
MicroRNA-3148 Modulates Allelic Expression of Toll-Like Receptor 7 Variant Associated with Systemic Lupus Erythematosus
We previously reported that the G allele of rs3853839 at 3′untranslated region (UTR) of Toll-like receptor 7 (TLR7) was associated with elevated transcript expression and increased risk for systemic lupus erythematosus (SLE) in 9,274 Eastern Asians [P = 6.5×10−10, odds ratio (OR) (95%CI) = 1.27 (1.17–1.36)]. Here, we conducted trans-ancestral fine-mapping in 13,339 subjects including European Americans, African Americans, and Amerindian/Hispanics and confirmed rs3853839 as the only variant within the TLR7-TLR8 region exhibiting consistent and independent association with SLE (Pmeta = 7.5×10−11, OR = 1.24 [1.18–1.34]). The risk G allele was associated with significantly increased levels of TLR7 mRNA and protein in peripheral blood mononuclear cells (PBMCs) and elevated luciferase activity of reporter gene in transfected cells. TLR7 3′UTR sequence bearing the non-risk C allele of rs3853839 matches a predicted binding site of microRNA-3148 (miR-3148), suggesting that this microRNA may regulate TLR7 expression. Indeed, miR-3148 levels were inversely correlated with TLR7 transcript levels in PBMCs from SLE patients and controls (R2 = 0.255, P = 0.001). Overexpression of miR-3148 in HEK-293 cells led to significant dose-dependent decrease in luciferase activity for construct driven by TLR7 3′UTR segment bearing the C allele (P = 0.0003). Compared with the G-allele construct, the C-allele construct showed greater than two-fold reduction of luciferase activity in the presence of miR-3148. Reduced modulation by miR-3148 conferred slower degradation of the risk G-allele containing TLR7 transcripts, resulting in elevated levels of gene products. These data establish rs3853839 of TLR7 as a shared risk variant of SLE in 22,613 subjects of Asian, EA, AA, and Amerindian/Hispanic ancestries (Pmeta = 2.0×10−19, OR = 1.25 [1.20–1.32]), which confers allelic effect on transcript turnover via differential binding to the epigenetic factor miR-3148.
Published in the journal:
. PLoS Genet 9(2): e32767. doi:10.1371/journal.pgen.1003336
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003336
Summary
We previously reported that the G allele of rs3853839 at 3′untranslated region (UTR) of Toll-like receptor 7 (TLR7) was associated with elevated transcript expression and increased risk for systemic lupus erythematosus (SLE) in 9,274 Eastern Asians [P = 6.5×10−10, odds ratio (OR) (95%CI) = 1.27 (1.17–1.36)]. Here, we conducted trans-ancestral fine-mapping in 13,339 subjects including European Americans, African Americans, and Amerindian/Hispanics and confirmed rs3853839 as the only variant within the TLR7-TLR8 region exhibiting consistent and independent association with SLE (Pmeta = 7.5×10−11, OR = 1.24 [1.18–1.34]). The risk G allele was associated with significantly increased levels of TLR7 mRNA and protein in peripheral blood mononuclear cells (PBMCs) and elevated luciferase activity of reporter gene in transfected cells. TLR7 3′UTR sequence bearing the non-risk C allele of rs3853839 matches a predicted binding site of microRNA-3148 (miR-3148), suggesting that this microRNA may regulate TLR7 expression. Indeed, miR-3148 levels were inversely correlated with TLR7 transcript levels in PBMCs from SLE patients and controls (R2 = 0.255, P = 0.001). Overexpression of miR-3148 in HEK-293 cells led to significant dose-dependent decrease in luciferase activity for construct driven by TLR7 3′UTR segment bearing the C allele (P = 0.0003). Compared with the G-allele construct, the C-allele construct showed greater than two-fold reduction of luciferase activity in the presence of miR-3148. Reduced modulation by miR-3148 conferred slower degradation of the risk G-allele containing TLR7 transcripts, resulting in elevated levels of gene products. These data establish rs3853839 of TLR7 as a shared risk variant of SLE in 22,613 subjects of Asian, EA, AA, and Amerindian/Hispanic ancestries (Pmeta = 2.0×10−19, OR = 1.25 [1.20–1.32]), which confers allelic effect on transcript turnover via differential binding to the epigenetic factor miR-3148.
Introduction
Systemic lupus erythematosus (SLE [OMIM 152700]) is a complex and heterogeneous autoimmune disease with a strong genetic component that is modified by environmental exposures. Although the detailed etiopathogenesis of SLE remains unknown, excessive innate immune activation involving toll-like receptors (TLRs, particularly TLR7/8/9) and type I interferon (IFN) has been recognized as an important pathogenic mechanism in the disease [1]. Therapeutics targeting the TLR/IFN pathway are in development for the treatment of SLE, with ongoing clinical trials investigating monoclonal antibodies against IFN-α and inhibitors for TLR7/TLR9 (reviewed in [2]). Recent genome-wide association (GWA) and follow-up studies have revealed the association of a number of polymorphic variants in genes encoding components of the TLR/type I IFN pathway with susceptibility to SLE (reviewed in [3], [4]), providing insights at the molecular level to refine our understanding of this dysregulated pathway in the predisposition to SLE.
Our previous study identified a single nucleotide polymorphism (SNP), rs3853839, in the 3′ UTR of an X-linked gene TLR7 to be associated with SLE in 4,334 cases and 4,940 controls of Eastern Asian descent [5], providing the first convincing evidence for the genetic contribution of TLR7 to human SLE. Individuals carrying the risk G allele exhibited increased TLR7 transcripts and a more robust IFN signature than non-risk C allele carriers [5]. In this study, by fine mapping the TLR7-TLR8 region, we confirmed that the previously reported functional SNP rs3853839, located within a predicted binding site of miR-3148, was most likely responsible for observed association with SLE in three populations of non-Asian ancestry. We demonstrated a differential miR-3148 modulation explaining the effect of allelic variation at rs3853839 on TLR7 expression.
