
Exome Sequencing Identifies Mutations in High Myopia
Myopia is the most common ocular disorder worldwide, and high myopia in particular is one of the leading causes of blindness. Genetic factors play a critical role in the development of myopia, especially high myopia. Recently, the exome sequencing approach has been successfully used for the disease gene identification of Mendelian disorders. Here we show a successful application of exome sequencing to identify a gene for an autosomal dominant disorder, and we have identified a gene potentially responsible for high myopia in a monogenic form. We captured exomes of two affected individuals from a Han Chinese family with high myopia and performed sequencing analysis by a second-generation sequencer with a mean coverage of 30× and sufficient depth to call variants at ∼97% of each targeted exome. The shared genetic variants of these two affected individuals in the family being studied were filtered against the 1000 Genomes Project and the dbSNP131 database. A mutation A672G in zinc finger protein 644 isoform 1 (ZNF644) was identified as being related to the phenotype of this family. After we performed sequencing analysis of the exons in the ZNF644 gene in 300 sporadic cases of high myopia, we identified an additional five mutations (I587V, R680G, C699Y, 3′UTR+12 C>G, and 3′UTR+592 G>A) in 11 different patients. All these mutations were absent in 600 normal controls. The ZNF644 gene was expressed in human retinal and retinal pigment epithelium (RPE). Given that ZNF644 is predicted to be a transcription factor that may regulate genes involved in eye development, mutation may cause the axial elongation of eyeball found in high myopia patients. Our results suggest that ZNF644 might be a causal gene for high myopia in a monogenic form.
Published in the journal:
. PLoS Genet 7(6): e32767. doi:10.1371/journal.pgen.1002084
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002084
Summary
Myopia is the most common ocular disorder worldwide, and high myopia in particular is one of the leading causes of blindness. Genetic factors play a critical role in the development of myopia, especially high myopia. Recently, the exome sequencing approach has been successfully used for the disease gene identification of Mendelian disorders. Here we show a successful application of exome sequencing to identify a gene for an autosomal dominant disorder, and we have identified a gene potentially responsible for high myopia in a monogenic form. We captured exomes of two affected individuals from a Han Chinese family with high myopia and performed sequencing analysis by a second-generation sequencer with a mean coverage of 30× and sufficient depth to call variants at ∼97% of each targeted exome. The shared genetic variants of these two affected individuals in the family being studied were filtered against the 1000 Genomes Project and the dbSNP131 database. A mutation A672G in zinc finger protein 644 isoform 1 (ZNF644) was identified as being related to the phenotype of this family. After we performed sequencing analysis of the exons in the ZNF644 gene in 300 sporadic cases of high myopia, we identified an additional five mutations (I587V, R680G, C699Y, 3′UTR+12 C>G, and 3′UTR+592 G>A) in 11 different patients. All these mutations were absent in 600 normal controls. The ZNF644 gene was expressed in human retinal and retinal pigment epithelium (RPE). Given that ZNF644 is predicted to be a transcription factor that may regulate genes involved in eye development, mutation may cause the axial elongation of eyeball found in high myopia patients. Our results suggest that ZNF644 might be a causal gene for high myopia in a monogenic form.
Introduction
Myopia is the most common ocular disorder worldwide, with a prevalence of 20–30% in North American, European and Australian populations [1], [2] and as high as 40–70% in the Asian population [3]–[5]. One type of myopia is high myopia, and it is prevalent in 1–2% in the general population [1]–[6].
In high myopia, affected patients' eyes have a spherical equivalent of less than or equal to −6.00 diopter sphere (DS) and an axial length longer than or equal to 26.0 mm. In some cases, high myopia may also show retinal pathological changes with progressive choroidal degeneration in the posterior pole and other complications, potentially resulting in severe vision loss. In such cases, high myopia is referred to as pathological or degenerative myopia, which is one of the leading causes of blindness in the world [1], [7], [8].
The exact pathogenesis of myopia remains unclear. There are indications that environmental factors (such as close working habits, higher education levels and higher socioeconomic class) [9], [10] and genetic predisposition both contribute to the development of myopia [10], [11], especially of high myopia [12]. The evidence that genetic variation plays a crucial role in the occurrence and development of myopia is based on studies showing different frequencies of myopia in different populations [2]–[5], [13], obvious family aggregation trends, twin studies [9], [14], [15], and the identification of 18 linked loci having an association with myopia (OMIM, 160700) [6], [16]–[19].
