
Plant-Symbiotic Fungi as Chemical Engineers: Multi-Genome Analysis of the Clavicipitaceae Reveals Dynamics of Alkaloid Loci
The fungal family Clavicipitaceae includes plant symbionts and parasites that produce several psychoactive and bioprotective alkaloids. The family includes grass symbionts in the epichloae clade (Epichloë and Neotyphodium species), which are extraordinarily diverse both in their host interactions and in their alkaloid profiles. Epichloae produce alkaloids of four distinct classes, all of which deter insects, and some—including the infamous ergot alkaloids—have potent effects on mammals. The exceptional chemotypic diversity of the epichloae may relate to their broad range of host interactions, whereby some are pathogenic and contagious, others are mutualistic and vertically transmitted (seed-borne), and still others vary in pathogenic or mutualistic behavior. We profiled the alkaloids and sequenced the genomes of 10 epichloae, three ergot fungi (Claviceps species), a morning-glory symbiont (Periglandula ipomoeae), and a bamboo pathogen (Aciculosporium take), and compared the gene clusters for four classes of alkaloids. Results indicated a strong tendency for alkaloid loci to have conserved cores that specify the skeleton structures and peripheral genes that determine chemical variations that are known to affect their pharmacological specificities. Generally, gene locations in cluster peripheries positioned them near to transposon-derived, AT-rich repeat blocks, which were probably involved in gene losses, duplications, and neofunctionalizations. The alkaloid loci in the epichloae had unusual structures riddled with large, complex, and dynamic repeat blocks. This feature was not reflective of overall differences in repeat contents in the genomes, nor was it characteristic of most other specialized metabolism loci. The organization and dynamics of alkaloid loci and abundant repeat blocks in the epichloae suggested that these fungi are under selection for alkaloid diversification. We suggest that such selection is related to the variable life histories of the epichloae, their protective roles as symbionts, and their associations with the highly speciose and ecologically diverse cool-season grasses.
Published in the journal:
. PLoS Genet 9(2): e32767. doi:10.1371/journal.pgen.1003323
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003323
Summary
The fungal family Clavicipitaceae includes plant symbionts and parasites that produce several psychoactive and bioprotective alkaloids. The family includes grass symbionts in the epichloae clade (Epichloë and Neotyphodium species), which are extraordinarily diverse both in their host interactions and in their alkaloid profiles. Epichloae produce alkaloids of four distinct classes, all of which deter insects, and some—including the infamous ergot alkaloids—have potent effects on mammals. The exceptional chemotypic diversity of the epichloae may relate to their broad range of host interactions, whereby some are pathogenic and contagious, others are mutualistic and vertically transmitted (seed-borne), and still others vary in pathogenic or mutualistic behavior. We profiled the alkaloids and sequenced the genomes of 10 epichloae, three ergot fungi (Claviceps species), a morning-glory symbiont (Periglandula ipomoeae), and a bamboo pathogen (Aciculosporium take), and compared the gene clusters for four classes of alkaloids. Results indicated a strong tendency for alkaloid loci to have conserved cores that specify the skeleton structures and peripheral genes that determine chemical variations that are known to affect their pharmacological specificities. Generally, gene locations in cluster peripheries positioned them near to transposon-derived, AT-rich repeat blocks, which were probably involved in gene losses, duplications, and neofunctionalizations. The alkaloid loci in the epichloae had unusual structures riddled with large, complex, and dynamic repeat blocks. This feature was not reflective of overall differences in repeat contents in the genomes, nor was it characteristic of most other specialized metabolism loci. The organization and dynamics of alkaloid loci and abundant repeat blocks in the epichloae suggested that these fungi are under selection for alkaloid diversification. We suggest that such selection is related to the variable life histories of the epichloae, their protective roles as symbionts, and their associations with the highly speciose and ecologically diverse cool-season grasses.
Introduction
Alkaloids play key roles in plant ecology by targeting the central and peripheral nervous systems of invertebrate and vertebrate animals, affecting their behavior, eliciting toxicoses, and reducing herbivory [1]. Alkaloids are very common in plants as well as certain plant-associated fungi, particularly those in the family Clavicipitaceae. Plant parasites such as Claviceps species often produce high levels of ergot alkaloids or indole-diterpenes, probably to defend their resting and overwintering structures (commonly called ergots) [2], [3]. A closely related group of fungi, the epichloae (Epichloë and Neotyphodium species) live as systemic symbionts of grasses, and produce a wide array of alkaloids that combat various herbivorous animals, a key determinant of mutualism in many grass-endophyte symbioses [4], [5].
Fungi of family Clavicipitaceae are generally biotrophs that grow in invertebrates, fungi, or plants. The major clade of plant-associated Clavicipitaceae [6] includes mutualistic symbionts as well as plant pathogens, many of which produce alkaloids with diverse neurotropic effects on vertebrate and invertebrate animals with important implications for human health, agriculture and food security [7], [8]. Most species of plant-associated Clavicipitaceae grow in or on grasses, but the group also includes systemic parasites of sedges or other plants, and heritable symbionts of morning glories [9]. The plant-associated Clavicipitaceae have very high chemotypic diversity, ecological significance [10], and agricultural impact [11]. Many produce abundant alkaloids such as ergot alkaloids and indole-diterpenes, which have potent neurotropic activities in mammals. The ergot alkaloids are named for the ergot fungi (Claviceps species), which are infamous for causing mass poisonings throughout much of human history, although ergot alkaloids also have numerous pharmaceutical uses [7], [12]–[14]. In contrast to the Claviceps species, the epichloae (Epichloë or Neotyphodium species) are systemic and often heritable, mutualistic symbionts of cool-season grasses (Poaceae, subfamily Poöideae)(Figure 1) [4]. Epichloae have diverse alkaloid profiles, and in addition to ergot alkaloids or indole-diterpenes, many produce lolines or peramine, which help to protect their grass hosts from insects [15], [16] and possibly other invertebrates [17].

The activities of alkaloids in animal nervous systems relates to their chemical similarities to biogenic amines [1]. Although poisoning of humans by alkaloids of clavicipitaceous fungi is now rare, toxicity to livestock is frequently observed [18]–[20]. Morning glories such as Ipomoeae asarifolia cause toxicity to livestock on ranges in Brazil, probably due to alkaloids produced by symbiotic Periglandula ipomoeae [9], [21]. Indole-diterpene or ergot alkaloids produced by epichloae in wild and cultivated grasses also can cause livestock toxicosis [7], [22]. For example, in 1993, losses to pastured U.S. beef production were estimated at $600 million due to widespread plantings of tall fescue symbiotic with ergot-alkaloid-producing strains of Neotyphodium coenophialum [23]. In addition to chemotypic variation [24], the epichloae also exhibit an extraordinary variety of host-interactions, whereby some are pathogenic and contagious, others are mutualistic and vertically transmitted (heritable), and others have both mutualistic and pathogenic characteristics [4], [25]. Relative benefits of epichloae and their alkaloids to host grasses are related to variations in life history [26], [27] and ecological contexts [28], [29], which may well explain why they have evolved such chemotypic diversity.
Even within an alkaloid class, structural variations can profoundly affect pharmacological spectra, as reflected for example in the diverse uses of ergot alkaloids in medicine [14], [30](Figure 2). Ergonovine ( = ergometrine) was long used to aid in childbirth, ergotamine is used for migraines, and, in recent years, 2-bromonated ergocryptine (bromocriptine) has been adopted for treatment of numerous disorders of the central nervous system, such as Parkinsonism and pituitary gland adenomas. In contrast, lysergic acid diethylamide (LSD), a semisynthetic ergot alkaloid originally developed as an antidepressant, is the most potent hallucinogen known [31], and was a major factor in the drug culture of the 1960's and 1970's. Historic episodes of mass poisoning in humans have resulted from contamination of grains with ergots (the resting structures of Claviceps species) [32], and the effects vary depending on which alkaloids are present. Symptoms range from the disfiguring dry gangrene of St. Anthony's fire to convulsions and hallucinations such as those associated with the Salem witch trials [33]. For example, outbreaks of convulsive ergotism in India in the late 1970's were due to Claviceps fusiformis producing mainly elymoclavine [34], while Ethiopia experienced a gangrenous ergotism outbreak in 1978 caused by C. purpurea producing ergopeptines [35].

Other alkaloids produced by Clavicipitaceae variously present hazards or benefits to agriculture. The indole-diterpenes (Figure 3) represent a broad diversity of bioactive compounds that exhibit mammalian and insect toxicity through activation of various ion channels [36], [37]. Livestock afflicted with indole-diterpene toxicity display symptoms of ataxia and sustained tremors [20]. For example, Paspalum staggers is caused by paspalitrems produced by Claviceps paspali and Claviceps cynodontis on seed-heads of dallisgrass (Paspalum dilatatum) and Bermuda grass (Cynodon dactylon), respectively [3], and common strains of Neotyphodium lolii symbiotic with perennial ryegrass (Lolium perenne) produce lolitrems, which cause ryegrass staggers [20]. In contrast, lolines (Figure 4) and peramine produced by many endophytic epichloae in forage grasses have not been linked to any toxic symptoms in grazing mammals, but instead provide potent protection from herbivorous insects [15], [16].


The discoveries of individual genes involved in biosynthetic pathways for each of the four alkaloid classes [16], [38]–[40] has led to elucidation of clusters of biosynthesis genes for ergot alkaloids (EAS) in C. purpurea [41], lolines (LOL) in Neotyphodium uncinatum [42], and lolitrems (a group of indole-diterpenes, IDT/LTM) in Neotyphodium lolii [43], as well as characterization of the perA gene of Epichloë festucae [16]. The identification of these genetic loci, elucidation of structural diversity within each alkaloid class, and new technologies for high-throughput DNA sequencing together provide an outstanding opportunity to investigate the genome dynamics governing chemotypic variation in fungi with diverse life histories and ecological functions. To that end, we sequenced genomes and compared alkaloid locus structures of 15 plant-associated Clavicipitaceae, including 10 epichloae, three Claviceps species, the nonculturable morning glory symbiont Periglandula ipomoeae, and the bamboo witch's broom pathogen Aciculosporium take (Table S1). We report that the alkaloid loci tend to be arranged with genes for conserved early pathway steps in their cores, and peripheral genes that vary in presence or absence, or in sequence, to diversify structures within each alkaloid class. Transposon-derived repeats, miniature inverted repeat transposable elements (MITEs), and telomeres were often associated with unstable loci or the variable peripheral genes, and were especially common in alkaloid clusters of the epichloae. We suggest that structures of the alkaloid loci, including distributions of repeat blocks, reflect selection on these fungi for niche adaptation.
Results
Genome sequences
Clusters of genes have been identified for the four alkaloid biosynthesis classes [16], [41]–[43], but in the absence of complete genome sequences it was unknown if the clusters had been fully characterized for any known producers in the Clavicipitaceae. Therefore, we sequenced 15 genomes of diverse species in the family with various alkaloid profiles (Figure 5, Table 1). The genomes were primarily sequenced by shotgun pyrosequencing, but paired-end and mate-pair reads were used to scaffold several assemblies. Notably, adding mate-pair pyrosequencing of C. purpurea DNA resulted in a 186-supercontig (scaffold) assembly of 32.1 Mb, and adding end-sequencing of fosmid clones E. festucae Fl1 DNA resulted in a 170-supercontig assembly of 34.9 Mb. Annotated genome sequences have been posted at www.endophyte.uky.edu, and (for C. purpurea 20.1) at http://www.ebi.ac.uk/ena/data/view/Project:76493, and GenBank and EMBL project numbers are listed in Table S2. Assembled genome sizes among the sequenced strains varied 2-fold from 29.2 to 58.7 Mb, with wide ranges even within the genera Claviceps (31–52.3 Mb) and Epichloë (29.2–49.3 Mb) (Table 1). The majority of genome size variation was due to repeat sequences, which ranged from 4.7–56.9% overall (excluding P. ipomoeae from consideration because contigs that lacked coding sequences may have been filtered from that assembly), and from 13.7–44.9% repeat DNA among the epichloae (Table 2). Also, the average GC contents of repeat sequences varied widely, from 22% in C. fusiformis PRL 1980 to 50% in C. purpurea 20.1 (Table 3). The sums of coding sequence lengths were estimated from ab initio gene predictions with FGENESH, and ranged from 9.4 Mb in A. take MAFF-241224 to 15.9 Mb in P. ipomoeae IasaF13 (Table 2). Most of the epichloae had approximately 11 Mb of coding sequence, with the exception of E. glyceriae E277, which had 14.9 Mb of coding sequence. Gene contents were not correlated with genome size, and although A. take had the largest genome at 58.7 Mb, it had the least coding sequence at 9.4 Mb.




