006 Lipofuscin-like fluorophores originated from malondialdehyde. Free Radic Res 40 1335 1338
32. GrayDA
WoulfeJ
2005 Lipofuscin and aging: a matter of toxic waste. Sci Aging Knowledge Environ 2005 re1
33. KellerJN
DimayugaE
ChenQ
ThorpeJ
GeeJ
2004 Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int J Biochem Cell Biol 36 2376 2391
34. CuervoAM
BergaminiE
BrunkUT
DrogeW
FfrenchM
2005 Autophagy and aging: the importance of maintaining “clean” cells. Autophagy 1 131 140
35. GerstbreinB
StamatasG
KolliasN
DriscollM
2005 In vivo spectrofluorimetry reveals endogenous biomarkers that report healthspan and dietary restriction in Caenorhabditis elegans. Aging Cell 4 127 137
36. RubinszteinDC
CuervoAM
RavikumarB
SarkarS
KorolchukV
2009 In search of an “autophagomometer”. Autophagy 5 585 589
37. LiuCL
ChenS
DietrichD
HuBR
2008 Changes in autophagy after traumatic brain injury. J Cereb Blood Flow Metab 28 674 683
38. AtwalRS
TruantR
2008 A stress sensitive ER membrane-association domain in Huntingtin protein defines a potential role for Huntingtin in the regulation of autophagy. Autophagy 4 91 93
39. MizushimaN
YoshimoriT
2007 How to interpret LC3 immunoblotting. Autophagy 3 542 545
40. YamamotoA
TagawaY
YoshimoriT
MoriyamaY
MasakiR
1998 Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 23 33 42
41. FassE
ShvetsE
DeganiI
HirschbergK
ElazarZ
2006 Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem 281 36303 36316
42. SarkarS
RavikumarB
RubinszteinDC
2009 Autophagic clearance of aggregate-prone proteins associated with neurodegeneration. Methods Enzymol 453 83 110
43. KumaA
HatanoM
MatsuiM
YamamotoA
NakayaH
2004 The role of autophagy during the early neonatal starvation period. Nature 432 1032 1036
44. NarainY
WyttenbachA
RankinJ
FurlongRA
RubinszteinDC
1999 A molecular investigation of true dominance in Huntington's disease. J Med Genet 36 739 746
45. SarkarS
PerlsteinEO
ImarisioS
PineauS
CordenierA
2007 Small molecules enhance autophagy and reduce toxicity in Huntington's disease models. Nature Chem Biol 3 331 338
46. RavikumarB
DudenR
RubinszteinDC
2002 Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 11 1107 1117
47. DuyaoMP
AuerbachAB
RyanA
PersichettiF
BarnesGT
1995 Inactivation of the mouse Huntington's disease gene homolog Hdh. Science 269 407 410
48. RavikumarB
VacherC
BergerZ
DaviesJE
LuoS
2004 Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nature Genet 36 585 595
49. CuervoAM
DiceJF
2000 When lysosomes get old. Exp Gerontol 35 119 131
50. HansenM
ChandraA
MiticLL
OnkenB
DriscollM
2008 A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet 4 e24 doi:10.1371/journal.pgen.0040024
51. SimonsenA
CummingRC
BrechA
IsaksonP
SchubertDR
2008 Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4 176 184
52. WangCE
ZhouH
McGuireJR
CerulloV
LeeB
2008 Suppression of neuropil aggregates and neurological symptoms by an intracellular antibody implicates the cytoplasmic toxicity of mutant huntingtin. J Cell Biol 181 803 816
53. TermanA
GustafssonB
BrunkUT
2006 The lysosomal-mitochondrial axis theory of postmitotic aging and cell death. Chem Biol Interact 163 29 37
54. HolzenbergerM
DupontJ
DucosB
LeneuveP
GeloenA
2003 IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421 182 187
55. BluherM
KahnBB
KahnCR
2003 Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 299 572 574
56. SelmanC
LingardS
ChoudhuryAI
BatterhamRL
ClaretM
2008 Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J 22 807 818
57. TaguchiA
WartschowLM
WhiteMF
2007 Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science 317 369 372
58. GerlandLM
PeyrolS
LallemandC
BrancheR
MagaudJP
2003 Association of increased autophagic inclusions labeled for beta-galactosidase with fibroblastic aging. Exp Gerontol 38 887 895
59. YoungAR
NaritaM
FerreiraM
KirschnerK
SadaieM
2009 Autophagy mediates the mitotic senescence transition. Genes Dev 23 798 803
60. ChuaKF
MostoslavskyR
LombardDB
PangWW
SaitoS
2005 Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab 2 67 76
61. WilliamsA
SarkarS
CuddonP
TtofiEK
SaikiS
2008 Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nature Chem Biol 4 295 305
62. LuoS
VacherC
DaviesJE
RubinszteinDC
2005 Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J Cell Biol 169 647 656
63. BjorkoyG
LamarkT
BrechA
OutzenH
PeranderM
2005 p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171 603 614
64. WeissA
KleinC
WoodmanB
SathasivamK
BibelM
2008 Sensitive biochemical aggregate detection reveals aggregation onset before symptom development in cellular and murine models of Huntington's disease. J Neurochem 104 846 858
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 2
Nejčtenější v tomto čísle
- Genome-Wide Association Study in Asian Populations Identifies Variants in and Associated with Systemic Lupus Erythematosus
- Nucleoporins and Transcription: New Connections, New Questions
- Nuclear Pore Proteins Nup153 and Megator Define Transcriptionally Active Regions in the Genome
- The Genetic Interpretation of Area under the ROC Curve in Genomic Profiling