Results
Confirmation of the association between rs3853839 and SLE susceptibility in European American, African American, and Hispanic ancestries
We conducted genotyping and imputation for genetic variants covering ∼80 kb of the TLR7-TLR8 region on Xp22.2. After applying quality control measures, 41 genotyped SNPs and 57–75 imputed SNPs/INDELs (insertion-deletion) (varying among different ancestries) were assessed for association with SLE in unrelated cases and healthy controls of European American (EA, 3,936 cases vs. 3,491 controls), African American (AA, 1,679 vs. 1,934) and Hispanic enriched for the Amerindian-European admixture (HS, 1,492 vs. 807) descent (Figure 1A).

The strongest association signal was consistently detected at rs3853839 in the three ancestries, including EA (minor allele frequency of 20.3% in cases vs. 17.2% in controls, P = 6.5×10−6, OR [95%CI] = 1.23 [1.13–1.35]), AA (19.8% vs. 16.7%, P = 1.1×10−3, OR = 1.24 [1.09–1.41]) and HS (44.8% vs. 37.3%, P = 7.5×10−4, OR = 1.26 [1.10–1.43]) (Figure 1B, Table 1). After Bonferroni correction for multiple comparisons, the association of rs3853839 with SLE remained significant in EA and HS, and reached a nominal significance in AA. Combining the EA, AA and HS datasets, the meta-analysis P value of rs3853839 (Pmeta = 7.5×10−11, OR = 1.24 [1.18–1.34]) exceeded the commonly used threshold of 5×10−8 for genome-wide significance (Figure 1C, Table 1). Thus, the association of rs3853839 with SLE previously identified in Eastern Asians was confirmed in three non-Asian ancestries.

Only six other SNPs within a relatively small interval of 5 kb spanning from TLR7 3′downstream to TLR8 intron 1 were consistently associated with SLE (P<0.05) in EA, AA and HS (Table S1), and remained significant trans-ancestral meta-analysis P values after Bonferroni correction (5.5×10−6≤Pmeta≤1.3×10−6, Table S1). Linkage disequilibrium (LD) analysis revealed low LD strength between rs3853839 and these SNPs across non-Asian ancestries (r2<0.26, 0.37, and 0.51 in EA, AA and HS, respectively), but these 6 SNPs are in strong LD with each other and could be defined as a block (Figure S1). Among them, non-synonymous SNP rs3764880 (Met1Val) located at TLR8 exon1 exhibited the strongest association (Pmeta = 1.3×10−6, OR = 1.15; Table S1). To distinguish whether the associations of these 6 SNPs with SLE were independent of rs3853839, we performed conditional haplotype-based association test. After conditioning on rs3853839, association signals detected at these 6 loci were completely eliminated in EA, AA and HS (Figure S1). In contrast, conditioning on rs3764880, a consistent association signal was detected at rs3853839 in EA and HS (Figure S1), indicating that the association signals detected at these 6 SNPs might be attributed to that of rs3853839.
Taken together, we confirmed rs3853839 as the only SNP in the TLR7-TLR8 region showing an independent association with SLE across all three non-Asian ancestries. A meta-analysis by combining all datasets of Asian and non-Asian ancestries showed compelling evidence of association with SLE at rs3853839 (Pmeta = 2.0×10−19, OR = 1.25 [1.20–1.32], Table 1). Given the location of TLR7 at X chromosome, we examined the allelic association of rs3853839 separately by gender. Of note, the sex-specific association of rs3853839 with SLE previously detected in Asian men [5] was not replicated in non-Asian ancestries (Table 1).
Regulation of TLR7 expression by rs3853839
Given the convincing evidence for the trans-ancestral association of rs3853839 with SLE susceptibility, we then evaluated its effect on regulation of TLR7/8 expression. Messenger RNA (mRNA) levels of TLR7 and the two alternative TLR8 isoforms were measured by real-time PCR in PBMCs from healthy EA individuals (n = 62). TLR7 mRNA levels were significantly different among women (n = 41) carrying different genotypes of rs3853839 [P = 0.003, one-way analysis of variance (ANOVA)], in which the GG and GC carriers exhibited notably increased TLR7 mRNA levels compared with the CC carriers [P = 0.02 for GG (n = 5) vs. CC (n = 18) and 0.02 for GC (n = 18) vs. CC, respectively, Student's t test; Figure 2A) and the number of rs3853839 risk G allele was significantly correlated with increased TLR7 mRNA levels (R2 = 0.26, P = 8×10−4, linear regression test). Consistently, male G allele carriers (n = 5) also had significantly higher TLR7 mRNA expression than male C allele carriers (n = 16) (P = 0.01, Figure 2A). There was no significant association of rs3853839 genotypes with mRNA levels of two TLR8 isoforms in either women or men (Figure 2A). No sex differences in TLR7 or TLR8 mRNA levels were observed between individuals carrying the same genotype [GG women vs. G men: P = 0.41 (TLR7), 0.63 (TLR8a) and 0.50 (TLR8b); CC women vs. C men: P = 0.10 (TLR7), 0.91 (TLR8a) and 0.65 (TLR8b)]. These results were in accordance with our previous observations in Chinese [5], supporting the importance of rs3853839 in regulating TLR7 rather than TLR8 gene expression.