Phylogenetic relationships
Phylogenetic analysis of aligned partial coding sequences for the RNA polymerase II largest subunit (rpbA) for all of the sequenced isolates, together with related fungi for which the sequence data are available (Figure 5), supported the relationships previously indicated for subsets of these fungi [44], [45]. The sequenced strains were contained in a clade that mainly included Clavicipitaceae associated with the plant families, Poaceae (grasses), and Convolvulaceae (morning glories). These had more distant relationships to the Clavicipitaceae associated with insects. The Epichloë and Neotyphodium species grouped in a single clade (epichloae), and until recently the sexual species were classified in genus Epichloë, and those with no known sexual state were classified in form genus Neotyphodium [25]. (This was in accord with the dual naming system for fungi, formerly specified in the botanical code of nomenclature.) The sister clade to the epichloae included the Claviceps, Aciculosporium and Neoclaviceps species. Outside of this clade grouped other plant associates and insect associates, including two Metarhizium species for which there are recently published genome sequences [46]. Metarhizium species are well-known insect pathogens, although some strains of Metarhizium anisopliae have recently been shown to be associated with plant rhizospheres [47].
Phylogenies of partial coding sequences for rpbA and two other housekeeping genes, rpbB (encoding RNA polymerase II second-largest subunit) and tefA (encoding translation elongation factor 1-α) (Figure S1) were compared by the Shimodaira-Hasegawa test (Table S3). The rpbA phylogeny was congruent with the rpbB phylogeny, but the tefA phylogeny was significantly incongruent with those of rpbA and rpbB. The tefA tree had a very different placement of M. anisopliae than did the other two phylogenies. Nevertheless, all three gene trees were in agreement with respect to the grouping of the epichloae in a single clade, with a sister clade that included Claviceps species and A. take. All trees also supported a relationship of P. ipomoeae (and, for rbpA, P. turbinae) with the fungal parasite, Verticillium epiphytum. Inclusion of another fungal parasite, Tyranicordyceps fratricidam, with Periglandula spp. and V. epiphytum was supported by the rpbA and rpbB trees, and not significantly contradicted by the tefA tree.
Alkaloid profiles
Plant-associated Clavicipitaceae generally produce alkaloids most consistently in association with host plants [16], [42], [43], [48], [49], so samples of plant material symbiotic with several epichloae were profiled for combinations of ergot alkaloids, indole-diterpenes, lolines and peramine, depending on which gene clusters were identified in the sequenced genomes. Symbiotic material was available for E. amarillans E57, E. elymi E56, E. festucae E2368 and Fl1, E. glyceriae E2772, E. typhina E8, N. gansuense E7080, N. gansuense var. inebrians E818, and N. uncinatum E167, and in limited amounts (sufficient for a loline alkaloid analysis) from E. brachyelytri E4804. Leaves and seeds of morning glory symbiotic with P. ipomoeae IasaF13 were assayed for ergot alkaloids and indole-diterpenes. Ergot alkaloids also were analyzed from ergots of C. purpurea 20.1, and the ergot alkaloid profile of C. fusiformis PRL 1980 is well established [50]. No infected plant material was available to assay the alkaloid profile of E. typhina E5819, and no ergots of C. paspali RRC-1481 were available. Alkaloid profiles listed in Table 4 indicated both interspecific and intraspecific variation.

Comparisons of ergot alkaloid profiles (Table 4) indicated likely presence, absence, or sequence variation in EAS genes among strains (Figure 2). For example, variations in lpsA were evident by the production of different ergopeptines, as previously demonstrated for C. purpurea [51]. More specifically, grass plants symbiotic with E. festucae Fl1 had ergovaline, morning glories symbiotic with P. ipomoeae IasaF13 had ergobalansine, and ergots of C. purpurea 20.1 had ergotamine and ergocryptine. Other strains lacked ergopeptines. The principal alkaloids in grass plants with N. gansuense var. inebrians E818 were simpler lysergyl amides, including high levels of ergonovine (EN), low levels of lysergic acid α-hydroxyethylamide (LAH), and intermediate levels of lysergic acid amide ( = ergine), which can result from breakdown of EN, LAH, or both. Morning glories with IasaF13 also had these simple lysergyl amides, which have been reported from C. paspali ergots as well [52]. Other strains produced compounds that were intermediates of the lysergic acid pathway; namely, elymoclavine (EC) produced by C. fusiformis PRL 1980, and chanoclavine I (CC) produced by E. elymi E56.
Each strain that produced indole-diterpenes had a different major pathway end product, although pathway intermediates were typically detected as well (Table 4). Different profiles were likely to be due to different specificities of idtP and idtQ, and the presence or absence of combinations of idtF, idtK, ltmE, and ltmJ (Figure 3) [53]. As apparent pathway end products, grass plants with E. festucae Fl1 had lolitrem B, plants with N. gansuense E7080 had paxilline, and morning glories with P. ipomoeae IasaF13 had terpendoles. Furthermore, C. paspali is reported to produce paspalitrem A [54].
Three different profiles of loline alkaloids (Figure 4) were evident among grass plants symbiotic with epichloae (Table 4). Grasses with E. festucae E2368 had primarily N-formylloline (NFL), but also the N-acetylloline (NAL), N-methylloline (NML), N-acetylnorloline (NANL), and loline. These alkaloids were also produced in planta by N. uncinatum E167. Plants with E. amarillans E57 and E. glyceriae E2772 accumulated NANL, and the plant material with E. brachyelytri E4804 accumulated 1-acetamidopyrrolizidine (AcAP).
Peramine, production of which is dependent upon the perA gene [16], was detected in grass plants symbiotic with E. festucae Fl1, but not with E. festucae E2368 (Table 4). This alkaloid was also detected in plants with symbiotic E. amarillans E57, E. elymi E56 and E. typhina E8.
Ergot alkaloid (EAS) loci
In the scaffolded assemblies of the C. purpurea and E. festucae Fl1 genomes, and the scaffolded E2368 assembly of 2010-06, the EAS genes were clustered within individual supercontigs (Figure 6). Also in the assemblies of C. fusiformis, C. paspali and P. ipomoeae genomes functional EAS genes were contained in single contigs. Other non-scaffolded assemblies had EAS genes in two or three contigs, but only in the case of E818 were the EAS genes unequivocally separated in two separate clusters. Long-range physical mapping of the EAS genes of E2368 confirmed that they were clustered (Figure S2).

Functions determined to date for enzymes in the ergot alkaloid biosynthetic pathway (Figure 2) [7], [55] were consistent with the presence or absence of specific EAS genes (Figure 6) in strains with particular ergot-alkaloid profiles (Table 4 and Table 5). Furthermore, genes without experimentally determined roles in the pathway could be linked with hypothesized steps on the basis of the functions predicted from their sequences, their presence in clusters among strains that produce specific ergot-alkaloid forms, and their absence from fungi lacking those forms. For example, easH was predicted to encode a nonheme-iron dioxygenase, and was present in all ergopeptine-producing strains and absent from most ergopeptine nonproducers, suggesting that EasH may catalyze oxidative cyclization of ergopeptams to ergopeptines. Likewise, easO and easP, were discovered within the EAS loci upon sequencing the genomes of the two LAH producers, P. ipomoeae and N. gansuense var. inebrians, and were absent from strains of species not known to produce LAH. These genes were also present in C. paspali, but the sequenced strain had a defective easE, and for this reason was not predicted to produce ergot alkaloids. Nevertheless, the fact that other C. paspali strains are reported to produce LAH [52] strengthens the association of easO and easP with LAH production.

The genome of P. ipomoeae was the only one sequenced that contained functional orthologs of all 14 EAS genes (Figure 6), and this was the only strain that produced EN, LAH, and an ergopeptine (ergobalansine). (The Metarhizium genomes described in [46] contained all of these genes, but some either had defects or sequencing errors.) Orthologs of twelve of these genes were clustered in the C. purpurea 20.1 genome, which had two lpsA genes consistent with production of two different ergopeptines (Figure 6). Also, based on gene content, C. purpurea 20.1 was predicted to produce EN, though this was not tested. The absence of a functional lpsB gene in C. fusiformis PRL 1980 accounts for termination of its ergot alkaloid pathway at an earlier position. This strain produces EC, although there was no obvious EAS cluster gene for a (mono)oxygenase to catalyze the final step from agroclavine to elymoclavine. The required enzyme seems likely to be encoded either by a non-cluster gene or the C. fusiformis isoform of cloA. The genome of N. gansuense var. inebrians E818 lacked only lpsA and easH in keeping with its chemotype as a producer of EN and LAH, but not of ergopeptines. In contrast, E. glyceriae E277, E. typhina E5819, and the two E. festucae isolates lacked lpsC, consistent with the observation that E. festucae Fl1 produced an ergopeptine (ergovaline) but not EN or LAH. The fact that no ergot alkaloids were detected in plants with E. festucae E2368 reflected the observation that most of the EAS genes in E2368 were not expressed (data not shown). Finally, E. brachyelytri E4804 and E. elymi E56 had functional copies of only the first four pathway genes, which accounted for the observed accumulation of CC in plants symbiotic with E56.
Indole-diterpene (IDT) loci
The IDT gene clusters in C. paspali , P. ipomoeae, N. gansuense var. inebrians and E. festucae Fl1 had conserved cores that contained the four genes for synthesis of paspaline (idt/ltmG, M, B, and C) (Figure 7). The gene cores also included the newly discovered gene idt/ltmS (discussed below) that was conserved in all indole-diterpene producers. Genes idt/ltmP, Q, K, and F, which by virtue of their presence, absence or sequence variation determine the particular forms of indole-diterpenes produced [56], were identified in the periphery of each cluster. Two more peripheral genes, ltmE and ltmJ, were present in the lolitrem producer, E. festucae Fl1, but not in the other sequenced genomes (Figure 7, Figure S3). Reciprocal blast analysis of inferred protein products, as well as identification of conserved intron locations, indicated that LtmJ was most closely related to LtmK with 36% overall identity (Figure 8). Furthermore, LtmE was most closely related to LtmC in its N-terminal region and to LtmF in its carboxy-terminal region. These relationships indicated duplications and neofunctionalizations of indole-diterpene modification genes, whereby ltmJ was probably derived from a duplication of ltmK, and ltmE was probably derived from a fusion of duplicated ltmC and ltmF genes.
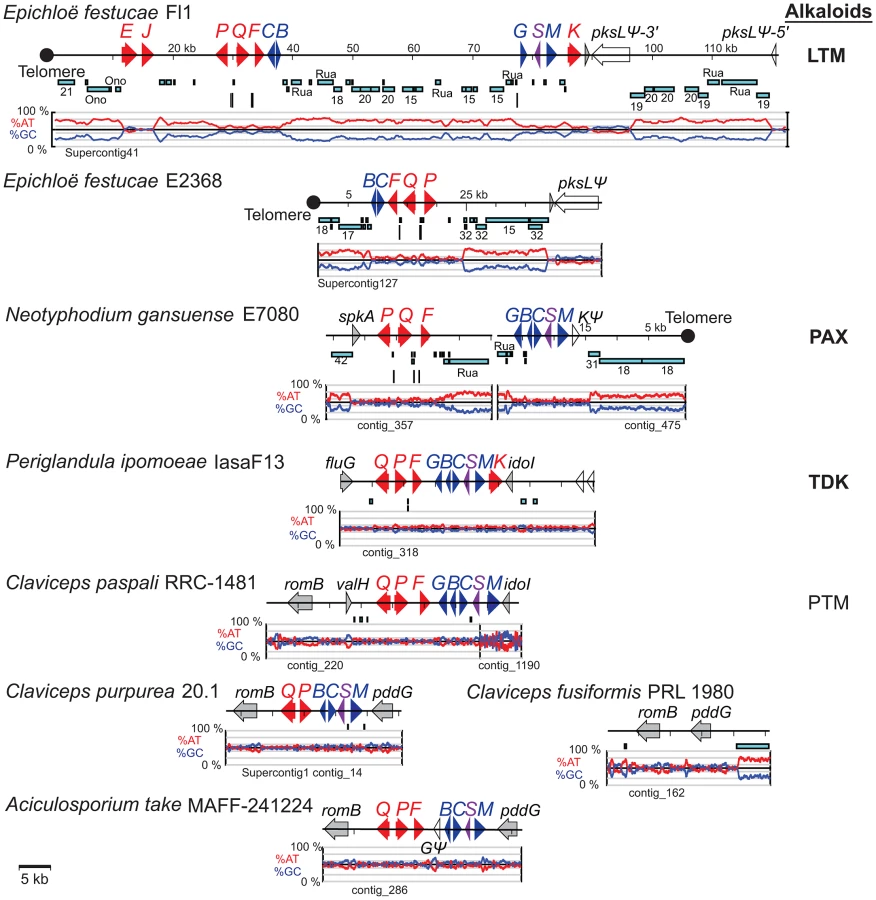

The gene arrangements in the IDT/LTM loci were conserved in Claviceps species, P. ipomoeae, and A. take MAFF-241224, and varied slightly in E. festucae Fl1 and N. gansuense E7080 (Figure 7, Figure S3). The gene order in N. gansuense E7080 differed by an inversion of the block containing peripheral genes itdP and idtQ. In turn, the gene order in E. festucae Fl1 differed from that of E7080 by an additional inversion of the segment containing three core genes, idt/ltmC, B, and G. Some strains had alterations that eliminated their potential to produce these alkaloids. Specifically, in C. purpurea 20.1 and A. take the idtG gene encoding the first pathway step was either absent or defective. This and several other IDT genes were absent from E. festucae E2368, and the remaining epichloae (E8, E56, E57, E167, E277, E2772, E818, E4804, and E5819) completely lacked IDT genes (Table 5), although E818 had two remnant IDT pseudogenes linked to its telomeric EAS locus (Figure 6).
