
Aconitase Causes Iron Toxicity in Mutants
The PTEN-induced kinase 1 (PINK1) is a mitochondrial kinase, and pink1 mutations cause early onset Parkinson's disease (PD) in humans. Loss of pink1 in Drosophila leads to defects in mitochondrial function, and genetic data suggest that another PD-related gene product, Parkin, acts with pink1 to regulate the clearance of dysfunctional mitochondria (mitophagy). Consequently, pink1 mutants show an accumulation of morphologically abnormal mitochondria, but it is unclear if other factors are involved in pink1 function in vivo and contribute to the mitochondrial morphological defects seen in specific cell types in pink1 mutants. To explore the molecular mechanisms of pink1 function, we performed a genetic modifier screen in Drosophila and identified aconitase (acon) as a dominant suppressor of pink1. Acon localizes to mitochondria and harbors a labile iron-sulfur [4Fe-4S] cluster that can scavenge superoxide to release hydrogen peroxide and iron that combine to produce hydroxyl radicals. Using Acon enzymatic mutants, and expression of mitoferritin that scavenges free iron, we show that [4Fe-4S] cluster inactivation, as a result of increased superoxide in pink1 mutants, results in oxidative stress and mitochondrial swelling. We show that [4Fe-4S] inactivation acts downstream of pink1 in a pathway that affects mitochondrial morphology, but acts independently of parkin. Thus our data indicate that superoxide-dependent [4Fe-4S] inactivation defines a potential pathogenic cascade that acts independent of mitophagy and links iron toxicity to mitochondrial failure in a PD–relevant model.
Published in the journal:
. PLoS Genet 9(4): e32767. doi:10.1371/journal.pgen.1003478
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003478
Summary
The PTEN-induced kinase 1 (PINK1) is a mitochondrial kinase, and pink1 mutations cause early onset Parkinson's disease (PD) in humans. Loss of pink1 in Drosophila leads to defects in mitochondrial function, and genetic data suggest that another PD-related gene product, Parkin, acts with pink1 to regulate the clearance of dysfunctional mitochondria (mitophagy). Consequently, pink1 mutants show an accumulation of morphologically abnormal mitochondria, but it is unclear if other factors are involved in pink1 function in vivo and contribute to the mitochondrial morphological defects seen in specific cell types in pink1 mutants. To explore the molecular mechanisms of pink1 function, we performed a genetic modifier screen in Drosophila and identified aconitase (acon) as a dominant suppressor of pink1. Acon localizes to mitochondria and harbors a labile iron-sulfur [4Fe-4S] cluster that can scavenge superoxide to release hydrogen peroxide and iron that combine to produce hydroxyl radicals. Using Acon enzymatic mutants, and expression of mitoferritin that scavenges free iron, we show that [4Fe-4S] cluster inactivation, as a result of increased superoxide in pink1 mutants, results in oxidative stress and mitochondrial swelling. We show that [4Fe-4S] inactivation acts downstream of pink1 in a pathway that affects mitochondrial morphology, but acts independently of parkin. Thus our data indicate that superoxide-dependent [4Fe-4S] inactivation defines a potential pathogenic cascade that acts independent of mitophagy and links iron toxicity to mitochondrial failure in a PD–relevant model.
Introduction
Parkinson's disease (PD) is the most frequent neurodegenerative movement disorder, but the pathways that explain disease pathology remain poorly understood [1], [2]. While the most recognized pathological feature of PD is the preferential loss of dopaminergic (DA) neurons, one of the earliest observations in post mortem PD brains was the accumulation of iron in the substantia nigra (SN) [3], [4]. Iron-mediated toxicity may thus contribute to DA neuron dysfunction but the mechanism has not been established.
Mitochondrial dysfunction is thought to be an important aspect of PD progression. Mitochondrial toxins have been linked to sporadic forms of the disease and mitochondrial defects have been described in many cell types, also in SN mitochondria of PD patients [5], [6]. Likewise some of the genetic factors linked to the disease also point to a role for mitochondria. PD-associated mutations in pink1 and parkin, both affect mitochondrial function in genetic model organisms and in mammalian cells [7], [8], but how mitochondrial dysfunction and iron toxicity are linked remains elusive.
Pink1 and Parkin have been implicated in the clearance of dysfunctional mitochondria, a process dubbed mitophagy. In support, loss of parkin or pink1 in different cell types in flies, results in the accumulation of swollen and clumped mitochondria [9], [10], believed to be the result of defective mitophagy [11]. Furthermore, expression of factors that promote mitochondrial fission and, as a consequence, also indirectly promote mitophagy (gain of drp1 or loss of opa1 or mfn) partially rescue defects seen in pink1 and parkin mutants [12]–[14]. Further studies indicate that mitochondrial depolarization triggers the recruitment Parkin to mitochondria in a Pink1-dependent manner, facilitating mitophagy [15]. In line with this idea, over expression of Parkin in pink1 mutants, alleviates pink1-associated defects [9]–[16]. Hence, Pink1 acts with Parkin to regulate mitophagy.
In parallel, pink1 may also harbor supplementary roles. Expression of Parkin or Drp1, or loss of opa1 or marf only partially rescue pink1-associated defects, suggesting additional pathways are contributing to the phenotype. Furthermore, loss of pink1 function causes defects in the electron transport chain in fly and mouse cells [17], [18] that are not [19] or only partially [20] rescued by expression of Drp1. Finally, bypassing Complex I dysfunction, by expressing a yeast Complex I equivalent protein Ndi1 partially rescues the defects in pink1 mutants, but not those seen in parkin mutants [19]. Hence, Pink1 may play multiple roles in mitochondria, but the relative contribution of these different pathways to the pink1-dependent phenotypes, including the accumulation of swollen, clumped mitochondria remains to be determined.
In an unbiased genetic screen for heterozygous suppressors of Drosophila pink1 [21] we identified mitochondrial aconitase (acon) that encodes an enzyme catalyzing the first step of the Krebs Cycle [22]. Acon harbors an iron-sulfur [4Fe-4S] cluster [23] and we show that oxidative inactivation of this cluster in pink1 mutants is a major cause of iron toxicity that contributes to mitochondrial swelling and clumping in pink1 mutants. Our data are most consistent with acon acting downstream of pink1 and affecting mitochondrial morphology independently of parkin-mediated mitophagy. Thus oxidative inactivation of Aconitase is a source of iron toxicity that leads to mitochondrial defects in pink1 mutants and we propose a model where different pathways controlled by Pink1, including mitophagy and the maintenance of ETC activity can contribute to mitochondrial failure in specific cell types.