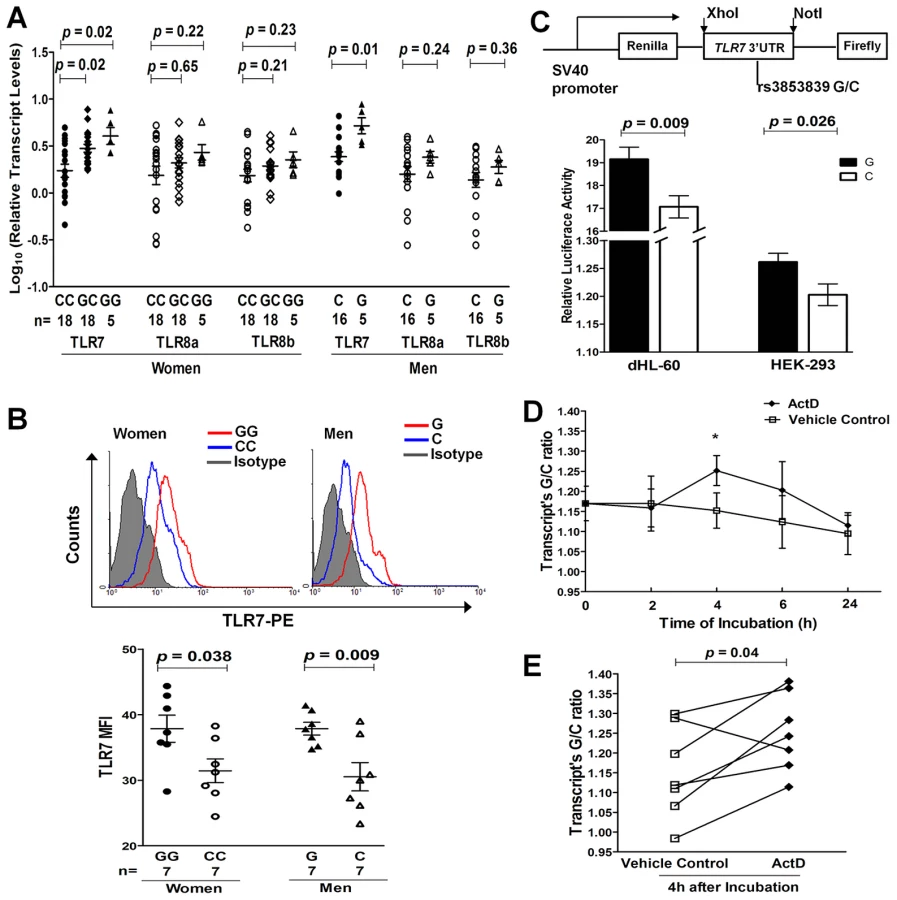
We assessed the intracellular expression of TLR7 and TLR8 proteins by flow cytometry in PBMCs from 7 pairs of healthy women (GG vs. CC) and men (G vs. C), respectively. Of the 7 pairs of individuals in each gender, 4 pairs were of EA descent and 3 pairs were Asians. Compared with C allele carriers, G allele carriers had significantly higher TLR7 protein levels in PBMCs (P = 0.038 and 0.009 in women and men, respectively; Figure 2B), especially in CD19+ B cells and CD14+ monocytes (Figure S2). No significant association between rs3853839 genotypes and TLR8 protein levels was observed in either total PBMCs or in specific cell subsets (Figure S3).
We next performed luciferase reporter assays to further confirm the functional effect of rs3853839 on TLR7 expression. PCR-amplified TLR7 3′UTR fragments with either the G or C allele of rs3853839 were cloned downstream of an SV40 promoter-driven Renilla luciferase gene in the psiCHECK-2 vector, which also contained a firefly luciferase gene to serve as an internal transfection normalization control (Figure 2C). Constructs were then transiently transfected into either HEK-293 or differentiated HL-60 (dHL-60, neutrophil-like cells) cells. After 24 hours, cell lysates transfected with the G-allele construct showed significantly higher luciferase activity than those transfected with the C-allele construct in both HEK-293 and dHL-60 cells (P = 0.026 and 0.009, respectively; Figure 2C). Taken together, consistent results from ex vivo and in vitro studies indicated that the SLE-risk G allele of rs3853839 conferred elevated TLR7 expression at the both mRNA and protein level.
Allelic differences of rs3853839 in TLR7 mRNA degradation rate
To explore the mechanism of rs3853839 in regulating TLR7 mRNA turnover, we assessed allelic difference in TLR7 mRNA degradation by pyrosequencing. We first determined the rs3853839 G/C allele ratio in genomic DNA (gDNA) and cDNA from healthy EA women (n = 7) carrying the GC genotype. The mean G/C allele ratio in cDNA was significantly higher than the theoretical ratio of 1 as detected in gDNAs (P = 0.02, Figure S4), indicating a higher expression of the G - than the C-allele containing TLR7 transcripts in heterozygous PBMCs. The allelic specific expression analysis in EA was similar to our previous findings in Chinese [5], and confirmed the result of real-time PCR that the G allele of rs3853839 is associated with increased TLR7 mRNA expression. Then, PBMCs were cultured in the absence or presence of the transcriptional inhibitor actinomycin D (ActD), and the G/C allele ratio in cDNA (normalized to that measured in gDNA) was determined after 0, 2, 4, 6, and 24 hours, respectively. As shown in Figure 2D and 2E, the G/C ratio in cDNA appeared to change over time when PBMCs were incubated with ActD and exhibited a statistical difference at the 4 hour point (P = 0.04), implicating slower degradation of the G allele - than the C allele-containing TLR7 transcript in heterozygous PBMCs. The inhibitory effect of ActD on RNA synthesis was corroborated by a decrease in total TLR7 mRNA level at increasing time points after the addition of ActD in PBMC aliquots measured by real-time PCR (Figure S5).
