
A Two-Stage Meta-Analysis Identifies Several New Loci for Parkinson's Disease
A previous genome-wide association (GWA) meta-analysis of 12,386 PD cases and 21,026 controls conducted by the International Parkinson's Disease Genomics Consortium (IPDGC) discovered or confirmed 11 Parkinson's disease (PD) loci. This first analysis of the two-stage IPDGC study focused on the set of loci that passed genome-wide significance in the first stage GWA scan. However, the second stage genotyping array, the ImmunoChip, included a larger set of 1,920 SNPs selected on the basis of the GWA analysis. Here, we analyzed this set of 1,920 SNPs, and we identified five additional PD risk loci (combined p<5×10−10, PARK16/1q32, STX1B/16p11, FGF20/8p22, STBD1/4q21, and GPNMB/7p15). Two of these five loci have been suggested by previous association studies (PARK16/1q32, FGF20/8p22), and this study provides further support for these findings. Using a dataset of post-mortem brain samples assayed for gene expression (n = 399) and methylation (n = 292), we identified methylation and expression changes associated with PD risk variants in PARK16/1q32, GPNMB/7p15, and STX1B/16p11 loci, hence suggesting potential molecular mechanisms and candidate genes at these risk loci.
Published in the journal:
. PLoS Genet 7(6): e32767. doi:10.1371/journal.pgen.1002142
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002142
Summary
A previous genome-wide association (GWA) meta-analysis of 12,386 PD cases and 21,026 controls conducted by the International Parkinson's Disease Genomics Consortium (IPDGC) discovered or confirmed 11 Parkinson's disease (PD) loci. This first analysis of the two-stage IPDGC study focused on the set of loci that passed genome-wide significance in the first stage GWA scan. However, the second stage genotyping array, the ImmunoChip, included a larger set of 1,920 SNPs selected on the basis of the GWA analysis. Here, we analyzed this set of 1,920 SNPs, and we identified five additional PD risk loci (combined p<5×10−10, PARK16/1q32, STX1B/16p11, FGF20/8p22, STBD1/4q21, and GPNMB/7p15). Two of these five loci have been suggested by previous association studies (PARK16/1q32, FGF20/8p22), and this study provides further support for these findings. Using a dataset of post-mortem brain samples assayed for gene expression (n = 399) and methylation (n = 292), we identified methylation and expression changes associated with PD risk variants in PARK16/1q32, GPNMB/7p15, and STX1B/16p11 loci, hence suggesting potential molecular mechanisms and candidate genes at these risk loci.
Introduction
Until the recent developments of high throughput genotyping and genome-wide association (GWA) studies, little was known of the genetics of typical Parkinson's disease (PD). Studies of the genetic basis of familial forms of PD first identified rare highly penetrant mutations in LRKK2 [1], [2], PINK1 [3], SNCA [4], PARK2 [5] and PARK7 [6]. Following these findings, GWA scans for idiopathic PD identified SNCA and MAPT as unequivocal risk loci [7], [8], [9], [10], [11] as well as implicated BST1 [8], GAK [12], and HLA-DR [13]. Using sequence based imputation methods [14], the meta-analysis of several GWA scans [7], [9], [10], [11] conducted by the International Parkinson's Disease Genomics Consortium (IPDGC) identified and replicated five new loci: ACMSD, STK39, MCCC1/LAMP3, SYT11, and CCDC62/HIP1R [15] and confirmed association at SNCA, LRRK2, MAPT, BST1, GAK and HLA-DR [15].
We conducted a two-stage association study. Combining stage 1 and stage 2, the data consist of 12,386 PD cases and 21,026 controls genotyped using a variety of platforms (Table 1). Stage 1 used genome-wide genotyping arrays and our initial analysis [15] focused on the subset of SNPs that passed genome-wide significance in stage 1. For stage 2 genotyping, we used a custom content Illumina iSelect array, the ImmunoChip and additional GWAS typing as previously described [15]. The primary content of the ImmunoChip data focuses on autoimmune disorders but, as part of a collaborative agreement with the Wellcome Trust Case Control Consortium 2, we included 1,920 ImmunoChip SNPs on the basis of the stage 1 GWA PD results.