Results
Aconitase downregulation suppresses pink1 mutant phenotypes
Pink1 mutants show a severe defect to fly caused by mitochondrial dysfunction [19], [21]. To identify genetic modifiers of pink1, we have tested a collection of 193 chemically induced (EMS) recessive lethal mutants that have been pre-selected for defects in mitochondrial function and neuronal communication [24]–[26], for their ability to modify the pink1 null mutant flight defect. At the 1% significance level we isolated 5 suppressors (p<0.01) [21] and to reveal mechanisms by which the modifiers affect Pink1, we mapped one of these recessive lethal suppressors to aconitase (acon) and named it acon1. This mutant fails to complement a deletion that uncovers acon as well as a lethal transposon insertion in acon that we named acon2 (Figure S1A). Sequence analysis of acon1 reveals a nonsense mutation in exon 2 (Figure S1A). In addition, semi-quantitative RT-PCR and Western blot analysis indicates severely reduced mRNA and protein levels in animals that are homozygous for either acon allele (Figure S1C and S1D), indicating that both are loss of function alleles. Moreover we can rescue the lethality and phenotypes associated with acon1/acon2 using a 20 kb genomic fragment encompassing the acon locus, yielding normal adult flies that do not show obvious behavioral abnormalities (Figure S1A, S1B). Likewise ubiquitous expression of acon cDNA is also able to rescue acon1/acon2-associated lethality (Figure S1B). Thus, one of the suppressors of pink1 harbors a lethal lesion in acon and the lethality in the mutants is solely due to disruption of acon.
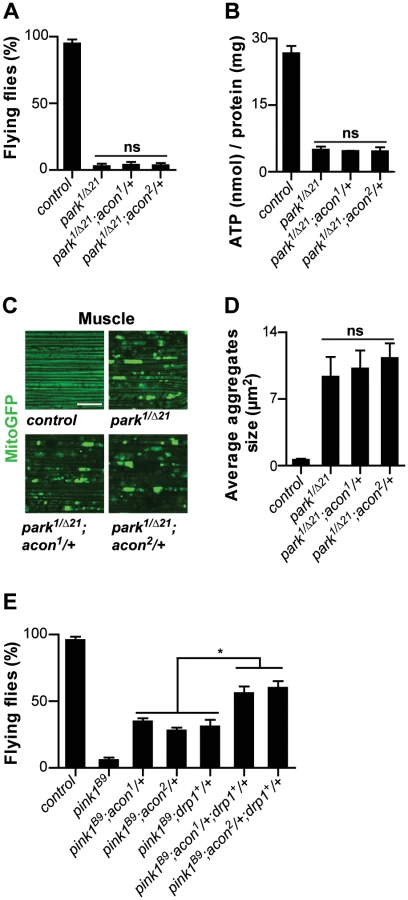
Heterozygosity of acon significantly suppresses the flight defect associated with pink1B9 mutants (Figure 1A, 1B). The extent of rescue we obtained by removing acon, is similar to previously reported conditions that suppress pink1 flight defects, including adding a copy of drp1 (drp1+) that facilitates mitochondrial fission, removing a copy of opa1 (opa1S3), reducing mitochondrial fusion (Figure S2A, S2B), expression of Parkin, expression of yeast NDI1 that bypasses Complex I of the electron transport chain (ETC), or feeding pink1 mutants ubiquinone or menaquinone that boost ETC function [9], [10], [13], [19], [21]. To test if the rescue that we observe is solely due to partial loss of acon (and not due to second site interactors on the chromosome), we determined flight but also ATP levels of pink1 mutants with one copy of a mutant acon allele. While heterozygous acon1 and acon2 mutants alone do not show defects (Figure 1C), we find that one copy of either acon1 or acon2 significantly rescue the reduced ATP levels in pink1 mutants (Figure 1D). This effect in pink1 mutants is specific to loss of acon as introduction of a genomic copy encompassing wild type acon in pink1B9;acon2/+ flies completely reverses both the flight and ATP level phenotypes to pink1B9 mutant levels (Figure 1B and 1D). Thus, pink1 mutant phenotypes are specifically rescued by partial loss of acon expression.

To further quantify the effect of acon on pink1 mutant phenotypes we also analyzed mitochondrial morphology in adult indirect flight muscles using transmission electron microscopy. As previously described [9], [10], the flight muscles of pink1 mutants exhibit swollen mitochondria with disorganized and fragmented cristae when compared to flight muscles from control flies or when compared to heterozygous acon mutants that do not show mitochondrial morphological defects (Figure 1E and Figure S1F). Partial loss of acon in pink1 mutant results in a substantial rescue of the mitochondrial morphological defects in flight muscles, displaying substantially more intact cristae and less swollen mitochondria compared to pink1 mutants (Figure 1E). Hence, also at the ultrastructural level, partial loss of acon significantly alleviates mitochondrial morphological defects in pink1 mutant muscles.
Pink1 mutants also show swollen and clumped mitochondria in dopaminergic neurons in the adult brain [9], [10]. To test if loss of acon can also rescue this defect, we expressed mitoGFP in pink1 mutant flies and in pink1 mutant animals heterozygous for acon. In line with the electron microscopy data of muscles, mitochondria in muscles labeled by mitoGFP (expressed using the ubiquitous da-GAL4) are spherical and aggregated in pink1 mutants and this defect is significantly rescued by partial loss of acon (Figure 1F and 1G). Next, we expressed mitoGFP in dopaminergic neurons using ple-Gal4 (also called TH-Gal4). While mitochondria are organized in a tubular network in wild type dopaminergic neurons, pink1 mutant mitochondria appear mostly as fragmented spherical aggregates in all dopaminergic neuron clusters analyzed (Figure S1H) [9], [10]. We quantified size and number of mitochondrial aggregates in the PPM3 cluster (Figure S1H and Methods). While heterozygous acon1 and acon2 mutants do not show defects compared to controls (Figure 1H, 1I), we find that both one copy of either acon1 or acon2 significantly rescue the increased size and number of mitochondrial aggregates in pink1 mutants (Figure 1H, 1I and Figure S2A). This rescue in pink1 mutants is specific to the partial loss of acon as introduction of a genomic copy encompassing wild type acon in pink1B9; acon1/+ flies reverses these phenotypes back to pink1B9 mutant levels (Figure 1F–1I). Furthermore, we confirm that protein levels are reduced by about 50% in pink1B9; acon1or2/+ compared to pink1 mutants and are restored in flies expressing a genomic copy of wild type acon (Figure S1E). Thus, together our data indicate that morphological defects of mitochondria in pink1 mutants are significantly rescued by partial loss of acon expression and the mitochondrial morphological defects in pink1 mutants are dependent on acon expression.
