
Signatures of Diversifying Selection in European Pig Breeds
Following domestication, livestock breeds have experienced intense selection pressures for the development of desirable traits. This has resulted in a large diversity of breeds that display variation in many phenotypic traits, such as coat colour, muscle composition, early maturity, growth rate, body size, reproduction, and behaviour. To better understand the relationship between genomic composition and phenotypic diversity arising from breed development, the genomes of 13 traditional and commercial European pig breeds were scanned for signatures of diversifying selection using the Porcine60K SNP chip, applying a between-population (differentiation) approach. Signatures of diversifying selection between breeds were found in genomic regions associated with traits related to breed standard criteria, such as coat colour and ear morphology. Amino acid differences in the EDNRB gene appear to be associated with one of these signatures, and variation in the KITLG gene may be associated with another. Other selection signals were found in genomic regions including QTLs and genes associated with production traits such as reproduction, growth, and fat deposition. Some selection signatures were associated with regions showing evidence of introgression from Asian breeds. When the European breeds were compared with wild boar, genomic regions with high levels of differentiation harboured genes related to bone formation, growth, and fat deposition.
Published in the journal:
. PLoS Genet 9(4): e32767. doi:10.1371/journal.pgen.1003453
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003453
Summary
Following domestication, livestock breeds have experienced intense selection pressures for the development of desirable traits. This has resulted in a large diversity of breeds that display variation in many phenotypic traits, such as coat colour, muscle composition, early maturity, growth rate, body size, reproduction, and behaviour. To better understand the relationship between genomic composition and phenotypic diversity arising from breed development, the genomes of 13 traditional and commercial European pig breeds were scanned for signatures of diversifying selection using the Porcine60K SNP chip, applying a between-population (differentiation) approach. Signatures of diversifying selection between breeds were found in genomic regions associated with traits related to breed standard criteria, such as coat colour and ear morphology. Amino acid differences in the EDNRB gene appear to be associated with one of these signatures, and variation in the KITLG gene may be associated with another. Other selection signals were found in genomic regions including QTLs and genes associated with production traits such as reproduction, growth, and fat deposition. Some selection signatures were associated with regions showing evidence of introgression from Asian breeds. When the European breeds were compared with wild boar, genomic regions with high levels of differentiation harboured genes related to bone formation, growth, and fat deposition.
Introduction
The domestic pig is an important livestock species and an important protein source worldwide. The pig originated from the wild boar, Sus scrofa, by multiple independent domestications, mainly in Asia Minor, Europe and East Asia [1], [2]. Domestication and subsequent selective pressures altered the behaviour and phenotypic characteristics of these animals [3]. Local pig types were developed in Europe and Asia after domestication, but the development of phenotypically distinct breeds chiefly occurred with the commencement of organised breeding in the 18th century [4].
Strict organised breeding was adopted to improve and develop livestock breeds and Britain in particular was a main centre of the early improvement of pig breeds [5], [6], as a reaction to increasing demand for meat in the wake of the industrial revolution. From the 18th century pig breeds were selectively bred for specific production traits such as early maturation, rapid growth and increased prolificacy. In addition, the coat colour phenotype (which includes both skin and hair pigmentation) was another morphological trait often used during the selective breeding process. Substantial morphological changes occurred in breeds over a short period of time, resulting in the development of numerous distinct pig breed phenotypes in Britain. Charles Darwin commented on the rapid morphological changes in pig breeds at that time: “Chiefly, in consequence of so much crossing, some well-known breeds have undergone rapid changes; thus, according to Nathusius […] the Berkshire breed of 1780 is quite different from that of 1810; and, since this latter period, at least two distinct forms have been borne the same name.” [4]. Although breeds tended to be formed by complex crossing with numerous other breeds, including a number from Asia, to introduce desirable traits [4]–[6], after improvement the breeds were kept distinct, resulting in highly specialised phenotypically distinct and genetically differentiated pig breeds [7]. From the 20th century, with the recognition of the benefits of genetic improvement and changing consumer preferences, certain pig breeds experienced further strong selection for lean meat content, muscularity and enhanced reproduction [5], [6].
To better understand the genetic basis for phenotypic variation in the pig, studies have focused on important traits relevant to the breed development process with the aim of identifying, characterising and mapping candidate genes, and subsequently identifying the underlying causal mutations and allelic differences between breeds [8], [9]. Studies mapping quantitative trait loci (QTL) have particularly focused on muscle growth. Fine mapping of one of these regions (SSC2) identified a causal mutation in the IGF2 gene, where a single nucleotide change is associated with high muscle content in some commercial pig populations [10]. The level of fat on the carcass is also a production trait of economic impact and QTL studies have mapped loci associated with fat deposition to various chromosomes, in particular SSC4 and SSC7 [11], [12]. Reproductive traits have received attention in pigs with several genes investigated in relation to litter size and the number of teats (ESR, PTHLH and PTHR1) [13]. Coat colour is considerably varied amongst breeds within domesticated animal species and investigations into the genetics of pigmentation have identified numerous loci influencing these traits [8], [9]. Variation at two genes, KIT and MC1R, is associated with a variety of pig breed colour types including red, black and white colouring and belted and spotted phenotypes [14]–[16].
With growing genomic resources, selection mapping approaches are increasingly being implemented to identify genetic variants that underlie the phenotypic diversity in domesticated animals. These approaches involve scanning the genome for levels of population differentiation and diversity [17]. Genome-wide scans for signatures of diversifying selection in livestock species have detected signals revealing candidate genes related to morphological variation such as body size, skeletal formation, cranial structure and coat patterns, and production traits such as muscle conformation and milk yield [18]–[25].
To further explore the genetic variation underlying the phenotypic diversity of pig breeds, a genome-wide scan of a diverse set of commercial and traditional British/European pig breeds was performed to identify genomic regions showing signatures of between-breed (diversifying) selection using levels of breed genetic differentiation (FST). Based on these results, sequence data from three candidate regions was analysed to investigate potential causative variants.
Results
Signatures of diversifying selection
A genome-wide scan for signatures of selection in 13 European pig breeds (Table 1) was carried out by estimating Wright's FST, a measure of population genetic differentiation, at each genetic marker. After adopting a sliding window approach, candidate regions that may have experienced diversifying selection were identified by taking the 99th percentile of the empirical distribution of FST–windows (Figure S1). A total of 491 FST–windows per breed were deemed as outlier regions and as many were adjacent SNPs that clustered together, a total of 446 genomic regions displayed strong breed differentiation.
Signatures of selection shared in multiple breeds
The genome-wide scan revealed five genomic regions of extremely high levels of differentiation that overlapped in five or more breeds; all of these regions contain biologically interesting candidate genes (Table 2). One such region was observed in eight breeds on SSC5 (32.32–34.06 Mb). In all but two of the breeds, the peak FST–window (∼32.6–32.8 Mb) overlapped with the genes WIF1 (32.66–32.72 Mb) and LEMD3 (32.77–32.89 Mb). This region is orthologous to a region in dogs associated with ear morphology [19], [24]. Another region was detected in five breeds on SSC7 (54.00–57.00 Mb), where at the 97.5th percentile a further four breeds also exhibited a signal. On SSC8, a region of high differentiation spanning 71.84–75 Mb was observed in nine breeds. More striking was the extended region of differentiation on SSC8 spanning 40–75 Mb observed in most breeds, with numerous overlapping and non-overlapping peaks of FST across a large genomic region on that chromosome (Figure S1), although fewer than five breeds overlapped directly in their peak FST–windows, except in the narrow interval mentioned above. Duroc was the only breed that did not show high levels of differentiation in this region, or even on that chromosome, at either the 99th or 97.5th percentile. Outlier regions were also found on SSC15 (139.60–142.10 Mb), observed in six breeds, and on SSC16 (18.72–20.63 Mb), observed in five breeds.

