
Identification of a Genomic Reservoir for New Genes in Primate Genomes
Tripartite Motif (TRIM) ubiquitin ligases act in the innate immune response against viruses. One of the best characterized members of this family, TRIM5α, serves as a potent retroviral restriction factor with activity against HIV. Here, we characterize what are likely to be the youngest TRIM genes in the human genome. For instance, we have identified 11 TRIM genes that are specific to humans and African apes (chimpanzees, bonobos, and gorillas) and another 7 that are human-specific. Many of these young genes have never been described, and their identification brings the total number of known human TRIM genes to approximately 100. These genes were acquired through segmental duplications, most of which originated from a single locus on chromosome 11. Another polymorphic duplication of this locus has resulted in these genes being copy number variable within the human population, with a Han Chinese woman identified as having 12 additional copies of these TRIM genes compared to other individuals screened in this study. Recently, this locus was annotated as one of 34 “hotspot” regions that are also copy number variable in the genomes of chimpanzees and rhesus macaques. Most of the young TRIM genes originating from this locus are expressed, spliced, and contain signatures of positive natural selection in regions known to determine virus recognition in TRIM5α. However, we find that they do not restrict the same retroviruses as TRIM5α, consistent with the high degree of divergence observed in the regions that control target specificity. We propose that this recombinationally volatile locus serves as a reservoir from which new TRIM genes arise through segmental duplication, allowing primates to continually acquire new antiviral genes that can be selected to target new and evolving pathogens.
Published in the journal:
. PLoS Genet 7(12): e32767. doi:10.1371/journal.pgen.1002388
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002388
Summary
Tripartite Motif (TRIM) ubiquitin ligases act in the innate immune response against viruses. One of the best characterized members of this family, TRIM5α, serves as a potent retroviral restriction factor with activity against HIV. Here, we characterize what are likely to be the youngest TRIM genes in the human genome. For instance, we have identified 11 TRIM genes that are specific to humans and African apes (chimpanzees, bonobos, and gorillas) and another 7 that are human-specific. Many of these young genes have never been described, and their identification brings the total number of known human TRIM genes to approximately 100. These genes were acquired through segmental duplications, most of which originated from a single locus on chromosome 11. Another polymorphic duplication of this locus has resulted in these genes being copy number variable within the human population, with a Han Chinese woman identified as having 12 additional copies of these TRIM genes compared to other individuals screened in this study. Recently, this locus was annotated as one of 34 “hotspot” regions that are also copy number variable in the genomes of chimpanzees and rhesus macaques. Most of the young TRIM genes originating from this locus are expressed, spliced, and contain signatures of positive natural selection in regions known to determine virus recognition in TRIM5α. However, we find that they do not restrict the same retroviruses as TRIM5α, consistent with the high degree of divergence observed in the regions that control target specificity. We propose that this recombinationally volatile locus serves as a reservoir from which new TRIM genes arise through segmental duplication, allowing primates to continually acquire new antiviral genes that can be selected to target new and evolving pathogens.
Introduction
The TRIM protein family constitutes a newly appreciated group of innate immune effectors involved in the response to viral infection [1]–[3]. TRIM5α, one of the best studied members of this family, is a pattern-recognition receptor for mammalian retroviruses including HIV [4], [5]. TRIM5α assembles into a hexameric lattice on the surface of retroviral cores as they enter the cytoplasm of a newly infected cell [6]. This interaction stimulates premature capsid disassembly [7], [8] and the formation of unanchored K63-linked polyubiquitin chains that trigger the production of chemokines and cytokines including interferon [4], [9]. The TRIM5 genetic locus has profound penetrance in determining viral titers in SIV (simian immunodeficiency virus) infected macaques [10]. TRIM5 also serves as a significant genetic barrier to the transmission of retroviruses between primate species [5], [10]–[13]. Other TRIM proteins have been linked to infection by different families of viruses altogether. TRIM25 interacts with the influenza protein NS1 [14], [15] and also activates the inflammatory response through the production of unanchored K63-linked polyubiquitin chains [16]. TRIM23 interacts with human cytomegalovirus [17], TRIM56 with pestivirus [18], while TRIM19/PML confers resistance to a broad range of DNA and RNA viruses [19]. In fact, more than one third of the approximately 70 known human TRIM genes have been shown to be transcriptionally upregulated in response to interferons [20]. Although the mechanistic details behind how TRIM proteins perform their antiviral roles remain elusive in most cases, their profound relevance to viral infection is made clear by the many viral antagonists that have been shown to target them. For example, influenza, herpes simplex virus-1, human cytomegalovirus, and adenovirus are all known to encode proteins that interact with, or alter the activity of, human TRIM proteins [15], [17], [19].
By definition, TRIM genes encode proteins with a conserved domain order: a RING zinc-coordinating domain, one or two zinc-coordinating B-boxes, followed by a coiled-coil domain (Figure 1A) [21]. These three domains constitute the “tripartite motif” that gives this family its name. Most TRIM genes also encode a variable C-terminal domain, and in many of them, this is a B30.2 domain [2]. The B30.2 is composed of a series of β-strands folded into a globular β-sandwich structure [22]. Different metazoan genomes contain different complements of TRIM genes. For example, Drosophila melanogaster has seven TRIM genes and Caenorhabditis elegans has eighteen [2]. In a previous comparison of the TRIM gene complements found in the human and mouse genomes, most were found to be strict 1∶1 orthologs [2]. This suggests that the majority of human TRIM genes are ancient, having originated more than 90 million years ago when human and mouse shared a last common ancestor. However, that study identified one phylogenetic clade of TRIM genes specific to the mouse genome, and two clades specific to the human genome. The clade of TRIM genes specific to the mouse genome (TRIM12/TRIM30 and related genes) was subsequently shown to be an expanded set of TRIM5 paralogs [23]. Based on this, we wished to describe the two phylogenetic clades of TRIM genes (TRIM50/73/74 and TRIM43/48/49/64) which are specific to the human genome (Figure 1B). We also wished to determine whether these genes have been maintained by neutral drift or by selection, potentially imposed by evolutionarily recent viral infections.