Here, we report the combined analysis for this full set of 1,920 SNPs. This step1+2 analysis identified seven new loci that passed genome-wide significance in the meta-analysis. During the process of analyzing these data and preparing for publication, we became aware that another group was also preparing a large independent GWA scan in PD for publication (Do et al, submitted). Following discussion with this group we agreed to cross validate the top hits from each study by exchanging summary statistics for this small number of loci.
To provide further insights into the molecular function of these associated variants, we tested risk alleles at these loci for correlation with the expression of physically close gene (expression quantitative trait locus, eQTL) and the methylation status (methQTL) of proximal DNA CpG sites in a dataset of 399 control frontal cortex and cerebellar tissue samples extracted post-mortem from individuals without a history of neurological disorders.
Results
In addition to eleven loci that passed genome-wide significance in stage 1 [15], we identified over 100 regions of interest defined as 10 kb windows containing at least one SNP associated at p<10−3. We submitted the most associated SNP in each region for probe design and follow-up genotyping using the ImmunoChip platform. For each region of interest, we also added four SNPs in high level of linkage disequilibrium (LD) to provide redundancy where the most associated SNP would not pass the Illumina probe design step or the assay for that SNP would fail. To complete the array design we also added all non-synonymous dbSNPs located in known PD associated regions [1], [2], [3], [4], [5], [6]. Out of these 2,400 submitted SNPs, 1,920 passed QC and were included in the final array design. For these 1,920 SNPs we combined stage 1 and stage 2 associated data in a meta-analysis of 12,386 cases and 21,026 controls (Table 1) from the IPDGC. We exchanged summary statistics for these most significant hits with an additional large, case-control replication dataset (3,426 PD cases and 29,624 controls) in an attempt to demonstrate independent replication.
On the basis of stage 1+2 results, seven new SNPs passed our defined genome-wide significance threshold (p<5×10−8, Table 2 and Figure 1). These loci are either novel or the previous evidence of association was not entirely convincing in individuals of European descent. We combined these results with the independent replication. Five of these seven loci replicated and showed strong combined evidence of PD association (p<10−10 overall). Taking either the nearest gene (or the strongest candidate when available) to designate these regions, these five loci are 1q32/PARK16 [7], 4q21/STBD1, 7p15/GPNMB, 8p22/FGF20 [16] and 16p11/STX1B.


rs708723/1q32 has been previously reported as PD associated (PARK16, [7], [8]) but this SNP lacked the unequivocal evidence of association in European samples (p = 9.47×10−10 in stage 2 only). To understand the potential biological consequences of risk variation at this locus we tested whether rs708723 was correlated with either gene expression or DNA methylation status of proximal transcripts or CpG sites respectively (Table 3). We found correlations with the expression of NUCKS1 (p = 1.8×10−7) and RAB7L1 (p = 7.2×10−4). We also found correlations with the methylation state of CpG sites located in the FLJ3269 gene (p = 3.9×10−22).

In the case of 16p11/STX1B, the proximal gene to the most associated SNP rs4889603 is SETD1A. However, STX1B is located 18 kb upstream of rs4889603 and is a more plausible PD candidate gene [17] owing to its synaptic receptor function. We therefore used this gene to designate this region. Our methQTL/eQTL dataset identified a correlation between the rs4889603 risk allele and increased methylation of a CpG dinucleotide in STX1B (Table 3).
The SNP rs591323 in the 8p22 region is located ∼150 kb downstream of the FGF20 gene (NCBI build 36.3), for which association with PD has been suggested previously in familial PD samples [16], [18] but which remained controversial [19]. Our findings provide further support for a PD association at this locus, but again, whether the functionally affected transcript is FGF20 or not remains unclear.