Myopia can be inherited as a complex trait or in a monogenic form. For the complex form, myopia appears to be the result of an interaction of multiple genes and environmental factors. Recently, several loci have been identified by genome-wide association study (GWAS) as being responsible for complex myopia [17]–[20]. On the other hand, high myopia in a monogenic form may be inherited in an autosomal dominant, autosomal recessive and X-linked recessive manner [15], [21]. In this study, we propose to use exome sequencing to identify a gene responsible for high myopia in a monogenic form in a Han Chinese population.
Results
Here we describe a Han Chinese family (951) from Chengdu, China, that has monogenic high myopia with a dominant inheritance model. The clinical features of the nineteen of 20 living family members who participated in this study are shown in Table 1. Ten patients within the family were diagnosed with high myopia; six were alive and available for this study. The four deceased patients were diagnosed based on available medical records (Figure 1A). The six living had refractive errors ranging from −6.27 to −20.00 diopter sphere (DS) for the left eye (OS) and from 7.51 to 11.49 DS for the right eye (OD), and eye globe axial length ranging from 26.0 to 31.1 mm for OS and 26.9 to 30.5 mm for OD. They also had pre-school age of onset. Three elderly patients showed typical fundus features of high myopia: a thinning of the RPE and the choriocapillaris, which gives what is described as a ‘tigroid’ or ‘tessellated’ fundus appearance (Figure 1B, Table 1).


For this analysis, we selected two affected family members (V:1 and III:2) (Figure 1A, Table 1). V:1, the proband, at age 8 in 2009, had refractive errors at −9.54 DS (OD) and −6.34 DS (OS) and normal fundus features. III:2, at age 68 in 2009, had refractive errors at −11.49 DS (OD) and −13.02 DS (OS) and tessellated or tigroid features (Table 1, Figure 1B).
We used exome sequencing to identify potential variants responsible for high myopia in this family. We generated an average of 2.4 Gb of sequence with 30× average coverage for each individual as single-end, 80-bp reads, and about 97% (∼32.98 Mb in length) of the targeted bases were covered sufficiently to pass our thresholds for calling SNPs and short insertions or deletions (indels) (Table 2). The bases with quality scores above 20 (99% accuracy of a base call) represent over 75% of total sequence data, while