In the process of characterizing these young human TRIM genes, we identified many additional, previously unidentified human TRIM genes to which they are closely related, bringing the total number of known human TRIM genes to approximately 100. We show that these novel genes have arisen from recent, and in some cases even human-specific, segmental duplication events. Specifically, we find that one locus on chromosome 11, containing nine tandemly situated TRIM genes, has spawned at least two separate segmental duplications of itself during the evolution of great apes, as well as having produced at least one other segmental duplication that is still polymorphic in the human population. This locus is therefore evolutionarily dynamic as well as copy number variable within the human population. In a fascinating example of trans-species copy number variation, this locus was recently annotated as one of 34 “hotspot” regions that are also copy number variable in the genomes of chimpanzees and rhesus macaques [24]. Trans-species copy number variation remains largely unstudied, and the evolutionary forces behind it remain unknown [25]. We propose that this locus is selected to remain recombinationally volatile so that it can serve as a reservoir from which new primate TRIM genes constantly arise. Theoretically, increased gene dosage of innate immunity genes, conveyed by increased copy number, could in itself provide protection against viral infection and disease progression. However, many of these genes are evolving under positive selection like other primate genes known to encode antiviral molecules [26]–[35]. Therefore, these duplicated genes also appear to be rapidly diversifying in function, possibly to expand the spectrum of antiviral affinities in response to new and evolving viruses.
Results
Young TRIM genes in the human genome
In a previous comparison of the TRIM genes found in the mouse and human genomes, several human-specific genes were noted (Figure 1B) [2]. Although these genes could have arisen anytime during the last 90 million years since human and mouse last shared a common ancestor, we were interested to know whether any of them have arisen during Catarrhini speciation (Figure 1C). This group constitutes our closest evolutionary kin, primates that have most likely faced pathogens similar to those that humans encounter. To address the evolutionary origins of these human-specific TRIM genes, we took advantage of the genome projects of several Catarrhini species, including chimpanzee and orangutan (both great apes), human, and rhesus macaque (an Old World monkey).
The first clade of human TRIM genes absent in the mouse genome contains TRIM50, TRIM73, and TRIM74 (Figure 1B). To investigate when these genes arose, orthologous sequences were identified in the other Catarrhini genomes and a phylogeny was constructed (Figure 1D). The most closely related human outgroup sequence, TRIM72, was also included. TRIM72 forms a clear orthogroup containing one gene from each species, with all nodes being consistent with speciation events (boxed in green). However, the TRIM50/TRIM73/TRIM74 clade has been more dynamic (boxed in yellow). This branching pattern is consistent with an ancestral TRIM50 that experienced two duplication events, each indicated by a star on the phylogeny. The first duplication, giving rise to TRIM73, occurred after the split between great apes and Old World monkeys, but before our last common ancestor with orangutan. It involved only the exons encoding the first three protein domains. The second duplication event occurred in the human lineage, less than 7 million years ago, giving rise to TRIM74. Consistent with two duplication events, TRIM50, TRIM73, and TRIM74 reside near each other on three segmental duplications on human chromosome 7 [36]. Spliced transcripts have been identified for all three genes, and while TRIM50 has been demonstrated to act as an E3 ubiquitin ligase, the biological functions of TRIM73 and TRIM74 remain uncharacterized [36]. Thus, this small TRIM clade has gained gene copies though segmental duplications that have occurred during recent primate evolution. One gene, TRIM74, is even specific to humans.
The second clade of human TRIM genes absent in the mouse genome contains TRIM43, TRIM48, TRIM49, and TRIM64 (Figure 1B). Using reciprocal best hit analysis, we failed to detect strict 1∶1 orthologs between human and the other primates being analyzed. In fact, reciprocal searches of the human genome with primate orthologs continually returned large numbers of mostly un-annotated human genes. In all, we identified a group of 31 human TRIM genes that form a single monophyletic clade to the exclusion of all other TRIM genes in the human genome (Figure 2A). The clade includes seven TRIM genes previously assigned standard TRIM names (including the four used as queries, bold type) and 24 uncharacterized paralogs. Uncharacterized genes were given temporary names reflecting their phylogenetic subclade (i.e. A1 and A2 are two genes in the ‘A’ subclade shown in Figure 2A), but actual locus identifiers for each gene are given in Table 1. Of these 31 genes, 20 have full-length open reading frames that are predicted to encode tripartite motifs either with or without a C-terminal B30.2 domain (Figures S1, S2). Using RT-PCR on testes mRNA, we were able to identify processed transcripts for 11 of these 20 full-length genes, and when combined with cDNA reads available in Genbank, 14 out of 20 genes have evidence for expression and splicing (Figure S2). Thus, we have identified a large set of previously undiscovered human TRIM genes, most of which appear to have protein coding potential.
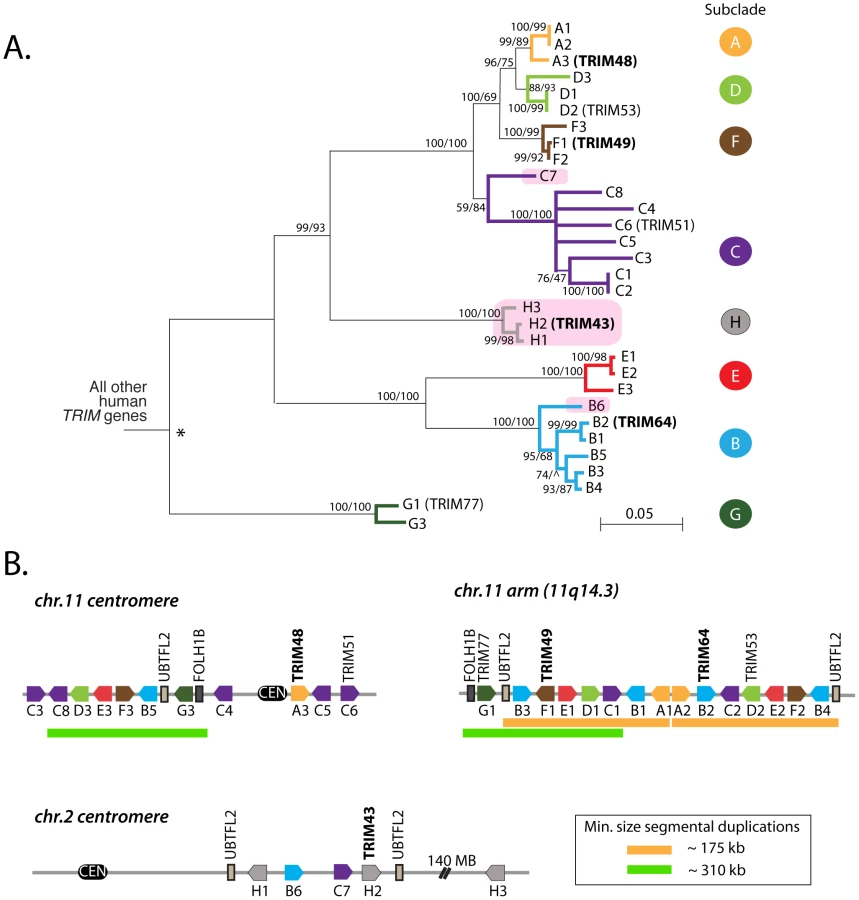

The 31 human TRIM genes in this clade are located at three genomic loci, one near the centromere of chromosome 2, one spanning the centromere of chromosome 11, and one on the arm of chromosome 11 at 11q14.3 (Figure 2B). When the genes in this schematic were color-coded to reflect the subclades in Figure 2A, it became clear that they arose through a series of segmental duplications. For example, the cluster of TRIM genes located on the chromosome 11 arm contains a mirror-image tandem inversion of 7 TRIM genes (denoted by two orange bars in Figure 2B). A second duplication event is evident in the region directly adjacent to the chromosome 11 centromere, where a stretch of 6 TRIM genes appears to be an inverted copy of part of the sequence located on the chromosome 11 arm (denoted by green bars in Figure 2B). The genes located near the chromosome 2 centromere cluster phylogenetically with those found on chromosome 11, suggesting that this may be yet another segmental copy, although the gene order is sufficiently degraded that we cannot draw any clear conclusions.