The regions 4q21/STBD1 and 7p15/GPNMD have not been previously implicated in PD etiology. We found that the risk allele of rs156429, the most associated SNP in the 7p15 region, is associated in our eQTL dataset with decreased expression of the proximal transcript encoded by NUPL2 (Table 3). The same risk allele is also associated with increased methylation of multiple CpG sites proximal to GPNMB itself (Table 3). Neither of these regions contains an obvious candidate gene.
Two additional loci (3q26/NMD3 and 8q21/MMP16) showed strong evidence of association in stage 1 and 2 but were not disease associated in the Do et al dataset. Further replication is required to clarify the role of variation at these loci in risk for PD.
The strongly associated G2019S variant in the LRRK2 gene [20] was included in the Immunochip design and we replicated the published association: control frequency: 0.045% case frequency 0.61%, estimated odds ratio: 13.5 with 95% confidence interval: 5.5–43. However, the case collections have been partially screened for this variant therefore its frequency in cases and the odds ratio is likely to be underestimated.
The ImmunoChip array design provides some power to detect whether multiple distinct association signals exist at individual loci. Indeed, if a SNP showed an independent and sufficiently strong association in stage 1, it would have been included in stage 2 provided that it was not located in the same 10 kb window as the primary SNP in the region. There is precedent for this in PD, with the previous identification of independent risk signals at the SNCA locus [11]. We therefore used the Immunochip data to test whether any of the seven loci in Table 2 showed some evidence of more than one independent signal. None of these seven loci showed any association (p>0.01) after conditioning on the main SNP in the region. In contrast, after conditioning on the most associated SNPs rs356182 in the SNCA region, several SNPs remained convincingly associated (p = 9.7×10−8 for rs2245801 being the most significant).
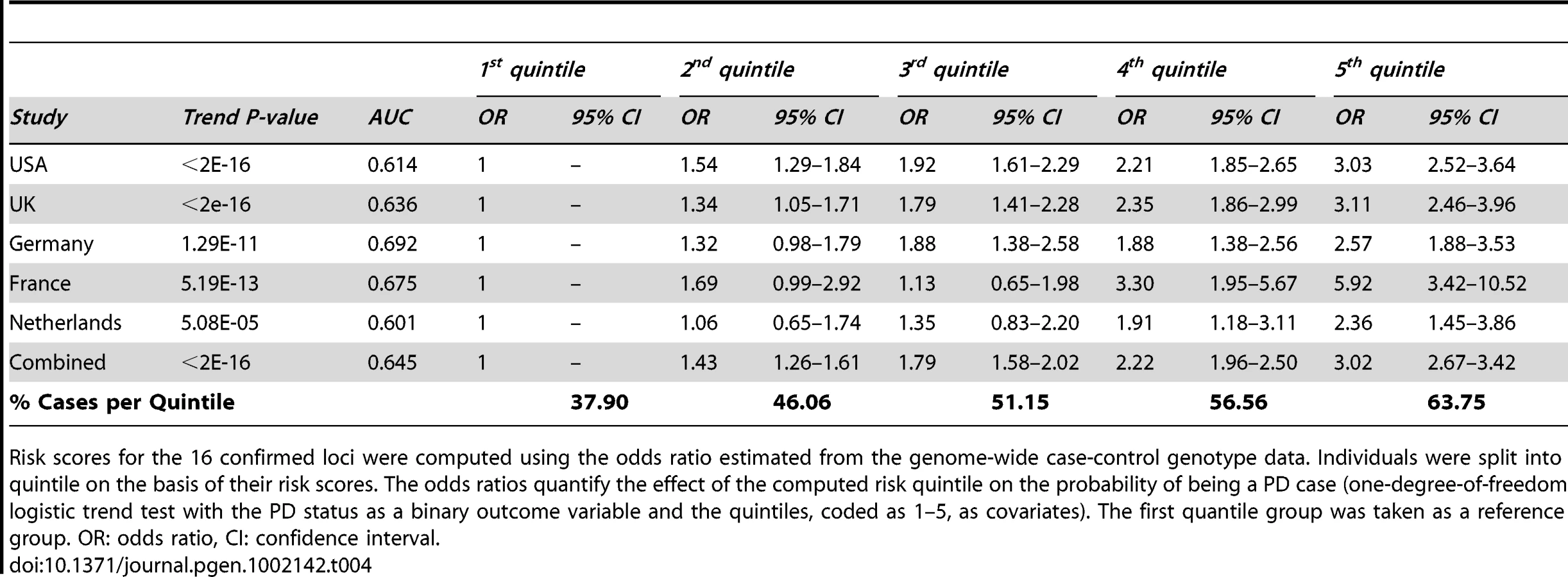
Lastly, we performed a risk profile analysis to investigate the power to discriminate cases and controls on the basis of the 16 confirmed common associated variants (Table 4). For each locus, we estimated the odds ratio on the basis of stage 1 data and we applied these estimates to compute for each individual in the ImmunoChip cohort a combined risk score. Solely based on these 16 common variants, and therefore not considering rare highly penetrant variants such as G2019S in LRKK2 [20], we found that individuals in the top quintile of the risk score have an estimated three-fold increase in PD risk compared to individuals in the bottom quintile (Table 4). We note however that the effect size of several of these associated variants could be over-estimated (an effect known as winner's curse, see [21]) but given the consistent estimates of odds ratio across studies (Table 4) we expect this bias to be minimal.