Alteration of microRNA–3148–mediated modulation of TLR7 expression by rs3853839
MicroRNAs (miRNAs) that bind to target sequences located within the 3′UTR of mRNAs by base pairing have been shown to result in accelerated mRNA turnover or translation repression [6]. Single nucleotide change either within or around the sequence of miRNA target sites can potentially alter the base-pairing patterns and affect miRNA-mediated regulation [7], [8]. The updated TargetScan database (Release 6.2; http://www.targetscan.org) indicates that rs3853839 is located within a binding site of miR-3148, where the non-risk allele (C), but not the risk allele (G), is predicted to match miR-3148 at the second position (Figure 3A). We hypothesized that the C to G variation of rs3853839 could reduce the binding and regulation incurred by miR-3148, therefore, leading to dysregulated TLR7 expression. We first showed that transcript levels of miR-3148 and TLR7 were inversely correlated in PBMCs from 16 patients with SLE and 21 healthy controls (R2 = 0.255, P = 0.001; Figure 3B), suggesting the possible regulation of TLR7 expression by miR-3148. Next, to verify whether allelic variation of rs3853839 affects the interaction of miR-3148 with TLR7 3′UTR, psiCHECK-2 vectors containing TLR7 3′UTR segment with either the C or G allele of rs3853839 were cotransfected with various doses of miR-3148 or nontarget control mimic into HEK-293 cells. As shown in Figure 3C, we observed significant dose-dependent miR-3148-mediated decrease in luciferase activity for the C-allele construct (P = 0.0003 over all miR-3148-treated C-allele vector groups, ANOVA test), but not for the G-allele construct (P = 0.14). Cotransfection with miR-3148 at a concentration of 6, 12, and 48 nM, respectively, led to greater than two-fold reduction of luciferase activity in the C-allele than the G-allele construct [reduction in C-allele vs. G-allele construct: 13.2% vs. 4.8%, P = 0.023 (6 nM); 22.5% vs. 9.9%, P = 0.0012 (12 nM); 21.4% vs. 8.5%, P = 0.0031 (48 nM)]. These data supported the bioinformatic prediction that miR-3148 directly targets TLR7 3′UTR and the C to G variation of rs3853839 within the binding site alters the inhibitory effect of miR-3148 on modulating TLR7 expression.

Discussion
Fine-mapping of the TLR7-TLR8 region with high-density genetic markers based on large scale genotyping and imputation confirmed SNP rs3853839 at TLR7 3′UTR as the most likely causal variant responsible for the association of TLR7-TLR8 region with SLE in populations of EA, AA and HS ancestry. In accordance with our previous observation in Asians [5], we detected elevated TLR7 expression at both mRNA and protein levels in PBMCs from EA homozygous risk G allele carriers, as well as a higher level of the risk than the non-risk allele-containing TLR7 transcripts in EA heterozygous PBMCs. The fact that two distinct ancestries share the same genotype-phenotype association implicates an important regulatory effect of rs3853839 on TLR7 expression. Toward this end, we have extended functional studies showing slower degradation of the risk allele-containing TLR7 transcripts in heterozygous PBMCs and regulation of TLR7 expression by miRNA-3148 that targets 3′UTR at the position of rs3853839. Finally, we showed that the presence of the risk G allele resulted in reduced suppression by miRNA-3148, suggesting a likely mechanism for increased TLR7 expression in risk-allele carriers.
The importance of TLR7 upregulation on mediating autoimmune responses has been addressed in murine models of SLE. The Y-linked autoimmune accelerator (Yaa) modifier, suggested mainly due to Tlr7 gene duplication, provides a prime example of TLR7 dysregulation leading to autoreactivity and inflammatory pathology [9]–[11]. Increasing Tlr7 gene dosage via generation of transgenic mice results in development of systemic autoimmunity, the severity of which directly correlates with the degree of Tlr7 overexpression [12]. Increased Tlr7 gene dosage promotes autoreactive lymphocytes activation, dendritic cells proliferation, and secretion of proinflammatory cytokines and IFN-α [12], which in turn upregulates TLR7 expression, leading to a feedback loop exacerbating autoimmunity [13]. In patients affected with SLE, up-regulated expression of TLR7 mRNA has been reported in PBMCs and B cells [14], [15]. Although a copy number variation (CNV) study in Mexican population showed increased TLR7 copies in childhood-onset SLE patients [16], no evidence for common CNVs at the TLR7-TLR8 region has been identified in individuals of diverse ancestries through our previous study by three independent methods including quantitative real-time PCR, PmeI pulsed-field gel electrophoresis and Southern blot [5], two recent studies using customized CGH platforms [17], [18] as well as other studies listed in the Database of Genomic Variants (http://projects.tcag.ca/variation; the latest version released in November 2010), suggesting that mutations similar to Yaa are not a frequent feature of human SLE. The current study identifying genetic variations conferred by a regulatory SNP in TLR7 expression and SLE susceptibility suggests that murine models provide profound clues to human genetics if we look beyond the specific mutations identified in the relevant pathways.
Unlike our findings in Asians that both sexes showed association [5], the impact of rs3853839 on risk for SLE was only observed for women in the non-Asian datasets (Table 1). Given the low prevalence of SLE in men, it is often challenging to collect a large enough number of affected men in a given population. Under the assumption that the associated G allele confers genetic risk with an odds ratio of 1.26 in EA, 1.22 in AA and 1.29 in HS subjects (ORs were determined in female datasets), and considering P<0.05 as the threshold of significance, the power estimate for female samples in each ancestry reaches more than 85%, whereas for male samples it is only 50% in EA, 19% in AA and 25% in HS dataset. Thus, there was clearly inadequate power to evaluate this association in AA and HS men. Despite a relatively robust sample size of EA men (344 SLE vs. 1,151 controls), a significantly higher G allele frequency was observed in male than female controls (20.0% vs. 16.5%, P = 0.005), contributing to the difficulty in assessing association with SLE in EA male subjects.
To our knowledge, the association of rs3853839 (or its tag SNP) with SLE has not been reported in four SLE GWA studies in European-derived populations [19]–[22] and three GWA studies in Asians [23]–[25]. According to the 1000 Genomes Project data, rs3853839 locates in a region with poor LD structure and cannot be tagged by any known SNP at the TLR7-TLR8 region with r2>0.65. The SNP rs850632, located at TLR7 3′downstream, shows the strongest LD with rs3853839 in Europeans (r2 = 0.38) and Asians (r2 = 0.65). However, neither rs3853839 nor rs850632 has been included in predesigned commercial genotyping arrays of those GWA studies, resulting in the absence of associations. Even if rs3853839 was genotyped, the published GWA studies might have inadequate statistical power to capture its association in the initial discovery analyses [5].