the error rate is below 3% (Figure 2). Table 3 presents the exome genetic variants identified from the exome sequencing analysis. The numbers in Table 3 are comparable to what was reported in two previously published results [4], [20]. The transition versus transversion ratio is 2.95 and 2.69 for the two samples respectively. The rate of heterozygous versus homozygous variants is 1.33 and 1.38 for the two samples respectively. For patients V:1 and III:2, respectively, we identified 10,156 and 10,358 SNPs (synonymous and non-synonymous) in coding regions; 447 and 501 variants (SNPs and indels) in introns that may affect splicing (within 5 bp of the intron/exon junction); and 2,370 and 2,642 indels in coding regions or introns. Given that these patients are related and they are expected to share the causal variant for high myopia, we filtered all the detected variations in these patients against each other and found that they shared 6,610 variants (SNPs and indels) (Table 3).
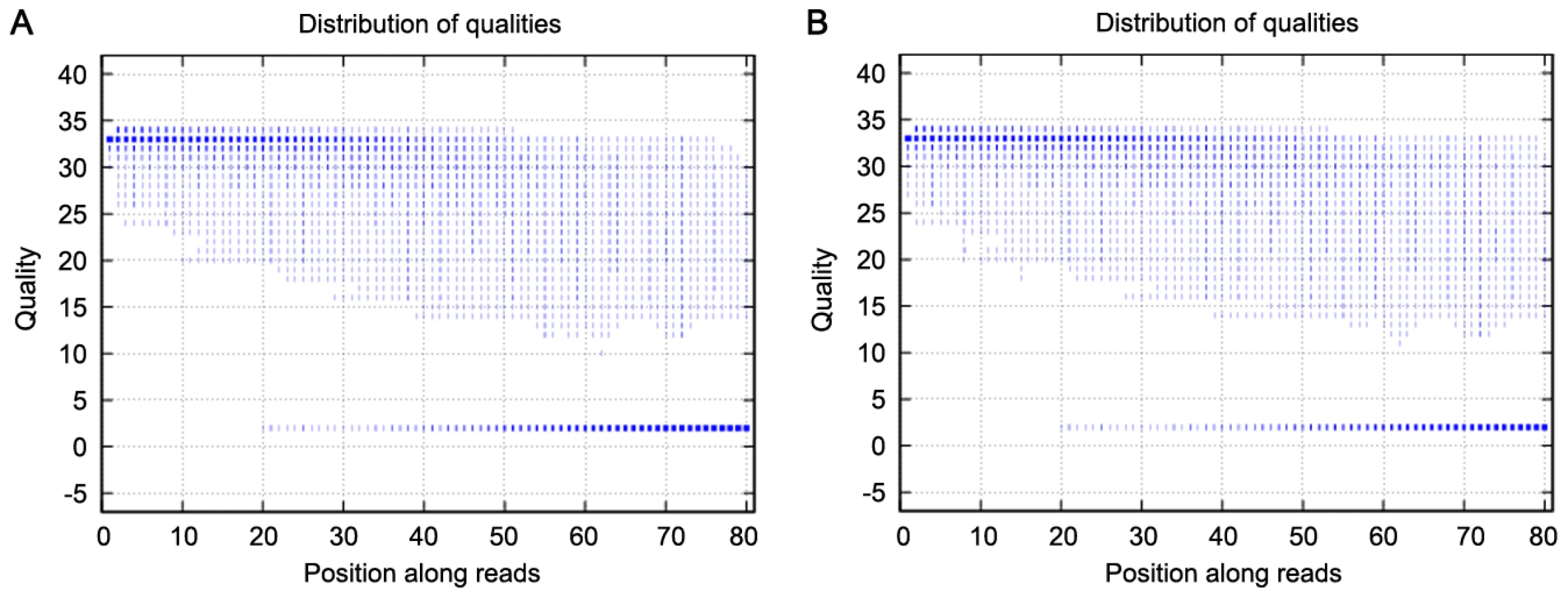


Because high myopia is a rare disorder but has a clear phenotype, there is a very low likelihood of the causal mutation in these patients being shared with a wider healthy population. We therefore compared the shared variants in these patients with the Han Chinese Beijing SNPs from dbSNP131 and the data from 30 genomes of Han Chinese Beijing recently available from the 1000 Genome Project (February 28, 2011 releases for SNPs and February 16, 2011 releases for indel fttp://www.1000genome.org). This left a total of 393 variants that were shared between these two patients. Of these, 332 genetic variants (including 62 non-synonymous SNPs, 5 splice acceptor and donor sites, and 265 indels) were predicted to potentially have a functional impact on the gene (Table 3). We carried out Sanger sequencing validation on these 332 variants, and obtained accuracy of 98% (66/67) for called SNPs and 96% (254/265) for indels, indicating the high quality of our variant calling method.
We then performed segregation analysis by Sanger sequencing on the 66 validated SNPs and 254 indels, using the available 19 members of family 951 (Figure 1). Only one variant co-segregated with the disease phenotype in this family: an A to G change in exon 3 (2156A>G), resulting in an S672G amino acid change, in the zinc finger protein 644 gene isoform 1 (ZNF644, located at 1p22.2) (Table 1, Figure 1, Figure 3). We obtained a LOD score of 