For discussion purposes, we denote the regions containing these apparent duplications on chromosome 11 as segment 1, segment 2, and segment 3, as illustrated in Figure 3. The duplicated chromosomal regions bearing these three segments are large, but we have focused only on the portion that contains TRIM genes. A careful inspection of the chimpanzee, orangutan, and rhesus macaque genomes in these regions was then performed (Figure 3). Segment 1 is found in all of these primate genomes, while segment 2 is found in the chimpanzee and human genomes only. In support of the young age of segment 2, segments 1 and 2 in the human genome are 96% identical along their length (calculated for 302 kilobase; Table S1). The identification of segment 2 in the genomes of orangutan and rhesus macaque is somewhat complicated by a large chromosomal inversion that has been reported in the region of the chromosome 11 centromere, which arose in the common ancestor of human, chimpanzee, and gorilla [37]. However, this segmental duplication has been previously analyzed by FISH, using BAC-derived probes that anneal in both segment 1 and segment 2 [37]. In that study, all primate species investigated showed a hybridization signal on the chromosome 11 arm at the location of segment 1. A second hybridization signal at the chromosome 11 centromere (segment 2) was observed in the genomes of human, chimpanzee, and gorilla, but not of orangutan, rhesus macaque, or any other primate tested, congruent with our conclusions made through comparative genomics. Therefore, segment 2 is a segmental duplication of segment 1 that is specific to humans, chimpanzees, gorillas, and presumably the final species of this monophyletic clade of African apes, bonobos. This is in agreement with a previous age estimate of 14 million years, based on sequence divergence, for the segmental duplication containing segment 2 [38].

Interestingly, segment 3 is found only in the human genome (Figure 3). The very recent acquisition of this segment as another copy of segment 1 is supported by the observation that segments 1 and 3 are 99.6% identical along their length (calculated for 170 kilobase; Table S1). In intra-chromosomal comparisons of all segmental duplications on chromosome 11, only 0.2 MB was found to have this level of identity [39], consistent with segment 3 being one of the newest segmental duplications on the entire chromosome. It is curious to note that genes A1 and A2, located in tail-to-tail fashion, are 100% identical along their length (over 6 kilobase) in the human genome, but only 96–98% identical in the genomes of orangutan or chimpanzee (Figure 3). A gene conversion event between A1 and A2 in the human genome may have accompanied or seeded the tandem inversion of segment 1 to create segment 3.
In summary, we have identified species - and human-specific TRIM genes on chromosome 11. The chromosomal region bearing segment 1 has been highly dynamic during the evolution of humans and African apes, seeding at least 2 segmental duplications in the last 18 million years since their last common ancestor with orangutan. There appears to be a gross dichotomy in the timing of TRIM gene acquisition by the human genome because, while the majority of human TRIM genes are ancient and arose >90 million years ago, the rest of them (approximately 20% of the TRIM genes in our genome) have arisen in very recent time, during the evolution of the great apes.
Copy number variation
Because the locus containing segment 1 has spawned multiple segmental duplications in recent primate history, we wished to determine whether there are also newer segmental duplications of this region that might be polymorphic in the human population. Genomic regions containing polymorphic segmental duplications or deletions greater than 1 kilobase in size are called ‘copy number variable’ (CNV) regions [40]. To characterize the population genetics of this locus, we employed the multiplex ligation-dependent probe amplification (MLPA) assay [41]. This is a PCR-based assay that utilizes a fragment analyzer to quantify the amount of product generated from different target regions in a genome (Figure 4A). Eighteen probe pairs were designed to tile across the length of segment 1 (Figure 4B and Table S4). Each probe in each pair anneals to 23–63 bases of genomic DNA, and is a perfect match to a target sequence in segment 1. Because of the high degree of similarity between segment 1 and segment 3, probes will also anneal to the cognate locus in segment 3 with perfect complementarily. However, probe pairs were carefully situated such that they have multiple mismatches to the corresponding sequence in segment 2, or to sequence anywhere else in the human genome (see materials and methods). Thus, each probe is expected to have four binding sites in a diploid genome because there will be two copies of each segment 1 and segment 3 target. The one exception is the probe pair “M-uniq,” which sits in a unique stretch of sequence between segments 1 and 3 and will have only two binding sites in a diploid genome. Control probe pairs that recognize standard single-copy genes distributed throughout the genome were also included (see materials and methods). Initially, 50 genomic DNA samples from individuals from around the world were analyzed. For each probe pair, the quantity of products produced from each sample was normalized to the quantity produced from a reference genome, as is standard for this assay. As the reference, we used the genome of a Caucasian male from Utah (NA10851) that has been previously used as the reference genome in several studies of CNV regions [40], [42]–[44].

The normalized fragment values for 12 representative genomes are graphed in Figure 4C, and values for all 50 surveyed genomes are presented in Table S5. We identified only one CNV, in the genome of a Han Chinese female (NA18573). In this individual, the 16 probe pairs spanning from GAP1-1 to M-uniq all yielded approximately 1.5 times the quantity of fragments as the reference genome (average enhancement across all 16 probe pairs = 1.6). This was verified in eight independent experiments (not shown). Therefore, this Han Chinese individual has two additional binding sites for each of these probe pairs, and one additional binding site for the M-uniq probe pair. This pattern is consistent with the segmental duplication scenario diagrammed in Figure 4D. A signal at this locus was previously detected in this same individual as part of a whole-genome array-based study that identified 1,447 human CNV regions [40]. Thus, we have identified a human individual of Chinese decent that has 43 TRIM genes belonging to this dynamic phylogenetic group instead of the 31 TRIM genes which most human individuals have.