Evidence of other TLR7 polymorphisms associated with SLE has been reported, including two intronic SNPs (rs179019 and rs179010) found in Japanese population [26] and an exonic SNP (rs179008) in individuals from Southern Brazil [27]. The reported associations were modest due to limited sample size of these studies (less than 400 cases and 450 controls), and none of them have been confirmed by the current fine-mapping study using a large collection of EA, AA and HS cases-controls (Table S1). TLR8 polymorphisms have been described in infectious diseases [28], [29] with a genetic effect localized to a functional variant at exon 1 (rs3764880, Met1Val). The G allele of rs3764880, which abolishes a putative start codon within the alternative TLR8 transcript isoform a (Figure 1A), conferred a protective effect on susceptibility to pulmonary tuberculosis in Indonesian and Russian men [28], as well as on HIV disease progression in Germans [29]. Our data showed a significantly increased frequency of rs3764880-G allele in SLE than healthy controls in the three non-Asian datasets; however, its association with SLE was dependent on that of TLR7 SNP rs3853839. Other variants at the TLR7-TLR8 region showed either weaker association than rs3853839 in trans-ancestral meta-analysis or association uniquely in EA or HS. Taken together, these data support rs3853839 as the most likely polymorphism associated with SLE shared by multiple ancestries. Although imputation facilitated our ability to capture common variants (MAF>1%), further refinement in genetic effects of rare variants (MAF<1%) is needed by deep sequencing of this locus, especially the intergenic region between TLR7 and TLR8 that was not well imputed in this study.
Variations in 3′UTR regions may be important in gene regulation. To date, expression quantitative trait loci (eQTL) mapping has been widely used for characterization of SNPs that affect gene expression [30]. Although the TLR7 expression has been measured in previous whole-genome eQTL studies, currently only those using EBV-transformed lymphoblastoid cell lines of 1000 Genomes Project individuals provide publically available genotyping data of rs3853839. Based on the study by Stranger et al [31], we found that CG carriers of rs3853839 showed elevated TLR7 expression compared with CC carriers in YRI women (P = 0.012). In male individuals, the G allele of rs3853839 showed a trend of association with elevated TLR7 expression in CHB+JPT, CEU and YRI men, and the association was significant when combining all male data (P = 0.014). These findings are consistent with our results that rs3853839 alleles are associated with differential TLR7 expression.
An important finding of this study is that the SLE-associated variant rs3853839 confers a genetic effect on modulation of TLR7 expression by an epigenetic factor miR-3148. Accumulating evidence suggests that miRNAs are fine tuners of TLR signaling pathways [32]. Regulation by miRNA may occur at various levels of TLR pathways by targeting adaptor molecules, downstream regulators and cytokines (reviewed in [32], [33]). However, few studies point to TLR themselves (e.g. TLR2 and TLR4) being directly targeted by miRNAs [34], [35]. Using algorithms from TargetScan, only the newly identified human miR-3148 [36], which is not evolutionarily conserved among mammals, is predicted to bind TLR7 3′UTR sequences at the position of rs3853839. The inverse correlation of miR-3148 and TLR7 levels in PBMCs, along with functional validation by reporter gene assay, confirms an inhibitory effect of miR-3148 on regulating TLR7 expression and allelic variation of rs3853839 affecting miRNA-mRNA interactions. Further study will focus on investigating miR-3148 expression patterns in specific immune cell types, assessing biological impacts of changes in miR3148-mediated TLR7 expression on downstream immune responses, and evaluating roles of other miRNAs that target sequences in the vicinity of rs3853839. Of interest, an unconventional role for miRNAs has been identified as endogenous activators for RNA-sensing receptors (TLR7/8) in a cell - or tissue-type specific manner [37], [38]. Therefore, miRNA regulation in TLR7 signaling is more complicated than we expected and further functional studies showing the exact effects of miRNAs on TLR7 responses are warranted.
In summary, we have advanced our previous study by showing rs3853839 (at TLR7 3′UTR) as the most likely polymorphism responsible for the association of TLR7-TLR8 region with SLE in individuals of EA, AA and HS ancestry, and have characterized a differential miR-3148 modulation which explains the effect of allelic variation of rs3853839 on TLR7 expression. Our study highlights the importance of TLR7 as a shared genetic contributor to SLE in multiple ancestries, and provides evidence that microRNA acts as a negative regulator to control TLR7 expression, suggesting the possibility of miRNA-based therapies for amelioration of autoimmune diseases such as SLE where excessive TLR7 activation exists.
Materials and Methods
Ethics statement
Written informed consent was obtained from all study participants and each participating institution had Institutional Review Board (IRB) approval to recruit samples. The overall study was approved by the IRB of the Oklahoma Medical Research Foundation (OMRF).
Subjects
To test the association of TLR7-TLR8 with SLE, we used a large collection of case-control subjects from the collaborative Large Lupus Association Study 2 (LLAS2), including European American (4,248 cases vs. 3,818 controls), African American (1,724 cases vs. 2,024 controls), and Hispanic enriched for the Amerindian-European admixture (1,622 cases vs. 887 controls). African Americans included 286 Gullahs (155 cases vs. 131 controls), who are subjects with African ancestry. Cases were defined by meeting at least four of the 1997 American College of Rheumatology (ACR) revised criteria for the classification of SLE [39].
SNP genotyping and quality control
DNA samples were processed at the Lupus Genetics Studies Unit of OMRF. SNP genotyping was performed using an Illumina custom bead array on the iSCAN instrument for 47 SNPs covering the TLR7-TLR8 region on Xp22.2 and 347 admixture informative markers (AIMs). SNPs meeting the following criteria were included in the association analysis: well-defined cluster scatter plots, SNP call rate >90%, minor allele frequency >1%, total proportion missing <5%, P>0.05 for differential missing rate between cases and controls, and Hardy-Weinberg proportion (HWP) test with a P>0.01 in controls and P>0.0001 in cases.