3.19 at theta = 0 given an autosomal dominant mode of inheritance with full penetrance and 0.0001 for the disease allele frequency. The power to obtain a LOD score greater than 3 was 88% when tested by SLINK, providing further support for this mutation being the disease-causing change for family 951. We then assessed the presence of the co-segregating mutation in the 600 matched normal controls using direct PCR sequencing of the ZNF644 exon 3, and did not find it in the 600 controls.

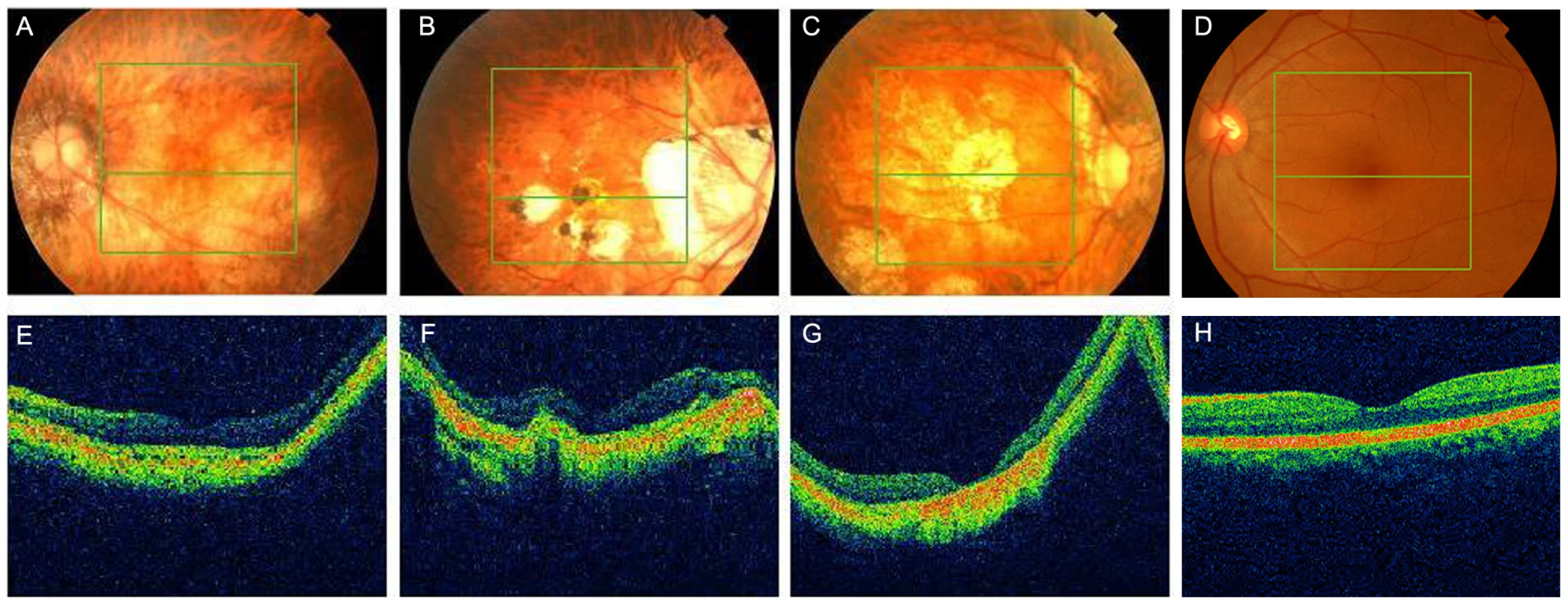
We further carried out direct PCR sequencing of the ZNF644 exons in an additional 300 unrelated (based on their self-identified geographical ancestry), sporadic high myopia patients. The 300 patients had refractive errors ranging from −6.0 to −29.0 DS for both eyes and an axial eye globe length from 26 to 33 mm for both eyes (Table 4). Some of these individuals also showed severe retinal pathological changes in the fundus appearance, an abnormal RPE, and photoreceptor layer alterations at the time of the OCT examination (Figure 4, Table 5). In the 300 sporadic patients, we identified a total of 8 variants when we sequenced the ZNF644 exons, and five out of these 8 variants (present in 11 unrelated individuals) were absent in all the 600 controls. Among these five mutations identified from the sporadic cases, three were in exon 3 (I587V, R680G and C699Y) and two were in the 3′ untranslated region (UTR) (+12 C>G and +592 G>A) (Figure 3, Table 5). The remaining three out of the 8 variants were found in both cases and controls, two (T404T and V444V) were synonymous changes which may not affect the biological function of ZNF644 and one was located in the 3′UTR (+1015 C>G). The P-value for the 17 potentially functional variants in 301 patients with high myopia (One mutation identified in one member of the high myopia family 951 plus the 6 mutations identified in the 16 unrelated patients from the 300 sporadic cases) compared with these variants being seen in 600 controls (3 in 600 controls) was 2.28×10−6 by Fisher's exact test. This data suggests that there are multiple rare variants in ZNF644 associated with high myopia.


To make sure that the ZNF644 gene is expressed in the eye, we examined ZNF644 expression in different human tissues using reverse transcript polymerase chain reaction (RT-PCR). The ZNF644 gene was expressed in the human retina and RPE as well as in the liver and placenta (Figure 5).