The newly discovered ltmS gene was identified within the LTM cluster of E. festucae Fl1 using RNA-seq data of Fl1 and its ΔsakA mutant [57] mapped back to the Fl1 genome. The ltmS gene followed the same expression pattern as the other LTM genes, being significantly down-regulated in the ΔsakA mutant. An ortholog of ltmS was identified in each IDT/LTM gene cluster from C. pupurea, C. paspali, A. take, P. ipomoeae and N. gansuense E7080 (Figure 7). However, homology search (BLASTp) against the nonredundant protein database identified no orthologs in non-clavicipitaceous fungi, and no protein domains were evident in InterPro analysis. Topology prediction tools HMMTOP [58], TMHMM [59] and TopPred [60] indicated that LtmS contains at least four transmembrane domains. The inferred LtmS peptide sequence was compared to the inferred product of the paxA gene, which is located in the P. paxilli indole-diterpene cluster gene in a similar orientation between the orthologs of ltmM (paxM) and ltmG (paxG) [61]. Although sequence similarity was not significant, hydrophobicity plots (data not shown) suggested a shared transmembrane domain structure. Currently, roles for paxA and ltmS remain to be elucidated, but their shared characteristics, common placement within orthologous IDT/LTM and PAX clusters, and co-regulation with other LTM genes suggested that they may be required for indole-diterpene production.
Loline alkaloid (LOL) loci
The loline alkaloid biosynthesis (LOL) genes were found only in the sequenced genomes of epichloae that produce lolines, and a remnant LOL cluster was identified in an additional epichloid strain. Figure 9 compares the LOL clusters with the two clusters previously characterized in the hybrid endophyte Neotyphodium uncinatum E167 [42]. In the periphery of the LOL locus of E. festucae E2368 were two divergently transcribed, newly discovered genes designated lolN and lolM. Orthologous lolN and lolM genes were also identified in survey sequencing of E167, which has a similar loline alkaloid profile to that of E2368, adding support to the hypothesis that these genes specify certain loline-decorating steps. Scaffolding and long-range physical mapping confirmed and extended previous analysis of large-insert clones [62], indicating that the LOL gene order in E2368 was similar to that in E167. In E2368, 10 of the 11 LOL genes were in pairs of divergently transcribed genes. In the other strains the precise LOL-gene orders were not completely elucidated, but no rearrangements within the cluster were evident. However, orientation of the LOL clusters relative to flanking housekeeping genes, nsfA and lteA, were not conserved. Also, several loline-alkaloid producers had missing or inactive decoration genes (lolN, lolM, and lolP). The LOL cluster of E. brachyelytri E4804, which accumulates AcAP without an ether bridge, had an inactive lolO gene due to an internal deletion, and also lacked functional lolN, lolM, and lolP genes.

No LOL genes were identified in E. typhina isolate E8 or E5819, E. festucae Fl1, N. gansuense var. inebrians E818, or E. elymi E56. Orthologs of the genes that flank LOL—namely, nsfA and lteA—were linked in the E5819 genome, with two additional hypothetical genes between them (Figure 9). The hypothetical genes were also associated with lteA in E8, E56, and E. amarillans E57, and nsfA in Fl1, although the orientation of the genes differed in E57 and Fl1. In genome assemblies of E8, Fl1, E56, and E818 linkage of nsfA and lteA was not established, and large repeat blocks were identified downstream of nsfA and upstream of lteA in E8 and Fl1. There was no indication of any LOL genes in the genomes of Claviceps species, A. take, or P. ipomoeae, and the nsfA and lteA orthologs were closely linked in all, although lteA was reoriented in C. fusiformis PRL 1980 (Figure 9).
Peramine (PER) loci
As was the case for the other alkaloid loci, the peramine (PER) locus was variable, containing the entire multifunctional perA gene in the peramine producers, no perA gene, or a partially deleted perA gene designated perA-ΔR* (Figure 10). The complete gene encodes a multifunctional protein with peptide synthetase, methyltransferase and reductase domains that together may be sufficient for synthesis of peramine [16]. The PER locus in E. festucae E2368 and E. typhina E5819 shared identical deletions of the C-terminal reductase domain. Nevertheless, perA-ΔR* was expressed in E2368, raising the possibility that this form also encodes a multifunctional protein, which may participate in the biosynthesis of a metabolite related to peramine if an appropriate thioesterase, condensation, cyclization or reduction domain is provided in trans.

Long, syntenous regions flanked both the 5′ and 3′ sides of the functional perA genes of E. typhina E8, E. festucae Fl1 and E. elymi E56 as well as the complete and probably functional perA genes in E. brachyelytri E4804 and E. amarillans E57 (Figure 10). The 5′ region included the divergently transcribed gene mfsA. The two genomes with perA-ΔR* shared synteny of the 3′ flanking region, but repeat blocks in the 5′ flanks apparently disrupted sequence assembly.
A possible perA ortholog was identified in P. ipomoeae, but it was a pseudogene, and was located in a different locus from the PER locus of the epichloae (Figure 10). The predicted gene product included all of the domains of PerA in the same order, with 47.6% amino acid sequence identity over 98.8% of the length of PerA.
Telomere positions relative to alkaloid loci
In the epichloae, EAS and IDT/LTM loci were almost always linked to telomeres but LOL and PER loci were not. In contrast, no telomere linkage of alkaloid loci was evident in Claviceps species, A. take or P. ipomoeae.
Out of the eight epichloae with EAS genes, seven had EAS clusters linked to terminal telomeres (Figure 6, Figure S2). Long-range mapping of EAS genes, telomeres, and other specialized (secondary) metabolism (SM) genes of E. festucae E2368 indicated that its EAS cluster was linked to a 6-module nonribosomal peptide synthetase (NRPS) gene located near a telomere (Figure S2). Other epichloae had terminal telomere repeat arrays on a contig or supercontig containing some or all of their EAS genes. The sole exception was E. brachyelytri E4804, which had a RecQ helicase pseudogene near an lpsA gene fragment, suggesting possible telomere linkage [63]. Interestingly, although the EAS cluster of N. gansuense var. inebrians E818 was arranged similarly to that of P. ipomoeae and the Claviceps species, it was broken into two clusters, one of which ended in a telomere located one bp from the cloA stop codon. In contrast, the C. purpurea EAS cluster clearly was not near a telomere, since it spanned positions 235,054 to 290,780 of the 464,384-bp Supercontig22, and that supercontig had no telomeric repeats at either end.
Among the IDT/LTM loci the functional clusters in N. gansuense E7080 and E. festucae Fl1, as well as the partial cluster in E. festucae E2368, were all telomere-linked (Figure 7). (The terminal telomere sequence adjacent to E2368 LTM was evident in the 2010-06 assembly, which is also posted at www.endophyte.uky.edu.) Like the EAS loci, these IDT/LTM loci had the telomeres at different relative positions. (Interestingly, although N. gansuense var. inebrians E818 lacked functional IDT genes, it had two remnant IDT pseudogenes adjacent to the telomere-linked EAS cluster, as indicated in Figure 6.) In contrast to the epichloid IDT/LTM clusters, the orthologous cluster in C. purpurea 20.1 was not telomere-linked. This cluster (which was predicted to be nonfunctional because it lacked idtG) extended from positions 574,647 to 587,656 on the 978,494-bp Supercontig1 of the C. purpurea assembly, and no telomere was present on this scaffold. Also, no telomeres were present on the contigs containing IDT genes in the genomes of A. take, C. paspali, or P. ipomoeae.
The LOL clusters were not near telomeres. Instead, in all loline-alkaloid producers the clusters were flanked on both sides by groups of housekeeping genes. Published analysis of E. festucae large-insert clones [62] indicated that lolF is linked to nsfA, and lolT and lolE are linked to lteA. The nsfA gene was near the end of the 148,125-bp Supercontig71, and the lteA gene was near the end of 217,442-bp Supercontig41, and neither of these scaffolds had a telomere end. Likewise, the PER loci with complete perA genes were not subtelomeric, and no terminal telomere repeats were present on contigs with perA-ΔR*.
Synteny with the Fusarium graminearum PH-1 genome
The genome of F. graminearum PH-I is almost completely assembled into its known linkage groups [64], and because this species is within the same order (Hypocreales), but a different family from the Clavicipitaceae, we considered it appropriate to compare regions of the alkaloid loci for synteny with the F. graminearum genome. None of the four alkaloid loci in the Clavicipitaceae was present in the F. graminearum genome. In cases where the alkaloid loci were subtelomeric, flanking genes on their centromeric sides were not orthologous to F. graminearum genes. Alkaloid loci that were not subtelomeric and had flanking genes orthologous to F. graminearum genes were the EAS loci of Claviceps species, IDT loci of Claviceps species and A. take, and LOL and PER loci of the epichloae. The genes flanking the EAS clusters of Claviceps species were linked and similarly oriented in a syntenous region of the F. graminearum genome (Figure S4A). In contrast to the EAS loci, the genes flanking Claviceps and A. take IDT clusters were not syntenous in the F. graminearum genome (Figure S4B). The F. graminearum orthologs of the LOL-flanking genes were contained in a syntenous block (Figure S4C). Likewise, as reported previously [16], perA of E. festucae Fl1 had apparently been inserted into a block of genes syntenous with their F. graminearum orthologs (Figure S4D). These observations raise the possibility that the non-terminal EAS, LOL and PER loci had inserted into their respective genome locations, but where they originally assembled cannot be discerned because no intermediate stages in the evolution of the alkaloid gene clusters have yet been identified.
Repeat blocks in alkaloid loci
The alkaloid loci of most Clavicipitaceae were associated with repeat DNA derived from transposable elements, which were often stacked and nested extensively into long blocks. The distribution of repeat blocks in alkaloid loci constituted a major and consistent structural difference distinguishing the epichloae from the other Clavicipitaceae. The epichloae typically had long and dynamic repeat blocks predominantly of transposon relics, interspersed throughout their alkaloid loci. This characteristic was not a reflection of overall repeat content of the genomes, considering that epichloae had proportionately less repeat content than C. fusiformis and A. take (Table 2).
Repeat blocks at alkaloid gene loci were usually very AT-rich. RIP-index analysis (Figure S5) indicated that this was most likely due to the repeat-induced point mutation (RIP) process of selective C to T transitions that is common in fungi [65]. The possibility of RIP was further substantiated by the identification of homologs of the Neurospora crassa rid-1 gene [66] in all of the sequenced genomes except that of C. purpurea 20.1, which paradoxically had one of the lowest repeat contents (Table 2). In most Clavicipitaceae the overall GC content of repeat sequences was very low (Table 3). An exception was C. purpurea 20.1, consistent with its lack of rid-1 homolog and therefore presumed inability to perform RIP. Among the epichloae, the GC contents of E. glyceriae repeats tended to be relatively higher, suggesting less history of RIP since the repeat blocks emerged in that lineage.
