
Charcot-Marie-Tooth–Linked Mutant GARS Is Toxic to Peripheral Neurons Independent of Wild-Type GARS Levels
Charcot-Marie-Tooth disease type 2D (CMT2D) is a dominantly inherited peripheral neuropathy caused by missense mutations in the glycyl-tRNA synthetase gene (GARS). In addition to GARS, mutations in three other tRNA synthetase genes cause similar neuropathies, although the underlying mechanisms are not fully understood. To address this, we generated transgenic mice that ubiquitously over-express wild-type GARS and crossed them to two dominant mouse models of CMT2D to distinguish loss-of-function and gain-of-function mechanisms. Over-expression of wild-type GARS does not improve the neuropathy phenotype in heterozygous Gars mutant mice, as determined by histological, functional, and behavioral tests. Transgenic GARS is able to rescue a pathological point mutation as a homozygote or in complementation tests with a Gars null allele, demonstrating the functionality of the transgene and revealing a recessive loss-of-function component of the point mutation. Missense mutations as transgene-rescued homozygotes or compound heterozygotes have a more severe neuropathy than heterozygotes, indicating that increased dosage of the disease-causing alleles results in a more severe neurological phenotype, even in the presence of a wild-type transgene. We conclude that, although missense mutations of Gars may cause some loss of function, the dominant neuropathy phenotype observed in mice is caused by a dose-dependent gain of function that is not mitigated by over-expression of functional wild-type protein.
Published in the journal:
. PLoS Genet 7(12): e32767. doi:10.1371/journal.pgen.1002399
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002399
Summary
Charcot-Marie-Tooth disease type 2D (CMT2D) is a dominantly inherited peripheral neuropathy caused by missense mutations in the glycyl-tRNA synthetase gene (GARS). In addition to GARS, mutations in three other tRNA synthetase genes cause similar neuropathies, although the underlying mechanisms are not fully understood. To address this, we generated transgenic mice that ubiquitously over-express wild-type GARS and crossed them to two dominant mouse models of CMT2D to distinguish loss-of-function and gain-of-function mechanisms. Over-expression of wild-type GARS does not improve the neuropathy phenotype in heterozygous Gars mutant mice, as determined by histological, functional, and behavioral tests. Transgenic GARS is able to rescue a pathological point mutation as a homozygote or in complementation tests with a Gars null allele, demonstrating the functionality of the transgene and revealing a recessive loss-of-function component of the point mutation. Missense mutations as transgene-rescued homozygotes or compound heterozygotes have a more severe neuropathy than heterozygotes, indicating that increased dosage of the disease-causing alleles results in a more severe neurological phenotype, even in the presence of a wild-type transgene. We conclude that, although missense mutations of Gars may cause some loss of function, the dominant neuropathy phenotype observed in mice is caused by a dose-dependent gain of function that is not mitigated by over-expression of functional wild-type protein.
Introduction
Charcot-Marie-Tooth disease (CMT) is a heterogeneous group of inherited neuropathies affecting approximately 1 in 2,500 people [1]. CMT is divided into type 1 forms, characterized by demyelination and decreased nerve conduction velocity, type 2 or axonal forms, characterized by axon loss and reduced evoked potential amplitudes, and intermediate forms having features of both demyelinating and axonal neuropathy [2], [3].
As the genetic causes of CMT have been identified, shared mechanisms have been elucidated that help explain the myelin degeneration in CMT type 1 and axon degeneration in CMT type 2. Disruption of myelin structure through mutation of proteins produced by peripheral Schwann cells is a recurring mechanism in type 1 CMTs [4]–[6]. Defects associated with vesicle trafficking [7], [8], mitochondrial morphology [9]–[11], and cytoskeletal integrity [12], [13] have each been implicated in multiple axonal forms of CMT.
Other axonal and intermediate CMTs have been linked to mutations in tRNA synthetase genes. The first to be identified and best characterized is CMT type 2D, caused by mutations in the glycyl-tRNA synthetase gene (GARS, MIM ID 601472) [14]. Subsequently, mutations in tyrosyl-tRNA synthetase (YARS, MIM ID 608323) were identified in patients with dominant intermediate CMTC [15]; a single missense change in the alanyl-tRNA synthetase gene (AARS, MIM ID 613287) was associated with axonal CMT2N [16]; and most recently compound heterozygous frame shift/missense mutations in the lysyl-tRNA synthetase gene (KARS, MIM ID 613641) were reported in a patient with recessive intermediate CMTB [17]. While human genetic studies have implicated mutations in multiple tRNA synthetase genes, the pathogenic mechanism for these neuropathies is still unclear, although a shared mechanism is an attractive hypothesis [18]. In particular, it is unclear whether the mutations result in a pathological gain of function, a partial loss of activity related to translation, an impact on an unknown, noncanonical activity, or a combination of these mechanisms.
tRNA synthetases covalently link tRNAs with their cognate amino acids to translate the genetic code. These enzymes serve an essential, nonredundant role in protein synthesis. It has been suggested that a reduction of this tRNA-charging function could result in neuropathy, and peripheral nerves with long, large-diameter axons may be especially vulnerable to decreased activity because of their unusual transport and metabolic demands [15], [19]. Consistent with this, several disease-causing mutations in GARS are associated with a reduction in charging function in cell-free assays, and this is roughly correlated with a decreased ability to rescue nonviability in yeast caused by deficiency of GRS1, the ortholog of GARS. However other mutations do not impair this function [19]–[21].
Structural studies have shown that many of the mutations alter dimer association, affecting homodimer formation that is essential for tRNA-charging activity [20], [22]. Furthermore, all mutations tested alter the subcellular distribution of GARS in transfected cells, also suggesting a possible loss of function at the cellular level through mislocalization, even if charging activity is preserved [19]–[21], [23], [24].
Two mouse models of CMT2D that share pathological features with the human disease, with differing severity, are caused by dominant amino acid substitutions in Gars. The GarsNmf249 allele (hereafter abbreviated Nmf249) causes reduced body weight and impaired mobility in heterozygous mice [25]. Axon number and neuromuscular junction (NMJ) morphology are normal at post-natal day 7, but subsequently axons are lost without a reduction in myelin thickness; NMJs show partial and sometimes complete denervation. The Nmf249 allele is an insertion in the Gars gene that substitutes lysine and tyrosine for proline at position 278 in the mouse GARS protein, equivalent to a P234KY change in human GARS (Note: numbering differences are because the human annotation does not consider the N-terminal mitochondrial localization signal appended through alternative start codon usage).
The GarsC201R allele (hereafter abbreviated C201R) is less severe and was identified in a chemical mutagenesis screen. In addition to impaired grip strength, these mice have impaired motor control, diminished muscle force, reduced weight, a shift towards smaller axon diameters, and some muscle denervation [26]. The mouse C201R substitution is equivalent to C157R in human GARS. These two alleles demonstrate the spectrum of phenotypic severity in mice and provide a range of tests that can be used to quantify the effects on neuromuscular function.