Discussion
For the past several decades, standard methods for identifying genes underlying disease in a monogenic form have primarily been through selecting candidate genes for testing or by using positional cloning. The candidate gene approach requires less work and costs less because only the candidate gene needs to be sequenced, but this method requires prior knowledge of the pathogenesis of a disease for gene selection. This fundamentally impedes the disease gene identification speed because the pathogeneses of many diseases have not yet been unmasked. Without pathogenesis information of a disease, the traditional positional cloning strategy can be used first to map the disease gene in the chromosome and then to identify the disease-causing gene within a specific interval. Thus the pathogenesis of a disease can be explored based on the identified disease gene. However, the positional cloning method requires marked locus heterogeneity and the availability of a large family. Focusing on the exome can be especially fruitful in disease gene identification given that previous studies have indicated that approximately 85% of causal mutations for human diseases are located within the coding region and canonical splice acceptor and donor sites (http://www.hgmd.cf.ac.uk/ac/index.php). Therefore, through sequencing and comparing the coding region of affected and unaffected individuals within a family and filtering the benign changes using a public database, such as 1000 Genomes Project and dbSNP databases, the mutation in the coding region can be identified even within small families and without knowing the pathway of a disease and marked locus heterogeneity. Currently, the cost of the exome sequencing method is even less than that of the positional cloning strategy. So, this method will not only speed up disease gene identification but will enable us to systematically tackle previously intractable monogenic disorders. In fact, exome sequencing approaches have been successfully used to identify disease genes for Mendelian disorders in recent studies [21]–[36]. Unfortunately, compared to the positional cloning strategy, the exome sequencing method may not identify the mutations in non-coding regions. This limitation promotes the use of the whole sequencing method to identify disease genes [26], [37]. Theoretically the whole gene sequencing will eventually become the best method of disease gene identification, because this method has the advantages of both positional cloning and exome sequencing methods. It can also identify disease genes caused by a large indel, inversion, translocation, and other chromosome structure aberrant. However, at the current stage, whole genome sequencing costs more and needs a lot of bioinformatics work, and this restricts its use in disease gene identification. Presently exome sequencing is a powerful tool with low cost for identifying genes that underlie disease. The whole genome sequencing method will very likely become the most powerful method for disease gene identification as the constant improvements to massively sequencing technologies and the impending massively parallel single-molecule sequencing technologies will reduce method costs and time barriers [38]. Practically, candidate gene approach, positional cloning strategy and exome sequencing or whole genome sequencing methods has been combined to identify the disease-causing genes in humans [39]–[43].
Our data here indicate again that exome sequencing can rapidly identify genes causing dominant Mendelian diseases, which can occur in a heterozygous form. We further were able to identify this gene by sequencing exomes of only two affected patients and using available public databases, such as dbSNP131 and the 1000 Genomes Project. Additionally, the use of second-generation sequencing produces a high level of coverage, with subsequent higher accuracy, and allows more regions of a genome to be sequenced in a very cost effective manner. The 30-fold average coverage we obtained here is a very high sequencing depth. It covered 97% of the target sequence with ∼96% accuracy rate, and thus allowed us to identify variants with high confidence.
Using this technique, we successfully identified a gene for high myopia in an affected family. Several lines of evidence provided support for the mutations in ZNF644, and thus the mutated ZNF644 gene, being the cause of high myopia: 1) only the S672G mutation identified in the two affected patients showed complete co-segregation with the disease phenotype in the family studied; 2) our analysis of the ZNF644 gene in 300 unrelated, sporadic high myopia patients identified an additional three missense mutations and two mutations in the 3′ UTR which may affect mRNA stability or microRNA interaction; 3) none of these identified mutations were present in the 30 genomes of Han Chinese Beijing in the 1000 Genomes Project database, the Han Chinese Beijing SNPs in the dbSNP131 database, or 600 normal ethnicity-matched controls; and 4) Comparative analyses of ZNF644 in other species showed that I587 is conserved and S672, R680, C699 are highly conserved among primates, placental animals, and other vertebrate species (http://genome.ucsc.edu/cgi-bin/hgPal). Based on protein structure, ZNF644 is predicted to be a transcription factor (http://www.genecards.org/cgi-bin/carddisp.pl?gene=ZNF644), and given that it has potentially deleterious mutations in patients with high myopia, it may play a role in gene expression regulation in the retina and retinal pigment epithelium (RPE). One important issue in the genetic study of high myopia is the age of disease onset. We would have an informative censoring problem if family members of 951 did not show the disease phenotype because their age was too young. However, the disease phenotype studied in family 951 is very special. The disease onset was at 3–4 years old for all affected patients in the family 951 with high myopia; all affected patients developed high myopia by the age of seven. The youngest unaffected member in the family (V:2) is 9 years old now; he does not show any signs of myopia at all. In addition, all affected patients in the family had severe high myopia, which allowed us identify the affected patients easily. Therefore, there is very little chance that an unaffected family member does not show the trait by virtue of being too young.
Although it clearly has a ubiquitous level of expression, this is common for other genes involved in eye diseases (for example, the retinitis pigmentosa disease-causing gene PRPC8 is ubiquitously expressed in human tissues [44]), and its expression in eye tissue allows for the ZNF644 gene having activity in the eye. Note that, given that less than 4% of the sporadic high myopia cases had mutations in ZNF644 (we identified 5 different mutations in 11 patients out of 300 cases), the ZNF644 gene is unlikely to play a major role in sporadic high myopia.