Subjects with genotype missing rate >10% (due to low quality), shared identical by descent >0.4 or showing mismatch between the reported and estimated gender were removed. The global ancestry of each subject was estimated based on genotype of AIMs using principal components analysis [40] and ADMIXMAP [41], as described in another LLAS2 study [42], and then genetic outliers were removed.
Finally, a total of 13,339 unrelated subjects, including European Americans (EA: 3,936 cases vs. 3,491 controls), African Americans (AA: 1,679 vs. 1,934; composed of 92.5% of African Americans and 7.5% Gullahs) and Hispanics enriched for the Amerindian-European admixture (HS: 1,492 vs. 807), were analyzed for 41 genotyped SNPs of TLR7-TLR8.
Imputation methods
Imputation was performed at 12.86–12.95 Mb on Xp22.2 using IMPUTE 2.1.2 [43], with SNP/INDEL genotypes of 381 Europeans, 246 Africans and 181 Americans from the 1000 Genomes Project (“version 3” of the Phase 1 integrated data, March 2012 release) as references in imputation for our EA, AA and HS subjects, respectively. Imputed genotypes had to meet information score of >0.9, as well as the quality control criteria as described above. After imputation, we obtained an additional 75 variants for EA, 57 for AA and 63 for HS (the number varied based on LD structure) for further analysis.
Real-time PCR
Total RNA was purified with TRIzol reagent (Invitrogen) from PBMCs and reverse-transcribed into cDNA with Superscript II Reverse Transcription kit (Invitrogen). The mRNA levels of TLR7 (NM016562.3) and TLR8 (isoform a: NM138636.4 and isoform b: AF246971.1) were measured by quantitative real-time PCR using TaqMan assays (TLR7 probe: Hs00152971_m1; TLR8 isoform a probe: Hs00607866_mH; TLR8 isoform b probe: Hs00152972_ml, Applied Biosystems). All samples were run in triplicate. Relative expression levels of TLR7 and TLR8 were normalized to the level of RPLP0, calculated by the 2−ΔΔCt method and Log10 transformed. The association of rs3853839 with mRNA levels of TLR7 or TLR8 was evaluated using ANOVA, Student's t and linear regression test.
To examine the correlation of miR-3148 and TLR7 mRNA levels, total RNA enriched in small RNAs were isolated from PBMCs using mirVanaTM miRNA isolation kit (Invitrogen), followed by reverse transcription with TaqMan MicroRNA Reverse Transcription kit (Applied Biosystems; for detecting miR-3148) and Superscript II Reverse Transcription kit (Invitrogen; for detecting TLR7), respectively. The miR-3148 level was quantified using Taqman MicroRNA Expression assay (Applied Biosystems), and the TLR7 level was measured using the same probe as described above. All samples were run in triplicate. Relative expression levels of miR-3148 and TLR7 were normalized to the level of snRNA U6 and RPLP0, respectively, calculated by the 2−ΔΔCt method and Log10 transformed. Association between transcript levels of TLR7 and miR-3148 was evaluated using linear regression test.
Flow cytometry
Four-color flow cytometry was performed to investigate intracellular expression of TLR7 and TLR8 in PBMCs from healthy EA and Asian individuals who were homozygous for rs3853839 (7 pairs of G-allele vs. C-allele carriers in each gender group). Freshly isolated PBMCs were incubated with 2% pooled human serum to block nonspecific binding to Fcγ receptors and then incubated with peridinin chlorophyll protein (PerCP)-conjugated anti-human CD3, allophycocyanin (APC)-conjugated anti-human CD19 and phycoerythrin (PE)-conjugated or fluorescein isothiocyanate (FITC)-conjugated anti-human CD14 (Miltenyi Biotec) to identify T cell, B cell and monocyte subpopulations, respectively. For intracellular staining, PBMCs were fixed in Fixation buffer (R&D Systems) for 10 minutes at room temperature, washed twice in Permeabilization/Wash buffer (R&D Systems) and stained with PE-conjugated mouse anti-human TLR7 mAb (R&D Systems) and FITC-conjugated mouse anti-human TLR8 mAb (Imgenex) for 1 hour at room temperature. Background fluorescence was assessed using appropriate isotype - and fluorochrome-matched control antibodies. Cells were collected and analyzed by FACSCalibur flow cytometer equipped with the manufacturer's software (CellQuest; BD Biosciences). Student's t test was used to compare protein levels of TLR7 or TLR8 in PBMCs from individuals of different genotypes.
Plasmid construction and luciferase reporter assay
The fragment of TLR7 3′-UTR bearing the G or C allele of rs3853839 was amplified by PCR from genomic DNA of subjects homozygous for the G or C allele using the following primers: 5′-TGTCTCGAGCCCTTCTTTGCAAAAC-3′ (forward) and 5′-AGAGCGGCCGCTAGTTGGCTCCAGCAAT-3′ (reverse). The PCR products were inserted into the downstream of the Renilla luciferase gene in the reporter vector psiCHECK-2 (Promega) by digestion using the restriction enzymes Not I and Xho I. The psiCHECK-2 vector also contained a firefly luciferase gene to serve as an internal transfection normalization control. All constructs were sequenced to assure proper orientation and authenticity in the vector.
HEK-293 (human embryonic kidney cell line) and HL-60 (human leukemic cell line) cells were obtained from the American Type Culture Collection (ATCC). HEK-293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS, seeded on a 24-well plate at a concentration of 2×105 cells/well, and transiently transfected using Lipofectamine 2000 (Invitrogen) with 1 µg of either rs3853839 G or C reporter construct. HL-60 cells are predominantly a neutrophilic promyelocyte (precursor) and can be induced to differentiate to neutrophil-like cells when grown in RPMI 1640 medium with 15% FBS plus 2 mM L-glutamine, 25 mM HEPES and 1.25% DMSO [44]. Differentiated HL-60 cells seeded on 24-well plates (2×106 cells/well) were electroporated with 3 µg of report construct on a nucleofector device (Amaxa). The luciferase activity in total cell lysates was measured after 24 hours using a dual luciferase reporter assay system (Promega). Renilla luciferase activities were normalized to firefly luciferase activities. Each transfection was performed in quadruplicates and triplicates for HEK-293 and HL-60 cells respectively, and luciferase assays were repeated four times.