Oxidative inactivation of [4Fe-4S] clusters results in increased H2O2 and Fe2+ levels in pink1 mutants
acon is predicted to encode mitochondrial Aconitase (Acon), an iron sulfur cluster containing protein, that catalyzes the formation of isocitrate in the first step of the Krebs cycle [22]. To assess whether Acon localizes to mitochondria we fractionated fly tissue in cytoplasmic and mitochondrially enriched fraction and performed Western blotting using anti-Acon antibodies. Acon is enriched in the mitochondrial fraction (Figure S1G).
Acon harbors a single unligated iron atom in its [4Fe-4S]2+ cluster, and the enzyme is in this respect unique in mitochondria. Such an unligated iron atom is particularly sensitive to superoxide (O2−)-dependent oxidation [27]–[29] that results in cluster instability. Oxidation is followed by the release of Fe2+ and H2O2 that may contribute to oxidative damage and mitochondrial morphological defects through the formation of the potent hydroxyl radical (OH.) by the Fenton reaction [30]. Thus, the specific configuration of the Acon [4Fe-4S]2+ cluster in combination with its proximity to mitochondrially generated superoxide place Acon as a major mediator of oxidative stress in mitochondria. We therefore wondered if O2− leaking from defective pink1 mutant mitochondria could be a source of Acon inactivation resulting in morphological defects. To test if also in fruit flies the loss of pink1 function results in increased O2− production, we incubated mitochondrial preparations from pink1 mutant flies and controls with Complex I substrates (pyruvate/malate) and used the fluorogenic probe dihydroethidium (DHE) to measure O2− production [31], [32]. Similar to wild type mitochondria in the presence of AntimycinA, known to induce O2− production (Figure 2A), pink1B9 mitochondria show a significant increase in DHE fluorescence compared to controls (Figure 2A). These data indicate that pink1 loss leads to increased O2− production.

If the increased O2− in pink1 mutants can act via the Acon [4Fe-4S] cluster to cause mitochondrial swelling, we expect (1) that partial loss of acon does not rescue the increased O2− production in pink1 mutants; (2) that Acon enzymatic activity normalized to total Acon protein is reduced in pink1 mutants; (3) that H2O2 and Fe2+ levels are increased in pink1 mutants as a result of Acon inactivation, and (4) that this defect is rescued by partial loss of acon. First we assessed O2− in pink1 mutants heterozygous for acon1 or acon2 that we showed rescues mitochondrial morphological defects in pink1B9. However, in line with our model, heterozygosity for acon does not reduce pink1B9-induced O2− production (Figure 2A), indicating that increased O2− production per se does not induce mitochondrial morphological defects. Next we measured Acon activity in pink1 mutant mitochondria and we find that Acon activity normalized to total Acon protein levels is significantly reduced compared to the controls. These data are in line with increased Acon inactivation in pink1 mutants (Figure 2B), likely as a result of the increased O2−.
Further testing our model, we also measured H2O2 and Fe2+ content. To measure H2O2 and its radical derivatives we incubated fly lysates with the fluorescent probe dichlorofluorescein diacetate (DHCF-DA) [33]. We find a 50% increase in fluorescence in pink1 mutant lysates compared to the control (Figure 2C). Thus, pink1 mutants accumulate H2O2 and/or derivatives thereof. We also measured mitochondrial Fe2+ content by incubating mitochondrial enriched fractions with Rhodamine B-[(1,10-phenanthrolin-5-yl)aminocarbonyl]benzyl ester (RPA) [34]. In the presence of Fe2+, RPA fluorescence quenches and in pink1B9 mitochondria, we observe a significant increase in RPA quenching compared to controls (Figure 2D). These data indicate increased mitochondrial Fe2+ levels in pink1B9 mutants. This effect is specific, as incubating mitochondria of controls and mutants in Rhodamine B 4-[(Phenanthren-9-yl)Aminocarbonyl]benzyl ester (RPAC) that consists of the same fluorophore as RPA but without iron-chelating properties, does not show quenching in pink1B9 or in controls (Figure 2D). Thus, our data indicate that pink1B9 mutants harbor increased levels of Fe2+ and of H2O2 and/or its radical derivatives.
Next we tested if increased mitochondrial Fe2+ and H2O2 accumulation in pink1 mutants is a consequence of Acon[4Fe-4S] inactivation by O2−. We therefore measured Fe2+ and H2O2 and its derivatives levels in mitochondria of pink1 mutants heterozygous for acon1 or acon2. While the increased O2− production in pink1B9 mutants was not reduced by heterozygous acon, as shown above (Figure 2A), we find that compared to pink1B9, mitochondrial Fe2+ and H2O2 levels are significantly lower in pink1B9 heterozygous for acon1 or acon2 (Figure 2C and 2D). Thus, these data are consistent with the possibility that mitochondrial Fe2+ and H2O2 and/or its radical derivatives-accumulation in pink1 mutants is caused by oxidative inactivation of Acon.
Mitochondrial morphological defects are critically dependent on the Acon dose
Our biochemical data support a model in which oxidative inactivation of Acon and ensuing Fe2+ and H2O2 accumulation contributes to the mitochondrial morphology defects in pink1 mutants. We reasoned that if partial loss of acon protects against mitochondrial stress in pink1 mutants, increased levels of Acon expression may predispose cells to develop mitochondrial morphological defects, provided sufficient O2− is around. We therefore created transgenic animals that overexpress wild type Acon (Figure 3A) resulting in increased Acon activity (Figure 3B). We then determined mitochondrial morphology using mito-GFP and the ple-GAL-4 driver upon expression of Acon in DA neurons. While mitochondria in DA neurons of control flies organize in a long tubular network, mitochondria in DA neurons that overexpress Acon form fragmented spherical aggregates (Figure 3C, 3D and Figure S2A). Hence, in contrast to partial loss of acon that rescues mitochondrial defects in pink1 mutants, overexpression of Acon causes mitochondrial morphological defects and swelling of mitochondria in DA neurons.
![Acon[4Fe-4S] cluster induces mitochondrial defect in DA neurons that is not rescued by increased mitophagy.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/48f4027eac8fc324338362fa577699fe.png)
Based on the finding that increased expression of Acon causes mitochondrial morphological defects we tested if pink1 mutant flies upregulate Acon expression. We measured acon mRNA and protein levels in pink1 mutants, but in contrast to our expectation, we find a significant downregulation of both acon mRNA and Acon protein levels in pink1 flies (Figure S2C, S2D) suggesting that an adaptive mechanism already acts in pink1 mutants to down regulate Acon expression. Thus, the pink1B9-induced stress response results in lower Acon levels and, as shown above, further reducing Acon expression (using heterozygous acon mutants) is protective against mitochondrial defects in pink1 mutants. Taken together, the data are consistent with Acon being a dosage sensitive modifier of morphological defects in mitochondria.