Discussion
The combination of GWA scans and imputation methods in large cohorts of PD cases and controls has enabled us to identify five PD associated loci in addition to the 11 previously reported by us. Two of these loci (1q32/PARK16, 8p22/FGF20) implicate regions that had been previously associated with PD risk [8], [16]. The 1q32/PARK16 showed convincing evidence of association in the Japanese population [8] but until now the association P-value had not passed a stringent genome-wide significance threshold in samples of European descent [7]. The 8p22/FGF20 locus had been previously reported in a study of familial PD [16] and we provide the first evidence of association in a case-control study. The remaining three loci (STX1B/16p11, STBD1/4q21 and GPNMB/7p15) are new.
Adding the eleven previously reported common variants [15] to the five convincingly associated loci identified in this study, common variants at 16 loci have now been associated with PD. Controlling for the risk score based on the 11 SNPs previously identified [15] in the risk profile analysis (Table 4), the addition of these five new loci provides a modest but significant (p = 2.2×10−3) improvement of our ability to discriminate PD cases from controls.
Combining eQTL/methylation and case-control data implicates potential mechanisms which could explain the increased PD risk associated some of these variants. In particular, the strong eQTL in the 1q32/PARK16 region with the RAB7L1 and NUCKS1 genes (Table 3) suggests that either one of these genes could be the biological effector of this risk locus. However, existing data show that eQTLs are widespread and this co-localization could be the result of chance alone [22]. Additional fine-mapping work will be required to assess whether the expression and case-control data are indeed fully consistent.
While we are unable to unequivocally pinpoint the causative genes underlying these associations, their known biological function can suggest likely candidates. At the 1q32/PARK16 loci our association and eQTL data indicate that RAB7L1 and NUCKS1 are the best candidates. The former is a GTP-binding protein that plays an important role in the regulation of exocytotic and endocytotic pathways [23]. Exocytosis is relevant for PD for two main reasons: firstly, since dopaminergic neurotransmission is mediated by the vesicular release of dopamine, i.e. dopamine exocytosis [24], and secondly because it has been shown that alpha-synuclein knock-out mice develop vesicle abnormalities [25], thus providing a potential direct link between genetic variability in the gene and a biological pathway involved in the disease. Less is known regarding NUCKS1; it has been described to be a nuclear protein, containing casein kinase II and cyclin-dependant kinases phosphorylation sites and to be highly expressed in the cardiac muscle [26]; but an involvement in PD pathogenesis has yet to be suggested.