Previous studies in mice are consistent with either a pathological gain-of-function for the mutant protein, or a partial loss-of-function, or both. Mouse and human mutations both cause dominantly inherited neuropathy. Moreover, mice heterozygous for a gene trap allele (GarsXM256/+, a presumed null allele, hereafter abbreviated XM256) do not have a phenotype, arguing against a simple loss-of-function and ruling out haploinsufficiency as a cause of the neuropathy phenotype [25]. As expected for a gene with such a critical activity, homozygous gene trap mice die as embryos. Mice homozygous for the C201R allele are more severely affected than heterozygotes of either mutant allele and die at about two weeks of age [26]. This increased severity may be the result of doubling a pathological gain of function associated with the C201R protein, or the inability of the mutant protein to actively perform its normal function, or a combination of these effects. Consistent with the inability of the mutant protein to sustain its normal function, both the C201R and the Nmf249 mutations fail to complement the loss-of-function XM256 allele, resulting in embryonic death in the absence of wild-type protein, without an increase in the genetic dosage of the mutant allele [25], [26]. The embryonic death of these mice may indicate that wild-type GARS is mitigating a toxic gain of function, or that the mutant proteins do not retain their normal function, or it may indicate a combination of these mechanisms.
We hypothesized that a partial loss-of-function could be corrected by transgenic over-expression of a functional wild-type GARS protein, whereas a pathological gain-of-function could be independent of wild-type activity and therefore would not necessarily be corrected by increased wild-type expression. In order to determine whether increased wild-type GARS rescues the Gars mutant mice, we crossed transgenic mice that over-express the wild-type human GARS gene to both the Nmf249/+ and C201R/+ mice, and analyzed the effects on the phenotype. We found a recessive reduction in perinatal viability in C201R as a homozygote or in combination with the XM256 gene trap allele, which could be rescued by wild-type GARS over-expression. However, wild-type over-expression did not substantially mitigate the neuropathy phenotype in heterozygous Nmf249/+ or C201R/+ mice, consistent with a predominant gain of function as the cause of peripheral neuropathy in these mice.
Results
Transgenic mice over-expressing human GARS
Wild-type human GARS was expressed in mice using a transgene construct with the full-length human GARS open reading frame under the control of a CAG promoter [27], which is a fusion of the CMV early enhancer and the chicken β actin promoter and leads to strong ubiquitous transcription of downstream elements (Figure 1A). Two independent transgene founder strains, designated Transgene A (TgA) and Transgene D (TgD), were identified and used in this study. These two lines over-express GARS in spinal cord and sciatic nerve at similar levels (>10 times endogenous levels), as measured by immunoblotting (Figure 1B).

Transgenic GARS is expressed in myelinating Schwann cells and sciatic nerve axons
In order to examine in situ expression of the transgenic protein in relevant cell types, we excised and fixed the sciatic nerve and teased the fibers apart to improve antibody penetration and to allow visualization of individual axons.
Endogenous GARS is present at low levels in the sciatic nerve (Figure 1C and [21]). In exposure-matched confocal images, anti-GARS staining was increased in animals with either TgA or TgD over the endogenous levels in strain matched FVB/N control mice without the transgene. The pattern of GARS immunoreactivity in transgenic mice was also similar to that of wild type mice, with staining found in both the axon and Schwann cells, suggesting the transgenic protein localizes normally (Figure 1C). Therefore, the transgenes produce protein that is present in Schwann cells and transported to peripheral axons, as indicated by overlapping neurofilament staining.
Over-expression of wild-type GARS does not reduce axon loss in Nmf249/+ mice
To examine the effect of over-expressing wild-type GARS on the neuropathy phenotype, the more severe Nmf249/+ mice were crossed with hemizygous transgenic mice and the four resulting F1 genotypes were analyzed: wild type, WT; Tg, Nmf249/+, and Nmf249/+; Tg. The mice were analyzed at post-natal day 30 (P30). Mice with the transgene had much stronger levels of GARS protein expression than non-transgenic littermates in spinal cord and sciatic nerve on immunoblots of animals with both wild-type and Nmf249/+ backgrounds (Figure 1D, 1E). Transgene expression was confirmed by immunoblot in mice with each genotype for crosses with both Nmf249/+ and C201R/+ (Figure S1). There was no difference overall in GARS levels between wild type and Nmf249/+ or C201R/+, however, both transgenic strains result in expression well above endogenous levels. Consistent with the increased GARS protein, aminoacylation activity was also increased, indicating the transgenic protein is enzymatically active. Homogenates from the spinal cord of wild type and WT;TgA and TgD mice were prepared and assayed for the activity of both GARS and alanyl tRNA synthetase (AARS) as an internal control. As anticipated, no increase in activity was seen for AARS (Figure 1F), whereas GARS activity was increased at least 10 fold (as judged by the initial rate of tRNA glycylation) in each transgenic strain (Figure 1G and data not shown).
To assess the effect of the transgenic GARS on peripheral neuropathy, we examined the femoral nerve, which consists of a primarily motor branch innervating the quadriceps, and a primarily sensory branch that becomes the saphenous nerve and innervates the skin of the lower leg. In Nmf249/+ mice, both branches had a reduction in myelinated axon number, whereas C201R/+ mice had normal axon numbers [25], [26]. We isolated motor and sensory branches of the femoral nerve from F1 progeny from our cross between Nmf249/+ mice and GARS wild-type transgenic mice at one month of age. The nerves were fixed and embedded, and semi-thin sections were stained with toluidine blue for quantification of axon number and size (Figure 2A–2L). In these cross sections, the size of the femoral nerves was reduced in the presence of the Nmf249/+ allele (Figure 2A–2F compared to Figure 2G–2L). This was quantified by counting myelinated axons, which showed on average a 26% reduction in axons in motor nerves with the Nmf249/+ allele compared to wild-type littermates. There was no significant improvement in the Nmf249/+ axon numbers in motor or sensory nerves in mice with TgA or TgD (Figure 2M).

We next examined the distribution of motor axon diameters to determine whether there was any improvement in motor nerve pathology that was not detected with the axon counts. A cumulative histogram of the frequency of observed axon diameters shows the distribution and range of axon diameters in this study. The plot shows a marked shift from large diameter axons (4–6 µm) to small diameter axons (0–3 µm) in Nmf249/+ mice (Figure 2N). Both TgA and TgD showed small but significant improvements in axon diameter in Nmf249/+ mice by a nested-ANOVA analysis (p = 0.006). There was no impact of either transgene on axon diameter in an otherwise wild-type background. With and without the transgene, the Nmf249 allele significantly reduced mean axon diameter, while the proportional observed decrease in myelin thickness is not as dramatic (Figure S2).