Signals unique to individual breeds
Most extreme genomic regions were observed in fewer breeds (1–4) (Figure S1) and we highlight examples of those found in the within-breed 99.9th percentile that overlapped QTLs and contained biologically interesting genes (Table S1). The Duroc breed exhibited several signatures of diversifying selection on two chromosomes. On SSC14 a highly differentiated region (123.08–123.41 Mb) overlapped with QTLs for fatty acid composition in Duroc [26], [27] and includes a gene involved in fatty acid biosynthesis, ELOVL3 (123.08–123.083 Mb) [28]. On SSC15 a highly differentiated genomic region (85.73–86.62 Mb) contained the MYO3B (Class III myosin B) gene (85.63–85.93 Mb), which directly overlapped the peak FST-window (85.83 Mb). An extended differentiated genomic region was observed in the Landrace breed on SSC13, with the highest FST–window occurring at 73.06 Mb, close to the GHRL gene (73.47–73.48 Mb). In addition, QTLs related to various reproductive traits in pigs have been mapped to SSC13 [29] and overlap with the extended differentiated genomic region.
Large, breed-specific signatures of diversifying selection were not limited to the commercial breeds, but also were observed in the traditional breeds (Table S1). Gloucestershire Old Spots displayed a signal of diversifying selection on SSC11, close to EDNRB (54.69–54.72 Mb), a gene implicated in coat colour pattern in mammals [30]. Near the peak FST–window (55.20 Mb) many SNPs in this region were fixed in this breed whereas alleles were segregating in all other pig breeds (Figure 1). A weaker signal in the region of this gene (seen in the 99th but not 99.9th percentiles) appeared in Mangalica and British Saddleback breeds (Figure S1). Another breed-specific signature of selection was observed on SSC5 at a different coat colour locus in the Berkshire. KITLG (KIT ligand, 98.74–98.78 Mb) was just upstream from a 99.9th percentile FST–window (98.84 Mb) on SSC5 and KITLG fell within the 99th percentile differentiation region. Many SNPs in the region of this gene were almost fixed for the same allele in Berkshire and the Asian breed, Meishan, whilst alleles were segregating in the other European pig breeds (Figure 1).

Phenotypic traits analysis
Ear
To identify genomic regions associated with ear morphology, we divided the breeds into three classes: prick (upright), intermediate (partly flopped down) and flat (completely flopped down) breeds (Figure S2 and Table S2). Comparisons between these classes revealed three highly differentiated regions on SSC5 and SSC7 (Tables S3, S4, S5; Figure 2A). When prick-eared breeds were contrasted with flat-eared breeds, there was a highly differentiated region on SSC5 (31.74–33.78 Mb) that overlapped with the region identified across eight breeds (see section FST–multiple breeds and Table 2). When prick-eared breeds were contrasted with intermediate-eared breeds, no signal was observed on SSC5 but two signals were observed on SSC7 (31.86–34.19, 55.43–58.19 Mb). When intermediate-eared breeds were contrasted with flat-eared breeds, the same signal was observed on SSC5 (32.28–33.80 Mb) and one of the two signals on SSC7 was present (55.41–58.20 Mb). The peak FST-window on SSC5 overlapped the LEMD3 gene. The peak FST-window on the second signal on SSC7 occurred at 56.75 Mb, at the ADAMTSL3 locus (56.50–56.85 Mb). Variation in allele frequencies was observed in the differentiated region on SSC5 overlapping the LEMD3 gene: alleles were near fixation in the flat-eared breeds, alleles were near fixation for the alternate allele or of intermediate allele frequency in the prick-eared breeds and alleles were segregating in intermediate-eared breeds (Figure 2B).

Coat colour
When red coat breeds were compared with non-red coat breeds, the observed outlier regions (Table S6) corresponded with the strong signals of diversifying selection on SSC14 and SSC15 detected in the Duroc from the individual breed comparison (see Figure S1 and Table S1). When black and partially black coat breeds (Large Black, Berkshire, Hampshire, British Saddleback) were compared against red coat breeds, outlier regions were found on 14 chromosomes (Table S7). No signals of selection were detected in the region of MC1R (SSC6 : 0.26 Mb) for either of these coat colour comparisons.
The next comparison was coat colour phenotypes known to be associated with allelic variation at the KIT gene (SSC8 : 43.55–43.59 Mb). When belted breeds were compared with non-belted breeds, a differentiated region was observed on SSC8 (41.18–43.08 Mb) near the KIT gene (Table S8). However, when non-belted breeds were compared with each other and when belted breeds were compared with each other, a signal of selection was again detected in the region of KIT. Although at the location of the KIT gene, FST-SNP estimates were higher in the belted vs non-belted breeds comparison than the within-belted breed comparison. When white-coated were compared against non-white-coated breeds a marked differentiation was again observed on SSC8 (43.46–43.73 Mb) in the region of the KIT gene, but this was also seen when white-coated breeds were compared against each other and when non-white-coated breeds were compared against each other (Table S9).
Teat number
Breeds that had a minimum breed standard of 14 teats were contrasted against breeds where 12 teats was the minimum breed standard. As a form of ‘control’, breeds with 14 teats were compared against one another and breeds with 12 teats were compared against one another. Outlying genomic regions from the 14 teat vs 12 teat comparison that did not overlap with those obtained from the ‘control’ analyses were found on 11 chromosomes (Figure S3 and Table S10). Only one region of several tightly clustered signals on SSC12 (26.83–26.96 Mb; 27.49–32.28 Mb) included genes that could be considered candidates for teat number.
European breeds versus wild boar
Levels of genetic differentiation were examined between the European pig breeds and wild boar (Table 1). None of the SNPs were found to be fixed for alternative alleles in the pig breeds and wild boar. The genome-wide distribution of FST for domestic pig breeds compared with wild boar is shown in Figure 3A. FST–windows falling into the 99th percentile were viewed as candidates of signatures of selection (Table S11) and contained some biologically interesting genes, as described below.

A genomic region on SSC1 showed high levels of differentiation (1.07–3.19 Mb, Table S11), homologous to a region of the canine genome associated with brachycephaly (broad and short skull shape) in dog breeds [31], [32]. This region contains, amongst seventeen characterised and uncharacterised genes, THBS2 (1.59–1.62 Mb) and SMOC2 (2.23–2.24 Mb), which were suggested as candidates for brachycephaly in the above-mentioned papers (Figure 3B). Pairwise FST–SNPs between wild boar and each breed in this region (48 SNPs) revealed maximum breed average FST values for Tamworth (0.42), Welsh (0.43) and Landrace (0.45), none of which have extremely brachycephalic skulls. A highly differentiated genomic region was also observed on SSC7 (31.30–38.89 Mb, Table S11). This region is close to the pig major histocompatibility complex: class I (∼24–26 Mb), class II (∼29 Mb) and class III (∼27 Mb). Within the differentiated region there are several genes of biological interest, including PPARD (36.14–36.22 Mb) (Figure 3C). Pairwise FST–SNPs (207) between wild boar and each breed in this region revealed highest breed average FST–SNPs in two commercial breeds, Duroc (0.50) and Landrace (0.37), and one traditional breed, Large Black (0.38); the minimum value of breed average FST was in Tamworth (0.09). Another interesting differentiation region observed between the domestic pigs and wild boar was on SSCX (Table S11). Amongst other genes, this region contained AR (60.31–60.50 Mb), the androgen receptor, previously suggested as a candidate gene for backfat thickness in pigs due to its proximity to mapped QTLs [33]. Other regions showing substantial differentiation between wild boar and pig breeds were found on SSC12, SSC13 and SSC14 but no clear candidate genes could be identified.