Based on this finding, we then screened 22 additional Han Chinese samples, but did not find another instance of this segmental duplication (Table S5). In all, 72 human genomes were analyzed by MLPA (human samples are listed in Table S6). The polymorphism thus seems to be rare, as it was detected in only 1/72 human individuals. However, CNVs have also been detected at this locus, using array-based platforms, in the genomes of several other Asians, including 2 Japanese, 2 Koreans, and 1 additional Chinese individual (Figure S5) [40], [42], [44]. There is one report of a CNV at this locus in the genome of a Yoruban from Africa [43]. Perhaps most interestingly of all, the region containing segment 1 is also copy number variable in chimpanzees and rhesus macaques [24]. In summary, the region containing segment 1 has been highly dynamic both during primate speciation, and also in current human and primate populations.
Positive selection of young TRIM genes
All of the findings described so far can potentially be explained as random events occurring in a dynamic genome. As segmental duplications arise, they may go to fixation through neutral drift even if there is no selection acting for or against them. A hallmark of genes that are being retained by neutral drift is that they accumulate equal rates of non-synonymous and synonymous mutations. Such genes have a characteristic signature of dN/dS = 1, where dN is the number of non-synonymous mutations per non-synonymous site, and dS is the number of synonymous mutations per synonymous site. In contrast, most functional genes accumulate non-synonymous mutations at a rate far slower than synonymous mutations (dN/dS<<1) due to the evolutionary constraint at play [45]. A third mode of evolution, recurrent positive selection, has influenced several TRIM genes in primate genomes, including Pyrin/TRIM20 [46], TRIM5 [28], [32], and TRIM22 [27]. Genes or gene regions subject to such a selective regime accumulate a characteristic signature of dN/dS>1 [47].
We analyzed the evolutionary pressures that have shaped these young TRIM genes at the sequence level in order to determine whether they have been neutrally or selectively retained. Usually, evolutionary datasets of orthologous sequences are used for such analyses, but because these genes are so new and dynamic, deep species sets of strictly orthologous sequences cannot be easily obtained. Instead, we looked at the patterns of nucleotide substitution that have occurred during the diversification of these genes by comparing human paralogs, all of which can be traced to a common ancestral gene (asterisk in Figure 2A). Of the 31 TRIM genes in the dynamic clade being investigated, 15 are predicted to encode proteins with the full TRIM-B30.2 structure (Figure S2). However, of these, two very recently diverged gene pairs (A1/A2 and C1/C2) are still identical along the length of their coding sequence (Table S2), leaving 13 unique sequences which can be analyzed.
Importantly, analysis of sequence evolution requires an accurate phylogenetic representation of the genes being analyzed [48]. One problem with understanding the phylogenetic relationship of paralogs from a single genome is the fact that gene conversion may have occurred. To detect phylogenetic incongruencies in our alignment, indicative of such events, we used the GARD program [49] as described in the materials and methods. Only one phylogenetic breakpoint was identified (p<0.05), located between the RING and B-box2 encoding domains of the first protein-coding exon (Figure 5A). The alignment of the 13 TRIM genes was subsequently divided at this point and the trees produced by each half are shown in Figure 5C. Only two branches differ between the trees (highlighted in red), suggesting that gene conversion has not been extensive. The tree for each half is highly supported, regardless of the phylogenetic method utilized (Figure 5C), or whether just sites at the third positions of codons are utilized (data not shown). With these trees, each half of the multiple alignment was analyzed separately using the codeml package in PAML (see materials and methods). We analyzed each half of the alignment separately, under variable models of selection and codon usage (Table S3). All models yielded strong support for positive selection acting on both halves of the gene (p<0.05). Each of the two trees has one poorly supported node (highlighted in green; Figure 5C). We confirmed that support for positive selection remains strong when each of these nodes is collapsed (p<0.05; Table S3). Therefore, there is convincing evidence that these new TRIM genes have not been retained by neutral drift alone.

dN/dS values were calculated for each branch on the two trees. On these trees, there are many branches where dN/dS>1 (text highlighted red; Figure 5C). In fact, the dN/dS values along some of the gene lineages are remarkable. For example, there have been 17 non-synonymous and 0 synonymous mutations that have accumulated during the divergence of the B5 gene (6 non-synonymous in the first half and 11 in the second half). The identical genes C1 and C2 have accumulated 25 non-synonymous changes and 0 synonymous changes since they shared a last common ancestor with the other genes of the ‘C’ clade, a stunningly intense episode of positive selection. Collapsing of the poorly supported node in each tree only marginally affects the results (Figure S4). Such extreme evolutionary patterns are unusual, but have been documented previously in other viral restriction factors due to the intense evolutionary arms races in which these genes are engaged [26], [28], [33], [34], [50].
These analyses can identify specific codon positions, and corresponding amino acid residues, that have repeatedly been subject to positive natural selection. Ten rapidly evolving codons were identified (Table S3), as illustrated with tick marks on the protein schematic in Figure 5A. Five of these fall in or near the RING domain (residues F48, V50, E54, E60, H69 in TRIM49/F1 coordinates), three in the coiled-coil domain (R166, C167, R222), and two in the B30.2 domain (Y320, A323).
Using secondary structure prediction and alignments to other TRIM proteins, we determined that rapidly evolving residues Y320 and A323 fall in a small loop (11–16 aa long) that lies between the second and third β-strands of the B30.2 domain (Figure S3). This surface-exposed “variable loop 1” (Figure 5B) has been rather well characterized, at least in the case of TRIM5α. In this protein, this loop is known to be a major determinant of recognition for retroviral capsids, and presumably constitutes the major binding interface with retroviruses. Sequence variation in this loop of TRIM5α accounts largely for the species-specific viral restriction patterns observed in various primate and mammalian species [28], [51]–[53]. It is hypothesized that the TRIM5 gene has been engaged in an arms race with retroviruses throughout the diversification of mammals, and that natural selection has driven rapid sequence evolution of this loop for improved recognition of constantly changing retroviruses [47], [54]. Accordingly, there are multiple sites of positive selection in the variable loop 1 region of TRIM5α from primates [28], cows [27], and rabbits and hares [55], all of which restrict mammalian retroviruses. It is intriguing that the young TRIM genes identified here should have two codons evolving under positive selection in the B30.2 domain, with both of them falling in this small surface loop known to interact with retroviral capsids.
Three rapidly evolving residues were also identified in the coiled-coil domain (Figure 5A). In TRIM5α, the coiled-coil domain is the second domain that participates in retroviral target specificity [51], [56]. The rapidly evolving residues identified here are in regions shown to be critical in defining virus-specificity in TRIM5α [56], and known to contain codons evolving under positive selection in the TRIM5 gene (Figure S3). In summary, positive selection has acted on these young TRIM genes in regions analogous to the major determinants of retroviral specificity in TRIM5α, suggesting that the novel genes could be retroviral restriction factors.