The EAS and IDT clusters of Claviceps species, P. ipomoeae and A. take had very little repeat DNA within them, although repeat blocks flanked the EAS clusters of C. fusiformis PRL 1980 and C. paspali RRC-1481 (Figure 6, Figure 7). These Claviceps strains had lost or inactivated one (C. paspali) or all (C. fusiformis) lysergyl peptide synthetase (lps) genes at the cluster periphery, but the conserved-gene cores of their EAS and IDT clusters remained nearly free of repeat sequences. In contrast, the epichloae all had long blocks of repeat sequences associated with their alkaloid gene loci, and (except for the EAS loci of E818, which were divided by a telomere) all EAS and IDT loci of epichloae were broken up further into subclusters by such long repeat blocks. Even within subclusters a large number of MITE insertions were evident in intergenic regions (Figure 11).

The positions and lengths of repeat blocks and arrangements of MITEs within EAS and IDT clusters, as well as the gene orders and orientations, widely varied among the sequenced epichloae (Figure 6, Figure 7). Expansions and losses of repeat blocks resulted in variation with respect to the grouping of genes within subclusters. The repeat blocks often extended well beyond the alkaloid gene loci. For example, they dominated the entire Fl1 267-kb scaffold, Supercontig41, extending from the telomere, separating the LTM genes into three clusters, and disrupting a polyketide synthase gene in an adjacent SM gene cluster (Figure 7).
In some cases, the order of repeat insertions could be identified within the gene clusters. For example, in E. festucae Fl1 the easD-lpsA intergenic region apparently had been invaded by Tahi, which in turn was invaded by repeat number 17 (Figure 6). However, many of the multiple repeat insertions were much more complex than this example and varied among the different species. Several differences in MITE positions relative to the genes also appeared to be related to insertions of one or more repeat elements. By example the Waru DNA-transposon relic adjacent to dmaW in E. typhina E5819 had displaced the proximity of the 3m MITE (Figure 11A), and MITEs adjacent to easE in E. elymi may have also been displaced by the insertion of a repeat (Figure 11B). Compared to the other epichloae, E. glyceriae E277 had fewer MITEs in its EAS cluster and throughout its genome.
The perA and LOL genes were only found in epichloae (Figure 9, Figure 10). Nevertheless, LOL loci resembled the other epichloid alkaloid gene clusters in that they contained multiple blocks of nested repeats. Furthermore, positions of the repeat blocks in LOL varied greatly between strains, even though the gene orders and orientations appeared to be stable. Repeats in the PER loci were associated with perA-ΔR* rather than perA (Figure 10). In those strains with the deleted R*domain, repeat blocks extended upstream of mfsA to the contig ends, leaving it unresolved whether mfsA and perA-ΔR* were linked. Also, MITE 3m was immediately downstream of the perA-ΔR* coding sequence, thus associated with the R*-domain deletion, as previously noted [67].
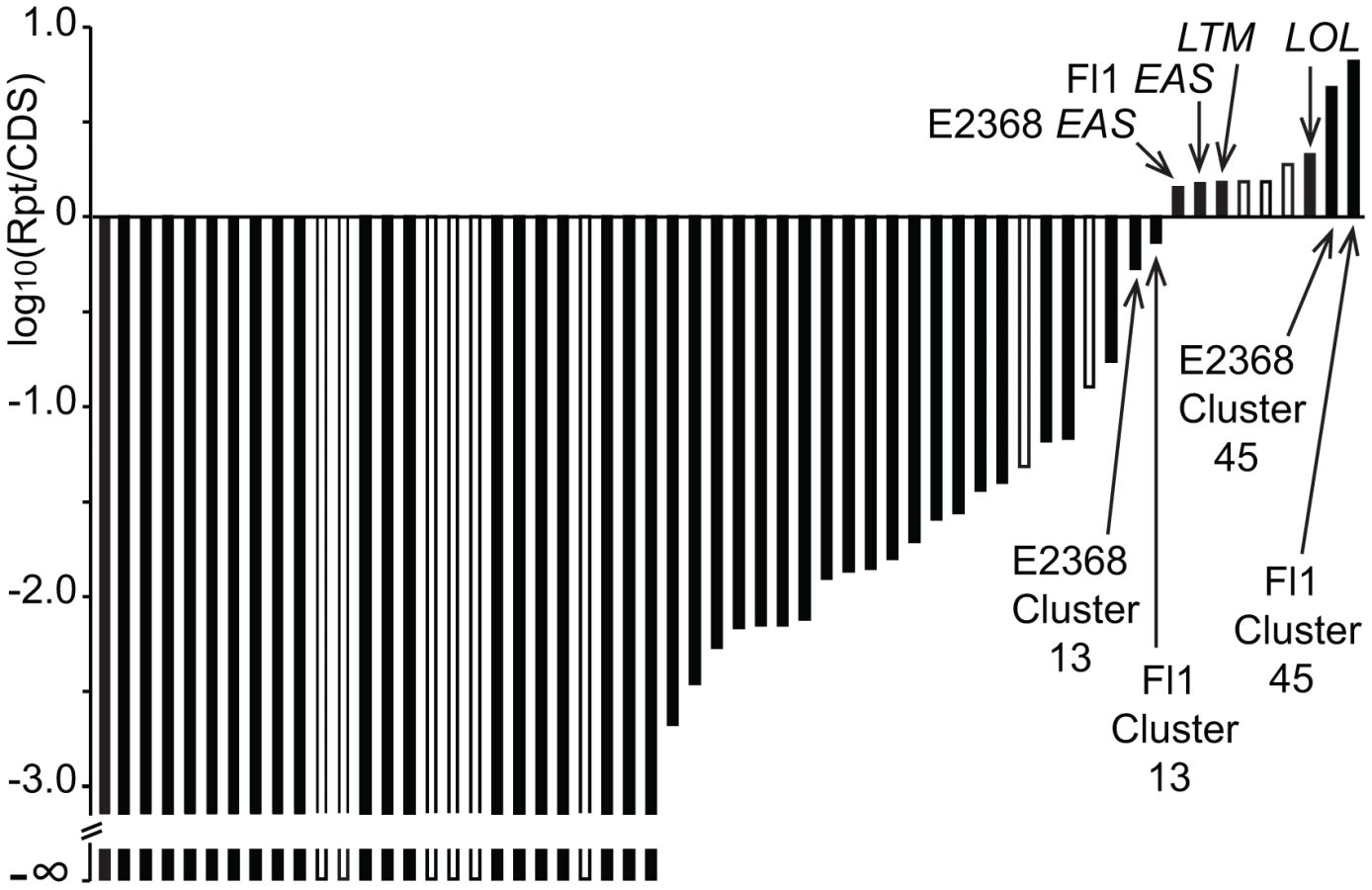
In order to assess whether large repeat blocks were mainly a feature of epichloid alkaloid clusters or, alternatively, a general feature of their SM clusters, the SM loci of both sequenced E. festucae strains as well as C. purpurea were manually identified and delineated (Table S4), and the proportions of repeat and coding sequences were determined. Each of these genome assemblies had been scaffolded by paired-end or mate-pair reads. In C. purpurea 20.1, repeat sequences within SM clusters were rare and small, though large repeat blocks flanked three SM clusters. For the SM clusters of the E. festucae strains, a logarithmic plot of total repeat sequence versus coding sequence lengths (Figure 12) demonstrated that only two active SM loci had comparable proportions of repeat sequence as the EAS, LTM, and LOL loci.
Discussion
Alkaloids play a major role in the ecology of many Clavicipitaceae, protecting seeds and foliage of host grasses and morning glories from herbivores, or protecting fungal structures (such as ergots) from fungivores. Typically the effects of alkaloids on animals (insects, mammals, etc.) are much more immediate than is the case for many other specialized metabolites because alkaloids target the nervous systems and directly affect behavior [1]. Systemic symbionts such as epichloae and Periglandula species supplement the diversity of protective metabolites in grasses and morning glories, respectively, and such diversification should serve an important role in bet-hedging [68], [69] to enhance overall fitness in populations of plant-fungus symbiota on an ecologically variable landscape. Alkaloid diversification occurs at two levels, one being the presence or complete absence of each of several different classes, and the other being variations within each class. Here we compared alkaloid profiles and total alkaloid gene contents among 15 Clavicipitaceae, and also compared the arrangements of those genes and their associations with telomeres and blocks of repeat sequences. Two noteworthy patterns emerged. First, in most alkaloid loci in most species, the periphery of each cluster was enriched in genes that by virtue of their presence, absence, or sequence variations determined the diversity of alkaloids within the respective chemical class. Second, alkaloid gene loci of the epichloae had extraordinarily large and pervasive blocks of AT-rich repeats derived from retroelements, DNA transposons, and MITEs. In the epichloae these gene clusters were clearly unstable, probably because of the repeat blocks and, in the cases of EAS and IDT/LTM clusters, nearby telomeres. This instability was manifested in strains that had lost complete clusters, strains that had lost large portions of clusters, and strains with variant alkaloids attributable to gene duplications and neofunctionalizations. Partial or complete losses of alkaloid gene clusters generated diversity both between and within species of epichloae, as was apparent in comparisons of two isolates from each of three species, N. gansuense, E. festucae, and E. typhina. Also, gene duplications and neofunctionalizations resulted in the two novel IDT genes, ltmE and ltmJ, required for lolitrem B biosynthesis in E. festucae. Here we discuss how the alkaloid locus architectures relate to chemical diversity for each class of alkaloids, and how different ecological contexts of these fungi might select for those architectural differences.
Comprehensive genome sequencing was necessary to identify, with high confidence, all biosynthesis genes for each class of alkaloids in each fungal strain. Every indication has been that, like many fungi, the Clavicipitaceae tend to cluster these genes [7], [42], [70]. However, traditional methods have proven slow and unreliable for complete characterization of each cluster. Cloning and genome walking through these regions is especially difficult when, as is typical of the epichloae, they contain very large blocks of repeat DNA sequence, most of which is highly AT-rich, and cloned fragments containing these sequences are unstable and underrepresented in most gene libraries [42], [43], [71]. Therefore, current genome sequencing technologies facilitated not only a more comprehensive analysis of the gene clusters (including flanking repeats), but also the identification of previously unknown genes in or near these loci. In this way, two genes were newly discovered in the peripheries of some EAS loci (easO and easP), and two new LOL genes were also discovered (lolN and lolM). Furthermore, transcriptome analysis revealed the ltmS gene, which eluded de novo gene prediction, but was present in all IDT/LTM loci of the clavicipitaceous fungi.
Although the role of ltmS is not yet apparent, reasonable hypotheses for roles of the newly discovered EAS and LOL genes could be formulated based on gene presence or absence, along with comparisons of alkaloid profiles. For example, easO and easP were associated with LAH production. The sequence of easO indicates that it encodes a flavin-binding monooxygenase that, in the context of LAH biosynthesis, may oxidize the α-carbon of the alanine-derived residue in ergonovine or the ergonovine precursor attached to the LpsB/LpsC peptide synthetase complex. Furthermore, the sequence of easP indicates that it encodes an α/β hydrolase-fold protein, which could be involved in subsequent hydrolysis to release LAH. Similarly, the presence of lolN and lolM, predicted to encode an acetamidase and an N-methyltransferase, respectively, fits well with late enzymatic steps needed for NFL biosynthesis. Evidence from the genomes and chemotypes of strains with different loline alkaloid profiles suggest that the first fully cyclized loline alkaloid is NANL, which has an acetylated 1-amine. In order to produce NFL from NANL, it would be necessary first to deacetylate, then di-methylate that amine to generate NML. These are the likely roles for LolN and LolM, respectively, and the previously characterized lolP gene encodes a cytochrome P450 involved in the final process of oxygenating NML to NFL [72].
The Clavicipitaceae are best known for their ergot alkaloids, and among species and strains there are dramatic differences in ergot-alkaloid profiles [2], [73]. This variation is due to the particular mid-pathway or late-pathway genes that they possess, as well as differences in substrate or product specificity due to gene sequence variations [51], [70], [74]. In this study we associated chemotypes of Claviceps species with presence or absence of the genes lpsA, lpsB, lpsC, easH, easO and easP. In the Claviceps and Periglandula species, these genes are all in the cluster periphery, in contrast to the early-pathway and most mid-pathway genes in the core. (The mid-pathway gene easA is an exception, but with a unique role in alkaloid diversification as discussed below.) Even in the extensively rearranged EAS clusters of the epichloae the lps genes are often in the periphery. This placement is interesting considering the key role of lps genes in much of ergot-alkaloid diversity [51], and the propensity we observed for transposon-derived repeats to flank the EAS clusters in most Clavicipitaceae. Indeed, long repeat blocks were generally evident whenever lps genes were partially or wholly deleted or inactivated by extensive mutation.