Mitochondrial morphological defects as a consequence of Acon inactivation depend on its [4Fe-4S] cluster
To test if the mitochondrial morphological defects in DA neurons following Acon overexpression are induced by increased Acon catalytic activity or by the presence of an [4Fe-4S] cluster we generated transgenic flies that either overexpress a catalytic inactive Acon (AconS677A) that still harbors its [4Fe-4S] cluster, or flies that overexpress an Acon without its [4Fe-4S] cluster (AconC459S) and is thus also catalytically inactive [22], [35].Western blotting indeed indicates overexpression of the mutant Acon proteins (Figure 3A), and as expected, Acon enzymatic activity measured in fly head lysates is only increased when wild type Acon is expressed, and not when AconS677A or AconC459S are expressed (Figure 3B). While overexpression AconS677A in DA neurons results in obvious mitochondrial morphological defects similar to the overexpression of wild type Acon, overexpression of AconC459S is inert (Figure 3C, 3D and Figure S2A). Hence, the Acon [4Fe-4S] cluster predisposes DA neurons to mitochondrial morphological defects.
Our data are in line with a model where oxidative inactivation of the Acon [4Fe-4S] cluster by O2− contributes to mitochondrial morphological defects. To find further evidence for this idea we expressed Drosophila mitochondrial Ferritin (Fer3HCH) [36] in DA neurons of pink1B9, using the ple-GAL4 driver and assessed mitochondrial morphology using mito-GFP. We find that expression of Fer3HCH significantly rescues defects in mitochondrial morphology in pink1B9 mutants (Figure 4E, Figure 2F, and Figure S2A), suggesting that iron toxicity causes mitochondrial defects in pink1 mutants. Consistent with this model, expression of Fer3HCH in flies that over express Acon also results in a significant rescue of the mitochondrial morphological defects in the DA neurons (Figure S2E, S2F). Hence, the mitochondrial swelling as a result of Acon overexpression is at least in part mediated by iron. Together these data indicate that Acon is a critical source of Fe2+-mediated mitochondrial toxicity.

Mitochondrial morphological defects upon Acon overexpression are not rescued by Drp1 or Parkin
Mitochondrial dynamics and mitophagy are critical processes in maintaining a healthy population of mitochondria. Pink1 has been implicated to regulate mitochondrial homeostasis via several mechanisms. Deregulation of these pathways may be a source of O2−, responsible for Acon inactivation. While Pink1 has been found to maintain the activity of Complex I in the ETC [17]–[20], the protein has also been linked to mitophagy in a pathway involving Drp1 and Parkin [11]–[15], [20], [37]–[40]. Dysfunctional mitochondrial parts may be segregated by the fission factor Drp1 [41], [42]. Pink1 stabilized on depolarized mitochondria then mediates Parkin recruitment causing the ubiquitination of mitochondrial proteins and activation of the autophagic machinery [41], [42]. To test if enlarged and swollen mitochondria upon Acon over expression are a consequence of defective remodeling or mitophagy we co-overexpressed Parkin, a protein that ubiquitinates mitochondrial targets, or Drp1, a mitochondrial fission factor, two conditions thought to facilitate mitophagy. While over expression of Parkin or Drp1 -as expected - result in fragmentation of mitochondria, these conditions do not rescue the defect in mitochondrial swelling and clumping induced by expression of Acon or AconS677A (Figure 3E, 3F and Figure S2A). Hence, our data suggest that the defects in mitochondrial morphology induced by Acon expression are at least in part caused independently from defects in remodeling and mitophagy.
Mitochondiral defects caused by Complex I dysfunction are rescued by partial loss of Acon and by mitoferritin
Given that pink1 mutants display reduced Complex I activity [17]–[20] and this feature may also be a source of increased O2− we tested if mitochondrial swelling and clumping seen in animals where we downregulated an evolutionary conserved Complex I component, NDUFA8, can be rescued by partial loss of acon. First, we confirm increased O2− production and find a concomitant inactivation of Acon activity upon RNAi-mediated downregulation of NDUFA8 (Figure 4A and 4B). Second, we believe that this O2− is produced at least partly independently from defects in mitochondrial remodeling because expression of Drp1 in DA neurons with reduced NDUFA8 function does not fully rescue the mitochondrial swelling and clumping phenotypes in PPM3 DA neurons (Figure 4C, 4D and Figure S2A). Next, we tested the ability of heterozygous acon to modulate the mitochondrial morphological defect induced by NDUFA8 RNAi and find that heterozygous acon is more effective than expression of Drp1 in rescuing the mitochondrial deficits in DA neurons (Figure 4C, 4D and Figure S2A). Likewise, and in line with our model, expression of mitoferritin (Fer3HCH) also alleviates mitochondrial defects in animals that express RNAi to NDUFA8 in DA neurons (Figure 4E, 4F and Figure S2A). Hence, our data suggest that Acon is inactivated by ETC-derived O2− causing oxidative stress.
Our work suggests that mitochondrial morphological defects in pink1 mutant DA cells can be of different origin: both O2−-dependent Acon inactivation or loss of Parkin-dependent mitophagy yield swollen and clumped mitochondria. Alleviating the defects induced by either pathway using heterozygous acon or expressing Drp1 or Parkin both rescue the mitochondrial morphological defects in pink1 mutants (this work; [12]–[14], [16]). To further support this notion, we first assessed if mitochondrial defects in parkin mutants can be rescued by partially removing acon function. parkin mutants display enlarged and swollen mitochondria in muscles and DA neurons, many of the flies also fail to fly and animals harbor lower ATP levels. In contrast to removing acon function in pink1 mutants, heterozygosity for acon fails to rescue the inability of parkin mutants to fly, their reduced ATP levels and their defects in mitochondrial morphology (Figure 5A–5D). Hence, our data suggest that acon acts independently from defects in Parkin-dependent mitophagy. Finally if our model is correct, we reasoned that the combination of Drp1 expression and acon heterozygosity in pink1 mutants should yield additive ‘super rescue’. We therefore tested the ability of these flies to fly and find that they fly significantly better than pink1 mutants or than pink1 mutants partially rescued by either Drp1 expression or by heterozygous acon (Figure 5E). Hence, these data are in line with Pink1 controlling different mitochondrial pathways that can be targeted largely independently. We speculate that increased O2− derived from a defective Complex I in pink1 mutants is an important contributor to Acon inactivation, but other sources of O2− may contribute to mitochondrial failing as well.