Nerve conduction and body weight are not improved by over-expression of wild-type GARS
Consistent with the slight improvement in axon diameters observed in Nmf249/+ mice with TgA and TgD (Figure 2N), there was trend towards improvement in nerve conduction velocity, but this was not significant. Nerve conduction in Nmf249/+ mice is significantly reduced to approximately 25% of wild-type velocities. Nmf249/+; TgA and Nmf249/+; TgD mice had NCVs that did not differ significantly from Nmf249/+ alone (Figure 3A).

Previous studies show that between 2 and 4 weeks of age, mice with the Nmf249/+ allele stop gaining weight and become significantly smaller than littermate controls [25]. Various factors are likely to contribute to the decreased weight. Denervation decreases muscle mass and probably compromises ability to feed. As a measure of overall health, we weighed the mice in this study. Consistent with measures of peripheral nerve function, mutant mice with TgD trended toward being heavier, but neither TgA nor TgD significantly improved the body weights of Nmf249/+ mice, and all mutant animals were still significantly lighter than littermate control mice (Figure 3B).
GARS over-expression does not improve neuromuscular junction occupancy in CMT2D mice
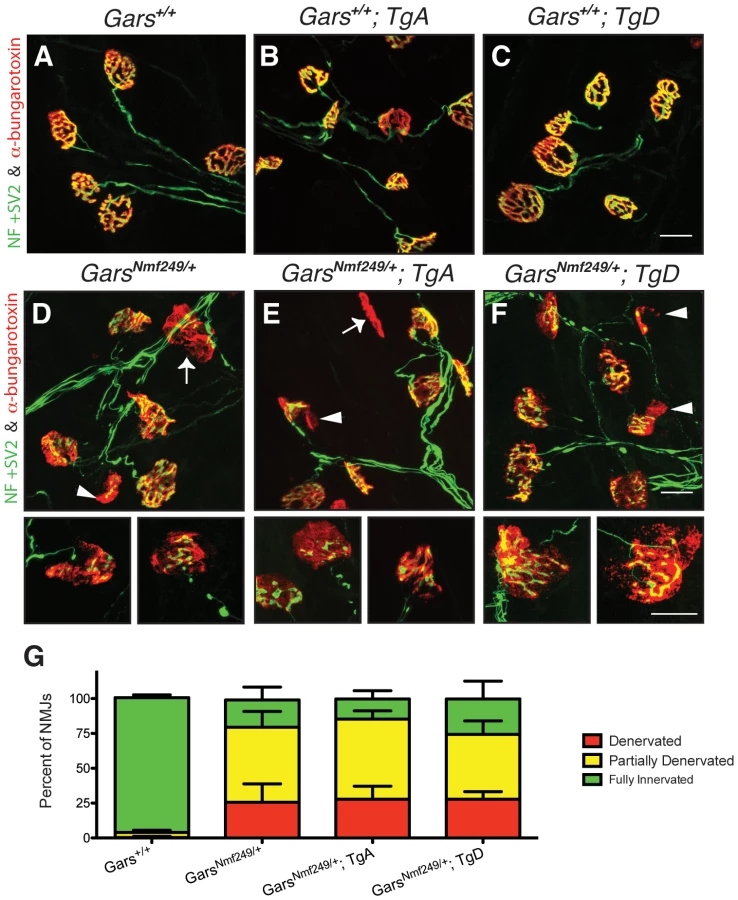
The loss of motor axons in peripheral nerves of Nmf249/+ mice also results in a disruption of the neuromuscular junction (NMJ). Mature, healthy motor neurons innervate muscle with pretzel-shaped presynaptic terminals. During development, post-synaptic regions mirror the shape of the axon terminal in regions of specialization where acetylcholine receptors concentrate to define the muscle side of the NMJ. The loss of distal motor axons is reflected in the NMJs of Nmf249/+ mice, which are severely dysmorphic. Many junctions in mutant mice are partially or completely denervated by post-natal day 30 [25]. To further study whether over-expression of GARS affects the motor phenotype in CMT2D mice, we looked at NMJ morphology and occupancy.
NMJs from wild-type mice with and without the transgene had normal “pretzel-like” morphology with full overlap between pre - and post-synaptic terminals (Figure 4A–4C). In mutant mice (Figure 4D), there are regions where the presynaptic markers are absent in areas where there is post-synaptic staining (arrowheads). There are also post-synaptic junctions that have no associated nerve terminal, indicating that the muscle fiber has been denervated (arrows). The morphology and occupancy of NMJs in Nmf249/+; TgA mice (Figure 4E) and Nmf249/+; TgD (Figure 4F) were not improved. In each animal, NMJs were scored based on the overlap between pre - and post-synaptic markers and classified as fully innervated, partially denervated, or fully denervated. This assessment of NMJ occupancy showed there was no significant improvement in mutant mice with transgenes over mutant mice without transgenes (Figure 4G). Therefore, with the exception of a slight increase in axon diameters, the over-expression of wild type GARS as a transgene did not lead to significant improvements in any phenotypic measure in the Nmf249/+ mice.
Neither wild-type transgene improves behavioral deficits in C201R/+ mice
To further substantiate these findings, we also tested the effects of both TgA and TgD in a second pathological Gars allele. The milder CMT2D disease model, C201R/+, has less severe overt neuromuscular phenotypes than the Nmf249/+ mice. In this model, behavioral measures of peripheral nerve dysfunction are useful to detect the subtler phenotype. One behavioral measure is a wire hang test of grip strength. Mice were placed on a wire grid that was then inverted and the time that mice were able to suspend themselves was recorded. At 3 and 6 weeks of age, wild-type mice are often able to hang for a full minute, when the test is stopped. Neither transgene significantly improved performance on this test at either age tested (Figure 5A–5D).

To look for subclinical improvements in motor unit function, we tested nerve conduction on mice from this cross as well. Nerve conduction in C201R/+ mice is significantly reduced to approximately 55% of wild-type levels. Neither TgA nor TgD significantly improved NCVs in the C201R/+ mice (Figure 5E, 5F).
In summary, there were trends towards slight and variable improvements in body weight, axon diameter, and NCV in CMT2D mouse models when wild-type GARS is ubiquitously over-expressed, but none of these improvements were statistically significant, and the mice still have compromised motor function assessed by behavioral, histological, and electrophysiological measures compared to control animals.
Wild-type GARS transgenes functionally substitute for an endogenous wild-type Gars allele
Although there was a robust increase in wild-type GARS protein detected by immunoblotting in both TgA and TgD, and animals with the transgenes had significantly higher tRNAGly charging activity, it remained to be demonstrated that the transgene products in these mice can substitute for a loss of wild-type Gars. Human GARS can functionally replace the fruit fly ortholog, Aats-gly [28], but does not rescue yeast lacking GRS1 (A.A. unpublished data). To confirm that our human GARS transgene could replace mouse Gars, we tested whether the transgenes could restore wild-type function in place of the XM256 loss-of-function gene trap allele [25]. The XM256 allele unambiguously causes a loss-of-function by intercepting splicing and reducing mRNA levels of Gars assayed by northern blot, as well as reducing enzymatic activity in tissue homogenates from heterozygous mice [25].