In addition to gene presence or absence, sequence variation of certain genes resulted in further diversification of ergot alkaloids. This was dramatically evident for the multi-module lysergyl peptide synthase subunit I encoded by lpsA. Variations in lpsA among genera, and even between the two copies found in C. purpurea 20.1 [51], dictate which three amino acids are added to lysergic acid, hence which of 19 known ergopeptines are produced [7]. In addition, easA, which encodes a mid-pathway step in synthesis of the first fully cyclized ergoline, is also one for which sequence variation results in diversification of ergot alkaloids. Different easA forms determine if ergolines or dihydroergolines are produced [74]. (None of the strains sequenced in this study produce dihydroergolines.) Conceivably, variation in cloA also plays a role in ergot-alkaloid diversity. The C. purpurea CloA cytochrome-P450 catalyzes oxygenation of elymoclavine to paspalic acid [75], which spontaneously rearranges to lysergic acid. The cloA gene from C. fusiformis PRL 1980, though expressed and without any apparent defect, fails to complement this role in a cloA-deleted strain of C. purpurea [70]. However, it is unknown whether the variant form of CloA in C. purpurea has another role, for example in the oxygenation of agroclavine to elymoclavine.
Clearly the EAS loci in the epichloae are unstable and subject to rearrangements and partial or complete elimination. We characterized genomes of several epichloae that have the 11 genes required to synthesize the complex ergopeptines, others that had only the four functional genes required for chanoclavine I biosynthesis, and still others that lacked any functional EAS genes. Extensive rearrangements of the epichloid EAS clusters contrasted with the gene arrangements conserved among Claviceps species, P. ipomoeae, and the published Metarhizium genomes [46]. Interestingly, although N. gansuense var. inebrians E818 had an EAS locus structure and chemotype more similar to that of P. ipomoeae IasaF13 than to other epichloae, the E818 EAS locus had been broken up with a telomere and had lost lpsA and easH. Therefore, a tendency for rearrangements and telomere associations was consistently evident in, and contributed to, the chemotypic diversity of the epichloae.
The organization of IDT/LTM genes showed an even more distinct and consistent positioning of early and late pathway genes compared to the EAS loci. Furthermore, sequence variations in essentially all of the peripheral IDT/LTM genes account for differences in specificities of the cytochromes P450 and prenyltransferases that they encode, resulting in broad diversity of alkaloids within this class [53]. Rearrangements of IDT/LTM genes in the epichloae associated with large repeat blocks and telomeres were probably also responsible for gene duplications and neofunctionalizations that generated two new peripheral genes (ltmE and ltmJ) in the LTM cluster, allowing E. festucae to produce an especially complex group of indole-diterpenes, the lolitrems. This is a dramatic illustration of chemical diversification by cluster rearrangements almost certainly facilitated by the blocks of transposon-derived repeats.
The LOL loci, which were found only in epichloae, had features similar to EAS and IDT/LTM. Two of the three decoration genes identified in E. festucae E2368 were at the locus periphery, and all functional LOL loci were riddled with large and dynamic blocks of transposon-derived repeats. One notable difference was that, unlike the EAS and IDT/LTM clusters, the LOL clusters were not subtelomeric. Nevertheless, like EAS and IDT/LTM, the LOL loci were subject to partial or complete loss, resulting in different loline alkaloid profiles. Even the PER locus of some strains contained repeat blocks, and the perA gene also exhibited instability.
It is noteworthy that ergot alkaloids and indole-diterpenes are known among diverse ascomycetes, for which they undoubtedly have a variety of ecological roles. For example, the presence of EAS and IDT genes in Metarhizium species could indicate that these neurotropic alkaloids contribute to their abilities to affect behavior of parasitized insects. In contrast, peramine and loline alkaloids are characteristic of the epichloae, but unknown among other fungi. Consequently, compared to other Clavicipitaceae the epichloae have an even more diverse pallet of alkaloids to draw on to protect host plants. As systemic, and often vertically transmitted symbionts, epichloae depend on host plants throughout their life cycles, so it is to be expected that such an arsenal of plant protectants greatly benefits the epichloae.
The dynamics of alkaloid loci in the Clavicipitaceae, and especially the epichloae, promote chemotypic diversification even within species, and with respect both to the classes of alkaloids as well as the particular structures within each class that are produced. Transposon-derived repeats such as typify these loci can promote both recombination and mutation, and their insertions or deletions can radically alter gene regulation [76], [77]. We suggest that selection for chemotypic diversification within epichloid species may be imposed by their exceptional variety of life histories and host interactions. Whereas most other Clavicipitaceae are either contagious parasites (A. take and Claviceps species) or vertically transmitted mutualists (P. ipomoeae), epichloae vary widely in relative mutualistic or parasitic effects on their hosts based largely on transmission mode, and many (e.g., E. amarillans, E. brachyelytri, E. elymi, E. festucae and, in some hosts, E. typhina) have the remarkable capability to exhibit both transmission modes simultaneously on different tillers of the same plant [25]. Variation in relative vertical or horizontal transmission is expected to impose variation in selection on the symbiont, whereby vertical transmission selects for enhancements of host fitness [78]. Alkaloids, which typically deter herbivores [1], can be major contributors to host fitness, but also expensive to produce [79]. We suggest that variation in life history traits among the epichloae, as well as variation in ecological settings of their hosts, selects for exceptionally dynamic alkaloid loci that ensure high interspecific and intraspecific chemotypic variability.
Materials and Methods
Biological materials
Fungal strains and their sources are listed in Table S1. The Epichloë and Neotyphodium species, Claviceps fusiformis, Claviceps paspali, and Aciculosporium take were cultured on potato dextrose agar (PDA) on a cellophane layer, or in potato dextrose broth (PDB) with shaking at 23°C. Mycelia were collected by centrifugation for 20 min at 5525× g, frozen and lyophilized prior to DNA isolation. Culture conditions for C. purpurea were as in Mey et al. [80].
Because Periglandula ipomoeae is so far nonculturable, the adaxial sides of the leaves of an infected host plant (Ipomoea asarifolia) were wetted with deionized water, and mycelia were picked off with a scalpel, placed into a vial with 70% ethanol, and stored at −20°C. The mycelium was harvested by centrifugation, frozen and lyophilized.
DNA was isolated by the method of Al-Samarrai et al. [81] or, for C. purpurea, by the method of Cenis [82].
Microscopic examination of Epichloë festucae in symbio
In order to document the stages of the life cycle of Epichloë festucae Fl1, the fungus was transformed with the plasmid, pCA49, which includes an enhanced cyan fluorescent protein (eCFP) coding sequence controlled by the Pyrenophora tritici-repentis TOXA gene promoter [83]. Fungal transformation was performed as previously described [72] and transformants were selected for resistance to hygromycin B. The transformants were introduced into seedlings of perennial ryegrass (Lolium perenne) [84], and the symbiotic fungus was detected by tissue-print immunoblot with antiserum raised against a protein extract from Neotyphodium coenophialum [85]. Plants were grown in the greenhouse, and vernalized to induce flowering and seed development [86]. Plant tissues were dissected manually with the aid of a dissecting scope, placed on a glass slide in a drop of 50% glycerol, and covered with a coverslip. Confocal micrographs were generated with an Olympus FV1000 point-scanning/point-detection laser scanning confocal microscope, equipped with a 440 nm laser. Emission fluorescence was captured and collected at 467±15 nm through the eCFP filter. Image acquisition was performed at a resolution of 512×512 pixels and a scan rate of 20 µs pixel−1. The objective, Olympus water immersion PLAN APO 20×-Water (NA 0.75), was used for observing and generating micrographs. FLUOVIEW 1.5 software (Olympus) was used to control the microscope and export images as TIFF files.
Alkaloid analyses
Methods of analyses were as described previously for ergot alkaloids [75], [87], lolines [88], peramine [89], and indole-diterpenes [90].
Clone libraries
For Sanger sequencing of the E. festucae E2368 genome, a clone library of randomly sheared genomic DNA was constructed as follows. Nuclear DNA was enriched by bisbenzimide-CsCl isopycnic ultracentrifugation, randomly sheared with a GeneMachines Hydroshear (DigiLab Genomic Solutions, Inc.), twice gel-fractionated to select DNA fragments of 3.5–4.5 kb, and cloned into pBCKS+ (Stratagene Cloning Systems, La Jolla, CA, USA). The library consisted of approx. 5 million clones, of which 2.5 million cfu were stored at −80°C as aliquots of transformed T1-phage resistant Escherichia coli cells (Electromax DH10B; Invitrogen Corp., Carlsbad, CA, USA), and the remainder as ligation mixture. The E. coli transformants were grown on LB agar with chloramphenicol (25 mg/L). Colonies were picked by a QPix robot (Genetix, Hampshire, UK) into 96-deep-well plates with 2× YT medium (1.5 ml per well), and grown overnight in a HiGro (GeneMachines, San Carlos, CA, USA) oxygenated shaking incubator for microtiter plates. The plasmids were purified robotically (Biomek FX, Beckman Coulter Inc, Fullerton, CA, USA) with the Perfect-Prep Plasmid 96 kit (Eppendorf AG, Hamburg Germany). Sequence reactions and capillary electrophoresis were conducted using vector primers and BigDye3.1 (Applied Biosystems, Foster City, CA, USA) at 1/16th reaction strength. The reactions were cleaned by ethanol precipitation and capillary electrophoresis was performed in a model 3730 DNA analyzer (Applied Biosystems). Both ends of each plasmid were sequenced. Sequencing results indicated that 99.8 of the clones contained genomic DNA inserts.
For the E. festucae Fl1 genome, a library was prepared in the fosmid vector pCC1FOS (Epicentre). DNA was fragmented with a Hydroshear equipped with the LARGE assembly, at speed setting 36, for 15 cycles. The fragments were end-repaired with the End-It kit (Epicentre), size-selected by electrophoresis in 0.4% agarose gel with Gelgreen stain (Biotium), imaged with blue light, purified from the agarose with Gelase (Epicentre), and blunt-end ligated to the fosmid arms using Fast-link (Epicentre). Escherichia coli Epi-300 T1R cells were transformed and selected for chloramphenicol resistance.
Genome sequencing
All DNA sequencing was conducted at the University of Kentucky Advanced Genetic Technologies Center. Most sequencing was conducted on a Roche/454 Titanium pyrosequencer. DNA was nebulized and size-selected to approximately 600 bp with AMPure beads (Agencourt), and subjected to shotgun pyrosequencing using the GS FLX Titanium General Library Preparation Kit, GS FLX Titanium LV emPCR Kit (Lib-L), and GS FLX Titanium Sequencing Kit XLR70 (Roche). Paired-end pyrosequencing was also conducted for E. festucae Fl1 (2-kb fragments 960,278 true paired end reads), E. festucae E2368 (3.0-kb fragments, 113,208 true paired end reads), and Claviceps purpurea (3.0-kb fragments, 1,128,137 true paired end reads). For paired ends, DNA was sheared with a Hydroshear with standard assembly, 20 cycles at speed setting of 12, then size selected with AMPure beads (Agencourt). The GS FLX Titanium Paired End adaptor set from Roche was used with Cre Recombinase, Exonuclease 1, and Bst polymerase from NEB according to the Roche GS FLX Titanium 3 kb Paired End Library Preparation Method Manual. Survey sequencing was conducted on and Ion Torrent PGM (Life Technologies) according to manufacturer's instructions. Sanger sequencing of the E. festucae E2368 genome was conducted on paired-ends of a library of cloned 3.8-kb (ave.) DNA fragments as described above (119,114 reads incorporated). In addition, the E2368 sequence assembly incorporated 235 reads of ca. 11-kb clones of genomic DNA in the pJAZZ system (Lucigen), as well as directly cloned telomere-containing fragments [91]. Sanger sequencing of the E. festucae Fl1 genome was conducted on paired-ends of the fosmid library of cloned 36-kb (ave.) DNA fragments (7259 read-pairs incorporated).