Discussion
Iron accumulation in the substantia nigra, systemic mitochondrial dysfunction and oxidative stress have all been implicated in PD pathology; however, a link between these factors remains elusive. Here we show that oxidative inactivation of Acon generates iron-mediated oxidative stress that contributes to mitochondrial swelling in Drosophila pink1 mutants (Figure 6). Inactivation of Acon[4Fe-4S] clusters could contribute to mediating O2− toxicity by simultaneous release of Fe2+ and H2O2 [43] that combine in the Fenton reaction to generate highly toxic hydroxyl radicals [30], [44] (Figure 6). Hydroxyl radicals can induce mitochondrial permeability transition and swelling [45]–[47], in line with electron microscopic analyses of pink1 mutants where mitochondria appear swollen and show disorganized cristae [9], [10] (Figure 1). Four major findings support that this iron-mediated toxic mechanism is an additional important aspect of mitochondrial dysfunction in pink1 mutants. First, we find increased O2− production, increased Acon inactivation and more Fe2+ and H2O2 accumulation in pink1 mutants (Figure 2). Second, partial loss of Acon reduces Fe2+ and H2O2 accumulation and alleviates pink1-associated phenotypes including mitochondrial morphological defects in muscle and DA neurons (Figure 1). Third, overexpression of wild type Acon in dopaminergic neurons produces a mitochondrial morphological defect and this effect is completely dependent on the presence of the [4Fe-4S] cluster in Acon (Figure 3). These data also indicate mitochondrial integrity is sensitive to Acon [4Fe-4S] cluster dosage. Finally, chelating iron by expressing mitochondrial Ferritin is sufficient to rescue pink1 mitochondrial morphological defects (Figure 4). Thus, our data suggest that inactivation of Acon and iron accumulation might be a pathogenic mechanism triggered by loss of pink1 and increased superoxide, linking iron accumulation and mitochondrial failure.

Acon inactivation is dependent on O2− that, amongst other sources (see below), may be produced in defective mitochondria. While various mitochondrial insults can result in increased O2− production, our work is most consistent with Parkin-dependent mitophagy being not the major source of Acon inactivation in pink1 mutants. The mitochondrial morphological defects induced by Acon overexpression were not strongly rescued by expressing Drp1, a condition that indirectly promotes mitophagy and parkin mutants were not majorly rescued by partial loss of acon (Figure 3 and Figure 5). In contrast, mitochondrial morphological defects in DA neurons of flies with reduced Complex I activity are significantly rescued when acon is heterozygous (Figure 4). Hence, Acon seems to act in a Pink1-dependent pathway that can operate largely independently of mitophagy (Figure 6).
Defects at the level of Complex I are often associated with increased leaking of the toxic O2− [48], [49], and likewise, systemic inhibition of Complex I mimics features of PD in animal models [50]–[53]. Previous work in flies or mice has indicated reduced ETC function [17]–[20] in pink1 mutants, and we show that this condition results in mitochondrial morphological defects in an Acon-dependent manner. Similar to pink1 mutants, RNAi-mediated knock down of an evolutionary conserved Complex I component, NDUFA8, also results in an increased production of superoxide as well as in Acon inactivation. We show that these biochemical changes correlate with mitochondrial morphology defects in dopaminergic neurons that can be rescued by partial loss of acon or by over-expression of mitoferritin that scavenges the released Fe2+ [36] (Figure 4).
It is interesting to note that increased O2− production per se is not sufficient to generate mitochondrial morphological defects, and that the presence of sufficient amounts of Acon is required. Indeed, our data indicate that pink1 mutants heterozygous for acon show increased levels of O2− but normal mitochondrial morphology. Our data also indicate that upstream events in pink1 mutants that result in increased O2− production contribute to mitochondrial morphological defects because of oxidative inactivation of Acon. In line with this, overexpression of the mitochondrial superoxide dismutase 2 (SOD2) that scavenges O2−, successfully rescues mitochondrial swelling phenotype of pink1 in DA neurons [54]. Given that both genetic forms of PD as well as sporadic cases of PD show ETC defects [5], [6], [17]–[19], our work may be relevant for idiopathic cases that suffer from mitochondrial dysfunction as well. Acon inactivation and iron-mediated toxicity might thus have a more general role in the pathogenesis of PD.
While pink1 loss affects numerous cell types, our data also start to provide insight as to why DA neurons in the substantia nigra are more vulnerable in PD. While overexpression of Acon or downregulation of Complex I produces mitochondrial morphological defects in DA neurons, in Drosophila flight muscles mitochondria appear morphologically largely normal (data not shown). These data suggest a tissue-specific response in that Acon inactivation has a stronger impact in DA neurons than in muscle cells. Each cell type is exposed to various sources of O2−, but DA neurons in particular are exposed to dopamine-induced oxidative stress that is a source of O2− [55]–[57]. Furthermore, the substantia nigra in humans is naturally rich in iron [58] and this feature may lower the threshold for hydroxyl radical production in the Fenton reaction that is facilitated by Acon inactivation. Pink1 mutations or environmental factors in some sporadic cases of PD already result in increased levels of O2−, but we hypothesize that in DA neurons, additional dopamine-induced oxidative stress may facilitate Acon inactivation and hydroxyl radical production providing insight into one of the pathways underlying mitochondrial failure in pink1 mutants.
Methods
Drosophila stocks and maintenance
Flies were raised on standard cornmeal and molasses medium at 25°C. w1118; UAS-mitoGFP, w1118; daGal, w1118; pleGal4, w; UAS-4EBP and w1118; Mi{ET1}AconMB09176/SM6a (acon2) and were obtained from Bloomington stock center (Indiana, USA). w1118 pink1B9 and w1118 pink1RV, parkin1 and parkinRV [59] were provided by Jongkyeong Chung (Advanced Institute of Science and Technology, Korea) [10]. parkinΔ21 mutant flies were a gift from Graeme Mardon (Baylor College of Medicine) [60] and drp1+ genomic rescue constructs were provided by Hugo Bellen (Baylor College of Medicine) [61] w1118; UAS-Fer3HCH was provided by Dr Fanis Missirlis (Qeen Mary University of London, UK). w1118; UAS-CG3683RNAi (w1118; P{GD16787}v46799/CyO) was from the Vienna Drosophila RNAi Center (VDRC) [62].