Signals of introgression from Asian pigs into European breeds
Consistent with previous studies [34], [35], genome-wide clustering results indicated substantial Asian ancestry for the European breeds. The clustering results indicated that the inferred ancestry of all Meishan individuals (a breed of Chinese origin, Table 1) to the first (“Asian”) cluster was high (92.3–93.9%). In contrast, the inferred ancestry of the European individuals to the second (“European”) cluster was lower (breed averages ranged from 69.6% for Large White up to 87.3% for Mangalica). With levels of ancestry varying across the genome, regions with particularly strong signals of Asian introgression into European breeds were identified according to two criteria: (1) high introgression probabilities (99th percentile) calculated by STRUCTURE software and (2) low differentiation based on FST (below the 1st percentile of individual European breeds versus Meishan) (Table S12). Two candidates of introgression overlapped with signals of selection associated with ear morphology. A genomic region on SSC5 (32–35 Mb), overlapping the region of differentiation detected when prick-eared breeds were contrasted with flat-eared breeds, was found in Gloucestershire Old Spots, Large Black and Mangalica (a signal of introgression in British Saddleback, the other flat-eared breed, was observed in this region only in the FST analysis). A genomic region on SSC7 (33–38 Mb), overlapping with one of the regions of differentiation detected when prick-eared breeds were contrasted with intermediate-eared breeds, was found in British Saddleback, Duroc, Landrace and Welsh. Another signal of introgression was detected on SSC11 (54–55 Mb) in Gloucestershire Old Spots, which overlapped with the differentiated region found in this breed and may be associated with coat pattern. The chromosome with the greatest number of regions showing evidence of Asian introgression was SSC14, where several regions overlapped across multiple breeds (81–85 Mb, eight breeds; 93–94 Mb, four breeds; 96–98 Mb, three breeds; 103–107 Mb, three breeds).
Sequencing of candidate regions
Based on the differentiation results, three genomic regions were further investigated using genome sequence data for 76 individuals from European and Asian breeds (Table S13).
SSC5 : 31.0–34.0 Mb
We identified 183 variants that were shared by the individuals from flat-eared breeds (British Saddleback, Gloucestershire Old Spots, Large Black and Mangalica) and differed from the individuals from prick-eared breeds (Berkshire, Hampshire, Large White, Middle White, Pietrain and Tamworth). All of these were either intergenic or intronic, with one located 504 bp upstream from a predicted precursor (ENSSSCG00000024846) of microRNA (miRNA) mir-584. However, no EST or RNA-seq evidence could be found in either ENSEMBL or NCBI gene expression data to suggest whether this SNP is located within the primary transcript of mir-584.
SSC5 : 98.0–99.0 Mb
Although the latest ENSEMBL annotation (release 69) predicted two genes in this 1 Mb region, a closer inspection showed that both are parts of the KIT-ligand gene (KITLG) but in opposite orientation, indicating probable errors or mis-assemblies here. We therefore blasted the porcine KITLG reference mRNA (NM_214269) [36] and an extended 5′-UTR (AB293552, [37]) sequences against the genome to first identify all the exons and the two flanking UTRs, before searching for variants within them.
A single SNP (C1089T), located on the 3′-UTR, was found in both Berkshire individuals but not in any other European breeds. In addition, the two Berkshires were found to harbour 11 other variants that were also present in one or more European breeds. Of these, two were non-synonymous (G548A, A919G) and the remaining nine were on the 5′ - or 3′ - UTR. The two non-synonymous SNPs resulted in R124K and T248A changes, respectively. The G548A variant was also found in three Pietrains (one a heterozygote) and one Tamworth individual. The A919G variant was also found in individuals of the following breeds: British Saddleback, Gloucestershire Old Spots, Large White, Mangalica, Middle White, Pietrain and Tamworth (two of these, a Pietrain and a Tamworth, also shared the G548 variant). We also examined the sequences of 24 individuals from eight Asian pig breeds and found that all three Jiangquhai individuals carried the C1089T found in the Berkshires, but none of the other Asian individuals carried this variant. The two non-synonymous variants were more common in the Asian than the European breeds: 16/24 Asian individuals carried both of them, compared to 3/50 of the European individuals (excluding the Berkshires).
SSC11 53.5–55.5 Mb
The analysed region encompasses 15 annotated genes (11 protein coding plus 4 non-coding RNA). We identified 474 variants in this region that were shared by the two Gloucestershire Old Spots individuals but differed from all other individuals in the European breeds. Of these, one was on the 3′ UTR of an uncharacterised protein-coding gene, three were synonymous variants (in the following genes: CLN5, MYCBP2 and KCTD12), and two variants resulted in non-synonymous changes (Table 3), both of which were found in the first exon of the endothelin receptor B (EDNRB) gene.

At residue 17 of EDNRB's signal peptide, the Gloucestershire Old Spots had a leucine (F17L), while the other individuals from European breeds carried a phenylalanine (Table 3, Figure 4). We also examined the EDNRB sequences for the Asian breeds and found three individuals (two Xiang, one Jiangquhai) that also carried the leucine, while the rest carried the phenylalanine. The Gloucestershire Old Spots leucine residue, however, was the most common among other mammalian reference genomes (e.g. mouse, cow, hedgehog and human) (Figure 4).

Within the N-terminal extracellular domain of EDNRB, the two Gloucestershire Old Spots individuals carried a phenylalanine at residue 68 (S68F), while a serine was found in the other individuals from European breeds (Table 3, Figure 4). One individual from the Asian Xiang breed also carried phenylalanine, while the other Asian individuals carried serine. There was substantial variability at this site in other mammalian reference proteins but none were found to carry phenylalanine.
Out of the 6928 variants in this region shared by the two Gloucestershire Old Spots, 897 were shared with all individuals from Asian breeds while only 29 were shared with all individuals from the other European breeds.
Discussion
Over the past 300 years, intense artificial selection in European pig breeds for production traits has led to the development of a number of pig breeds with well-defined, specialised phenotypic traits. In this study a number of regions showing between-breed signatures of selection have been identified. Various genes found within these regions can be considered as candidates under selection based on function or previous association with traits that are known to be favoured in pig breeds.
Breed standard traits (ear and coat colour)
Signatures of diversifying selection were found for traits related to morphological variation described by breeding criteria. Ear morphology is one trait that plays a major role in pig breed standards with strict conditions over ear form. By grouping breeds based on this phenotypic trait, the genome-wide scan suggested that the genetic basis of ear variation in pigs involves at least three genomic regions, located on SSC5 and SSC7. The region on SSC5 was associated with the difference between prick or intermediate ears and large, flat ears and the signals on SSC7 were associated primarily with the differences between prick - and intermediate-eared breeds. Our results from an introgression analysis also suggest that the SSC5 region of flat-eared breeds derives from Asian pigs.
The signatures of selection associated with ear morphology concurred with an earlier QTL study of the trait in pigs [38]. The SSC7 QTL of Wei et al [38] overlaps directly with the first differentiated region (31.82–34.19 Mb) on that chromosome. The suggestion that PPARD located on SSC7 plays a role in ear variation in pig breeds could not be supported as it was not positioned near either of the two signals of selection identified on this chromosome. However, as PPARD is involved in many biological processes and is located next to major QTLs for fat deposition and growth, its role in ear morphology warrants further investigation [39]. The QTL peak on SSC5 reported by Wei et al [38] is located approximately 10 Mb upstream of the peak FST signal but the confidence interval for the QTL location could overlap this position. Genome-wide association studies (GWAS) on ear morphology in dog breeds identified a region underlying this trait that was syntenic to the region on SSC5 in this study [19], [24]. Both these studies suggest MSRB3 and HGMA2 as candidate genes due to the proximity of the associated SNP. However, in the pig breeds the peak signal was located closer to LEMD3, which is involved in bone morphogenetic protein (BMP) signalling. Recently, a fine mapping study in pigs has suggested HMGA2 as a candidate locus for this QTL [40]. Mutations in the human version of this gene are associated with disorders involving increased bone density, suggesting a possible role in bone development [41]. However, analysis of coding sequences of these genes in this region of SSC5 for prick - and flat-eared pig breeds did not reveal any shared non-synonymous differences between the two groups, suggesting that changes in regulatory elements or miRNA genes may be responsible. Expression studies are required to test this hypothesis.