Based on the evolutionary signatures observed, we next tested whether these genes might encode retroviral restriction factors. We chose some of the genes with the highest branch-specific dN/dS values for functional testing, ones which could also be amplified in their complete form from human mRNA samples. These candidates (A1, B1, B5, F1/TRIM49, F2, and F3) are indicated with stars in Figure 5C. Except for F1/TRIM49, none of these genes have ever been previously studied. We tested the ability of these genes to restrict cellular entry of three different mammalian retroviruses which are known to be restricted by human and/or rhesus macaque TRIM5α: feline immunodeficiency virus (FIV), HIV, and murine leukemia virus (MLV). Interestingly, the young human TRIM genes did not restrict these retroviruses (Figure S6). If these genes do have anti-retroviral activity, these data suggest that their specificity is different than that of TRIM5α, consistent with the high degree of divergence observed in the regions that control target specificity. Perhaps these TRIM genes were honed to target retroviruses that are now extinct [57], [58]. Alternately, these TRIM genes may target other virus families altogether. For instance, TRIM22 has experienced positive selection in these same retroviral targeting motifs, and while this gene may be relevant to retroviral infection [59], it also has activity against hepatitis B [60] and picornaviruses [61].
Discussion
Here we identify and characterize what are likely to be the youngest TRIM genes in the human genome. While the ∼100 human TRIM genes are for the most part ancient, we now show that a substantial number of them (approximately 20%, not counting additional TRIM gene copies that are polymorphic in the human population) have arisen in recent evolutionary time, during the speciation of the great apes. Many of these genes have full-length open reading frames and produce spliced transcripts. Almost all have arisen from segmental duplications that can be traced to a single locus on the arm of chromosome 11. We propose that the segment 1 region on the arm of chromosome 11 is a TRIM gene “factory,” producing copies of the genes that it contains by spawning segmental duplications around the genome. Increased dosage of these genes may, in itself, be adaptive. Further, depending on the chromosomal context of new segmental duplications, the genes that they contain may be expressed in different tissues or at different developmental stages. However, we also find that positive selection is rapidly shaping the sequence of these genes, such that new copies may quickly become specialized for new functions or specificities. It will be important to determine what role, if any, these young TRIM genes play in innate immunity, particularly because these genes are copy number variable in human and primate populations.
Several insights into the evolutionary dynamics of large gene families can be gained from this study. TRIM genes are found throughout the human genome and the segmental duplications described here may illustrate one mechanism by which this family has expanded over time. Because all gene duplicates start out as polymorphisms segregating in populations, the observed copy number variation suggests that this gene family is still growing. Several lines of evidence support the idea that the fixation of new TRIM paralogs in the human genome has, at least in part, been an adaptive rather than a neutral process. First, TRIM5 [28], TRIM22 [27], Pyrin/TRIM20 [46], and many of the TRIM genes described herein, have all evolved under positive selection, accumulating unexpectedly high numbers of non-synonymous mutations. New genes are especially prone to positive selection, probably because redundant gene copies provide templates for the evolution of new functions [24], [62]. Second, some TRIM genes have been highly dynamic in terms of species-specific gene gain and loss during mammalian evolution. For instance, cows and rodents possess independent, species-specific expansions of tandemly situated TRIM5 paralogs, while dogs and cats have independently lost the function of this gene [27], [54], [63]. Likewise, the TRIM genes in segment 1 on chromosome 11 are also highly dynamic, having spawned at least two segmental duplications in African apes and another that is now polymorphic in the human population. Immunity genes are, in general, overrepresented amongst mammalian gene families that show rapid gene gain and loss, suggesting that these events are often adaptive [64]. Third, the acquisition of the young TRIM genes has not come without a cost to the human genome. Unequal crossing-over and aberrant homologous recombination between the tandem segments that contain TRIM50, TRIM73, and TRIM74 causes Williams-Beuren syndrome in 1/7,500 to 1/25,000 newborns [65]. This fitness consequence might be expected to be offset by a fitness advantage, otherwise these regions would be selectively lost from the genome.
Historically, studies of human genetic variation have focused almost exclusively on single nucleotide polymorphism (SNP) differences between individuals. Recently, it has become apparent that large DNA segments are also commonly polymorphic between individuals, resulting from recent segmental duplications and deletions. CNV regions can be associated with disease, usually related to the altered gene dosage that they convey [66]. Another negative consequence of CNV duplications is that blocks of nearly identical sequence interspersed in a genome can create a volatile landscape for recombination. However, positive attributes can also be imagined for CNV regions, such as benefits that might be gained from increased dosage of certain genes. Such a fitness advantage has been suggested for the salivary amylase gene (AMY1), which is found in higher copy number in populations with higher starch diets [67]. Here we propose that CNV regions can also be a positive, adaptive force in genomes by driving the generation and diversification of gene families important to human immunity. For all of these reasons, studies of CNV regions are important for understanding individual disease susceptibility.
Materials and Methods
Gene sequences and phylogenetic analysis
Refseq annotated human coding sequences of genes of interest were downloaded from Genbank. Analysis of human – chimpanzee – orangutan – rhesus macaque orthogroups was performed by reciprocal-best hit analysis performed in the UCSC genome database [68]. Briefly, each human gene was used as a BLAT query [69] against the genome projects of the other species investigated. The top hits from these queries were then used to reciprocally query the human genome. All related sequences were then compiled and subjected to phylogenetic analysis. cDNA or full-gene sequences were aligned using MUSCLE as implemented in MEGA5 [70]. Alternate trees (neighbor joining, maximum likelihood, and maximum parsimony) were constructed within MEGA, with gapped positions excluded. Tree nodes were critically evaluated by performing 1,000 bootstrap replicates. The physical locations of the TRIM genes in the human (hg19), chimpanzee (panTro2), orangutan (ponAbe2), and rhesus macaque (rheMac2) genome assemblies were determined by manual inspection in the UCSC genome browser [68]. It was not possible to determine the structure of the chromosomal 11 loci in the marmoset genome assembly (calJac3), due to poor sequence quality.
Gene expression analysis
Primers were designed to recognize novel TRIM genes (primer sequences are given in Table S7). Each primer set was designed to span at least one intron so that products resulting from processed transcripts could be differentiated from those potentially resulting from contaminating genomic DNA. SuperScript first-strand synthesis system for RT-PCR (Invitrogen) was used to synthesize cDNA from human testis total RNA (Clontech, 636533). PCR Supermix (Invitrogen) or Ex Taq polymerase (Takara) was used to amplify from cDNA. Individual PCR amplicons were cloned into vectors using the TOPO-TA Cloning kit (Invitrogen). For each sample, at least ten different colonies were randomly selected and were sequenced. These sequences have been submitted as records to Genbank (JF968445-JF968463).