Molecular biology and biochemistry
The genomic clone CH322-18I04 was obtained from BACPAC Resources (Children's Hospital Oakland).
UAS-Aconwt was generated by PCR amplification of BDGP cDNA clone LD24561 using primers: AconcDNA.F (5′ ATGGCTGCGAGATTGATGAACG) and AconcDNA.F (5′ TTACTGGGCCAGCTCCTTCATGC). The S677A and C459S mutations were introduced in the primers and the mutated cDNAs were generated by overlap extension PCR using the following primers: S677A.F (5′GAcgCACCCTCGCCGTAGTTCTCATC) S677A.R (5′AACTACGGCGAGGGTGcgTC), C459.F (5′GGTCCCtCcATTGGACAGTGGGATCG) and C459.R (5′CGATCCCACTGTCCAATgGaGGGACC). All constructs were cloned into the EcoRI and NotI restriction sites of pUAST-attB [63]. Following sequencing, transgenic flies were created at GenetiVision Inc. (Houston, USA) using PhiC31 mediated transgenesis in the VK1 docking site (2R, 59D3) [64].
For quantitative RT-PCR, total RNA was isolated using TRI Reagent (Sigma-Aldrich) according to the manufacturer's protocol. Subsequently, the RNA samples were cleaned up using the RNeasy Mini Kit with the on-column DNAse treatment (Qiagen). 1 µg of total RNA was used as a template for synthesis of oligodT-primed double stranded cDNA using the SuperScriptIII First-Strand Synthesis System (Invitrogen). 20 ng cDNA of each sample was used for acon SYBR Green PCR Master mix (Applied Biosystems) and the following primers were used: aconRT-F (5′ TCGTGCCATTATCGTCAAGTC) and aconRT-F (5′ AGGTTGAGCAGGGAGATTTTG). All experiments were performed in triplicate and run on a Roche LC480 system. The data were normalized utilizing RP-49, a ribosomal gene, using following primers: RP-49-F (5′ ATCGGTTACGGATCGAACAA) and RP-49-R (5′ GACAATCTCCTTGCGCTTCT).
For Western blots, flies were homogenized in cold T-PER buffer (ThermoScientific) with complete protease inhibitor mixture (Roche). Protein concentration was determined by BCA protein quantification kit (Pierce). Samples were diluted in 2-mercaptoethanol 10% SDS loading buffer and boiled for 5 min and 15 µg of proteins were separeted on pre-cast 4–12% NuPage Bis-Tris gels (Invitrogen). Following transfer to nitrocellulose, blots were probed with primary antibodies: 1∶5000 Anti-ACO2 (AbGent), 1∶1000 anti-Tubulin (B5–12, Sigma) and 1∶1000 HRP coupled secondary antibodies (Jackson immunolabs). Blots were developed with Western-Lightning-ECL (PerkinElmer) and imaged. Quantification was performed using gel analyzer tool in ImageJ software from the US National Institute of Health (http://rsb.info.nih.gov/ij/).
Flight assay
Batches of 5 days old male flies were transferred to an empty vial (5 cm D, 10 cm H). Flies were allowed to climb above a marked line at 9 cm height; the vial was gently tapped and visually scored for flying flies. Flies at the bottom were removed and the remaining flies were retested. Flies that fly twice were assigned a score of 1, the others a score of 0.
ATP measurements
ATP content was determined as described [10]. 5 days-old flies with abdomen dissected out were homogenized in 50 µl of 6 M guanidine-HCl 100 mM Tris and 4 mM, EDTA, pH 7.8. These homogenates were snap-frozen in liquid nitrogen and then boiled for 3 min. Samples were then centrifuged and the supernatant was diluted (1/50) in extraction buffer, mixed with luminescent solution (ATP Determination Kit, Invitrogen) and luminescence was measured on an EnVision Multilabel Reader (Perkin Elmer). ATP (nmol) was determined using a standard curve and normalized to protein content (mg) measured by BCA assay (Pierce).
Muscle section and TEM
Thoraxes were fixed in paraformaldehyde/glutaraldehyde, postfixed in osmium tetroxide, dehydrated and embedded in Epon. Sections 80 nm thick were stained with uranyl acetate and lead citrate and subjected to TEM analysis.
H2O2 levels
H2O2 was measured as described [33]. 4–5 adult flies were homogenized in 50 µl cold lyses buffer T-PER (Thermo scientific) and the homogenate were cleared by centrifugation at 1000×g for 5 min at 4°C. 140 µL of PBS containing 50 µM of DCFH-DA (molecular probe) were added to 10 µL of lysate in a 96-well plate format and incubated at 25°C for 10 minutes in the dark. DCFH-DA fluorescence (485exc/530em) was measured using Wallac Victor2 1420 (Perkin Elmer). Fluorescence intensity was normalized to the protein amount (BCA, Pierce) and expressed as relative to the control.
Mitochondria isolation
Fifty flies were gently crushed in 1 ml chilled mitochondrial isolation medium (Mitosciences) by using a porcelain mortar and pestle, then spun twice at 1,000×g for 5 min at 4°C to remove debris. The supernatant was then spun at 12,000×g, for 15 min at 4°C. The pellet, containing the mitochondria, was washed with 1 ml of isolation medium and resuspended in 40 µl of isolation medium supplemented with complete protease inhibitor mixture without EDTA (Roche).
Superoxide production
Mitochondrial Superoxide production was measured as described [32]. 10 µg of mitochondria were incubated in experimental buffer (EB: 125 mM KCl, 10 mM Tris-MOPS, 1 mM KPi, 10 µM EGTA-Tris, pH 7.4, 25°C) supplemented with 1.25 mM Pyruvate/1.25 mM malate and 5 µM DHE (Molecular probe) in a 96-well plate format for 10 min. The fluorescence was measured (485exc/590em) using Wallac Victor2 1420 (Perkin Elmer). Fluorescence intensity was normalized to the initial value and expressed as relative to the control. 10 µM antimycin A was used to induce superoxide production in control mitochondria.
Fe2+ measurements
For mitochondrial ferrous iron level measurements, 10 µg of mitochondria were resuspended in isolation buffer (Mitosciences) and incubated with 20 µM of RPA or RPAC (Squarix Biotechnology) in a 96-well plate format at room temperature for 10 min. RPA/RAPC fluorescence (560 exc/600 em) was measured using Wallac Victor2 1420 (Perkin Elmer). Quenching was calculated as percent of initial fluorescence.