Compound heterozygotes of the gene trap allele with either mutant allele (C201R/XM256 and Nmf249/XM256) die embryonically [25], [26]. We crossed XM256/+; TgA or TgD mice with C201R/+ mice, and the offspring included both viable C201R/XM256; TgA and C201R/XM256; TgD mice. This shows that both TgA and TgD were able to restore viability by substituting for the endogenous wild-type Gars allele. We expected C201R/XM256; Tg mice to have a mild neuropathy because of the pathogenic C201R allele, and “full rescue” would therefore constitute viable mice with a phenotype comparable to C201R/+.
Chi-square analysis showed that C201R/XM256; TgA and C201R/XM256; TgD mice were born at expected Mendelian ratios (6/48, p = 1.0, and 4/63, p = 0.14, respectively), while C201R/XM256 mice without the transgenes were not viable, as expected, and represented a significantly absent class (p = 0.009, p = 0.003, respectively).
In addition to restoration of viability, the C201R/XM256 mice with the transgenes were restored to a C201R/+-like neuropathy. The rescued mice had femoral nerve pathology and axon numbers that were not significantly different from C201R/+ mice or controls at eight weeks of age (Figure 6A–6J, 6L). As previously reported, neither the XM256/+ nor the C201R/+ mice had a decrease in myelinated axon number in the motor or sensory branch of the femoral nerve, whereas the Nmf249/+ mice do have reduced axon numbers (Figure 6 and [25], [26]). Therefore, the rescued mice do not show an increased severity in their neuropathy by this measure. We also tested this functionally by measuring nerve conduction velocities, which are normal in XM256/+ mice (Figure 6K), but are significantly reduced in C201R/+ mice (Figure 5E and 5F, Figure 6K). Conduction velocities in rescued mice were significantly below control values, and were not significantly different than C201R/+ values.

To further verify that the transgenes fully rescued the neuropathy to the severity anticipated for C201R/+, we also examined NMJ morphology and occupancy at 8 weeks of age. The NMJs of XM256/+ mice are indistinguishable in morphology and occupancy from wild-type mice (Figure 7A and not shown), and form complex pretzel structures with complete overlap between pre and post-synaptic markers. NMJs in C201R/+ mice (Figure 7B) have regions of immature morphology or denervation that are noticeably different from wild-type, but are not as dysmorphic as the NMJs in Nmf249/+ mice. C201R/XM256 mice with TgA or TgD had neuromuscular junction morphology similar to that of C201R/+ mice (Figure 7C, 7D). No significant difference in the proportion of NMJs that were fully occupied was observed between C201R/+, C201R/XM256; TgA, and C201R/XM256; TgD mice (Figure 7E).

While the severity of the neuropathy was the same in C201R/+ and C201R/XM256; Tg mice, the rescued mice had significantly lower body weights than wild type or XM256/+ animals (Figure S3A). The muscle weight/body weight ratio was not reduced in these mice, suggesting that the lower body weight was not a result of neuropathic muscle degeneration (Figure S3B). Therefore, the reduced body weight probably reflects some insufficiency in transgene expression in other tissues. However, the viability of compound heterozygous C201R/XM256 mice with TgA and TgD indicates that both produce functional protein and both are capable of substituting for a wild type allele of Gars.
Increasing the genetic dosage of mutant Gars increases the severity of neuropathy
To examine the effects of increasing the genetic dose of the mutant alleles of Gars and to further parse if there is a loss-of-function component to the amino acid substitutions, we generated C201R homozygotes and C201R/Nmf249 compound heterozygotes, both with and without TgD.
C201R homozygous mice have a more severe neuropathy phenotype than C201R/+ heterozygotes, and typically die at about two weeks of age [26]. It is unclear whether the more severe phenotype in homozygotes is due to a double-dose of a toxic mutant protein, to a compounded loss-of-function, or a combination of these effects. Based on the ability of TgD to substitute for a wild type Gars allele in rescuing viability in combination with XM256, we hypothesized that a loss of function would be corrected by the presence of the transgene, whereas increased toxicity from the higher dose of mutant protein would not be corrected, based on the inability of the transgene to improve the neuropathy phenotype in C201R/+ mice.
Consistent with previous reports, we found that C201R/C201R mice were subviable (X2 p = 0.016) [26]. Only two homozygous C201R mice were recovered in 69 offspring (1/8 anticipated), and these died by P12 before they could be analyzed. However, C201R/C201R; TgD mice were born at expected Mendelian ratios (11/69, X2 p = 0.39), and consistently lived to post-natal day 17, when tissues were collected and analyzed. The maximum observed lifespan for these mice was to P20. Despite the restoration of wild-type GARS function with TgD, these mice had a more severe neuropathy than C201R/+ heterozygotes. The C201R/C201R;TgD mice had approximately a 50% reduction in axon numbers in the motor branch of the femoral nerve (Figure 8A, 8B, 8D, 8G), and the mice were <5 grams in weight at P17 (Figure 8H). Neuromuscular junction defects were similarly increased in severity (Figure 8I, 8J, 8L, 8M, 8P). At P17, wild-type NMJs have developed into mature pretzel shapes, but C201R/+ NMJs are arrested at a less-mature, plaque-like morphology, and show an increased incidence of partially innervated junctions. The C201R/C201R;TgD mice demonstrated frank denervation at almost half of the postsynaptic sites. In agreement with this, we could not obtain nerve conduction velocities from these mice, because muscle action potentials were too small to reliably record (n = 8). Thus, the reduced viability embryonically and in neonates seen in C201R homozygous mice was corrected by the TgD, as demonstrated by the recovery of C201R/C201R;TgD mice at the expected Mendelian ratios and their survival to P17. However, the increased genetic dosage of the C201R allele leads to a more severe neuropathy.

To further examine this effect of genetic dosage and apparent recessive partial-loss-of-function, we also made compound heterozygous C201R/Nmf249 mice. Interestingly, both C201R/Nmf249 and C201R/Nmf249; TgD mice were viable and born at expected Mendelian frequencies (5/33, X2 p = 0.95 and 4/33, p = 0.65, respectively). Considering that both C201R/XM256 and Nmf249/XM256 are nonviable in the absence of a wild-type transgene, the viability of C201R/Nmf249 mice suggests that there is intragenic complementation between the two mutant alleles that alleviates the loss-of-function and subsequent reduction in viability at birth.
Like homozygous C201R/C201R; TgD mice, the motor branch of the femoral nerve is noticeably smaller and axon numbers are significantly reduced in C201R/Nmf249 and C201R/Nmf249; TgD mice compared to wild-type or heterozygous littermate controls (Figure 8A–8G). No significant difference in axon numbers was seen between the C201R/C201R; TgD, C201R/Nmf249, and C201R/Nmf249; TgD genotypes. The same qualitative and quantitative differences were seen in the sensory nerves of these animals (Figure S4).