ZNF644 belongs to the Krüppel C2H2-type zinc-finger protein family, which contains 7 C2H2-type zinc fingers. Among the six identified mutations, four missense mutations were found clustered in exon 3 of the ZNF644 gene, suggesting that this exon may code for important protein domain structures or have regulatory functions. The other two mutations were located in the 3′ UTR of ZNF644 gene, which is a region often important for RNA degradation. The main feature of high myopia is axial elongation of the eye globe. Given that ZNF644 is predicted to be a transcription factor that may regulate genes involved in eye development, a mutant ZNF644 protein may impact the normal eye development and therefore underlie the axial elongation of the eye globe in high myopia patients. However, the exact mechanism of ZNF644 action and its role in high myopia pathogenesis remains unclear, and future functional studies will be important. To date, there have been no documented studies on the ZNF644 gene, and the data here indicating its involvement in a devastating eye disease provide excellent motivation for future investigation of the ZNF644 gene, which in turn should enable dissection of its relationship with high myopia pathogenesis.
Materials and Methods
Ethics statement
All procedures used in this study conformed to the tenets of the Declaration of Helsinki. The Institutional Review Board and the Ethics Committee of Sichuan Academy of Medical Sciences & Sichuan Provincial People's Hospital approved the protocols used. Informed consent was obtained from all participants.
Study population
We undertook exome sequencing and validation studies between September 1, 2009 and December 2, 2009. This study included a Han Chinese family (designated as 951) with high myopia that had 30 (20 living) family members (Figure 1, Table 1); 300 sporadic patients with high myopia; and 600 matched, normal controls. The 300 sporadic patients and the 600 controls were non-related, were of Han Chinese ethnicity (Figure 4, Table 4), came from the Chengdu region of Sichuan Province, China, and were recruited at the ophthalmic clinic at Sichuan Academy of Medical Sciences & Sichuan Provincial People's Hospital, Chengdu, China. All participants underwent an extensive, standardized examination by ophthalmologists, including visual acuity (VA) testing, a detailed clinical examination, optical coherence tomography (OCT), and ocular imaging prior to genetic testing. Refractive error and the radius of corneal curvature in the horizontal and vertical meridian were measured using an autorefractor (KR8800, Topcon, Tokyo, Japan). Final refractive error status was established with subjective visual acuity testing by trained and certified optometrists. The diagnosis for high myopia in this study required a spherical equivalent of ≤−6.0 DS for both eyes and an axial length of the eye globe of ≥26.0 mm for both eyes. For controls, the spherical equivalent for both eyes had to be in the range of −0.50 to +1.0 DS and show no evidence of disease in either eye.
Targeted capture and exome sequencing
The exome sequencing approach was used to identify the disease-causing genetic variant for the high myopia family in the study (951). Genomic DNA was extracted from peripheral white blood cells, using Gentra Systems PUREGENE DNA purification kit (Minneapolis, MN, USA). Fifteen µg of genomic DNA from each of the two selected individuals (V:1 and III:2, Figure 1A) with high myopia from family 951 were separately sheared into about 200-bp DNA fragments by sonication. Exome capture was performed to collect the protein coding regions of human genome DNA using a NimbleGen 2.1M HD array as described in the manufacturer's instructions (Roche NimbleGen, Inc., Madison, WI, USA). The array was able to capture 18,654 (92%) of the 20,091 genes. The gene sequences for this array are available in the Consensus Coding Sequence Region (CCDS) database (http://www.ncbi.nlm.nih.gov/projects/CCDS/). The exon-enriched DNA libraries were then subjected to a second library construction in preparation for Illumina GA sequencing and were sequenced using the Illumina Genome Analyzer II platform, following the manufacturer's instructions (Illumina, San Diego, USA) [23]. We obtained a mean exome coverage of 30×, which allows each selected region of the genome to be checked, on average, 30 times. Such deep coverage provided sufficient depth to accurately call variants at ∼97% of each targeted exome.
Read mapping and variant analysis
The human reference genome, together with its gene annotation, was downloaded from the UCSC database (http://genome.ucsc.edu/), version hg18 (build36). Alignment of the sequences from the two affected individuals was performed using SOAPaligner after we removed the duplicated reads [32], and SNPs were called using SOAPsnp set with the default parameters [33]. Indels affecting coding sequence or splicing sites were identified as described previously [34]. The thresholds for calling SNPs and short insertions or deletions (indels) included the following: 1) the number of unique mapped reads supporting a SNP had to be ≥4 and ≤100; and 2) the consensus quality score had to be ≥20 (The quality score is a Phred score, generated by the program SOAPsnp 12, quality score 20 represents 99% accuracy of a base call). The shared changes of the two affected individuals were obtained by further comparison of the variants of each of the two affected individuals. All changes were filtered against exome data of 30 genomes from ethnic Han Chinese individuals from Beijing available in the 1000 Genomes Project (February 28, 2011 releases for SNPs and February 16, 2011 releases for indel fttp://www.1000genome.org), and against the Han Chinese Beijing SNPs in the dbSNP131. Sanger sequencing was then used to validate the identified potential disease-causing variants. SIFT (http://sift.jcvi.org) was used to predict whether an amino acid substitution affects protein function.