At the 16p11/STX1B locus, notwithstanding the fact that other genes are in the associated region, STX1B is the most plausible candidate. It has been previously shown to be directly implicated in the process of calcium-dependent synaptic transmission in rat brain [17], having been suggested to play a role in the excitatory pathway of synaptic transmission. Since parkin, encoded by PARK2, negatively regulates the number and strength of excitatory synapses [27] , it makes STX1B a very interesting candidate from a biologic perspective.
FGF20 at 8p22 has been suggested to be involved in PD [16], albeit negative results in smaller cohorts have followed the original finding [28]. FGF20 is a neurotrophic factor that exerts strong neurotrophic properties within brain tissue, and regulates central nervous development and function [29]. It is preferentially expressed in the substantia nigra [30], and it has been reported to be involved in dopaminergic neurons survival [30].
The ImmunoChip data provide limited resolution for the detection of multiple independent association signals in these regions. A previous study [31] reported some evidence of allelic heterogeneity at the 1q32/PARK16 locus but the ImmunoChip data do not support this result. A previous study [11] also reported two independent associations at the 4q22/SNCA locus and our data are consistent with this scenario. However, the newly reported secondary association (rs2245801) is in low LD (r2 = 0.21) with rs2301134, the SNP reported in [11] as an independent association. Taken together, these findings suggest that at least three independent associations exist at SNCA/4q22. A more exhaustive fine-mapping analysis using either sequencing of large cohorts or targeted genotyping arrays will also be required to fully explore this locus.
As yet, we do not know which of the variants and which genes within each region are exerting the pathogenic effect. We cannot exclude that some of the currently reported variants are in fact tagging high penetrance, but rare, mutations [32]. Nevertheless, the successful identification of these 16 risk loci further demonstrates the power of the GWA study design, even in the context of disorders like PD that have a complex genetic component. We therefore expect that further and larger association analyses, perhaps using dedicated high-throughput genotyping arrays like the ImmunoChip, will continue to yield new insights into PD etiology.
Material and Methods
Genotyping and case control cohorts
Participating studies were either genotyped using the ImmunoChip as part of a collaborative agreement with the ImmunoChip Consortium, or as part of previous GWA studies provided by members of the IPDGC or freely available from dbGaP [7], [9], [10], [11]. Genotyping of the UK cases using the Immunochip was undertaken by the WTCCC2 at the Wellcome Trust Sanger Institute which also genotyped the UK control samples. The constituent studies comprising the IPDGC have been described in detail elsewhere [15], although a summary of individual study quality control is available as part of Table S1. In brief all studies followed relatively uniform quality control procedures such as: minimum call rate per sample of 95%, mandatory concordance between self-reported and X-chromosome-heterogeneity estimated sex, exclusion of SNPs with greater than 5% missingness, Hardy Weinberg equilibrium p-values at a minimum of 10−7, minor allele frequencies at a minimum of 1%, exclusion of first degree relatives, and the exclusion of ancestry outliers based on either principal components or multidimensional scaling analyses using either PLINK [33] or EIGENSTRAT [34] to remove non-European ancestry samples. All GWAS studies utilized in this analysis (and in the QTL analyses) were imputed using MACHv1.0.16 [14] to conduct a two-stage imputation based on the August 2009 haplotypes from initial low coverage sequencing of 112 European ancestry samples in the 1000 Genomes Project [35], filtering the data for a minimum imputation quality of (RSQR>0.3) [14]. Logistic regression models were utilized to quantify associations with PD incorporating allele dosages as the primary predictor of disease. Imputed data was analyzed using MACH2DAT, and genotyped SNPs were analyzed using PLINK. All models were adjusted for covariates of components 1 and 2 from either principal components or multidimensional scaling analyses to account for population substructure and stochastic genotypic variation (except in the UK-GWAS data which were not adjusted for population substructure).
Association test statistics
Single SNP test statistics were combined across datasets using a score test methodology, essentially assuming equal odds ratio across cohorts. In addition, fixed and random effects meta-analyses were implemented in R (version 2.11) to confirm that the score test approximation does not affect the interpretation of the results. We also tested the relevant SNPs heterogeneity across cohorts and no significant heterogeneity was detected (Table S2).