The overall health and overt appearance of the compound heterozygous mice was better than that of homozygous C201R/C201R;TgD mice. The body weights of C201R/Nmf249 and C201R/Nmf249; TgD mice were significantly lower than C201R/+ mice (Figure 8H), and C201R/C201R; TgD weighed significantly less than C201R/Nmf249 mice.
Morphological changes seen in the neuromuscular junctions of C201R/Nmf249 and C201R/Nmf249; TgD mice were also more severe than either allele as a heterozygote and reflected the axon loss seen in the femoral nerves. Like C201R/+ mice at P17, Nmf249/+ mice have partially denervated NMJs (Figure 8K) with areas of disorganized post-synaptic staining, but also more examples of complete denervation. Like the C201R/C210R;TgD mice, compound heterozygotes with or without TgD had even more dysmorphic NMJ morphology than Nmf249/+ heterozygotes, with a majority of NMJs that were partially or fully denervated and very few that were fully innervated (Figure 8N–8P). There were significantly more denervated NMJs in C201R/C201R; TgD, C201R/Nmf249, and C201R/Nmf249; TgD mice compared to Nmf249/+ mice (Figure 8P). Compound muscle action potentials were again too small to reliably record in C201R/Nmf249 (n = 4), and C201R/Nmf249; TgD (n = 5) mice.
Therefore, the presence of TgD did not alter viability, axon number, body weight, or NMJ occupancy of the compound heterozygous mice, consistent with intragenic complementation improving the recessive loss-of-function sub-viability phenotype observed in C201R homozygotes. In both C201R/Nmf249 compound heterozygotes and C201R homozygotes, the severity of the neuropathy increases as the genetic dosage of the mutant Gars increases, even in the presence of the wild-type transgene.
Discussion
To summarize our results, the over-expression of functional, wild-type GARS does not suppress the neuropathy caused by dominant amino acid substitutions, but does rescue reduced viability seen with homozygous point mutations or in point mutations combined with a null allele. Furthermore, the neuropathy is worsened with homozygous or compound heterozygous amino acid substitution alleles (Table 1). From these results we conclude that the dominant mechanism underlying GARS mutations and the degeneration of peripheral axons is a toxic gain of function by the mutant GARS protein. This toxicity depends on the dosage of the mutant protein and is not mitigated or out-competed by the over-expression of wild-type GARS, suggesting that the neuropathy is not likely to be caused by a loss-of-function or a simple dominant-negative mechanism. There is also a loss of GARS function with the mutations, but this is only seen when alleles with amino-acid changes are made homozygous or placed over a loss-of-function allele created by a gene trap insertion. The recessive loss of function reduces viability, but is corrected by the transgenic over-expression of wild-type GARS, leaving these mice with only the dose-dependent neuropathy caused by expression of the mutant protein. Interestingly, compound heterozygous mice carrying two different alleles with pathological amino-acid changes show “additive” neuropathy that is more severe than either allele alone as a heterozygote, and is comparable in severity to a point mutation as a homozygote. However, compound heterozygous mice do not show reduced neonatal viability associated with a loss of function, and the phenotype is not improved by over-expression of wild-type GARS, indicating that there is intragenic complementation and little or no loss of function in the compound heterozygotes. The basis for these conclusions and the implications for CMT are discussed below.

Peripheral neuropathy phenotype is caused by a gain of function
Overall, no significant improvement in axonopathy phenotypes was seen in CMT2D model mice over-expressing wild-type GARS. Two transgenes with similar levels of expression were used in these crosses and yielded effectively the same results. Neither transgene prevented axon degeneration or improved nerve conduction velocity or NMJ morphology and occupancy in the Nmf249/+ mice at one month of age. The only positive effect of the transgenes was a minor improvement in axon diameter. Similarly, neither transgene improved behavioral measures at three or six weeks of age, or NCVs at eight or twelve weeks of age in C201R/+ mice. Thus, over-expression of wild-type GARS did not suppress the dominant neuropathy phenotype. It is also notable that the over-expression of wild-type GARS did not cause any phenotype on its own in either transgenic line.
These crosses, and the observation that XM256/+ mice do not have a neuropathy, show that the mutant protein itself is toxic and causes peripheral nerve degeneration. The toxicity can be intensified, and the neuropathy worsened, by increasing the dose of mutant protein in either homozygotes or compound heterozygotes. Lifespan, NMJ occupancy, axon numbers and body weight are significantly reduced in C201R/Nmf249, C201R/Nmf249; TgD, and C201R/C201R; TgD mice below the levels of Nmf249/+ and C201R/+ mice, which only have one copy of the mutant protein. This is consistent with experiments with YARS in Drosophila, which also indicate that the mutant tRNA synthetase has a dose-dependent toxicity [29].
Loss-of-function component is responsible for reduced viability
Although the peripheral axon degeneration is caused by toxicity of the mutant protein, the mutations do have loss-of-function characteristics. Homozygous C201R mice are subviable, and neither allele is viable in combination with the gene trap allele, despite the Nmf249/+ P278KY protein being fully active in cell-free activity assays of amino acylation and tRNA charging [25].
The embryonic lethality observed in these Gars alleles is probably not related to the neuropathy phenotype. In fact, a functioning nervous system is not required during embryonic development. Mice deficient for proteins that are crucial for the function of the central and peripheral nervous system are born in the anticipated numbers, but do not survive independently after birth [30]–[32]. GARS is necessary for translation in every cell, and loss of function in non-neurological tissue is a likely cause of the embryonic lethality.
With transgenic over-expression of wild-type protein, we were able to restore full viability to C201R/XM256 mice, which otherwise die embryonically, and C201R/C201R mice, which are otherwise subviable. It is notable that we were unable to rescue XM256 as a homozygote with either transgene. This may be due to a closely linked second mutation on the gene trap chromosome, but is more likely due to an insufficiency of transgene expression at some critical point in development. This is supported by the reduced body weight of XM256/C201R mice with transgene rescue despite full rescue of viability and neurological function equivalent to a wild type allele. Presumably, the C201R allele provides sufficient GARS activity in whatever tissue may be lacking transgene expression, allowing rescue of the point mutation over the gene trap. Thus, over-expression of wild-type GARS can rescue postnatal viability, but does not improve the neuropathic phenotype, suggesting the loss of viability is caused by a loss of function for which the transgene can compensate, whereas the neuropathy is a gain of function that the transgene cannot correct.
Intragenic complementation confirms that toxicity is the major determinant of disease severity
C201R homozygous mice have decreased viability, and when they are born they have a severe neuropathy. We expected C201R/Nmf249 mice to be at least as severe because the Nmf249 allele is more pathogenic than the C201R allele despite its normal enzymatic activity. Surprisingly, the compound heterozygous mice were as healthy as or even healthier than C201R homozygotes with TgD. The C201R/Nmf249 mice were fully viable and weighed significantly more than C201R/C201R; TgD mice, although their neuropathy was markedly more severe than either allele as a heterozygote. Therefore, the C201R and Nmf249 alleles are capable of intragenic complementation in regard to the loss-of-function phenotype, but still display “additive” severity for the gain-of-function neuropathy phenotype. Over-expression of wild-type GARS did not improve the phenotype in the compound heterozygous mice, which are severely affected because they express two mutant versions of the protein. This suggests that the loss-of-function aspect of the C201R homozygotes that is rescued by the transgene is not a factor in the compound heterozygotes, and that the dose of the mutant protein determines the severity of the neuropathy. It also suggests that this loss of function, perhaps in enzyme charging function, differs between the alleles, because C201R/Nmf249 mice are more viable than C201R/C201R mice even though the Nmf249 allele is more neurotoxic than the C201R allele.