Mutation validation
All shared variants of the two affected individuals after filtering against the 30 Han Chinese Beijing genomes of 1000 Genomes Project and the Han Chinese Beijing SNPs in the dbSNP131 were then confirmed by direct polymerase chain reaction (PCR)-product sequencing using Bigdye terminator v3.1 cycle sequencing kits (ABI, Foster City, CA, USA) and analyzed on an ABI 3130XL Genetic Analyzer. We used Sanger sequencing to determine whether any of the remaining variants co-segregated with the disease phenotype in family 951 and used MLINK of the LINKAGE program to calculate a two-point LOD score for the detected variants to assess the locus position of the predicted disease gene in family 951 [35]. The primers flanking all exons of ZNF644 were designed using primer 3 (http://frodo.wi.mit.edu/primer3/) (Table S1), and all exons of the 300 sporadic patients with high myopia were analyzed using the same method as above.
ZNF644 expression
We looked at expression of the ZNF644 gene in the human liver, retina, RPE, and placenta. The human liver, retina, and RPE were donated by a deceased 55-year-old Han Chinese male and the human placenta was donated by a 29-year-old Han Chinese female. Total RNA from the human liver, placenta, retina, and RPE was extracted by trizol (Invitrogen, Carlsbad, CA, USA), and reverse transcription was performed using a reverse transcription kit (Invitrogen, Carlsbad, CA, USA). The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control (Table S2). All RT-PCR products were confirmed by direct sequencing.
Supporting Information
Zdroje
1. SperdutoRDSeigelDRobertsJRowlandM 1983 Prevalence of myopia in the United States. Arch Ophthalmol 101 405 407
2. KempenJHMitchellPLeeKETielschJMBromanAT 2004 The prevalence of refractive errors among adults in the United States, Western Europe, and Australia. Arch Ophthalmol 122 495 505
3. SawadaATomidokoroAAraieMIwaseAYamamotoT 2008 Refractive errors in an elderly Japanese population: the Tajimi study. Ophthalmology 115 363 370 e363
4. HeMZhengYXiangF 2009 Prevalence of myopia in urban and rural children in mainland China. Optom Vis Sci 86 40 44
5. WongTYFosterPJHeeJNgTPTielschJM 2000 Prevalence and risk factors for refractive errors in adult Chinese in Singapore. Invest Ophthalmol Vis Sci 41 2486 2494
6. LiYJGuggenheimJABulusuAMetlapallyRAbbottD 2009 An international collaborative family-based whole-genome linkage scan for high-grade myopia. Invest Ophthalmol Vis Sci 50 3116 3127
7. XuLWangYLiYCuiTLiJ 2006 Causes of blindness and visual impairment in urban and rural areas in Beijing: the Beijing Eye Study. Ophthalmology 113 1134 e1131-1111
8. BuchHVindingTLa CourMAppleyardMJensenGB 2004 Prevalence and causes of visual impairment and blindness among 9980 Scandinavian adults: the Copenhagen City Eye Study. Ophthalmology 111 53 61
9. DiraniMShekarSNBairdPN 2008 The role of educational attainment in refraction: the Genes in Myopia (GEM) twin study. Invest Ophthalmol Vis Sci 49 534 538
10. LopesMCAndrewTCarbonaroFSpectorTDHammondCJ 2009 Estimating heritability and shared environmental effects for refractive error in twin and family studies. Invest Ophthalmol Vis Sci 50 126 131
11. TeikariJMO'DonnellJKaprioJKoskenvuoM 1991 Impact of heredity in myopia. Hum Hered 41 151 156
12. YoungTLMetlapallyRShayAE 2007 Complex trait genetics of refractive error. Arch Ophthalmol 125 38 48
13. KatzJTielschJMSommerA 1997 Prevalence and risk factors for refractive errors in an adult inner city population. Invest Ophthalmol Vis Sci 38 334 340
14. HammondCJSniederHGilbertCESpectorTD 2001 Genes and environment in refractive error: the twin eye study. Invest Ophthalmol Vis Sci 42 1232 1236
15. DiraniMChamberlainMShekarSNIslamAFGaroufalisP 2006 Heritability of refractive error and ocular biometrics: the Genes in Myopia (GEM) twin study. Invest Ophthalmol Vis Sci 47 4756 4761
16. SchwartzMHaimMSkarsholmD 1990 X-linked myopia: Bornholm eye disease. Linkage to DNA markers on the distal part of Xq. Clin Genet 38 281 286
17. YoungTLRonanSMDrahozalLAWildenbergSCAlvearAB 1998 Evidence that a locus for familial high myopia maps to chromosome 18p. Am J Hum Genet 63 109 119