Characterizing patterns of molecular evolution
A sequence alignment was created from TRIM genes that have a full-length open reading frame (RING through B30.2). The tree length of this dataset is approximately 5 [73]. First, this alignment was checked for the signatures of gene conversion. If gene conversion of one paralog by another has occurred along the entire length of the two genes, this will not present a problem because a gene phylogeny will correctly reflect the fact that these two genes now have a very recent common ancestor and have been diverging only since the gene conversion event (although the record of previous evolutionary adaptations will have been erased in the converted gene). Problems occur when a gene conversion event has affected only part of a gene, as each gene half will then have a different location on the phylogenetic tree and no single tree will accurately represent the evolutionary history of the entire gene. To detect such events, the alignment was checked for phylogenetic incongruencies with the GARD program [49] implemented in Datamonkey [71]. Once the breakpoint had been identified, the tree structure for each half of the alignment was checked with multiple bootstrapping algorithms using MEGA5 as described above. Using the two halves of the alignment and the corresponding trees, maximum likelihood analysis was performed with codeml in the PAML 3.14.1 software package [72]. The multiple alignments were fitted to the NSsites models M1a, M7, M8a (null models) and M2a, M8 (positive selection models). Simulations were run with alternate models of codon frequencies (f3x4 and f61), and with multiple seed values for dN/dS (ω). Likelihood ratio tests were performed to assess whether positive selection models provide a significantly better fit to the data than null models. In situations where the null model could be rejected (p<0.05), posterior probabilities were assigned to individual codons belonging to the class of codons with dN/dS>1 with the Naive Empirical Bayes (NEB) algorithm implemented in codeml. The free ratio model (model 1, one dN/dS per branch) was also run in codeml to assess branch-specific values of dN/dS.
Generation of stable cell lines
HA-tagged versions of human and rhesus TRIM5 in the LPCX retroviral vector were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program. TRIM A1, B1, F1, F2, and F3 open reading frames were amplified from human cDNA using primers shown in Table S7. We were unable to amplify B5 in its full length form, so in this case we fused the B30.2 domain of this gene to the tripartite domains of rhesus TRIM5. HA tags were fused to the C-terminus of each gene using PCR and these products were cloned into the LPCX retroviral vector (Clontech). Retroviruses containing these vectors were packaged in 293T cells by co-transfecting them along with the NB-MLV packing plasmid pCS2-mGP [74] and pC-VSV-G (provided by Hyeryun Choe). Supernatants were collected and used to infect CRFK cells purchased from American Type Culture Collection (ATCC) and grown in DMEM supplemented with 10% FBS. After 24 hours, media containing 8 µg/ml puromycin was added to select for transduced cells. Expression of TRIM proteins was detected by Western blot of 30 µg total protein using an anti-HA antibody (3F10, Roche, catalog 1867431).
Viral entry assays
Viruses for single-cycle infection assays were packaged in 293T cells by co-transfection of plasmids encoding viral proteins and VSV-G, along with a transfer vector, as follows: N-MLV (pCIG3-N [75], pC-VSV-G, pLXCG:GFP), HIV-1 (pMDLg/pRRE, pRSV-Rev, pMD2.G, pRRLSIN.cPPT.PGK-GFP.WPRE; all available on Addgene), FIV (pFP93 [76], pC-VSV-G, pGINSIN:GFP [77]). After 48 hours, supernatant containing viruses was harvested, filtered, and frozen. For infection assays, CRFK stable cell lines were plated at a concentration of 5×104 cells/well in a 12-well plate and infected with N-MLV, FIV, or HIV-1 the following day. Two days post-infection, cells were analyzed by flow cytometry for expression of GFP.
Multiplex ligation-dependant probe amplification (MLPA) assay
We utilized the SALSA MLPA kit P200 Human DNA reference-1 and associated protocol (MRC-Holland, Amsterdam, The Netherlands). This kit includes the human control probes utilized. Our custom probe set was designed to contain eighteen pairs of MLPA probes spanning segment 1 (Table S4). These probes also perfectly match paralogous regions in segment 3, due to the fact that these segments are nearly identical, but are designed to contain at least two mismatches to all other paralogous sequences located on chromosomes 2 or 11 (or anywhere else in the human genome, as determined by BLAT searching on the UCSC human genome browser). Probes were positioned both in genes (8 probe pairs) and in intergenic regions (10 probe pairs). The average distance between probe pairs is 9304 bp. PCR primers supplied with this kit were fluorescently labeled with FAM, and FAM-labeled fragments obtained from each experiment were analyzed with an Applied Biosystems 3730 DNA analyzer. Peak spectra were checked for quality in two ways. First, the spectra were analyzed with the ABI software Peak Scanner (v.1.0) to evaluate the fragment size quality using a size standard that was included during fragment analysis (500ROX, ABI). If the quality flag indicated “pass,” the samples including their fragment size information were exported as a combined table. Second, the MRC-Holland software Coffalyzer (v.8) was used to evaluate the signals of the control probes supplied with the MLPA kit. Controls are designed to confirm sufficient amounts of template DNA and completion of DNA denaturation and ligation steps. Finally, GeneMarker (v.1.7) software was used to normalize and analyze MLPA experiments that passed both of these quality control steps. Advanced population normalization was used and MLPA analysis settings were as follows: MLPA ratio (analysis method), adjustment by control probes, and quantification by peak height. After normalization of fragment data to the reference genome (sample NA10851 from Utah), duplications and deletions were defined as probes that gave a signal intensity of >1.35 (duplication) or <0.65 (deletion) that of the reference genome. Because the samples analyzed were a mixture of male and female samples, control probes on the X and Y chromosomes were used to show that these enrichment and depletion thresholds are robust in predicting gain and loss of control targets located on sex chromosomes (Table S5). Because false signals may be caused by unknown SNPs at the target locus or elsewhere, signals of enrichment or depletion seen only with a single probe pair were disregarded.