Data sets consisted of 2.3 M to 6.0 M reads per genome. Assemblies of E. festucae Fl1 and E2368 genomes incorporated paired-end data from Sanger sequencing in addition to 454 paired-end and single-end pyrosequencing data from a Roche/454 Titanium sequencer. The C. purpurea assembly used both single-end and paired-end pyrosequencing reads. All other genome assemblies used single-end pyrosequencing reads, some supplemented with sequences obtained on an Ion Torrent PGM (Life Technologies). Pyrosequencing reads that were duplicates, very short (<80 nt), or very long (>650 nt), or that had more than 1% of uncalled bases, were purged using utility program prinseq-lite-1.5 (http://prinseq.sourceforge.net) as suggested in Huse et al. [92]. Ion Torrent reads were trimmed of all base-calls after the first 230 bases. All genomes were assembled using Newbler Assembler ver. 2.5.3 (Roche/4540) with default parameters and the -sio option to ensure proper order of input data, with single-end reads preceding any paired-end data, and paired-end read libraries (Sanger and pyrosequencing) ordered by increasing insert size. Assemblies were uploaded to GenBank (Table S2), and are provided with annotations on GBrowse web sites (www.endophyte.uky.edu). The annotated assembly of the C. purpurea genome sequence can be viewed at http://www.ebi.ac.uk/ena/data/view/Project:76493.
Annotation of repetitive DNA elements
Repetitive DNA families in the genome sequences were defined by processing a self-BLASTN report from each genome using a custom PERL script (Amyotte S.G. et.al Manuscript in preparation) that identified sequences with multiple genome copies and classified these repeats into non-redundant families. The repeat families were then manually curated to correct or remove families misidentified in the automated process above. The genome distribution of repeated sequences was characterized using RepeatMasker version 3.2.9 [93], with Cross_Match [94] version 0.990329, with the final set of repetitive families serving as a custom library. Results have been included in the GBrowse web sites (www.endophyte.uky.edu). All unique repeats identified from the genome custom libraries were compared by reciprocal BLASTn to identify conserved sequences within and between each species. Repeat sequences with BLAST scores greater than 100 were used to develop a matrix table of corresponding repeat numbers. A common number was given to each repeat association to rapidly identify repeat families conserved across species. The matrix table was used to label repeats within each gene cluster with the universal repeat numbers (Figure 6, Figure 7, Figure 9, Figure 10, Figure 11). Repeats were assigned to putative superfamilies, and families where possible, based on BLASTx analysis and the presence and orientation of terminal repeats (Table S5).
Miniature inverted repeat transposable elements (MITEs) previously characterized in E. festucae [67] were identified in other genomes by BLASTn using a personal database. To determine whether short repeats found in non-epichloid clusters were MITEs they were analyzed for terminal inverted repeats using einverted (EMBOSS; http://emboss.bioinformatics.nl/cgi-bin/emboss/einverted). Individual repeat-containing loci were aligned using MUSCLE and manually analyzed for evidence of recombination.
RIP-index analysis
RIP indices were calculated using a sliding window analysis with a 200 bp window and a step size of 20-bp (in the centromere-to-telomere direction). RIP indices (ApT/TpA) [95] were calculated for each window. The process was repeated until the window met the end of the sequence (i.e partial windows were not counted). These operations were performed automatically using a perl script (Protocol S1).
Gene identification and orthology and phylogenetic analyses
Gene predictions were conducted by various methods available in MAKER version 2.0.3 [96]. In the MAKER runs, assembled contigs were filtered against RepBase [97] model organism “fungi,” using RepeatMasker [93] version open-3.2.8. Our MAKER runs used the predictors AUGUSTUS 2.3.1 (Fusarium model) [98], FGENESH 3.1.1 (Fusarium model) [99], GeneMark-ES 2.3a (self-trained), and SNAP 2006-07-28 (trained with C. purpurea gene predictions for genus Claviceps, and with E. festucae E2368 gene predictions for other genera). These ab initio predictions were supplemented with evidence from Clavicipitaceae proteins in the NCBI non-redundant protein database and from assembled E. festucae ESTs (unigenes). Relationships of predicted proteins to known protein families were assessed by running InterProScan [100] on the inferred protein sequences inferred from the predicted genes. Results, including MAKER and FGENESH predictions and subsequent analyses, have been included in a collection of web sites based on GBrowse 1.70 [101], [102], posted at www.endophyte.uky.edu.
Gene modeling for C. purpurea was done similarly, by applying three different gene prediction programs: 1) FGENESH [99] with different matrices (trained with Aspergillus nidulans, Neurospora crassa and a mixed matrix based on different species); 2) GeneMark-ES [103] and 3) AUGUSTUS [98] with available ESTs as hints. The different gene structures were displayed in GBrowse [101], [102], allowing manual validation of all coding sequences (CDSs). Annotation was aided by BLASTx hits between the C. purpurea genome and those from Blumeria graminis, Neurospora crassa, Fusarium graminearum and Ustilago maydis, respectively. For the cluster regions and selected genes of interest the best fitting model per locus was selected manually and gene structures were adjusted by manually splitting them or redefining exon-intron boundaries based on EST data where necessary.
Orthology analysis was conducted on FGENESH-predicted proteins with length 10 amino acids or greater. Each inferred protein sequence was assigned a unique label with a prefix indicating its source genome. The predicted genes were first compared to the curated ortholog groups in OrthoMCL-DB [104] version 4 using the OrthoMCL web service (http://orthomcl.org/cgi-bin/OrthoMclWeb.cgi?rm=proteomeUploadForm), to which each predicted proteome was submitted independently. Next, the combined set of inferred proteins from all of the sequenced Clavicipitaceae was analyzed as described in the OrthoMCL algorithm document (http://docs.google.com/Doc?id=dd996jxg_1gsqsp6). The software versions used for this procedure were: OrthoMCL version 2.0.2 [105], MCL-bio [100], MCL version 10–201 (http://micans.org/mcl/) [106], and NCBI BLAST version 2.2.25 [107].
As noted by Li et al. [105], an OrthoMCL-derived “ortholog group” may contain paralogs as well as orthologs. We used the COCO-CL [108] software distribution to recursively divide the ortholog groups obtained from OrthoMCL into sub-groups. A division was accepted if it had a bootstrap score of 0.75 or greater and a split-score (number of taxa common to both sub-groups, divided by the number of taxa represented in the smaller sub-group) of 0.5 or greater; a high split score indicates that the group is likely to be the result of an ancient duplication event, as many taxa have representative protein sequences on both sides of the split. For these analyses, COCO-CL was slightly modified as follows. The multiple amino-acid-sequence alignments employed MUSCLE version 3.8.31 [106] instead of ClustalW (version 1.83) [109] because rigorous experiments with simulated protein data sets, have shown that MUSCLE is comparable or superior in speed and average accuracy to the best current methods, such as CLUSTALW, MAFFT, and T-Coffee [106]. ClustalW was still used to compute distance matrices from MUSCLE's alignments. The remaining calls to ClustalW in the COCO-CL source code were converted to non-interactive mode, avoiding freezes that can occur when ClustalW prompts the user unexpectedly. Finally, we addressed a potential infinite loop generated by the COCO-CL clustering program when a cluster cannot be partitioned into precisely two subclusters. In our version this situation terminates the clustering program, leaving the cluster unpartitioned.
Results of OrthoMCL and COCO-CL have been included in the genome browser web sites (www.endophyte.uky.edu). Clicking an FGENESH prediction in the browser opens a data page that lists and hotlinks the prediction's homologs and orthologs, as well as a link to download the multiple sequence alignment for that cluster. A patch file is provided in Supporting Information (Protocol S2).
For phylogenetic analysis, the following steps were performed on the Phylogeny.fr site [110]: Sequences were aligned with MUSCLE [111], the phylogenetic tree was inferred with PhyML [112], and branch support was estimated by the approximate likelihood-ratio test [113] with the SH-like option. Trees were compared by the Shimodaira-Hasegawa test [114] implemented in phangorn [115] with the default parameters and 10,000 bootstrap replications.
mRNA sequence analysis
Sequences of cDNA generated by reverse transcription of mRNA provided the information required for manual annotation to refine models of alkaloid biosynthesis genes. Using the Qiagen RNeasy Plant Mini kit, RNA was isolated from symbiota composed of Lolium pratense ( = Schedonorus pratensis) with E. festucae (see Figure 1D). Tissues analyzed were newly emerged stromata and pre-anthesis inflorescences. RNA quality and quantity was checked on Bioanalyzer 2100 (Agilent) using plant RNA nano chip. A clone library was constructed by cDNA synthesis with the SMART kit (ClonTech) [116], normalization, and cloning into the λTriplex2 vector (ClonTech). Transfected E. coli BM25.8 cells were grown with ampicillin to select clones, and plasmid DNA was isolated and sequenced by standard Sanger sequencing using BigDye version 3.0 and an Applied Biosystems (Life Technologies) model 3730 xl DNA sequencer.
For deep cDNA sequencing (RNA-seq), 10 µg of high quality total RNA was used for cDNA library preparation according to the mRNA sequencing sample preparation guide (Illumina, Cat# RS 930-1001). Sources of RNA were inflorescences and stromata of L. pratense-E. festucae symbiota, laboratory id numbers 2194 and 2352. The libraries were validated on a 2100 Bioanalyzer using a DNA-1000 chip (Agilent). These libraries were used for bridge-PCR (SR cluster generation kit v4, Illumina) and 82-cycle single-read sequencing was conducted on a Genome Analyzer IIx (Illumina) in the DNA Facility at Iowa State University.
RNA-seq data from stromata and inflorescences, as well as RNA-seq data previously obtained from wild type E. festucae and a sakA mutant [57] were used for genome-wide identification and annotation of the expressed protein-coding genes of the Epichloë festucae strains. The RNA-seq data were combined with previously generated RNA-seq data and assembled into TAC contigs (defined as a continuous exonic region with contiguous read coverage) on both E2368 and Fl1 assemblies by MapSplice [117], which performs both spliced and unspliced alignment of RNA-seq reads to the reference genome. Then the combined read alignment coverage of the 6 tissues was used to detect exons. Exon boundaries are determined by splice junctions or the absence of the read coverage. Two TACs can be merged together by a splice junction connecting them. A TAC-contig is a maximum set of TACs that are linked together by splice junctions. If alternative splicing events exist, the alternative splice junction with more read alignment support is preferred. Because intergenic transcription or overlapping transcription of convergent genes sometimes led to merged gene models, junctions that crossed two FGENESH genes were filtered out, and TAC contigs that overlapped with more than two FGENESH-predicted genes were split according to the predicted gene boundaries. The 5′ and 3′ boundaries of gene structures were also trimmed based on the predicted genes.
Manual gene annotation
All of the genes for ergoline, peramine, indole-diterpene and loline alkaloid biosynthesis, as well as genes used for phylogenetic analysis, were manually annotated. Many of the genes were previously characterized in the same or related species and strains by targeted reverse transcription of their mRNAs followed by cDNA sequencing [21], [39], [41]–[43], [70], [71], [118]–[121]. Transcriptome information from E. festucae, including reads from cloned cDNAs and assembled TAC contigs, was used to model the gene exons. Cross-species comparisons, for example by using tBLASTn or tBLASTx, were employed to refine models in species for which transcript data were unavailable. In some cases, such as the newly discovered easO and easP genes, mRNA segments were amplified by reverse-transcription-PCR, and sequenced.
Identification and delimitation of specialized metabolism (SM) gene clusters
BLASTx and InterproScan [100] were employed to identify genes encoding enzymes that are signatures of SM gene clusters in the Ascomycota; namely, nonribosomal peptide synthetases (NRPS; IPR010071, IPR006163, IPR001242), polyketide synthases (PKS; IPR013968), DMATS-family aromatic prenyltransferases (IPR017795, Pfam PF11991), and terpene synthases/cyclases (IPR008949). The probable functions of proteins encoded by nearby genes were similarly assessed, as SM gene clusters contain various families of biosynthetic genes including mono - and dioxygenases, dehydrogenases, reductases, pyridoxal-phosphate (PLP)-cofactor enzymes, hydrolases, prenyltransferases and methyltransferases, as well as ABC or MFS efflux pumps and transcription regulators. However, many members of these enzyme families are involved in primary metabolism. Considering that most Clavicipitaceae can grow on minimal salts medium with sugars and inorganic nitrogen, those genes that had orthologs (identified by COCO-CL) among all of the sequenced genomes were considered probable primary metabolism genes. This interpretation was validated by the observation that most apparently active SM signature genes were flanked on one or both sides by ortholog groups with limited distribution among the 12 sequenced genomes. (Note that even after COCO-CL analysis, NRPS and PKS ortholog groups usually had several members in each genome, making it difficult to discern the distribution of their true orthologs, but this was not generally a problem for nearby genes.).