Therapies for tRNA–synthetase–linked neuropathies should selectively target the mutant allele
Dominant mutations in GARS, YARS, and AARS all cause CMT [14]–[16]. Our results suggest that mutant forms of GARS adopt a pathological function that may be impacting the normal role of GARS in translation, but that the loss-of-function effects are recessive, and would not be an important factor in CMT patients with dominantly inherited disease. Instead, the neuropathy is a caused by a gain of function, demonstrated by the inability of functional wild-type transgenic GARS to rescue the phenotype either by restoring lost activity or outcompeting this pathological function. One patient reported to carry compound heterozygous mutations in KARS may be the exception to this, but it is notable that this genotype was associated with peripheral neuropathy with additional neurological and non-neurological symptoms [17].
The interplay between normal protein function and pathogenic function has been dissected in other neurodegenerative diseases. For example, hereditary sensory and autonomic neuropathy (HSAN) type 1A is caused by mutations in the serine palmitoyl transferase gene (SPTLC1), which alter the protein's amino acid substrate specificity so that the mutant produces two neurotoxic atypical deoxysphingoid bases [33]–[35]. Over-expression of wild-type SPTLC1 rescued a transgenic mouse model of the disease by outcompeting mutant forms of SPTLC1 in heterodimer formation with SPTLC2, restoring the substrate specificity, lowering levels of neurotoxic sphingoid bases, and demonstrating that mutant SPTLC1 is not inherently toxic [36]. Our work suggests that mutant GARS protein is toxic independent of wild-type protein levels. These results are more like what has been observed in SOD1-linked amyotrophic lateral sclerosis, which is caused by dominant mutations and is not improved with wild-type over-expression [37].
These conclusions have implications for therapeutic approaches. To reduce the effect of mutant GARS, the expression of the mutant allele must be diminished. Future therapeutic approaches should focus on allele specific knockdown of the mutant gene expression; alternatively, the specific pathways affected by mutant GARS toxicity could be identified and targeted.
Materials and Methods
Generation of transgenic mice
The Ins-CMV-C-B-A vector [38] was modified to include a Gateway conversion cassette (Invitrogen) in the multiple cloning site, which includes a ccdB cassette flanked by attR1 and attR2 recombination sequences. A Gateway LR clonase reaction was used to insert a PCR amplicon of the GARS cDNA (human) into the pINS2-Gateway vector. The cDNA amplicon that was inserted into pINS2 included 47 basepairs of the endogenous GARS 5′ UTR upstream of the start site of the mitochondrial isoform, and 19 basepairs of endogenous 3′UTR flanked by 5′ and 3′ untranslated regions (UTR) from rabbit β globin also containing a polyadenylation signal at the 3′ end. The cDNA and control elements are flanked by two chicken β-globin 5′HS4 insulators, which have been shown to reduce variability in transgene expression caused by position effects of insertion site [39]. Pronuclear injection of this construct into single cell FVB/N embryos was performed in the NHGRI Transgenic Mouse Core according to existing protocols [40].
Mouse strains, husbandry, and genotyping
All mouse husbandry and procedures were conducted according to the NIH Guide for Care and Use of Laboratory Animals and were approved by NINDS or The Jackson Laboratory Animal Care and Use Committee. Tail, toe or ear tissue was lysed with proteinase K incubation at 55°C and boiled for 10 min to inactivate the proteinase K. DNA derived from the mouse tissue was then used in PCR to determine genotype.
Primers GARS WT Tg F (5′-CCCATTACTGGAAATGATCTA-3′) and GARS WT Tg R (5′-TTTCCGAGCGGACTGTCCGC-3′), which anneal to exons 7 and 9, respectively, were used to determine if the wild-type transgene was present. Primers targeting Gars intron 6F (5′-GCCTTGTTCTGTAACGTTTGCAC-3′) and a primer specific to the Nmf249 allele (5′-CCAGGCATATTTCCTCCATATTT-3′) were used to identify mice with the Nmf249 mutation [25]. Primers Gars C201R F (5′-CACGTGCTTGCTCTAGCAAGA-3′) and Gars C201R R (5′-GTCTACCACTGAACACAGTCC-3′) were used in PCR reaction, and the product was digested with HhaI restriction enzyme. PCR products containing the pathogenic mutation are digested into two smaller bands [26]. Primers BgeoR (5′-CGCCAGGGTTTTCCCAGT-3′) and En2-i1F (5′-AATGCCCAACACTTGTATGG-3′) were used to determine if the XM256 gene trap allele was present. To screen for XM256/XM256 mice, primers flanking the gene trap insertion 519 bp into intron 2 of Gars, GarsXMvsWT2F (5′-GCTTCCGCACTACCTGAACCCAAACT-3′) and GarsXMvsWT2R (5′-TGAATTCAGCAGCCCCCTCTGTACCC-3′), were used. No XM256/XM256 mice were identified, with or without the transgene. PCR amplification indicated the wild-type allele was present. All genotyping products were resolved on 2% agarose gels with ethidium bromide (Sigma, St. Louis, MO). Nmf249 mice are maintained on a C57BL/6 background and are occasionally outcrossed to CAST/Ei to improve their ability to breed [21]. C201R mice were originally on a mixed C57BL/6 and C3H background and subsequently bred into a C57BL/6 background. The TgA and TgD alleles were maintained on a pure FVB/N background with one exception: the TgA mice that were used in the cross with C201R mice were on a FVB/N X C57BL/6 F1 hybrid background. Therefore, experiments were done primarily in a [FVB/N X BL/6] F1 background unless two matings were required to generate homozygotes or compound heterozygotes, in which case mice were effectively in an [N2] C57BL/6 background. All control animals were siblings of the experimental genotypes.
Tissue lysate preparation
Spinal cord and sciatic nerve were isolated from animals immediately after they were euthanized by CO2 inhalation. The tissues were frozen in liquid nitrogen and stored at −80°C. The tissues were then homogenized in 1% NP-40 in phosphate buffered saline (PBS) supplemented with Protease Inhibitor Cocktail Tablets (Roche, Basal, Switzerland) using a PowerGen Model 125 Homogenizer (Fisher Scientific, Pittsburgh, PA) then centrifuged at 14,000 g for 10 min at 4°C. Cleared homogenates were then sonicated at 4°C and centrifuged again at 14,000 g for 10 min. Protein concentrations were assessed using a Bradford assay (BioRad, Hurcules, CA). 20 µg of protein was then analyzed by immunoblot.