MicroRNA hsa-miR-3148 and nontarget control (NC) mimics were synthesized by Thermo Fisher Scientific. To test the effect of miR-3148, HEK-293 cells plated in 96-well plates were transiently cotransfected with 100 ng of each reporter construct (psiCHECK-2 empty vector, rs3853839-G or -C allele constructs) and increasing concentrations (1, 6, 12 and 48 nM) of miR-3148 or nontarget control mimic using Lipofectamine 2000 reagent (Invitrogen), and luminescence was measured 24 hours after transfection. Each transfection was performed in quadruplicates and repeated three times. Luciferase activity of reporter vectors was compared using Student's t test.
Assessment of allelic difference in RNA degradation rate
PBMCs isolated from EA healthy women with the GC genotype of rs3853839 (n = 7) were cultured in the absence or presence of 5 µg/mL ActD for 0, 2, 4, 6 and 24 hours. Using real-time PCR, we detected a decrease in total TLR7 mRNA levels over time with ActD incubation, which confirmed the transcriptional inhibition by ActD and allowed for detection of allelic differences in mRNA degradation. The G/C allelic ratio in the cDNA and gDNA after treatment of PBMCs with or without ActD were determined by pyrosequencing and calculated using software PSQMA 2.1 (Biotage) as previously described [5]. The G/C allele ratio obtained in TLR7 transcripts was normalized to that measured from gDNA of the same sample. A paired t test was used to compare the mean G/C allele ratio in TLR7 transcripts in PBMCs treated with ActD or vehicle control at each time point.
Statistical analysis
Associations of SNPs with SLE were assessed in each ancestral group under a logistic regression model adjusted for gender and the first three principal components estimated using AIMs. Conditional haplotype-based association tests were also performed by adjusting for gender and the first three principal components. The trans-ancestral meta-analysis was conducted on 40 genotyped and 14 imputed SNPs that were shared by the three ancestries with both a fixed and random-effects model. Homogeneity of odds ratios was evaluated using Cochrane's Q test. For each SNP, if the Cochran's Q test showed no evidence of genetic heterogeneity (P>0.05), a fixed-effects model was implemented; otherwise, a random-effects model was used. The Bonferroni corrected P-value threshold was adjusted to P<9.1×10−4 on the basis of the maximum number of tests across all populations (55 independent variants with r2<0.8). All analyses described above were performed using PLINK v1.07. Pairwised LD values shown in Figure 1 and Figure S1 were calculated using Haploview 4.2. Other data were analyzed using GraphPad Prism 4.0 software. A P value<0.05 was considered to be statistically significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. KontakiE, BoumpasDT (2010) Innate immunity in systemic lupus erythematosus: sensing endogenous nucleic acids. J Autoimmun 35 : 206–211.
2. LichtmanEI, HelfgottSM, KriegelMA (2012) Emerging therapies for systemic lupus erythematosus–focus on targeting interferon-alpha. Clin Immunol 143 : 210–221.
3. DengY, TsaoBP (2010) Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol 6 : 683–692.
4. BronsonPG, ChaivorapolC, OrtmannW, BehrensTW, GrahamRR (2012) The genetics of type I interferon in systemic lupus erythematosus. Curr Opin Immunol [Epub ahead of print].
5. ShenN, FuQ, DengY, QianX, ZhaoJ, et al. (2010) Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci U S A 107 : 15838–15843.
6. BushatiN, CohenSM (2007) microRNA functions. Annu Rev Cell Dev Biol 23 : 175–205.
7. SaundersMA, LiangH, LiWH (2007) Human polymorphism at microRNAs and microRNA target sites. Proc Natl Acad Sci U S A 104 : 3300–3305.
8. ZhangW, EdwardsA, ZhuD, FlemingtonEK, DeiningerP, et al. (2012) miRNA-mediated relationships between Cis-SNP genotypes and transcript intensities in lymphocyte cell lines. PLoS ONE 7: e31429 doi:10.1371/journal.pone.0031429
9. PisitkunP, DeaneJD, DiffilipantonioMJ, TarasenkoT, SatterthwaiteAB, et al. (2006) Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 312 : 1669–1672.
10. SubramanianS, TusK, LiQZ, WangA, TianXH, et al. (2006) A TLR7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci USA 103 : 9970–9975.
11. FairhurstAM, HwangSH, WangA, TianXH, BoudreauxC, et al. (2008) Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur J Immunol 38 : 1971–1978.
12. DeaneJA, PisitkunP, BarrettRS, FeigenbaumL, TownT, et al. (2007) Control of Toll-like Receptor 7 Expression Is Essential to Restrict Autoimmunity and Dendritic Cell Proliferation. Immunity 27 : 801–810.
13. RonnblomL, ElorantaML, AlmGV (2006) The type I interferon system in systemic lupus erythematosus. Arthritis Rheum 54 : 408–420.
14. KomatsudaA, WakuiH, IwamotoK, OzawaM, TogashiM, et al. (2008) Up-regulated expression of Toll-like receptors mRNAs in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin Exp Immunol 152 : 482–487.
15. MidgleyA, ThorbinsonC, BeresfordMW (2012) Expression of Toll-like receptors and their detection of nuclear self-antigen leading to immune activation in JSLE. Rheumatology (Oxford) 51 : 824–832.
16. Garcia-OrtizH, Velazquez-CruzR, Espinosa-RosalesF, Jimenez-MoralesS, BacaV, et al. (2010) Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Ann Rheum Dis 69 : 1861–1865.