Supporting Information
Zdroje
1. McNabFWRajsbaumRStoyeJPO'GarraA 2011 Tripartite-motif proteins and innate immune regulation. Curr Opin Immunol 23 46 56
2. SardielloMCairoSFontanellaBBallabioAMeroniG 2008 Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol Biol 8 225
3. NisoleSStoyeJPSaïbA 2005 TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Micro 3 799 808
4. PertelTHausmannSMorgerDZügerSGuerraJ 2011 TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472 361 365
5. StremlauMOwensCMPerronMJKiesslingMAutissierP 2004 The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427 848 853
6. Ganser-PornillosBKChandrasekaranVPornillosOSodroskiJGSundquistWI 2011 Hexagonal assembly of a restricting TRIM5 protein. Proc Natl Acad Sci USA 108 534 539
7. StremlauMPerronMLeeMLiYSongB 2006 Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc Natl Acad Sci USA 103 5514 5519
8. ZhaoGKeDVuTAhnJShahVB 2011 Rhesus TRIM5a Disrupts the HIV-1 Capsid at the Inter - Hexamer Interfaces. PLoS Pathog 7 e1002009 doi:10.1371/journal.ppat.1002009
9. TareenSUEmermanM 2011 Human Trim5α has additional activities that are uncoupled from retroviral capsid recognition. Virology 409 113 120
10. KirmaierAWuFNewmanRMHallLRMorganJS 2010 TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol 8 e1000462 doi:10.1371/journal.pbio.1000462
11. HatziioannouTPrinciottaMPiatakMYuanFZhangF 2006 Generation of simian-tropic HIV-1 by restriction factor evasion. Science 314 95
12. NewmanRMJohnsonWE 2007 A brief history of TRIM5alpha. AIDS Rev 9 114 125
13. KaiserSMMalikHSEmermanM 2007 Restriction of an Extinct Retrovirus by the Human TRIM5 Antiviral Protein. Science 316 1756 1758
14. KuoR-LZhaoCMalurMKrugRM 2010 Influenza A virus strains that circulate in humans differ in the ability of their NS1 proteins to block the activation of IRF3 and interferon-β transcription. Virology 408 146 158
15. GackMUAlbrechtRAUranoTInnK-SHuangI-C 2009 Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5 439 449
16. ZengWSunLJiangXChenXHouF 2010 Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 141 315 330
17. PooleEGrovesIMacdonaldAPangYAlcamiA 2009 Identification of TRIM23 as a Cofactor Involved in the Regulation of NF - B by Human Cytomegalovirus. J Virol 83 3581 3590
18. WangJLiuBWangNLeeY-MLiuC 2011 TRIM56 is a virus - and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection. J Virol 85 3733 3745
19. GeoffroyM-CChelbi-AlixMK 2011 Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res 31 145 158
20. CarthagenaLBergamaschiALunaJMDavidAUchilPD 2009 Human TRIM gene expression in response to interferons. PLoS ONE 4 e4894 doi:10.1371/journal.pone.0004894
21. MeroniGDiez-RouxG 2005 TRIM/RBCC, a novel class of “single protein RING finger” E3 ubiquitin ligases. Bioessays 27 1147 1157
22. MastersSLYaoSWillsonTAZhangJ-GPalmerKR 2006 The SPRY domain of SSB-2 adopts a novel fold that presents conserved Par-4-binding residues. Nat Struct Mol Biol 13 77 84
23. TareenSUSawyerSLMalikHSEmermanM 2009 An expanded clade of rodent Trim5 genes. Virology 385 473 483
24. GokcumenOBabbPLIskowRZhuQShiX 2011 Refinement of primate CNV hotspots identifies candidate genomic regions evolving under positive selection. Genome Biol 12 R52
25. PerryGHTchindaJMcGrathSDZhangJPickerSR 2006 Hotspots for copy number variation in chimpanzees and humans. Proc Natl Acad Sci USA 103 8006 8011
26. SawyerSLEmermanMMalikHS 2004 Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol 2 e275 doi:10.1371/journal.pbio.0020275
27. SawyerSLEmermanMMalikHS 2007 Discordant evolution of the adjacent antiretroviral genes TRIM22 and TRIM5 in mammals. PLoS Pathog 3 e197 doi:10.1371/journal.ppat.0030197
28. SawyerSLWuLIEmermanMMalikHS 2005 Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci USA 102 2832 2837
29. GuptaRKHuéSSchallerTVerschoorEPillayD 2009 Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog 5 e1000443 doi:10.1371/journal.ppat.1000443
30. LimESMalikHSEmermanM 2010 Ancient adaptive evolution of Tetherin shaped Vpu and Nef functions in human immunodeficiency virus and primate lentiviruses. J Virol 1 38
31. McNattMWZangTHatziioannouTBartlettMFofanaIB 2009 Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog 5 e1000300 doi:10.1371/journal.ppat.1000300
32. LiuH-LWangY-QLiaoC-HKuangY-QZhengY-T 2005 Adaptive evolution of primate TRIM5alpha, a gene restricting HIV-1 infection. Gene 362 109 116
33. EldeNCChildSJGeballeAPMalikHS 2009 Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457 485 489
34. KernsJAEmermanMMalikHS 2008 Positive selection and increased antiviral activity associated with the PARP-containing isoform of human zinc-finger antiviral protein. PLoS Genet 4 e21 doi:10.1371/journal.pgen.0040021
35. WlasiukGNachmanMW 2010 Adaptation and constraint at Toll-like receptors in primates. Mol Biol Evol 27 2172 2186
36. MicaleLFuscoCAugelloBNapolitanoLMRDermitzakisET 2008 Williams-Beuren syndrome TRIM50 encodes an E3 ubiquitin ligase. Eur J Hum Genet 16 1038 1049
37. CardoneMFLomientoMTetiMGMisceoDRobertoR 2007 Evolutionary history of chromosome 11 featuring four distinct centromere repositioning events in Catarrhini. Genomics 90 35 43
38. ZhangJQinSSaitSNHaleyLLHenryWM 2001 The pericentromeric region of human chromosome 11: evidence for a chromosome-specific duplication. Cytogenet Cell Genet 94 137 141
39. TaylorTDNoguchiHTotokiYToyodaAKurokiY 2006 Human chromosome 11 DNA sequence and analysis including novel gene identification. Nature 440 497 500
40. RedonRIshikawaSFitchKRFeukLPerryGH 2006 Global variation in copy number in the human genome. Nature 444 444 454
41. SchoutenJPMcElgunnCJWaaijerRZwijnenburgDDiepvensF 2002 Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 30 e57
42. ParkHKimJ-IJuYSGokcumenOMillsRE 2010 Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing. Nat Genet 42 400 405
43. PerryGHBen-DorATsalenkoASampasNRodriguez-RevengaL 2008 The fine-scale and complex architecture of human copy-number variation. Am J Hum Genet 82 685 695