Accession numbers
Genome and sequence accession numbers are listed in Table S2.
Supporting Information
Zdroje
1. Wink M (2000) Interference of alkaloids with neuroreceptors and ion channels. In: Atta-ur-Rahman, editor. Bioactive Natural Products (Part B): Elsevier. pp. 3–122.
2. PažoutováS, OlšovskáJ, LinkaM, KolínskáR, FliegerM (2000) Chemoraces and habitat specialization of Claviceps purpurea populations. Appl Environ Microbiol 66 : 5419–5425.
3. UhligS, BothaCJ, VrålstadT, RolénE, MilesCO (2009) Indole-diterpenes and ergot alkaloids in Cynodon dactylon (Bermuda grass) infected with Claviceps cynodontis from an outbreak of tremors in cattle. J Agric Food Chem 57 : 11112–11119.
4. SchardlCL, LeuchtmannA, SpieringMJ (2004) Symbioses of grasses with seedborne fungal endophytes. Annu Rev Plant Biol 55 : 315–340.
5. IannoneLJ, NovasMaV, YoungCA, De BattistaJP, SchardlCL (2012) Endophytes of native grasses from South America: Biodiversity and ecology. Fungal Ecology 5 : 357–363.
6. SpataforaJW, SungGH, SungJM, Hywel-JonesNL, WhiteJF (2007) Phylogenetic evidence for an animal pathogen origin of ergot and the grass endophytes. Mol Ecol 16 : 1701–1711.
7. SchardlCL, PanaccioneDG, TudzynskiP (2006) Ergot alkaloids–biology and molecular biology. Alkaloids Chem Biol 63 : 45–86.
8. ClayK, SchardlC (2002) Evolutionary origins and ecological consequences of endophyte symbiosis with grasses. Am Nat 160: S99–S127.
9. SteinerU, LeibnerS, SchardlCL, LeuchtmannA, LeistnerE (2011) Periglandula, a new fungal genus within the Clavicipitaceae and its association with Convolvulaceae. Mycologia 103 : 1133–1145.
10. RudgersJA, KoslowJM, ClayK (2004) Endophytic fungi alter relationships between diversity and ecosystem properties. Ecol Lett 7 : 42–51.
11. MalinowskiDP, BeleskyDP (2000) Adaptations of endophyte-infected cool-season grasses to environmental stresses: Mechanisms of drought and mineral stress tolerance. Crop Sci 40 : 923–940.
12. TudzynskiP, CorreiaT, KellerU (2001) Biotechnology and genetics of ergot alkaloids. Appl Microbiol Biotechnol 57 : 593–605.
13. WiesemullerW (2005) Present and historical significance of ergot. Ernährungs Umschau 52 : 147–148.
14. GigerRKA, EngelG (2006) Albert Hofmann's pioneering work on ergot alkaloids and its impact on the search of novel drugs at Sandoz, a predecessor company of Novartis. CHIMIA International Journal for Chemistry 60 : 83–87.
15. SchardlCL, GrossmanRB, NagabhyruP, FaulknerJR, MallikUP (2007) Loline alkaloids: currencies of mutualism. Phytochemistry 68 : 980–996.
16. TanakaA, TapperBA, PopayA, ParkerEJ, ScottB (2005) A symbiosis expressed non-ribosomal peptide synthetase from a mutualistic fungal endophyte of perennial ryegrass confers protection to the symbiotum from insect herbivory. Mol Microbiol 57 : 1036–1050.
17. BacettyAA, SnookME, GlennAE, NoeJP, HillN, et al. (2009) Toxicity of endophyte-infected tall fescue alkaloids and grass metabolites on Pratylenchus scribneri. Phytopathology 99 : 1336–1345.
18. BoutonJH, LatchGCM, HillNS, HovelandCS, McCannMA, et al. (2002) Reinfection of tall fescue cultivars with non-ergot alkaloid-producing endophytes. Agron J 94 : 567–574.
19. LyonsPC, PlattnerRD, BaconCW (1986) Occurrence of peptide and clavine ergot alkaloids in tall fescue grass. Science 232 : 487–489.
20. GallagherRT, HawkesAD, SteynPS, VleggaarR (1984) Tremorgenic neurotoxins from perennial ryegrass causing ryegrass staggers disorder of livestock: structure elucidation of lolitrem B. J Chem Soc Chem Commun 1984 : 614–616.
21. MarkertA, SteffanN, PlossK, HellwigS, SteinerU, et al. (2008) Biosynthesis and accumulation of ergoline alkaloids in a mutualistic association between Ipomoea asarifolia (Convolvulaceae) and a clavicipitalean fungus. Plant Physiol 147 : 296–305.
22. Tor-AgbidyeJ, BlytheLL, CraigAM (2001) Correlation of endophyte toxins (ergovaline and lolitrem B) with clinical disease: fescue foot and perennial ryegrass staggers. Vet Hum Toxicol 43 : 140–146.
23. ThompsonRW, FribourgHA, WallerJC, SandersWL, ReynoldsJH, et al. (1993) Combined analysis of tall fescue steer grazing studies in the eastern United States. J Anim Sci 71 : 1940–1946.
24. SchardlCL, YoungCA, FaulknerJR, FloreaS, PanJ (2012) Chemotypic diversity of epichloae, fungal symbionts of grasses. Fungal Ecol 5 : 331–344.
25. SchardlCL (2010) The epichloae, symbionts of the grass subfamily Poöideae. Ann Mo Bot Gard 97 : 646–665.
26. ZhangD-X, NagabhyruP, BlankenshipJD, SchardlCL (2010) Are loline alkaloid levels regulated in grass endophytes by gene expression or substrate availability? Plant Signal Behav 5 : 1419–1422.
27. LeuchtmannA, SchmidtD, BushLP (2000) Different levels of protective alkaloids in grasses with stroma-forming and seed-transmitted Epichloë/Neotyphodium endophytes. J Chem Ecol 26 : 1025–1036.
28. SaariS, HelanderM, LehtonenP, WalliusE, SaikkonenK (2010) Fungal endophytes reduce regrowth and affect competitiveness of meadow fescue in early succession of pastures. Grass and Forage Science 65 : 287–295.
29. AfkhamiME, RudgersJA (2009) Endophyte-mediated resistance to herbivores depends on herbivore identity in the wild grass Festuca subverticillata. Environ Entomol 38 : 1086–1095.
30. CrosignaniPG (2006) Current treatment issues in female hyperprolactinaemia. Eur J Obstet Gynecol Reprod Biol 125 : 152–164.
31. Nichols DE (2001) LSD and its lysergamide cousins. The Heffter Review of Psychedelic Research. Santa Fe, New Mexico: Heffter Research Institute. pp. 80–87.
32. EadieMJ (2003) Convulsive ergotism: epidemics of the serotonin syndrome? Lancet Neurol 2 : 429–434.
33. CaporaelLR (1976) Ergotism: the Satan loosed in Salem? Science 192 : 21–26.
34. ScottP (2009) Ergot alkaloids: extent of human and animal exposure. World Mycotoxin Journal 2 : 141–149.
35. UrgaK, DebellaA, W'MedihnY, NA, BayuA, et al. (2002) Laboratory studies on the outbreak of gangrenous ergotism associated with consumption of contaminated barley in Arsi, Ethiopia. Ethiopian Journal of Health and Development 16 : 317–323.
36. SmithMM, WarrenVA, ThomasBS, BrochuRM, ErtelEA, et al. (2000) Nodulisporic acid opens insect glutamate-gated chloride channels: identification of a new high affinity modulator. Biochemistry 39 : 5543–5554.
37. KnausHG, McManusOB, LeeSH, SchmalhoferWA, Garcia-CalvoM, et al. (1994) Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry 33 : 5819–5828.
38. YoungC, McMillanL, TelferE, ScottB (2001) Molecular cloning and genetic analysis of an indole-diterpene gene cluster from Penicillium paxilli. Mol Microbiol 39 : 754–764.
39. TsaiH-F, WangH, GeblerJC, PoulterCD, SchardlCL (1995) The Claviceps purpurea gene encoding dimethylallyltryptophan synthase, the committed step for ergot alkaloid biosynthesis. Biochem Biophys Res Commun 216 : 119–125.
40. SpieringMJ, WilkinsonHH, BlankenshipJD, SchardlCL (2002) Expressed sequence tags and genes associated with loline alkaloid expression by the fungal endophyte Neotyphodium uncinatum. Fungal Genet Biol 36 : 242–254.
41. LorenzN, HaarmannT, PažoutováS, JungM, TudzynskiP (2009) The ergot alkaloid gene cluster: Functional analyses and evolutionary aspects. Phytochemistry 70 : 1822–1832.
42. SpieringMJ, MoonCD, WilkinsonHH, SchardlCL (2005) Gene clusters for insecticidal loline alkaloids in the grass-endophytic fungus Neotyphodium uncinatum. Genetics 169 : 1403–1414.
43. YoungCA, FelittiS, ShieldsK, SpangenbergG, JohnsonRD, et al. (2006) A complex gene cluster for indole-diterpene biosynthesis in the grass endophyte Neotyphodium lolii. Fungal Genet Biol 43 : 679–693.
44. SungGH, SungJM, Hywel JonesNL, SpataforaJW (2007) A multi-gene phylogeny of Clavicipitaceae (Ascomycota, Fungi): Identification of localized incongruence using a combinational bootstrap approach. Mol Phylogenet Evol 44 : 1204–1223.
45. TanakaE, TanakaC (2008) Phylogenetic study of clavicipitaceous fungi using acetaldehyde dehydrogenase gene sequences. Mycoscience 49 : 115–125.
46. GaoQ, JinK, YingS-H, ZhangY, XiaoG, et al. (2011) Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet 7: e1001264 doi:10.1371/journal.pgen.1001264
47. Pava-RipollM, AngeliniC, FangW, WangS, PosadaFJ, et al. (2011) The rhizosphere-competent entomopathogen Metarhizium anisopliae expresses a specific subset of genes in plant root exudate. Microbiology 157 : 47–55.
48. FleetwoodDJ, ScottB, LaneGA, TanakaA, JohnsonRD (2007) A complex ergovaline gene cluster in epichloë endophytes of grasses. Appl Environ Microbiol 73 : 2571–2579.
49. SteinerU, LeistnerE (2012) Ergoline alkaloids in convolvulaceous host plants originate from epibiotic clavicipitaceous fungi of the genus Periglandula. Fungal Ecol 5 : 316–321.
50. Gröger D, Floss HG (1998) Biochemistry of ergot alkaloids – achievements and challenges. In: Cordell GA, editor. Alkaloids Chem Biol. New York: Academic Press. pp. 171–218.
51. HaarmannT, LorenzN, TudzynskiP (2008) Use of a nonhomologous end joining deficient strain (Δku70) of the ergot fungus Claviceps purpurea for identification of a nonribosomal peptide synthetase gene involved in ergotamine biosynthesis. Fungal Genet Biol 45 : 35–44.
52. CastagnoliNJr, CorbettK, ChainEB, ThomasR (1970) Biosynthesis of N-(α-hydroxyethyl) lysergamide, a metabolite of Claviceps paspali Stevens and Hall. Biochem J 117 : 451–455.
53. SaikiaS, TakemotoD, TapperBA, LaneGA, FrazerK, et al. (2012) Functional analysis of an indole-diterpene gene cluster for lolitrem B biosynthesis in the grass endosymbiont Epichloë festucae. FEBS Lett (in press)..
54. ColeRJ, DornerJW, LansdenJA, CoxRH, PapeC, et al. (1977) Paspalum staggers: isolation and identification of tremorgenic metabolites from sclerotia of Claviceps paspali. J Agric Food Chem 25 : 1197–1201.
55. LiSM, UnsoldIA (2006) Post-genome research on the biosynthesis of ergot alkaloids. Planta Med 72 : 1117–1120.
56. SaikiaS, ParkerEJ, KoulmanA, ScottB (2007) Defining paxilline biosynthesis in Penicillium paxilli: functional characterization of two cytochrome P450 monooxygenases. J Biol Chem 282 : 16829–16837.