Like ear morphology, variation in coat colour patterns occurred post-domestication and signals of selection related to the traits indicate strong historic selection for the different phenotypes. Molecular studies have already identified the major coat colour loci in pigs, KIT and MC1R, for which allelic variation is associated with many of the coat colour variants (see references in [9], [17]). However, in this study signals of selection were not observed at or near MC1R (SSC6) for individual breeds that have an allele associated with a particular coat colour or when breeds were grouped by coat colour traits. The other locus, KIT (SSC8), is found ∼1 Mb downstream from a differentiated region shared by three breeds (British Saddleback, Hampshire, Pietrain). Several possible explanations could account for weak and absent signals of diversifying selection at KIT and MC1R, respectively. The differentiated region on SSC8 was quite extensive in genomic size and KIT may have been one of several targets of selection in that region, thus dampening any KIT-specific signals. Furthermore, allelic variation at both KIT and MC1R is associated with a large variety of coat colours and patterns for many breeds. With the breed set analysed in this study, there is no simple dichotomous division of the breeds based on coat type for these two genes, which could have weakened the power of this approach. Lastly, the inter-SNP distances in the MC1R region of SSC6 were particularly high (the distance between the flanking markers was in the 99th percentile of the genome-wide distribution of inter-SNP distances). Thus it appears that the MC1R region was not well covered by the PorcineSNP60 chip, which may explain why no signals of diversifying selection were detected there.
In contrast to the weak or absent signals of selection at the two major coat colour loci, KIT and MC1R, strong breed-specific signals of diversifying selection were observed near other coat colour loci. Two non-synonymous mutations were found in the endothelin receptor B (EDNRB) gene, in a region exhibiting substantial differentiation unique to Gloucestershire Old Spots. EDNRB encodes a G protein-coupled receptor that binds to the different isoforms of endothelins. The EDNRB-endothelin interaction plays a role in a range of critical physiological processes including the formation of enteric nerves and melanocytes (pigment-producing cells), both of which are neural crest derivatives [42], [43].
Mutations in EDNRB, leading to a reduced expression of the gene and partial or complete loss-of-function, have been shown to be associated with changes in pigmentation due to its role in melanocyte development [43], [44]. The piebald phenotype in mouse, characterised by white coat spotting [43], results from the insertion of a large retrotransposon in the first intron of EDNRB [45]. Several different mutations in humans are associated with a loss of pigmentation in the hair, skin and iris (Hirschsprung's disease/Waardenburg syndrome) [43] while a missense mutation gives rise to the Lethal White Foal Syndrome [46], where homozygous foals are completely white (and die early due to intestinal blockage) while heterozygous animals usually have distinctive white patches.
The mechanism(s) by which point mutations in EDNRB could be associated with (partial) loss of function is not yet known. The amino acid changes at residues 64 (Jinhua) and 68 (Gloucestershire Old Spots and Xiang) are both located in the N-terminal extracellular domain of the protein. One of the non-synonymous EDNRB mutations associated with Hirschsprung's disease is located in the same domain, at residue 57. This domain has been suggested to be important for stable ligand binding [47]–[49]. Furthermore, human EDNRB is believed to be cleaved by a metalloprotease at R64|S65 (R65|S66 in pig) and a truncated EDNRB (missing the first 64 residues) was found to be functional but had significantly reduced cell surface expression [50]. Using a program that predicts cleavage sites by membrane-type metalloproteases (SitePrediction, [51]), the reference pig EDNRB with S68 was, like its human homologue, found more likely to be cleaved at the R65|S66 site than the Gloucestershire Old Spots protein with F68 (unpublished results). The SNPs that alter residues 64 and/or 68 may result in an incomplete or uncleaved EDNRB and hence altered expression on the cell surface.
Black spotting in the Gloucestershire Old Spots has been previously associated with the EP allele at the melanocortin receptor 1 (MC1R locus): a 2-bp insertion in MC1R causes a frameshift mutation which results in a premature stop codon further downstream [15]. That study also demonstrated irregular somatic reversion to the black form of MC1R in two spotted breeds, Pietrain and Linderod, such that some regions of the body (black spots) expressed the form of the protein that enables black pigment production, whereas other (white) regions mainly expressed the mutated (non-functional) form of the protein. However, as breeds with various spotted and non-spotted patterns carry the 2-bp insertion, it is likely that additional loci also influence coat pattern and colour in these breeds. A recent paper demonstrated the complex interactions between melanocortin and endothelin signalling in determining coat patterns in cats [52] and similar interactions may also influence coat pattern diversity in pigs. We propose that the variant MC1R, resulting from the 2-bp insertion (and somatic reversion), may interact with partial loss of function in EDNRB such that only part of the body is populated by melanocytes which have the potential to revert and become pigmented. This in turn could give the Gloucestershire Old Spots its characteristic spotting pattern of relatively few and small spots compared to those observed, for example, in Pietrain. Functional analyses are required to characterize the effects of the Gloucestershire Old Spots variants on EDNRB function and on pigmentation patterns.
Although the variants at EDNRB were unique to the Gloucestershire Old Spots in the analysis of European breeds, they were shared by the Asian breed Xiang. We do not have phenotypic information for the Xiang individual who shares the Gloucestershire Old Spots variants but one of the most common Xiang subtypes is two-end black with a white middle body, akin to the familiar piebald mouse (http://www.viarural.com.pe/ganaderia/a-porcinos/exteriorcerdos/paises/china.htm). The Jinhua breed, which carries a proline to serine change at nearby residue 64 (Figure 4; [53]), has a similar phenotype. The difference in the phenotypes between the Asian breeds and Gloucestershire Old Spots is likely to be related to their different MC1R genotypes. The Asian breeds with EDNRB mutations do not carry the MC1R insertion (unpublished results), consistent with previous studies that show a low frequency or absence of this allele in Asian pigs [54], [55]. The two Gloucestershire Old Spots individuals are substantially more similar to the Asian breeds than the European ones in the EDNRB region. This finding, the shared EDNRB genotypes of Gloucestershire Old Spots and Xiang, and the introgression results described above together suggest an Asian origin for the Gloucestershire Old Spots mutations.
A putatively selected region identified in the Berkshire breed includes the KITLG locus and further sequence analysis revealed several non-synonymous variants in this breed. KITLG binds to the KIT receptor and plays a role in the melanocyte production pathway. Variation at the locus has been implicated in different skin pigmentation phenotypes in mice (i.e. steel mutant) [44], [56] and humans [57], [58], including hypo - and hyper-pigmentation, and has been investigated previously for its role in pig colouration [59]. The breed standard for Berkshire is a black animal with six white points (on the snout, tip of the tail and tips of each of the legs). The Berkshire was allegedly highly variable in coat colour until introgression of Asian genetic material and selection for breed homogeneity led to its contemporary coat pattern [5], [6]. Our tests using PorcineSNP60 data did not detect evidence of Asian introgression for Berkshire in the KITLG region (as assessed using comparisons with Meishan), although Berkshire shared the C1089T variant with Jiangquhai, another Asian breed, but not with any other European or Asian individuals. Furthermore, the two non-synonymous variants found in Berkshire were more common in the Asian than the European breeds. Similarly, Okumura and colleagues [37], [60] found evidence for an Asian origin of KITLG in Berkshire, as the breed shared haplotypes similar to Asian breeds at the locus whilst differing from other European breeds.