Western blots
Protein lysates were resolved on Novex 10% Tris-Glycine Gels (Invitrogen, Carlsbad, CA) and transferred to an Invitrolon PVDF membrane for western blot analysis. Membranes were blocked with 5% skim milk in TBST (1× Tris-buffered saline, 0.1% Tween-20), and incubated overnight with GARS rabbit polyclonal antibody ab42905 (1∶2,000) (Abcam, Cambridge, MA) and β actin mouse monoclonal antibody clone AC-74 (1∶10,000)(Sigma Aldrich, St. Louis, MO) diluted in blocking solution at 4° C. Following three 10 min washes in TBST, the blots were incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) diluted in blocking solution. After three 10-min washes in TBST, the blots were developed using Western Lightening Plus-ECL, Enhanced Chemiluminescence Substrate (Perkin Elmer, Waltham, MA).
Teased nerve immunohistochemistry
Teased nerve fibers were stained as described in the methods of [21], [41]. In brief, sciatic nerves were excised, immediately placed in fresh 4% paraformaldehyde, and fixed on ice for 15 minutes. Nerves were then transferred to ice cold PBS for dissection and teasing. Using #5 forceps and 30 gauge needles, nerve sheaths were removed and the nerves were cut into 1 cm segments and teased apart at one end. The nerve segments were then transferred to a fresh Superfrost Plus Gold slides (Fisher Scientific, Pittsburgh, PA) and pulled from a drop of PBS onto a dry section of the slide to straighten the fibers for imaging. The slides were then dried overnight at room temperature and incubated in acetone at −20°C for 10 min. The samples were then rehydrated with two 5 min incubations in PBS and blocked with 5% normal goat serum in PBS with 0.5% Triton X-100 for 1 h at room temperature. Primary antibodies rabbit anti-GARS (1∶500) (Abcam), mouse anti-neurofilament with the 2H3 antibody (1∶500)(Developmental Studies Hybridoma Bank, Iowa City, IA) were gently placed on the slide, covered with Parafilm coverslips (Pechinery Plastic, Chicago, IL) and stored in a humidified chamber overnight at 4° Celsius. After three 5-min washes in PBS, the samples were incubated in the following secondary antibodies diluted 1∶1,000 in blocking solution: AlexaFluor 555 goat anti-rabbit, and AlexaFluor 488 goat anti-mouse IgG1 (y1) (Invitrogen, Carlsbad, CA). The samples were covered with Parafilm coverslips and incubated for 2 h at room temperature. The samples were then washed three times in PBS for 5 min each in watch glasses and mounted with Vectashield (Vector Labs, Burlingame, CA).
Aminoacylation assays
Aminoacylation assays were performed at room temperature in a reaction mixture containing 50 mM HEPES (pH 7.5), 20 mM KCl, 2 mM ATP, 5 mM MgCl2, 1 mM DTT, 19 µM L-glycine (or L-alanine), 1 µM 3H-L-glycine (or 3H-L-alanine), 2 µM transcribed human tRNAGlyCCC [for GlyRS activity] or 120 µM yeast total tRNA (Roche Diagnostics, Indianapolis, IN) [for AlaRS activity] and 30 µg total protein from tissue homogenates. Reactions were initiated by addition of reaction mixture and tRNA to tissue homogenates. Aliquots were quenched at different time points and precipitated in 96-well Multiscreen filter plates (Millipore) as described previously [42]. After washing and elution by NaOH, samples were counted in a MicroBeta plate reader (PerkinElmer Life Sciences). Initial rates were measured from slopes obtained by fitting the data to a non-linear regression curve using GraphPad Prism 5 software. For TgA, 3 wild type and 4 transgenic littermates were sampled at 9 weeks of age: for TgD, 4 wild type and 3 transgenic littermates were tested at 6 weeks of age.
Motor and sensory nerve analysis
The sensory and motor branches of the femoral nerve were isolated and fixed overnight in 2% glutaraldehyde and 2% paraformaldehyde in a 0.1 M cacodylate buffer. The tissue was then processed for transmission electron microscopy and embedded in plastic before 0.5 µm sections were cut and stained with toluidine blue. For more information see [43]. For axon counting and axon diameter measurement the images were captured using a Nikon Eclipse E600 microscope with 40× and 100× objectives. Axon counts were done using the Cell Counter Plug-in in ImageJ. Left and right nerves were averaged. Axon diameters were measured using the Measure and Label Plugin, also in ImageJ.
NMJ imaging and analysis
Mouse plantaris muscles were surgically removed and fixed in freshly prepared 2% paraformaldehyde in PBS for four hours. The samples were then transferred to a blocking and permeabilizing solution of 5% normal goat serum and 0.5% Triton-X 100 in PBS for 1 h before they were pressed between two glass slides using a binder clip for 15 min, after which they were returned to the blocking and permeabilizing solution. The samples were then incubated overnight at 4°C with 1∶1,000 dilutions of anti-SV2 and anti-neurofilament (2H3) primary antibodies (Developmental Studies Hybridoma Bank, Iowa City, IA). After at least three 1 h washes in PBS with 0.5% Triton-X 100, the samples were transferred to blocking and permeabilizing solution with AlexaFluor 488 goat anti-mouse IgG1 (y1) (Invitrogen, Carlsbad, CA) and α-bungarotoxin conjugated with Alexa Fluor 594. After incubation overnight at 4°C, the samples were washed three times for 1 h each and mounted with Vectashield mounting media (Vector Labs, Burlingame, CA) and imaged using a confocal microscope.
Confocal microscopy
Confocal images were gathered using a Carl Zeiss LSM 710 or Leica SP5 laser-scanning confocal microscope with a 63× objective. Z stacks were collapsed into projected images and merged using ImageJ (NIH, http://rsb.info.nih.gov/ij/). The color balance of the NMJ images was adjusted for clarity.
Nerve conduction studies
Sciatic nerve conduction velocity was calculated by measuring the latency of compound motor action potentials recorded in the muscle of the left rear paw. The mice were anesthetized with 1% isofluorane and placed on a thermostatically regulated heating pad to maintain normal body temperature. Action potentials were produced by subcutaneous stimulation at the sciatic notch and at the ankle. For recording, the active needle electrode was inserted in the center of the paw and a reference electrode was placed in the skin between the first and second digits.