57. EatonCJ, CoxMP, AmbroseB, BeckerM, HesseU, et al. (2010) Disruption of signaling in a fungal-grass symbiosis leads to pathogenesis. Plant Physiology 153 : 1780–1794.
58. TusnádyGE, SimonI (2001) The HMMTOP transmembrane topology prediction server. Bioinformatics 17 : 849–850.
59. KroghA, LarssonBr, von HeijneG, SonnhammerELL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305 : 567–580.
60. MasamiI, MasafumiA, DemeloML, ToshioS (2002) Transmembrane topology prediction methods: a re-assessment and improvement by a consensus method using a dataset of experimentally-characterized transmembrane topologies. In Silico Biol 2 : 19–33.
61. SaikiaS, ParkerEJ, KoulmanA, ScottB (2006) Four gene products are required for the fungal synthesis of the indole-diterpene, paspaline. FEBS Lett 580 : 1625–1630.
62. KutilBL, GreenwaldC, LiuG, SpieringMJ, SchardlCL, et al. (2007) Comparison of loline alkaloid gene clusters across fungal endophytes: Predicting the co-regulatory sequence motifs and the evolutionary history. Fungal Genet Biol 44 : 1002–1010.
63. GaoWM, KhangCH, ParkSY, LeeYH, KangSC (2002) Evolution and organization of a highly dynamic, subtelomeric helicase gene family in the rice blast fungus Magnaporthe grisea. Genetics 162 : 103–112.
64. CuomoCA, GuldenerU, XuJ-R, TrailF, TurgeonBG, et al. (2007) The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317 : 1400–1402.
65. ClutterbuckAJ (2011) Genomic evidence of repeat-induced point mutation (RIP) in filamentous ascomycetes. Fungal Genet Biol 48 : 306–326.
66. FreitagM, WilliamsRL, KotheGO, SelkerEU (2002) A cytosine methyltransferase homologue is essential for repeat-induced point mutation in Neurospora crassa. Proc Natl Acad Sci U S A 99 : 8802–8807.
67. FleetwoodDJ, KhanAK, JohnsonRD, YoungCA, MittalS, et al. (2011) Abundant degenerate miniature inverted-repeat transposable elements in genomes of epichloid fungal endophytes of grasses. Genome Biol Evol 3 : 1253–1264.
68. PhilippiT, SegerJ (1989) Hedging one's evolutionary bets, revisited. Trends Ecol Evol 4 : 41–44.
69. SvardalH, RuefflerC, HermissonJ (2011) Comparing environmental and genetic variance as adaptive response to fluctuating selection. Evolution 65 : 2492–2513.
70. LorenzN, WilsonEV, MachadoC, SchardlCL, TudzynskiP (2007) Comparison of ergot alkaloid biosynthesis gene clusters in Claviceps species indicates loss of late pathway steps in evolution of C. fusiformis. Appl Environ Microbiol 73 : 7185–7191.
71. YoungC, BryantM, ChristensenM, TapperB, BryanG, et al. (2005) Molecular cloning and genetic analysis of a symbiosis-expressed gene cluster for lolitrem biosynthesis from a mutualistic endophyte of perennial ryegrass. Mol Genet Genomics 274 : 13–29.
72. SpieringMJ, FaulknerJR, ZhangD-X, MachadoC, GrossmanRB, et al. (2008) Role of the LolP cytochrome P450 monooxygenase in loline alkaloid biosynthesis. Fungal Genet Biol 45 : 1307–1314.
73. BlaneyBJ, MaryamR, MurrayS-A, RyleyMJ (2003) Alkaloids of the sorghum ergot pathogen (Claviceps africana): assay methods for grain and feed and variation between sclerotia/sphacelia. Aust J Agric Res 54 : 167–175.
74. CoyleCM, ChengJZ, O'ConnorSE, PanaccioneDG (2010) An old yellow enzyme gene controls the branch point between Aspergillus fumigatus and Claviceps purpurea ergot alkaloid pathways. Appl Environ Microbiol 76 : 3898–3903.
75. HaarmannT, OrtelI, TudzynskiP, KellerU (2006) Identification of the cytochrome P450 monooxygenase that bridges the clavine and ergoline alkaloid pathways. Chembiochem 7 : 645–652.
76. KidwellMG (2002) Genome evolution - Lateral DNA transfer mechanism and consequences. Science 295 : 2219–2220.
77. RouxelT, GrandaubertJ, HaneJK, HoedeC, van de WouwAP, et al. (2011) Effector diversification within compartments of the Leptosphaeria maculans genome affected by Repeat-Induced Point mutations. Nat Commun 2 : 202.
78. EwaldPW (1987) Transmission modes and evolution of the parasitism-mutualism continuum. Ann N Y Acad Sci 503 : 295–306.
79. ZhangD-X, NagabhyruP, SchardlCL (2009) Regulation of a chemical defense against herbivory produced by symbiotic fungi in grass plants. Plant Physiol 150 : 1072–1082.
80. MeyG, HeldK, SchefferJ, TenbergeKB, TudzynskiP (2002) CPMK2, an SLT2-homologous mitogen-activated protein (MAP) kinase, is essential for pathogenesis of Claviceps purpurea on rye: evidence for a second conserved pathogenesis-related MAP kinase cascade in phytopathogenic fungi. Molecular Microbiology 46 : 305–318.
81. Al-SamarraiTH, SchmidJ (2000) A simple method for extraction of fungal genomic DNA. Lett Appl Microbiol 30 : 53–56.
82. CenisJL (1992) Rapid extraction of fungal DNA for PCR amplification. Nucleic Acids Res 20 : 2380.
83. AndrieRM, MartinezJP, CiuffettiLM (2005) Development of ToxA and ToxB promoter-driven fluorescent protein expression vectors for use in filamentous ascomycetes. Mycologia 97 : 1152–1161.
84. LatchGCM, ChristensenMJ (1985) Artificial infections of grasses with endophytes. Ann Appl Biol 107 : 17–24.
85. AnZ-q, SiegelMR, HollinW, TsaiH-F, SchmidtD, et al. (1993) Relationships among non-Acremonium sp. fungal endophytes in five grass species. Appl Environ Microbiol 59 : 1540–1548.
86. ChungK-R, SchardlCL (1997) Sexual cycle and horizontal transmission of the grass symbiont, Epichloë typhina. Mycol Res 101 : 295–301.
87. PanaccioneDG, CipolettiJR, SedlockAB, BlemingsKP, SchardlCL, et al. (2006) Effects of ergot alkaloids on food preference and satiety in rabbits, as assessed with gene-knockout endophytes in perennial ryegrass (Lolium perenne). J Agric Food Chem 54 : 4582–4587.
88. FaulknerJR, HussainiSR, BlankenshipJD, PalS, BrananBM, et al. (2006) On the sequence of bond formation in loline alkaloid biosynthesis. Chembiochem 7 : 1078–1088.
89. SpieringMJ, DaviesE, TapperBA, SchmidJ, LaneGA (2002) Simplified extraction of ergovaline and peramine for analysis of tissue distribution in endophyte-infected grass tillers. J Agric Food Chem 50 : 5856–5862.
90. RasmussenS, LaneGA, MaceW, ParsonsAJ, FraserK, et al. (2012) The use of genomics and metabolomics methods to quantify fungal endosymbionts and alkaloids in grasses. Methods in Molecular Biology 860 : 213–226.
91. FarmanML (2011) Targeted cloning of fungal telomeres. Methods in Molecular Biology 722 : 11–31.
92. HuseS, HuberJ, MorrisonH, SoginM, DW (2007) Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biology 8: R143.
93. Smit AFA, Hubley R, Green P (1996–2010) RepeatMasker Open-3.0. 3.0 ed: Institute for Syst Biol.
94. EwingB, HillierL, WendlMC, PG (1998) Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res 8 : 175–185.
95. MargolinBS, Garrett-EngelePW, StevensJN, FritzDY, Garrett-EngeleC, et al. (1998) A methylated Neurospora 5S rRNA pseudogene contains a transposable element inactivated by repeat-induced point mutation. Genetics 149 : 1787–1797.
96. CantarelBL, KorfI, RobbSMC, ParraG, RossE, et al. (2008) MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18 : 188–196.
97. JurkaJ, KapitonovVV, PavlicekA, KlonowskiP, KohanyO, et al. (2005) Repbase Update, a database of eukaryotic repetitive elements. Cytogentic and Genome Res 110 : 462–467.
98. StankeM, KellerO, GunduzI, HayesA, WaackS, et al. (2006) AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34: W435–W439.
99. SalamovAA, SolovyevVV (2000) Ab initio gene finding in Drosophila genomic DNA. Genome Res 10 : 516–522.
100. EnrightAJ, Van DongenS, OuzounisCA (2002) An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 30 : 1575–1584.
101. SteinLD, MungallC, ShuS, CaudyM, MangoneM, et al. (2002) The generic genome browser: a building block for a model organism system database. Genome Res 12 : 1599–1610.
102. Donlin MJ (2007) Chapter 9, Unit 9.9: Using the generic genome browser (GBrowse). Current Protocols in Bioinformatics: Wiley Online Library.
103. Ter-HovhannisyanV, LomsadzeA, ChernoffYO, BorodovskyM (2008) Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res 18 : 1979–1990.
104. ChenF, MackeyAJ, StoeckertCJ, RoosDS (2006) OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res 34: D363–D368.
105. LiL, StoeckertCJJr, RoosDS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13 : 2178–2189.
106. EdgarRC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32 : 1792–1797.
107. AltschulSF, MaddenTL, SchäfferAA, ZhangJ, ZhangZ, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25 : 3389–3402.
108. JothiR, ZotenkoE, TasneemA, PrzytyckaTM (2006) COCO-CL: hierarchical clustering of homology relations based on evolutionary correlations. Bioinformatics 22 : 779–788.
109. ChennaR, SugawaraH, KoikeT, LopezR, GibsonTJ, et al. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31 : 3497–3500.
110. DereeperA, GuignonV, BlancG, AudicS, BuffetS, et al. (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36: W465–W469.
111. EdgarR (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5 : 113.
112. GuindonS, GascuelO (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52 : 696–704.
113. AnisimovaM, GascuelO (2006) Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol 55 : 539–552.
114. ShimodairaH, MasamiH (1999) Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Molecular Biology and Evolution 16 : 1114–1116.
115. SchliepKP (2011) phangorn: Phylogenetic analysis in R. Bioinformatics 27 : 592–593.
116. ZhuYY, MachlederEM, ChenchikA, LiR, SiebertPD (2001) Reverse transcriptase template switching: A SMART (TM) approach for full-length cDNA library construction. Biotechniques 30 : 892–897.
117. WangK, SinghD, ZengZ, ColemanSJ, HuangY, et al. (2010) MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 38: e178.
118. YoungCA, TapperBA, MayK, MoonCD, SchardlCL, et al. (2009) Indole-diterpene biosynthetic capability of epichloë endophytes as predicted by ltm gene analysis. Appl Environ Microbiol 75 : 2200–2211.
119. LorenzN, OlšovskáJ, ŠulcM, TudzynskiP (2010) Alkaloid cluster gene ccsA of the ergot fungus Claviceps purpurea encodes chanoclavine I synthase, a flavin adenine dinucleotide-containing oxidoreductase mediating the transformation of N-methyl-dimethylallyltryptophan to chanoclavine I. . Appl Environ Microbiol 76 : 1822–1830.
120. PanaccioneDG, JohnsonRD, WangJH, YoungCA, DamrongkoolP, et al. (2001) Elimination of ergovaline from a grass-Neotyphodium endophyte symbiosis by genetic modification of the endophyte. Proc Natl Acad Sci U S A 98 : 12820–12825.
121. WangJ, MachadoC, PanaccioneDG, TsaiH-F, SchardlCL (2004) The determinant step in ergot alkaloid biosynthesis by an endophyte of perennial ryegrass. Fungal Genet Biol 41 : 189–198.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 2
Nejčtenější v tomto čísle
- Complex Inheritance of Melanoma and Pigmentation of Coat and Skin in Grey Horses
- Coordination of Chromatid Separation and Spindle Elongation by Antagonistic Activities of Mitotic and S-Phase CDKs
- Autophagy Induction Is a Tor- and Tp53-Independent Cell Survival Response in a Zebrafish Model of Disrupted Ribosome Biogenesis
- Assembly of the Auditory Circuitry by a Genetic Network in the Mouse Brainstem