We identified the same two non-synonymous variants (A919G, G458A) in Berkshire and several Asian breeds as Okumura and colleagues [37], [60]. However, these variants cannot on their own explain the Berkshire phenotype because they were also found in three European individuals, including a Pietrain and a Tamworth (both homozygous), the latter breed which is red. Alternatively, the Berkshire phenotype might be attributed to differential regulation of KITLG, in conjunction with variation at other pigmentation genes (e.g. MC1R—Berkshire also carries the black spotting allele discussed above—and KIT). This could be related to the C1089T 3′-UTR variant that was only seen in Berkshire and Jiangquhai (also a black breed) or another regulatory element. Cis-regulatory differences in KITLG expression have been associated with pigmentation differences in stickleback fish [61] and a SNP located 350 Kb upstream of the KITLG gene was found to be associated with human hair colour, suggesting a possible regulatory role [62]. However, we were unable to search for variants in either proximal or distant enhancer/repressor elements due to errors in this region of the current pig genome assembly.
Pig production traits
Signatures of diversifying selection were found that may be associated with important pig production traits. Teat number is an important reproductive trait because with increased litter size, which is often selected for in pig breeds, a sufficient number of teats are required to support the litter [13]. Although the FST teat-trait analysis results had some ambiguity, the signal on SSC12 seen in the 14 vs 12 teats comparison but not the ‘control’ comparison (breeds with 14 teats compared with one another and breeds with 12 teats compared with one another) overlapped with documented QTL. Both Hirooka et al [63] and Rodriguez et al [64] reported a significant QTL for teat number on this chromosome, with the latter study suggesting that the most likely position of the QTL was between markers SW874 (23.67 Mb) and SW1956 (40.77 Mb), which overlapped with the region of high differentiation observed in the current study. The NME1 gene, which is found in this region (27.46–27.50 Mb), plays a role in mammary gland development. NME1-deficient mice, although they reproduce normally, have delayed mammary gland development [65] and incomplete maturation of the lactiferous duct in the nipple [66].
Amongst the production characteristics that commercial pig breeds share, they also possess breed-specific characteristics. Duroc pigs are known for their high intramuscular fat content (IMF) in comparison to other commercial pig breeds [67] and for their higher concentrations of saturated and mono-unsaturated fatty acids (and lower concentrations of poly-unsaturated fatty acids) [68], characteristics that play key roles in meat quality. Uemoto et al [27] found a significant QTL for fatty acid composition in Duroc on SSC14 that has not been reported for other breeds. This QTL region overlaps with an extreme differentiation region observed only in the Duroc breed and contains ELOVL3, a gene involved in the synthesis of fatty acids; in mice a lack of ELOVL3 resulted in decreased levels of certain fatty acids due to an inability to convert saturated fatty acyl-CoAs into very long chain fatty acids [28]. In addition, SCD (stearoyl-CoA desaturase), a gene located close to the peak differentiation region, encodes a key enzyme in the synthesis of fatty acids and has thus been proposed as a candidate gene for the fatty acid composition QTL [27].
Landrace also exhibited high levels of differentiation, in this case in an extended region of SSC13. The peak differentiation values were found close to the grehlin (GHRL) gene, which is a candidate for associations with appetite and feeding behaviour. The regulation of voluntary food intake is controlled by a biological cascade of chemical signals that controls appetite and satiation, where various hormones are involved in the starting and/or termination of an eating episode. Grehlin has been specifically proposed in prompting hunger feelings and therefore initiating eating [69]. Its involvement in regulating feeding behaviour in pigs has only recently been considered [70].
Genetic signatures underlying domestication
By comparing pig breeds with their ancestral species, the wild boar, we sought to identify genomic regions and genes that could be involved in the domestication process. The largest differentiated genomic region between the domestic pig breeds and wild boar was observed on SSC7. Numerous QTLs have previously been mapped to this chromosome for traits such as growth, carcass length, skeletal morphology and backfat depth using several types of crosses [11], [12]. Several genes located in the region of differentiation have been investigated for possible physiological roles: PPARD and CDKN1A have been considered candidates for fat deposition [71] and, as mentioned above, PPARD has also been considered a candidate gene for ear structure variation [39]. In addition, the genomic signal of selection is close to the MHC region, a complex that is crucial in vertebrate immunity, making it a potential source of evolutionary change on the chromosome. The large differentiated region on SSC7 may reflect strong diversifying selection in domestic pig breeds as this chromosome appears to influence many pig production traits.
Domestic pig breeds are also different from wild boar in skeletal morphology. Substantial changes have occurred in the body and cranial dimensions following domestication [72]. In the comparison of pig breeds with wild boar, a region of genetic differentiation identified on SSC1 is syntenic to a region associated with cranial dimensions in dog breeds [32]. The cranial trait under investigation in the dog studies, brachycephaly, is characterised by a strong alteration of the facial bone structure through shortening of the muzzle and shortening and widening of the skull [31]. Pig breeds possess variable skull morphology ranging from a long snout (Tamworth) to shorter wider faces (Berkshire, Gloucestershire Old Spots, Large Black) to very short faces with upturned snouts, similar to brachycephaly in dogs (Middle White) (see Figure S1). However, Middle White, the most brachycephalic-like breed, did not show significant differentiation from wild boar in the SSC1 region. Incidentally, it has been suggested that Middle White acquired its ‘dished’ face from Asian pigs [6]. However, there was no evidence of Asian introgression into the Middle White in the regions orthologous to the dog brachycephaly regions, suggesting that if it did indeed acquire its squashed face from Asian pigs, there has been independent evolution for this trait in dogs and pigs. As various skeletal and cranial changes occurred after domestication of the wild boar [72], the region of high differentiation overlapping the brachycephaly region in dogs could be associated with other bone alterations.
Evolutionary perspectives on the development of pig breeds
The putative genomic signatures of selection for breed-defining phenotypic traits and levels of breed genetic differentiation reflect the historical development of the pig breeds. The Duroc had the strongest signals of diversifying selection, evidenced by the levels of genomic differentiation, which were observed to be unique to this breed and unlike the other breeds, no signals of diversifying selection were observed on SSC8 for the Duroc, indicating that this breed may have a distinct genetic origin, as previously noted from microsatellite and sequence data [35], [73]. Some of the clearest signals of both diversifying selection and introgression from Asian pigs were associated with highly visible phenotypes such as coat pattern and ear morphology, suggesting that these traits have been under particularly strong selection during the development of European pig breeds. In particular, selection associated with flat ears was detected in breeds that do not appear to share recent ancestry [7], [73], which may reflect convergent evolution through independent selection for that trait. In contrast, although microsatellite markers indicate a common ancestry for Berkshire and Gloucestershire Old Spots [7], [73], shared differentiation signals were not seen, illustrating differing breed development trajectories. Signatures of selection were also observed in regions associated with certain quantitative traits in pig production, but there was a paucity of signals at loci associated with those related to reproduction. The lack of differentiation signals associated with such traits may reflect their control by many genes of small effect, as suggested by Boyko and colleagues [19].
The genomic regions identified in this study using the genetic differentiation approach generally did not overlap with those identified in a scan for extreme homozygosity in European pig breeds: none of the regions identified in five or more breeds overlapped with the regions reported by Rubin and colleagues [25] and only two out of 109 regions identified in individual breeds overlapped (SSC1 : 172.13 Mb and SSC15 : 115.17–115.77 Mb). The Rubin study used more dense genomic data so it is possible that the Porcine SNP60 chip did not contain variants close to the regions they identified. However, in our study we have detected what appear to be genuine signals of selection in pig breed development. Another explanation for the lack of overlap between the studies is that, by pooling genomic data across several breeds, Rubin and colleagues [25] identified regions of homozygosity that were shared amongst the breeds, arguably picking out candidates more likely to be involved in the domestication process and early, post-domestication pig development. In contrast, our methodological approach searched for between-breed differences, thus revealing candidates arising from diversifying selection that occurred during breed development.