Aconitase activity
Aconitase enzyme activity microplate kit (Mitosciences) was used according to the manufacturer's protocol to measure Aconitase activity. 20 µg of mitochondria were incubated with assay buffer and the activity was measured by following conversion of isocitrate to cis-aconitate as in increased in 240 nm UV absorbance. Measurements were recorded over 30 min. at 1 min intervals and aconitase activity were calculated from the linear increase in absorbance and normalized to the amount of aconitase, determined by western blot, in the same mitochondrial preparation. Values were reported as relative activity to the control.
Mitochondrial morphology in DA neurons
Brain dissection and whole-mount immunohistochemistry for tyrosine hydroxylase (TH) was performed as described [65]. Primary 1∶100 antibody against TH (Chemicon) and secondary alexa 555 (Invitrogen) were used. Brains were imaged on a Zeiss LSM 510 META confocal microscope using a 63xoil NA 1.4 lens. Mitochondrial tagged GFP (mito-GFP) was visualized using 488 nm laser and 500–530 band pass emission filter. Because mitochondrial morphology is sensitive to environmental conditions, variations did occur from batch to batch. We only compared flies of different genotypes if normal mitochondrial morphology was observed in the control samples (Figure S1H–S1H″) in the same batch. For quantification of mitochondrial aggregates size and numbers, DA neurons of PPM3 cluster (Figure S1H–S1H″) were scored. Quantification of aggregate size was done using “analyzing particles” plugin in ImageJ (http://rsb.info.nih.gov/ij/): rounded particles were automatically detected and the average surface area of aggregates in each neuron was determined as total area occupied by aggregates/number of aggregates.
Mitochondrial morphology in flight muscles
Adult flies were fixed in PBS with 5% formaldehyde and 0.4% Triton for 3 hours. Thoraxes were dissected in PBS and mounted in vectashield (Vector Laboratories) and were imaged on a Zeiss LSM 510 META confocal microscope using a 63xoil NA 1.4 lens. Mitochondrial tagged GFP (mito-GFP) was visualized using 488 nm laser and 500–530 band pass emission filter. For muscle section with same area were scored and quantification of mitochondrial aggregates was performed as described above.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. DawsonTM, DawsonVL (2003) Molecular pathways of neurodegeneration in Parkinson's disease. Science 302 : 819–822.
2. ThomasB, BealMF (2007) Parkinson's disease. Hum Mol Genet 16 Spec No. 2: R183–194.
3. HorowitzMP, GreenamyreJT (2010) Mitochondrial iron metabolism and its role in neurodegeneration. J Alzheimers Dis 20 Suppl 2: S551–568.
4. SoficE, PaulusW, JellingerK, RiedererP, YoudimMB (1991) Selective increase of iron in substantia nigra zona compacta of parkinsonian brains. J Neurochem 56 : 978–982.
5. LestienneP, NelsonJ, RiedererP, JellingerK, ReichmannH (1990) Normal mitochondrial genome in brain from patients with Parkinson's disease and complex I defect. J Neurochem 55 : 1810–1812.
6. SchapiraAH, CooperJM, DexterD, JennerP, ClarkJB, et al. (1989) Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1 : 1269.
7. DawsonTM, KoHS, DawsonVL (2010) Genetic animal models of Parkinson's disease. Neuron 66 : 646–661.
8. JonesR (2010) The roles of PINK1 and Parkin in Parkinson's disease. PLoS Biol 8: e1000299 doi:10.1371/journal.pbio.1000299.
9. ClarkIE, DodsonMW, JiangC, CaoJH, HuhJR, et al. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441 : 1162–1166.
10. ParkJ, LeeSB, LeeS, KimY, SongS, et al. (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441 : 1157–1161.
11. JinSM, YouleRJ (2012) PINK1 - and Parkin-mediated mitophagy at a glance. J Cell Sci 125 : 795–799.
12. DengH, DodsonMW, HuangH, GuoM (2008) The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A 105 : 14503–14508.
13. PooleAC, ThomasRE, AndrewsLA, McBrideHM, WhitworthAJ, et al. (2008) The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A 105 : 1638–1643.
14. YangY, OuyangY, YangL, BealMF, McQuibbanA, et al. (2008) Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A 105 : 7070–7075.
15. NarendraDP, JinSM, TanakaA, SuenDF, GautierCA, et al. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298 doi:10.1371/journal.pbio.1000298.
16. YangY, GehrkeS, ImaiY, HuangZ, OuyangY, et al. (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A 103 : 10793–10798.
17. GautierCA, KitadaT, ShenJ (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A 105 : 11364–11369.
18. MoraisVA, VerstrekenP, RoethigA, SmetJ, SnellinxA, et al. (2009) Parkinson's disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol Med 1 : 99–111.
19. VilainS, EspositoG, HaddadD, SchaapO, DobrevaMP, et al. (2012) The yeast complex I equivalent NADH dehydrogenase rescues pink1 mutants. PLoS Genet 8: e1002456 doi:10.1371/journal.pgen.1002456.
20. LiuW, Acin-PerezR, GeghmanKD, ManfrediG, LuB, et al. (2011) Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc Natl Acad Sci U S A 108 : 12920–12924.
21. VosM, EspositoG, EdirisingheJN, VilainS, HaddadDM, et al. (2012) Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science 336 : 1306–1310.
22. BeinertH, KennedyMC (1993) Aconitase, a two-faced protein: enzyme and iron regulatory factor. FASEB J 7 : 1442–1449.
23. LaubleH, KennedyMC, BeinertH, StoutCD (1992) Crystal structures of aconitase with isocitrate and nitroisocitrate bound. Biochemistry 31 : 2735–2748.
24. HiesingerPR, FayyazuddinA, MehtaSQ, RosenmundT, SchulzeKL, et al. (2005) The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell 121 : 607–620.
25. UytterhoevenV, KuenenS, KasprowiczJ, MiskiewiczK, VerstrekenP (2011) Loss of skywalker reveals synaptic endosomes as sorting stations for synaptic vesicle proteins. Cell 145 : 117–132.
26. VerstrekenP, OhyamaT, HaueterC, HabetsRL, LinYQ, et al. (2009) Tweek, an evolutionarily conserved protein, is required for synaptic vesicle recycling. Neuron 63 : 203–215.
27. FlintDH, TuminelloJF, EmptageMH (1993) The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J Biol Chem 268 : 22369–22376.
28. GardnerPR, FridovichI (1991) Superoxide sensitivity of the Escherichia coli 6-phosphogluconate dehydratase. J Biol Chem 266 : 1478–1483.
29. GardnerPR, FridovichI (1991) Superoxide sensitivity of the Escherichia coli aconitase. J Biol Chem 266 : 19328–19333.