Data exchange
We communicated to our colleagues in charge of the independent study (Do et al) the seven SNPs listed in Table 2. For this subset of SNPs they selected the marker with the highest r2 value on their genotyping platform and provided us with the following summary statistics: odds ratio, direction of effect, standard error for the estimated odds ratio and one degree-of-freedom trend test P-value.
eQTL analysis and methylation analysis
Quantitative trait analyses were conducted to infer effects of risk SNPs on proximal CpG methylation and gene expression. For the five replicated SNP associations (Table 2), all available CpG probes and expression probes within +/−1 MB of the target SNP were investigated as candidate QTL associations in frontal cortex and cerebellar tissue samples. 399 samples were assayed for genome-wide gene expression on Illumina HumanHT-12 v3 Expression Beadchips and 292 samples were assayed using Infinium HumanMethylation27 Beadchips, both per manufacturer's protocols in each brain region. A more in depth description of the sample series comprising the QTL analyses, relevant laboratory procedures and quality requirements may be found in [15]. The QTL analysis utilized multivariate linear regression models to estimate effects of allele dosages per SNP on expression and methylation levels adjusted for covariates of age at death, gender, the first 2 component vectors from multi-dimensional scaling, post mortem interval (PMI), brain bank from where the samples were provided and in which preparation/hybridization batch the samples were processed. A total of 670 candidate QTL associations were tested: 87 expression QTLs in the cerebellum samples, 85 expression QTLs in the frontal cortex samples, 249 methylation QTLs in the cerebellum samples and 249 methylation QTLs in the frontal cortex samples. Multiple test correction was undertaken using false discovery rate adjusted p-values<0.05 to dictate significance, with the p-value adjustment undertaken in each series separately, stratified by brain region and assay. A complete list of all QTL associations tested is included in Table S3.
Supporting Information
Zdroje
1. ZimprichAMüller-MyhsokBFarrerMLeitnerPSharmaM 2004 The PARK8 locus in autosomal dominant parkinsonism: confirmation of linkage and further delineation of the disease-containing interval. American journal of human genetics 74 11 19
2. Paisán-RuízCJainSEvansWGilksWSimónJ 2004 Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44 595 600
3. ValenteEMAbou-SleimanPCaputoVMuqitMHarveyK 2004 Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304 1158 1160
4. PolymeropoulosMHLavedanCLeroyEIdeSEDehejiaA 1997 Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276 2045 2047
5. KitadaTAsakawaSHattoriNMatsumineHYamamuraY 1998 Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392 605 608
6. BonifatiVRizzuPvan BarenMSchaapOBreedveldG 2003 Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299 256 259
7. Simón-SánchezJSchulteCBrasJSharmaMGibbsR 2009 Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nature Genetics 41 1308 1312
8. SatakeWNakabayashiYMizutaIHirotaYItoC 2009 Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nature Genetics 41 1303 1307
9. SaadMLesageSSaint-PierreACorvolJ-CZelenikaD 2011 Genome-wide association study confirms BST1 and suggests a locus on 12q24 as risk loci for Parkinson's disease in the European population. Human Molecular Genetics 20 615 627
10. Simon-SanchezJvan HiltenJvan de WarrenburgBPostBBerendseH 2011 Genome-wide association study confirms extant PD risk loci among the Dutch. European Journal of Human Genetics aop
11. 2011 Dissection of the genetics of Parkinson's disease identifies an additional association 5′ of SNCA and multiple associated haplotypes at 17q21. Human Molecular Genetics 20 345 353
12. PankratzNWilkJLatourelleJDeStefanoAHalterC 2009 Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Human genetics 124 593 605
13. HamzaTZabetianCTenesaALaederachAMontimurroJ 2010 Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nature Genetics 42 781 785