Materials and Methods
Ethics statement
DNA samples were obtained from blood samples collected by veterinarians according to national legislation, from tissue samples from animals obtained from the slaughterhouse or, in the case of wild boar, from animals culled within wildlife management programs.
DNA samples, SNP genotyping, and data preparation
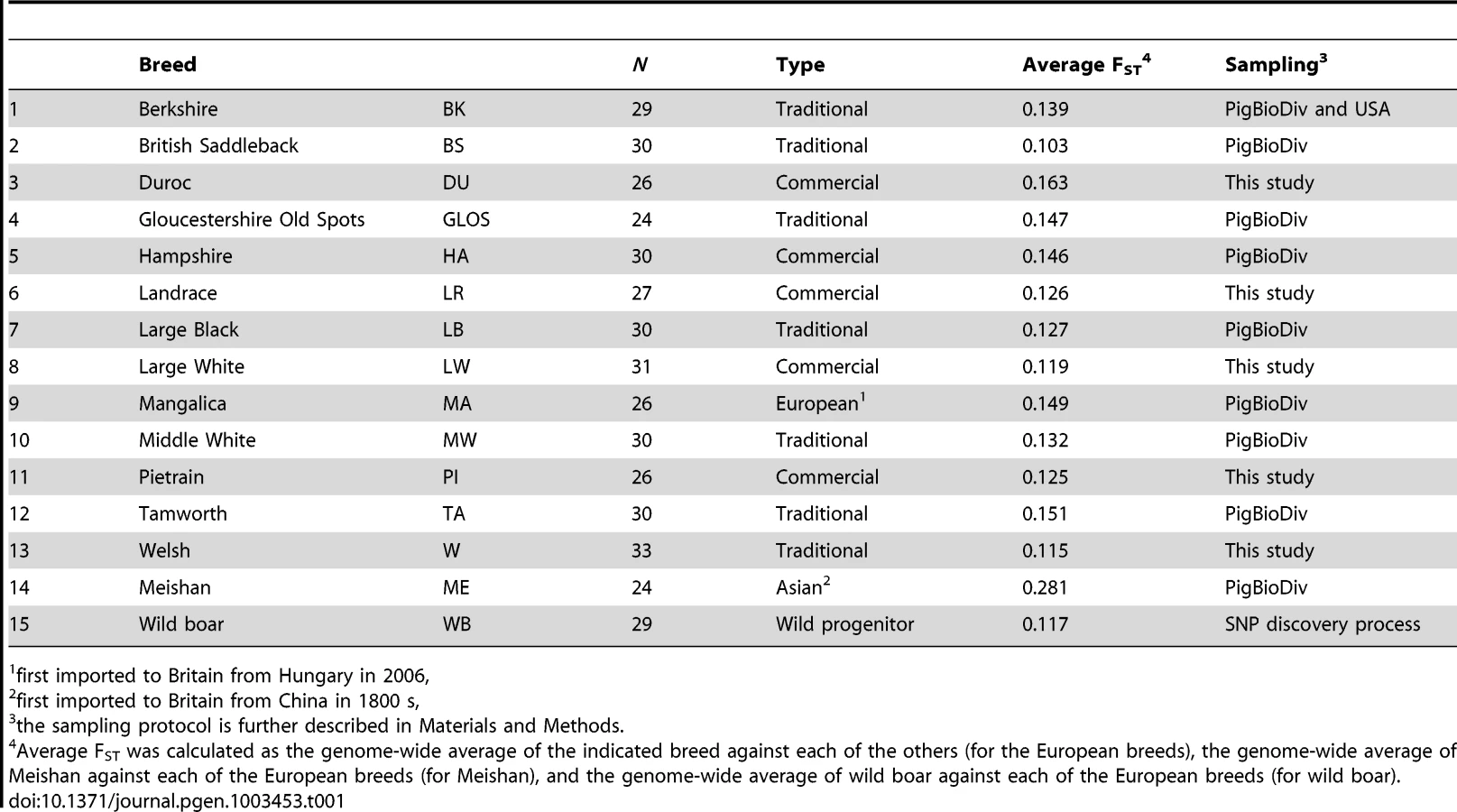
DNA samples were obtained from blood samples collected by veterinarians according to national legislation, from tissue samples from animals obtained from the slaughterhouse or in the case of wild boar, from animals culled within wildlife management programs. Samples for SNP genotyping were obtained from between 24 and 34 individuals for 14 pig breeds, described in Table 1, and were genotyped using the PorcineSNP60 chip assay [74]. Most breed samples (including the Asian breed, Meishan) were from the PigBioDiv study whereby a maximum of two individuals were sampled from a litter from as many herds as possible, so as to have as few related individuals as possible in the sample set [75]. For the four commercial breeds (Duroc, Landrace, Large White and Pietrain), the data was from individual commercial populations, which were found to be good representatives of the breeds based on clustering analysis of multiple populations (unpublished results). Welsh samples were provided by the Pedigree Welsh Pig Society. Wild boar samples were those used in the original SNP discovery procedure [74]. Genotype data are deposited in the Dryad repository (http://dx.doi.org/10.5061/dryad.c2124).
All analyses were carried out in R ([76], http://www.r-project.org/). A series of quality control measures were applied to the dataset to filter out any possible genotyping anomalies. First, SNP markers that had greater than 10% missing genotypes were discarded. Second, markers that were monomorphic across all the breeds (i.e. MAF<0.01) were also discarded from further analysis. Third, SNP markers were tested for deviations from Hardy-Weinberg equilibrium within each breed using an exact test [77]. At a critical rejection region of 8.33×10−7 (0.05/60,000) a total of 66 SNPs did not conform to HWE expectations in one or more breeds and were removed from the analysis. Of these, 46 deviated from HWE due to excess of heterozygote genotypes in one or more breeds. The other 20 SNPs deviated from HWE due to heterozygote deficit in one or more breeds. Fourth, markers that were not mapped to the porcine genome were removed, based on the current pig genome assembly, Sus scrofa (SSC) Build 10.2. For the remaining markers, SNPs that were not yet mapped to a specific location on a specific chromosome of the pig genome were also filtered out. Following quality control, 49 260 markers were considered for the majority of analyses (see below for one exception). After QC, average individual genotype coverage was 99.20% across all breeds and average individual genotype coverage in individual breeds ranged from 96.09% in the Mangalica breed to 99.96% in the Hampshire breed.
Statistical analysis
Pairwise Wright's FST [78], the classical measure of population genetic differentiation, was used to detect signatures of diversifying selection. We previously showed [79] that pairwise measures of differentiation were better at identifying markers that distinguished breeds than global measures and that Wright's estimate of FST was highly correlated to that of Weir & Cockerham's [80]. The use of population (breed) differentiation to identify candidate selected regions, as implemented in the current study, was originally suggested by Akey and colleagues [81]. This approach was justified by use of simulations in a follow-up study on dogs [18] and has subsequently been implemented in various empirical studies [22], [24], [82].
The PorcineSNP60 chip assay was designed to include SNPs evenly distributed across the genome, with per-chromosome average inter-SNP distances ranging from 30 to 40 kb (except for SSCX) (based on builds 7 and 8) [74], with a median of 30 kb for the genome-wide distribution. Across the genome, the majority (80%) of inter-SNP distances were less than 70 kb in this study. Recent studies (e.g. Ref. [83]) show high linkage disequilibrium across commercial pig genomes (r2∼0.4 between adjacent SNPs on the PorcineSNP60 chip), suggesting that our study is likely to detect most signals. To account for stochasticity in locus-by-locus variation, for all of the FST analyses a 13-SNP sliding window was implemented on the estimated values, with the mid-SNP determining the genomic location of the window (hereafter designated as FST-window). To allow 13-SNP sliding windows across a whole chromosome, the first window on a chromosome was centred at the 7th SNP position and the last window on a chromosome was centred at the 7th from last SNP position. Candidate selected regions were defined as the 99th percentile of the empirical distributions of FST-windows, except where indicated otherwise.