17. ConradDF, PintoD, RedonR, FeukL, GokcumenO, et al. (2010) Origins and functional impact of copy number variation in the human genome. Nature 464 : 704–712.
18. ParkH, KimJI, JuYS, GokcumenO, MillsRE, et al. (2010) Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing. Nat Genet 42 : 400–405.
19. HomG, GrahamRR, ModrekB, TaylorKE, OrtmannW, et al. (2008) Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med 358 : 900–909.
20. HarleyJB, Alarcon-RiquelmeME, CriswellLA, JacobCO, KimberlyRP, et al. (2008) Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 40 : 204–210.
21. KozyrevSV, AbelsonAK, WojcikJ, ZaghloolA, Linga ReddyMV, et al. (2008) Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 40 : 211–216.
22. GrahamRR, CotsapasC, DaviesL, HackettR, LessardCJ, et al. (2008) Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet 40 : 1059–1061.
23. HanJW, ZhengHF, CuiY, SunLD, YeDQ, et al. (2009) Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet 41 : 1234–1237.
24. YangW, ShenN, YeDQ, LiuQ, ZhangY, et al. (2010) Genome-Wide Association Study in Asian Populations Identifies Variants in ETS1 and WDFY4 Associated with Systemic Lupus Erythematosus. PLoS Genet 6: e1000841 doi:10.1371/journal.pgen.1000841
25. OkadaY, ShimaneK, KochiY, TahiraT, SuzukiA, et al. (2012) A genome-wide association study identified AFF1 as a susceptibility locus for systemic lupus eyrthematosus in Japanese. PLoS Genet 8: e1002455 doi:10.1371/journal.pgen.1002455
26. KawasakiA, FurukawaH, KondoY, ItoS, HayashiT, et al. (2011) TLR7 single-nucleotide polymorphisms in the 3′ untranslated region and intron 2 independently contribute to systemic lupus erythematosus in Japanese women: a case-control association study. Arthritis Res Ther 13: R41.
27. dos SantosBP, ValverdeJV, RohrP, MonticieloOA, BrenolJC, et al. (2012) TLR7/8/9 polymorphisms and their associations in systemic lupus erythematosus patients from southern Brazil. Lupus 21 : 302–309.
28. DavilaS, HibberdML, Hari DassR, WongHE, SahiratmadjaE, et al. (2008) Genetic association and expression studies indicate a role of toll-like receptor 8 in pulmonary tuberculosis. PLoS Genet 4: e1000218 doi:10.1371/journal.pgen.1000218
29. OhDY, TaubeS, HamoudaO, KuchererC, PoggenseeG, et al. (2008) A functional toll-like receptor 8 variant is associated with HIV disease restriction. J Infect Dis 198 : 701–709.
30. CooksonW, LiangL, AbecasisG, MoffattM, LathropM (2009) Mapping complex disease traits with global gene expression. Nat Rev Genet 10 : 184–194.
31. StrangerBE, MontgomerySB, DimasAS, PartsL, StegleO, et al. (2012) Patterns of cis regulatory variation in diverse human populations. PLoS Genet 8: e1002639 doi:10.1371/journal.pgen.1002639
32. O'NeillLA, SheedyFJ, McCoyCE (2011) MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol 11 : 163–175.
33. ContrerasJ, RaoDS (2012) MicroRNAs in inflammation and immune responses. Leukemia 26 : 404–413.
34. BenakanakereMR, LiQ, EskanMA, SinghAV, ZhaoJ, et al. (2009) Modulation of TLR2 protein expression by miR-105 in human oral keratinocytes. J Biol Chem 284 : 23107–23115.
35. ChenXM, SplinterPL, O'HaraSP, LaRussoNF (2007) A cellular micro-RNA, let-7i, regulates Toll-like receptor 4 expression and contributes to cholangiocyte immune responses against Cryptosporidium parvum infection. J Biol Chem 282 : 28929–28938.
36. StarkMS, TyagiS, NancarrowDJ, BoyleGM, CookAL, et al. (2010) Characterization of the Melanoma miRNAome by Deep Sequencing. PLoS ONE 5: e9685 doi:10.1371/journal.pone.0009685
37. LehmannSM, KrugerC, ParkB, DerkowK, RosenbergerK, et al. (2012) An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci 15 : 827–835.
38. FabbriM, PaoneA, CaloreF, GalliR, GaudioE, et al. (2012) MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A 109: E2110–2116.
39. HochbergMC (1997) Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40 : 1725.
40. PriceAL, PattersonNJ, PlengeRM, WeinblattME, ShadickNA, et al. (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics 38 : 904–909.
41. HoggartCJ, ParraEJ, ShriverMD, BonillaC, KittlesRA, et al. (2003) Control of confounding of genetic associations in stratified populations. Am J Hum Genet 72 : 1492–1504.
42. LessardCJ, AdriantoI, KellyJA, KaufmanKM, GrundahlKM, et al. (2011) Identification of a systemic lupus erythematosus susceptibility locus at 11p13 between PDHX and CD44 in a multiethnic study. Am J Hum Genet 88 : 83–91.
43. HowieBN, DonnellyP, MarchiniJ (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5: e1000529 doi:10.1371/journal.pgen.1000529
44. BirnieGD (1988) The HL60 cell line: a model system for studying human myeloid cell differentiation. Br J Cancer Suppl 9 : 41–45.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 2
Nejčtenější v tomto čísle
- Complex Inheritance of Melanoma and Pigmentation of Coat and Skin in Grey Horses
- Coordination of Chromatid Separation and Spindle Elongation by Antagonistic Activities of Mitotic and S-Phase CDKs
- Autophagy Induction Is a Tor- and Tp53-Independent Cell Survival Response in a Zebrafish Model of Disrupted Ribosome Biogenesis
- Assembly of the Auditory Circuitry by a Genetic Network in the Mouse Brainstem