30. Vasquez-VivarJ, KalyanaramanB, KennedyMC (2000) Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J Biol Chem 275 : 14064–14069.
31. BenovL, SztejnbergL, FridovichI (1998) Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med 25 : 826–831.
32. HorakP, CrawfordAR, VadysirisackDD, NashZM, DeYoungMP, et al. (2010) Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc Natl Acad Sci U S A 107 : 4675–4680.
33. WangYC, LeeCM, LeeLC, TungLC, Hsieh-LiHM, et al. (2011) Mitochondrial dysfunction and oxidative stress contribute to the pathogenesis of spinocerebellar ataxia type 12 (SCA12). J Biol Chem 286 : 21742–21754.
34. PetratF, WeisheitD, LensenM, de GrootH, SustmannR, et al. (2002) Selective determination of mitochondrial chelatable iron in viable cells with a new fluorescent sensor. Biochem J 362 : 137–147.
35. PhilpottCC, KlausnerRD, RouaultTA (1994) The bifunctional iron-responsive element binding protein/cytosolic aconitase: the role of active-site residues in ligand binding and regulation. Proc Natl Acad Sci U S A 91 : 7321–7325.
36. MissirlisF, HolmbergS, GeorgievaT, DunkovBC, RouaultTA, et al. (2006) Characterization of mitochondrial ferritin in Drosophila. Proc Natl Acad Sci U S A 103 : 5893–5898.
37. GeislerS, HolmstromKM, SkujatD, FieselFC, RothfussOC, et al. (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12 : 119–131.
38. GeislerS, HolmstromKM, TreisA, SkujatD, WeberSS, et al. (2010) The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 6 : 871–878.
39. KohH, ChungJ (2012) PINK1 as a molecular checkpoint in the maintenance of mitochondrial function and integrity. Mol Cells 34 : 7–13.
40. YuW, SunY, GuoS, LuB (2011) The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum Mol Genet 20 : 3227–3240.
41. ImaiY, LuB (2012) Mitochondrial dynamics and mitophagy in Parkinson's disease: disordered cellular power plant becomes a big deal in a major movement disorder. Curr Opin Neurobiol 21 : 935–941.
42. TwigG, ShirihaiOS (2011) The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal 14 : 1939–1951.
43. CantuD, SchaackJ, PatelM (2009) Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS ONE 4: e7095 doi:10.1371/journal.pone.0007095.
44. LiochevSI, FridovichI (1994) The role of O2. - in the production of HO.: in vitro and in vivo. Free Radic Biol Med 16 : 29–33.
45. SakuraiK, StoyanovskyDA, FujimotoY, CederbaumAI (2000) Mitochondrial permeability transition induced by 1-hydroxyethyl radical. Free Radic Biol Med 28 : 273–280.
46. SrivastavaS, ChanC (2007) Hydrogen peroxide and hydroxyl radicals mediate palmitate-induced cytotoxicity to hepatoma cells: relation to mitochondrial permeability transition. Free Radic Res 41 : 38–49.
47. VercesiAE, KowaltowskiAJ, GrijalbaMT, MeinickeAR, CastilhoRF (1997) The role of reactive oxygen species in mitochondrial permeability transition. Biosci Rep 17 : 43–52.
48. PitkanenS, RobinsonBH (1996) Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J Clin Invest 98 : 345–351.
49. TurrensJF (1997) Superoxide production by the mitochondrial respiratory chain. Biosci Rep 17 : 3–8.
50. BetarbetR, ShererTB, MacKenzieG, Garcia-OsunaM, PanovAV, et al. (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 3 : 1301–1306.
51. CannonJR, TapiasV, NaHM, HonickAS, DroletRE, et al. (2009) A highly reproducible rotenone model of Parkinson's disease. Neurobiol Dis 34 : 279–290.
52. CoulomH, BirmanS (2004) Chronic exposure to rotenone models sporadic Parkinson's disease in Drosophila melanogaster. J Neurosci 24 : 10993–10998.
53. DauerW, PrzedborskiS (2003) Parkinson's disease: mechanisms and models. Neuron 39 : 889–909.
54. KohH, KimH, KimMJ, ParkJ, LeeHJ, et al. (2012) Silent information regulator 2 (Sir2) and Forkhead box O (FOXO) complement mitochondrial dysfunction and dopaminergic neuron loss in Drosophila PTEN-induced kinase 1 (PINK1) null mutant. J Biol Chem 287 : 12750–12758.
55. BermanSB, HastingsTG (1999) Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson's disease. J Neurochem 73 : 1127–1137.
56. ZhangF, DryhurstG (1994) Effects of L-cysteine on the oxidation chemistry of dopamine: new reaction pathways of potential relevance to idiopathic Parkinson's disease. J Med Chem 37 : 1084–1098.
57. ZoccaratoF, ToscanoP, AlexandreA (2005) Dopamine-derived dopaminochrome promotes H(2)O(2) release at mitochondrial complex I: stimulation by rotenone, control by Ca(2+), and relevance to Parkinson disease. J Biol Chem 280 : 15587–15594.
58. SnyderAM, ConnorJR (2009) Iron, the substantia nigra and related neurological disorders. Biochim Biophys Acta 1790 : 606–614.
59. ChaGH, KimS, ParkJ, LeeE, KimM, et al. (2005) Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc Natl Acad Sci U S A 102 : 10345–10350.
60. PesahY, PhamT, BurgessH, MiddlebrooksB, VerstrekenP, et al. (2004) Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 131 : 2183–2194.
61. VerstrekenP, LyCV, VenkenKJ, KohTW, ZhouY, et al. (2005) Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47 : 365–378.
62. DietzlG, ChenD, SchnorrerF, SuKC, BarinovaY, et al. (2007) A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448 : 151–156.
63. BischofJ, MaedaRK, HedigerM, KarchF, BaslerK (2007) An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A 104 : 3312–3317.
64. VenkenKJ, HeY, HoskinsRA, BellenHJ (2006) P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 314 : 1747–1751.
65. WuJS, LuoL (2006) A protocol for dissecting Drosophila melanogaster brains for live imaging or immunostaining. Nat Protoc 1 : 2110–2115.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 4
Nejčtenější v tomto čísle
- The G4 Genome
- Neutral Genomic Microevolution of a Recently Emerged Pathogen, Serovar Agona
- The Histone Demethylase Jarid1b Ensures Faithful Mouse Development by Protecting Developmental Genes from Aberrant H3K4me3
- The Tissue-Specific RNA Binding Protein T-STAR Controls Regional Splicing Patterns of Pre-mRNAs in the Brain