Individual pig breeds
A breed average FST was first calculated. FST was estimated between pairs of European breeds at each SNP marker using the breed allele frequencies. For each breed this produced 12 breed-pairwise FST comparisons at each SNP marker. The FST at each SNP marker for all of these pairwise comparisons were averaged to produce an overall FST for each SNP marker in each breed (here after designated as FST-SNP).
Phenotypic traits
The FST analysis was extended to compare groups with different phenotypic traits. For each trait classes were formed, based on the observed phenotypic variation between breeds (see below), and breeds were placed into one of the classes. For each trait, FST was estimated between each breed in one class compared against each breed in the next class and averaged across the pairwise comparisons to obtain a FST-SNP estimate. A summary table of the different traits, the phenotypic classes and the class designation of each breed is shown in Table S2.
Ear morphology in European pigs is variable, ranging from upright or prick ears that may be slightly inclined forwards (the ancestral state as seen in wild boar), to a medium sized ear that points forwards and downwards but is not too heavy, to a completely dropped ear that is long, thin and lies relatively flat against the face slightly curbing vision of the animal (see Figure S2). Ear morphology was grouped into the following classes: prick-eared breeds, intermediate-eared breeds and flat-eared breeds.
Coat colour in European pigs is a highly variable phenotypic trait including from black, red, brown and white, with and without spots and belts. The coat colour was grouped into the following classes: red coat breeds compared with non-red coat breeds; saddleback breeds compared with non-saddleback breeds; white coat breeds compared with non-white coat breeds; red coat breeds compared with black coat breeds.
Amongst the breed standard requirements set by the British Pig Association (BPA), the number of teats is one listed criterion. Using this trait, breeds were grouped in the following classes: breeds where the BPA standards required a minimum of 14 displayed teats compared with breeds where the BPA standards required a minimum of 12 displayed teats, Berkshire and Middle White were removed from this trait comparison because there was not a definitive breed standard requirement (breed standards suggested a “minimum of 12 but preferably 14 teats”) and Mangalica was also removed because the breed standard number of teats was unknown.
Pig breeds versus wild boar
Levels of genetic differentiation between the domestic pig breeds and wild boar were estimated. The SNPs that were monomorphic in the pig breeds were compared with wild boar genotypes to determine if some were segregating in the wild boar. The (mapped) breed-monomorphic SNPs that were segregating in the wild boar were added to the set of polymorphic SNPs described above, giving a total of 49 556 markers. FST was estimated between wild boar and each pig breed, which produced 13 pairwise comparisons at each SNP marker. The FST at each SNP marker for each of these pairwise comparisons were averaged to produce an overall FST for each SNP marker (here after designated as FST-SNP).
Signals of Asian introgression into European breeds
Two methods were employed to infer signals of Asian introgression in European breeds. First, an FST analysis, as described above, was used to quantify differentiation between the Asian Meishan breed and each of the 13 European breeds. Regions of particularly low differentiation (below 1st percentile) were interpreted as showing evidence of Asian introgression.
Second, a Bayesian analysis was performed using the site-by-site linkage model in STRUCTURE software [84]. This model was designed to infer the ‘population-of-origin’ assignment of genomic regions and has been used to determine levels of introgression between populations (e.g. Ref. [85]). Each of the 13 European breeds was compared with the Meishan breed, using no a priori population information: at a pre-defined number of clusters, K = 2, the linkage model was run five times for 20,000 iterations after a burn-in of 40,000 iterations (which included 20,000 iterations with the admixture model). Due to computer memory limitations, for the analysis 15 individuals per breed (approximately half of the total dataset) were chosen at random and every second marker across each chromosome was removed from the input data set leaving a total of 24 630 markers.
Ancestry proportions across the two clusters (“Asian” and “European”) were estimated for each of the European individuals. Estimates of Asian ancestry for each European animal for each SNP were obtained from the probability of assignment to the Asian cluster and then averaged across the individuals within each breed. As described above, a sliding window average of Asian ancestry values across each chromosome was calculated, with windows composed of 7 SNPs (half the number used for the analyses of the full set of SNPs). The average value for the window was assigned to the position of the central SNP. These values were interpreted as probabilities of introgression from Asian to European breeds.
In order to identify genomic regions with clear signals of Asian introgression, we identified SNP positions (to the closest Mb) that met two criteria: (1) values below the 1st percentile of the Meishan-European breed FST-windows distribution and (2) found in the 99th percentile of STRUCTURE-calculated introgression probabilities for that breed.
Sequencing strategy
DNA samples for sequencing were obtained as described above for SNP genotyping. Individual samples (52) from 12 of the European breeds analysed above (no Welsh pigs were included) as well as 24 samples from eight Asian breeds (Table S13) were sequenced using the Illumina HiSeq2000 platform, with library preparation and sequence generation per manufacturers protocols. Sequence mapping and variant calling were carried out as described previously [25], [34]. Briefly, Illumina (v. 1.3–1.8) formatted fastq files, with sequence reads of 100 bp were subject to quality trimming prior to sequence alignment. The trimming strategy involved a 3 bp sliding window, running from 5′ to 3′, with sequence data upstream being discarded if the 3 bp window average quality dropped below 13 (i.e. average error probability equal to 0.05). Only sequences of 45 bp or more in length were retained. In addition, sequences with mates <45 bp after trimming were also discarded. During trimming, quality scores were re-coded to follow the Sanger fastq format to standardize downstream processing.
Sequences were aligned against the Sscrofa10.2 reference genome using Mosaik 1.1.0017. Alignment was performed using a hash-size of 15, with a maximum of 10 matches retained, and 7% maximum mismatch score, for all pig populations and outgroup species. Alignment files were then sorted using the MosaikSort function, which entails removing ambiguously mapped reads that are either orphaned or fall outside a computed insert-size distribution. Alignment archives were converted to BAM format using the Mosaiktext function. Manipulations of BAM files, such as merging of alignments archives pertaining the same individual, were conducted using SAMtools v. 1.12a [86].
Variant allele calling was performed per individual using the pileup function in SAMtools, and variations were initially filtered to have minimum quality of 50 for indels, and 20 for SNPs. In addition, all variants showing higher than 3x the average read density, estimated from the number of raw sequence reads, were also discarded to remove false positive variant calling originating from off-site mapping as much as possible. Heterozygous variants and those with minimal SNP/indel qualities were further inspected manually to ensure that they were true variants.
We examined the sequence variation in three genomic regions that showed extreme differentiation in one or more breeds (Table S1) for the individuals from the 12 European breeds: (1) SSC5 : 31.0–34.0, (2) SSC5 : 98.0–99.0 and (3) SSC11 : 53.5–55.5 Mb. Information for the relevant regions was excised from the BAM files using SAMtools v. 1.12a [86]. Alignment files and variants called in these regions for all animals considered in this manuscript are deposited in the Dryad repository (http://dx.doi.org/10.5061/dryad.c2124). For the first region, we identified all variants that were shared by the individuals from flat-eared breeds but differed from all individuals from the prick-eared breeds (Table S2); for the second region, we identified all variants that were shared by the two Berkshire individuals but differed from the other individuals; and for the third region, we identified all variants that were shared by the two Gloucestershire Old Spots individuals but differed from the other individuals. Data for the individuals from Asian breeds was then used to examine specific sequence variants, as described in the Results.
Supporting Information
Zdroje
1. LarsonG, AlbarellaU, DobneyK, Rowley-ConwyP, SchiblerJ, et al. (2007) Ancient DNA, pig domestication, and the spread of the Neolithic into Europe. Proceedings of the National Academy of Sciences 104 : 15276–15281.