44. KimJ-IJuYSParkHKimSLeeS 2009 A highly annotated whole-genome sequence of a Korean individual. Nature 460 1011 1015
45. HurstLD 2002 The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet 18 486
46. SchanerPRichardsNWadhwaAAksentijevichIKastnerD 2001 Episodic evolution of pyrin in primates: human mutations recapitulate ancestral amino acid states. Nat Genet 27 318 321
47. MeyersonNRSawyerSL 2011 Two-stepping through time: mammals and viruses. Trends Microbiol 19 286 294
48. ShrinerDNickleDJensenMMullinsJ 2003 Potential impact of recombination on sitewise approaches for detecting positive natural selection. Genet Res 81 115 121
49. PondSLPosadaDGravenorMBWoelkCHFrostSDW 2006 Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol 23 1891 1901
50. OhAinleMKernsJAMalikHSEmermanM 2006 Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J Virol 80 3853 3862
51. YapMWNisoleSStoyeJP 2005 A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr Biol 15 73 78
52. StremlauMPerronMWelikalaSSodroskiJ 2005 Species-specific variation in the B30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiency virus restriction. J Virol 79 3139 3145
53. Perez-CaballeroDHatziioannouTYangACowanSBieniaszPD 2005 Human tripartite motif 5alpha domains responsible for retrovirus restriction activity and specificity. J Virol 79 8969 8978
54. JohnsonWESawyerSL 2009 Molecular evolution of the antiretroviral TRIM5 gene. Immunogenetics 61 163 176
55. FletcherAJHuéSSchallerTPillayDTowersGJ 2010 Hare TRIM5α restricts divergent retroviruses and exhibits significant sequence variation from closely related lagomorpha TRIM5 genes. J Virol 84 12463 12468
56. MaillardPVEccoGOrtizMTronoD 2010 The specificity of TRIM5 alpha-mediated restriction is influenced by its coiled-coil domain. J Virol 84 5790 5801
57. YohnCTJiangZMcGrathSDHaydenKEKhaitovichP 2005 Lineage-specific expansions of retroviral insertions within the genomes of African great apes but not humans and orangutans. PLoS Biol 3 e110 doi:10.1371/journal.pbio.0030110
58. LanderESLintonLMBirrenBNusbaumCZodyMC 2001 Initial sequencing and analysis of the human genome. Nature 409 860 921
59. SinghRGaihaGWernerLMcKimKMlisanaK 2011 Association of TRIM22 with Type 1 Interferon Response and Viral Control during Primary HIV-1 Infection. Journal of Virology 85 208 216
60. GaoBDuanZXuWXiongS 2009 Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology 50 424 433
61. EldinPPaponLOteizaABrocchiELawsonTG 2009 TRIM22 E3 ubiquitin ligase activity is required to mediate antiviral activity against encephalomyocarditis virus. J Gen Virol 90 536 545
62. HanMVDemuthJPMcGrathCLCasolaCHahnMW 2009 Adaptive evolution of young gene duplicates in mammals. Genome Res 19 859 867
63. McEwanWASchallerTYlinenLMHosieMJTowersGJ 2009 Truncation of TRIM5 in the Feliformia explains the absence of retroviral restriction in cells of the domestic cat. J Virol 83 8270 8275
64. BarreiroLBQuintana-MurciL 2010 From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet 11 17 30
65. BayésMMaganoLFRiveraNFloresRPérez JuradoLA 2003 Mutational mechanisms of Williams-Beuren syndrome deletions. Am J Hum Genet 73 131 151
66. LupskiJRStankiewiczP 2005 Genomic Disorders: Molecular Mechanisms for Rearrangements and Conveyed Phenotypes. PLoS Genet 1 e49 doi:10.1371/journal.pgen.0010049
67. PerryGHDominyNJClawKGLeeASFieglerH 2007 Diet and the evolution of human amylase gene copy number variation. Nat Genet 39 1256 1260
68. KentWJSugnetCWFureyTSRoskinKMPringleTH 2002 The Human Genome Browser at UCSC. Genome Res 12 996 1006
69. KentWJ 2002 BLAT—The BLAST-Like Alignment Tool. Genome Res 12 656 664
70. TamuraKPetersonDPetersonNStecherGNeiM 2011 MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol in press
71. DelportWPoonAFYFrostSDWKosakovsky PondSL 2010 Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26 2455 2457
72. YangZ 1997 PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13 555 556
73. AnisimovaMBielawskiJPYangZ 2001 Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol 18 1585 1592
74. YamashitaMEmermanM 2004 Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J Virol 78 5670 5678
75. BockMBishopKNTowersGStoyeJP 2000 Use of a transient assay for studying the genetic determinants of Fv1 restriction. J Virol 74 7422 7430
76. BarrazaRAPoeschlaEM 2008 Human gene therapy vectors derived from feline lentiviruses. Vet Immunol Immunopathol 123 23 31
77. LoewenNBarrazaRWhitwamTSaenzDTKemlerI 2003 FIV Vectors. Methods Mol Biol 229 251 271
78. PerelmanPJohnsonWERoosCSeuánezHNHorvathJE 2011 A molecular phylogeny of living primates. PLoS Genet 7 e1001342 doi:10.1371/journal.pgen.1001342
79. HedgesSB 2002 The origin and evolution of model organisms. Nat Rev Genet 3 838 849
80. ParkEYKwonO-BJeongB-CYiJ-SLeeCS 2010 Crystal structure of PRY-SPRY domain of human TRIM72. Proteins 78 790 795
81. CuffJAClampMESiddiquiASFinlayMBartonGJ 1998 JPred: a consensus secondary structure prediction server. Bioinformatics 14 892 893
82. JamesLCKeebleAHKhanZRhodesDATrowsdaleJ 2007 Structural basis for PRYSPRY-mediated tripartite motif (TRIM) protein function. Proc Natl Acad Sci USA 104 6200 6205
83. GrütterCBriandCCapitaniGMittlPREPapinS 2006 Structure of the PRYSPRY-domain: implications for autoinflammatory diseases. FEBS Lett 580 99 106
84. SongBJavanbakhtHPerronMParkDHStremlauM 2005 Retrovirus restriction by TRIM5alpha variants from Old World and New World primates. J Virol 79 3930 3937
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 12
Nejčtenější v tomto čísle
- Targeted Proteolysis of Plectin Isoform 1a Accounts for Hemidesmosome Dysfunction in Mice Mimicking the Dominant Skin Blistering Disease EBS-Ogna
- The RNA Silencing Enzyme RNA Polymerase V Is Required for Plant Immunity
- The FGFR4-G388R Polymorphism Promotes Mitochondrial STAT3 Serine Phosphorylation to Facilitate Pituitary Growth Hormone Cell Tumorigenesis
- Target Site Recognition by a Diversity-Generating Retroelement