14. LiYWillerCDingJScheetPAbecasisG 2010 MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genetic epidemiology 34 816 834
15. NallsMPlagnolVHernandezDSharmaMSheerinU-M 2011 Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet 377 641 649
16. van der WaltJNoureddineMKittappaRHauserMScottW 2004 Fibroblast growth factor 20 polymorphisms and haplotypes strongly influence risk of Parkinson disease. American journal of human genetics 74 1121 1127
17. SmirnovaTStinnakreJMalletJ 1993 Characterization of a presynaptic glutamate receptor. Science 262 430 433
18. WangGvan der WaltJMayhewGLiY-JZüchnerS 2008 Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha-synuclein. American journal of human genetics 82 283 289
19. WiderCDachselJSotoAHeckmanMDiehlN 2009 FGF20 and Parkinson's disease: no evidence of association or pathogenicity via alpha-synuclein expression. Movement disorders 24 455 459
20. GilksWAbou-SleimanPGandhiSJainSSingletonA 2005 A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet 365 415 416
21. ZollnerSPritchardJ 2007 Overcoming the winner's curse: estimating penetrance parameters from case-control data. American journal of human genetics 80 605 615
22. PlagnolVSmythDToddJClaytonD 2008 Statistical independence of the colocalized association signals for type 1 diabetes and RPS26 gene expression on chromosome 12q13. Biostatistics (Oxford, England)
23. ShimizuFKatagiriTSuzukiMWatanabeTKOkunoS 1997 Cloning and chromosome assignment to 1q32 of a human cDNA (RAB7L1) encoding a small GTP-binding protein, a member of the RAS superfamily. Cytogenetics and cell genetics 77 261 263
24. KoshimuraKOhueTAkiyamaYItohAMiwaS 1992 L-dopa administration enhances exocytotic dopamine release in vivo in the rat striatum. Life sciences 51 747 755
25. CabinDShimazuKMurphyDColeNGottschalkW 2002 Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. The Journal of neuroscience 22 8797 8807
26. GrundtKHagaIVAleporou-MarinouVDrososYWanvikB 2004 Characterisation of the NUCKS gene on human chromosome 1q32.1 and the presence of a homologous gene in different species. Biochemical and biophysical research communications 323 796 801
27. HeltonTOtsukaTLeeM-CMuYEhlersM 2008 Pruning and loss of excitatory synapses by the parkin ubiquitin ligase. Proceedings of the National Academy of Sciences of the United States of America 105 19492 19497
28. ClarimonJXiromerisiouGEerolaJGourbaliVHellströmO 2005 Lack of evidence for a genetic association between FGF20 and Parkinson's disease in Finnish and Greek patients. BMC neurology 5
29. JeffersMShimketsRPrayagaSBoldogFYangM 2001 Identification of a novel human fibroblast growth factor and characterization of its role in oncogenesis. Cancer research 61 3131 3138
30. OhmachiSMikamiTKonishiMMiyakeAItohN 2003 Preferential neurotrophic activity of fibroblast growth factor-20 for dopaminergic neurons through fibroblast growth factor receptor-1c. J Neurosci Res 72 436 443
31. TucciANallsMHouldenHReveszTSingletonA 2010 Genetic variability at the PARK16 locus. European journal of human genetics : EJHG 18 1356 1359
32. DicksonSWangKKrantzIHakonarsonHGoldsteinD 2010 Rare Variants Create Synthetic Genome-Wide Associations. PLoS Biol 8 e1000294 doi:10.1371/journal.pbio.1000294
33. PurcellSNealeBTodd-BrownKThomasLFerreiraM 2007 PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 81 559 575
34. PriceAPattersonNPlengeRWeinblattMShadickN 2006 Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics 38 904 909
35. 2010 A map of human genome variation from population-scale sequencing. Nature 467 1061 1073
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 6
Nejčtenější v tomto čísle
- Recurrent Chromosome 16p13.1 Duplications Are a Risk Factor for Aortic Dissections
- Statistical Inference on the Mechanisms of Genome Evolution
- Genome-Wide Association Study of White Blood Cell Count in 16,388 African Americans: the Continental Origins and Genetic Epidemiology Network (COGENT)
- Chromosomal Macrodomains and Associated Proteins: Implications for DNA Organization and Replication in Gram Negative Bacteria