Statistical analysis
Statistical tests were performed using GraphPad's Prism 5 software. Statistical significance was determined using a one-way ANOVA and a post hoc Tukey test for individual differences when appropriate. A threshold of p<0.05 was considered significant. The use of other tests is noted in the text. All results are presented as means ± SD.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. SkreH 1974 Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clinical Genetics
2. Jani-AcsadiAKrajewskiKShyME 2008 Charcot-Marie-Tooth neuropathies: diagnosis and management. Semin Neurol 28 185 194
3. NicholsonGMyersS 2006 Intermediate forms of Charcot-Marie-Tooth neuropathy: a review. Neuromolecular Med 8 123 130
4. TimmermanVNelisEVan HulWNieuwenhuijsenBWChenKL 1992 The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-Tooth disease type 1A duplication. Nat Genet 1 171 175
5. HayasakaKHimoroMSatoWTakadaGUyemuraK 1993 Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat Genet 5 31 34
6. BergoffenJSchererSSWangSScottMOBoneLJ 1993 Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262 2039 2042
7. VerhoevenKDe JonghePCoenKVerpoortenNAuer-GrumbachM 2003 Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet 72 722 727
8. ZhaoCTakitaJTanakaYSetouMNakagawaT 2001 Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell 105 587 597
9. ZuchnerSMersiyanovaIVMugliaMBissar-TadmouriNRochelleJ 2004 Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36 449 451
10. BaxterRVBen OthmaneKRochelleJMStajichJEHuletteC 2002 Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet 30 21 22
11. CuestaAPedrolaLSevillaTGarcía-PlanellsJChumillasMJ 2002 The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet 30 22 25
12. EvgrafovOVMersiyanovaIIrobiJVan Den BoschLDierickI 2004 Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet 36 602 606
13. JordanovaADe JonghePBoerkoelCFTakashimaHDe VriendtE 2003 Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain 126 590 597
14. AntonellisAEllsworthRESambuughinNPulsIAbelA 2003 Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 72 1293 1299
15. JordanovaAIrobiJThomasFVan DijckPMeerschaertK 2006 Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet 38 197 202
16. LatourPThauvin-RobinetCBaudelet-MeryCSoichotPCusinV 2010 A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 86 77 82
17. McLaughlinHMSakaguchiRLiuCIgarashiTPehlivanD 2010 Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet 87 560 566
18. MotleyWWTalbotKFischbeckKH 2010 GARS axonopathy: not every neuron's cup of tRNA. Trends Neurosci 33 59 66
19. AntonellisALee-LinSWasterlainALeoPQuezadoM 2006 Functional Analyses of Glycyl-tRNA Synthetase Mutations Suggest a Key Role for tRNA-Charging Enzymes in Peripheral Axons. J Neurosci 26 10397 10406
20. NangleLZhangWXieWYangXLSchimmelP 2007 Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc Natl Acad Sci USA 104 11239 11244
21. StumMGMcLaughlinHMKleinbrinkELMiersKEAckermanS 2010 An assessment of mechanisms underlying peripheral axonal degeneration caused by aminoacyl-tRNA synthetase mutations. Mol Cell Neurosci 46 432 443
22. XieWNangleLAZhangWSchimmelPYangXL 2007 Long-range structural effects of a Charcot-Marie-Tooth disease-causing mutation in human glycyl-tRNA synthetase. Proc Natl Acad Sci U S A 104 9976 9981
23. XieWSchimmelPYangXL 2006 Crystallization and preliminary X-ray analysis of a native human tRNA synthetase whose allelic variants are associated with Charcot-Marie-Tooth disease. Acta Crystallogr Sect F Struct Biol Cryst Commun 62 1243 1246
24. CaderMZRenJJamesPABirdLETalbotK 2007 Crystal structure of human wildtype and S581L-mutant glycyl-tRNA synthetase, an enzyme underlying distal spinal muscular atrophy. FEBS Lett 581 2959 2964
25. SeburnKLNangleLCoxGASchimmelPBurgessRW 2006 An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron 51 715 726
26. AchilliFBros-FacerVWilliamsHPBanksGTAlqatariM 2009 An ENU-induced mutation in mouse glycyl-tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot-Marie-Tooth type 2D peripheral neuropathy. Dis Model Mech 2 359 373
27. NiwaHYamamuraKMiyazakiJ 1991 Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108 193 199
28. ChiharaTLuginbuhlDLuoL 2007 Cytoplasmic and mitochondrial protein translation in axonal and dendritic terminal arborization. Nat Neurosci 10 828 837
29. StorkebaumELeitao-GoncalvesRGodenschwegeTNangleLMejiaM 2009 Dominant mutations in the tyrosyl-tRNA synthetase gene recapitulate in Drosophila features of human Charcot-Marie-Tooth neuropathy. Proc Natl Acad Sci U S A 106 11782 11787
30. GautamMNoakesPGMoscosoLRuppFSchellerRH 1996 Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 85 525 535
31. VerhageMMaiaASPlompJJBrussaardABHeeromaJH 2000 Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287 864 869
32. MisgeldTBurgessRWLewisRMCunninghamJMLichtmanJW 2002 Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron 36 635 648
33. DawkinsJLHulmeDJBrahmbhattSBAuer-GrumbachMNicholsonGA 2001 Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet 27 309 312
34. BejaouiKWuCSchefflerMDHaanGAshbyP 2001 SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat Genet 27 261 262
35. PennoAReillyMMHouldenHLauraMRentschK 2010 Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem 285 11178 11187
36. EichlerFSHornemannTMcCampbellAKuljisDPennoA 2009 Overexpression of the wild-type SPT1 subunit lowers desoxysphingolipid levels and rescues the phenotype of HSAN1. J Neurosci 29 14646 14651
37. BruijnLIHouseweartMKKatoSAndersonKLAndersonSD 1998 Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281 1851 1854
38. DevlinEEDacostaLMohandasNElliottGBodineDM 2010 A transgenic mouse model demonstrates a dominant negative effect of a point mutation in the RPS19 gene associated with Diamond-Blackfan anemia. Blood 116 2826 2835
39. PottsWTuckerDWoodHMartinC 2000 Chicken beta-globin 5′HS4 insulators function to reduce variability in transgenic founder mice. Biochem Biophys Res Commun 273 1015 1018
40. NagyA 2003 Manipulating the mouse embryo : a laboratory manual Cold Spring Harbor, N.Y. Cold Spring Harbor Laboratory Press viii 764
41. ArroyoEJSirkowskiEEChitaleRSchererSS 2004 Acute demyelination disrupts the molecular organization of peripheral nervous system nodes. J Comp Neurol 479 424 434
42. BeebeKWaasWDruzinaZGuoMSchimmelP 2007 A universal plate format for increased throughput of assays that monitor multiple aminoacyl transfer RNA synthetase activities. Anal Biochem 368 111 121
43. BurgessRWCoxGASeburnKL 2009 Neuromuscular disease models and analysis. Methods Mol Biol 602 347 393
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 12
Nejčtenější v tomto čísle
- Targeted Proteolysis of Plectin Isoform 1a Accounts for Hemidesmosome Dysfunction in Mice Mimicking the Dominant Skin Blistering Disease EBS-Ogna
- The RNA Silencing Enzyme RNA Polymerase V Is Required for Plant Immunity
- The FGFR4-G388R Polymorphism Promotes Mitochondrial STAT3 Serine Phosphorylation to Facilitate Pituitary Growth Hormone Cell Tumorigenesis
- Target Site Recognition by a Diversity-Generating Retroelement