18. NakanishiHYamadaRGotohNHayashiHYamashiroK 2009 A genome-wide association analysis identified a novel susceptible locus for pathological myopia at 11q24.1. PLoS Genet 5 e1000660 doi:10.1371/journal.pgen.1000660
19. AndrewTManiatisNCarbonaroFLiewSHLauW 2008 Identification and replication of three novel myopia common susceptibility gene loci on chromosome 3q26 using linkage and linkage disequilibrium mapping. PLoS Genet 4 e1000220 doi:10.1371/journal.pgen.1000220
20. LiYVinckenboschNTianGHuerta-SanchezEJiangT 2010 Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet 42 969 972
21. NgSBTurnerEHRobertsonPDFlygareSDBighamAW 2009 Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461 272 276
22. NgSBBuckinghamKJLeeCBighamAWTaborHK 2010 Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 42 30 35
23. ChoiMSchollUIJiWLiuTTikhonovaIR 2009 Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A 106 19096 19101
24. HoischenAvan BonBWGilissenCArtsPvan LierB 2010 De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet 42 6 483 5
25. ShoubridgeCTarpeyPSAbidiFRamsdenSLRujirabanjerdS 2010 Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nat Genet 42 486 488
26. LupskiJRReidJGGonzaga-JaureguiCRio DeirosDChenDC 2010 Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med 362 1181 1191
27. SunYAlmomaniRAtenECelliJvan der HeijdenJ 2010 Terminal osseous dysplasia is caused by a single recurrent mutation in the FLNA gene. Am J Hum Genet 87 146 153
28. PierceSBWalshTChisholmKMLeeMKThorntonAM 2010 Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrome. Am J Hum Genet 87 282 288
29. WalshTShahinHElkan-MillerTLeeMKThorntonAM 2010 Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am J Hum Genet 87 90 94
30. KrawitzPMSchweigerMRRodelspergerCMarcelisCKolschU 2010 Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet 42 827 829
31. JohnsonJOMandrioliJBenatarMAbramzonYVan DeerlinVM 2010 Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68 857 864
32. BilguvarKOzturkAKLouviAKwanKYChoiM 2010 Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467 207 210
33. ZuchnerSDallmanJWenRBeechamGNajA 2011 Whole-Exome Sequencing Links a Variant in DHDDS to Retinitis Pigmentosa. Am J Hum Genet 88 201 206
34. GilissenCArtsHHHoischenASpruijtLMansDA 2010 Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet 87 418 423
35. MusunuruKPirruccelloJPDoRPelosoGMGuiducciC 2010 Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med 363 2220 2227
36. NgSBBighamAWBuckinghamKJHannibalMCMcMillinMJ 2010 Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 42 790 793
37. RoachJCGlusmanGSmitAFHuffCDHubleyR 2010 Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 328 636 639
38. TeerJKMullikinJC 2010 Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet 19 R145 151
39. SobreiraNLCirulliETAvramopoulosDWohlerEOswaldGL 2010 Whole-genome sequencing of a single proband together with linkage analysis identifies a Mendelian disease gene. PLoS Genet 6 e1000991 doi:10.1371/journal.pgen.1000991
40. AnastasioNBen-OmranTTeebiAHaKCLalondeEAliR 2010 Mutations in SCARF2 are responsible for Van Den Ende-Gupta syndrome. Am J Hum Genet 87 553 559
41. WangJun LingYangXuXiaKunHuZheng MaoWengLing 2010 TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain 133 3510 3518
42. CaliskanMChongJXUricchioLAndersonRChenP 2011 Exome sequencing reveals a novel mutation for autosomal recessive non-syndromic mental retardation in the TECR gene on chromosome 19p13. Hum Mol Genet [Epub ahead of print]
43. KalayEYigitGAslanYBrownKEPohlE 2011 CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet 43 23 26
44. McKieABMcHaleJCKeenTJTarttelinEEGoliathR 2001 Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum Mol Genet 10 1555 1562
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 6
Nejčtenější v tomto čísle
- Recurrent Chromosome 16p13.1 Duplications Are a Risk Factor for Aortic Dissections
- Statistical Inference on the Mechanisms of Genome Evolution
- Genome-Wide Association Study of White Blood Cell Count in 16,388 African Americans: the Continental Origins and Genetic Epidemiology Network (COGENT)
- Chromosomal Macrodomains and Associated Proteins: Implications for DNA Organization and Replication in Gram Negative Bacteria