2. LarsonG, DobneyK, AlbarellaU, FangMY, Matisoo-SmithE, et al. (2005) Worldwide phylogeography of wild boar reveals multiple centers of pig domestication. Science 307 : 1618–1621.
3. FAO (2007) The State of the World's Animal Genetic Resources for Food and Agriculture – in brief. Commission on Genetic Resources for Food and Agriculture: FAO, Rome, Italy.
4. Darwin C (1868) The Variation of Animals and Plants under Domestication. London: John Murray.
5. BPA (2002) British Pig Breeds. Trumpington, Cambridge: British Pig Association.
6. Porter V (1993) Pigs. A Handbook to the Breeds of the World. Mountfield, East Sussex: Helm Information, Ltd.
7. WilkinsonS, HaleyC, AldersonL, WienerP (2011) An empirical assessment of individual-based population genetic statistical techniques: application to British pig breeds. Heredity 106 : 261–269.
8. AnderssonL (2001) Genetic dissection of phenotypic diversity in farm animals. Nature Reviews Genetics 2 : 130–138.
9. AnderssonL, GeorgesM (2004) Domestic-animal genomics: deciphering the genetics of complex traits. Nat Rev Genet 5 : 202–212.
10. Van LaereA-S, NguyenM, BraunschweigM, NezerC, ColletteC, et al. (2003) A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature 425 : 832–836.
11. AnderssonL, HaleyCS, EllegrenH, KnottSA, JohanssonM, et al. (1994) Genetic-mapping of quantitative trait loci for growth and fatness in pigs. Science 263 : 1771–1774.
12. KnottSA, MarklundL, HaleyCS, AnderssonK, DaviesW, et al. (1998) Multiple marker mapping of quantitative trait loci in a cross between outbred wild boar and large white pigs. Genetics 149 : 1069–1080.
13. BuskeB, SternsteinI, BrockmannG (2006) QTL and candidate genes for fecundity in sows. Animal Reproduction Science 95 : 167–183.
14. GiuffraE, EvansG, TörnstenA, WalesR, DayA, et al. (1999) The Belt mutation in pigs is an allele at the Dominant white locus. Mammalian Genome 10 : 1132–1136.
15. KijasJMH, MollerM, PlastowG, AnderssonL (2001) A frameshift mutation in MC1R and a high frequency of somatic reversions cause black spotting in pigs. Genetics 158 : 779–785.
16. KijasJMH, WalesR, TörnstenA, ChardonP, MollerM, et al. (1998) Melanocortin receptor 1 (MC1R) mutations and coat color in pigs. Genetics 150 : 1177–1185.
17. WienerP, WilkinsonS (2011) Deciphering the genetic basis of animal domestication. Proceedings of the Royal Society B: Biological Sciences 278 : 3161–3170.
18. AkeyJM, RuheAL, AkeyDT, WongAK, ConnellyCF, et al. (2010) Tracking footprints of artificial selection in the dog genome. Proceedings of the National Academy of Sciences 107 : 1160–1165.
19. BoykoAR, QuignonP, LiL, SchoenebeckJJ, DegenhardtJD, et al. (2010) A simple genetic architecture underlies morphological variation in dogs. PLoS Biol 8: e1000451 doi:10.1371/journal.pbio.1000451.
20. FloriL, FritzS, JaffrezicF, BoussahaM, GutI, et al. (2009) The genome response to artificial selection: a case study in dairy cattle. PLoS ONE 4: e6595 doi:10.1371/journal.pone.0006595.
21. GibbsRA, TaylorJF, Van TassellCP, BarendseW, EversoieKA, et al. (2009) Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 324 : 528–532.
22. KijasJW, LenstraJA, HayesB, BoitardS, Porto NetoLR, et al. (2012) Genome-wide analysis of the world's sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol 10: e1001258 doi:10.1371/journal.pbio.1001258.
23. QanbariS, PimentelECG, TetensJ, ThallerG, LichtnerP, et al. (2010) A genome-wide scan for signatures of recent selection in Holstein cattle. Animal Genetics 41 : 377–389.
24. VaysseA, RatnakumarA, DerrienT, AxelssonE, Rosengren PielbergG, et al. (2011) Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLoS Genet 7: e1002316 doi:10.1371/journal.pgen.1002316.
25. RubinC-J, MegensH-J, Martinez BarrioA, MaqboolK, SayyabS, et al. (2012) Strong signatures of selection in the domestic pig genome. Proceedings of the National Academy of Sciences of the United States of America 109 : 19529–19536.
26. SanchezM-P, IannuccelliN, BassoB, BidanelJ-P, BillonY, et al. (2007) Identification of QTL with effects on intramuscular fat content and fatty acid composition in a Duroc x Large White cross. BMC Genetics 8 : 55.
27. UemotoY, SomaY, SatoS, IshidaM, ShibataT, et al. (2012) Genome-wide mapping for fatty acid composition and melting point of fat in a purebred Duroc pig population. Animal Genetics 43 : 27–34.
28. WesterbergR, ManssonJE, GolozoubovaV, ShabalinaIG, BacklundEC, et al. (2006) ELOVL3 is an important component for early onset of lipid recruitment in brown adipose tissue. Journal of Biological Chemistry 281 : 4958–4968.
29. OnteruSK, FanB, DuZQ, GarrickDJ, StalderKJ, et al. (2012) A whole-genome association study for pig reproductive traits. Animal Genetics 43 : 18–26.
30. CieslakM, ReissmannM, HofreiterM, LudwigA (2011) Colours of domestication. Biological Reviews 86 : 885–899.
31. BannaschD, YoungA, MyersJ, TruveK, DickinsonP, et al. (2010) Localization of canine brachycephaly using an across breed mapping approach. PLoS ONE 5: e9632 doi:10.1371/journal.pone.0009632.
32. QuilezJ, ShortAD, MartinezV, KennedyLJ, OllierW, et al. (2011) A selective sweep of >8 Mb on chromosome 26 in the Boxer genome. BMC Genomics 12 : 339.
33. HarliziusB, RattinkAP, de KoningDJ, FaivreM, JoostenRG, et al. (2000) The X Chromosome harbors quantitative trait loci for backfat thickness and intramuscular fat content in pigs. Mammalian Genome 11 : 800–802.
34. GroenenMAM, ArchibaldAL, UenishiH, TuggleCK, TakeuchiY, et al. (2012) Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491 : 393–398.
35. AmaralAJ, FerrettiL, MegensH-J, CrooijmansRPMA, NieH, et al. (2011) Genome-wide footprints of pig domestication and selection revealed through massive parallel sequencing of pooled DNA. PLoS ONE 6: e14782 doi:10.1371/journal.pone.0014782.
36. ZhangZ, AnthonyRV (1994) Porcine stem-cell factor/c-kit ligand: its molecular-cloning and localization within the uterus. Biology of Reproduction 50 : 95–102.
37. OkumuraN, MatsumotoT, HamasimaN, AwataT (2008) Single nucleotide polymorphisms of the KIT and KITLG genes in pigs. Animal Science Journal 79 : 303–313.
38. WeiWH, de KoningDJ, PenmanJC, FinlaysonHA, ArchibaldAL, et al. (2007) QTL modulating ear size and erectness in pigs. Animal Genetics 38 : 222–226.
39. RenJ, DuanY, QiaoR, YaoF, ZhangZ, et al. (2011) A missense mutation in PPARD causes a major QTL effect on ear size in pigs. PLoS Genet 7: e1002043 doi:10.1371/journal.pgen.1002043.
40. LiP, XiaoS, WeiN, ZhangZ, HuangR, et al. (2012) Fine mapping of a QTL for ear size on porcine chromosome 5 and identification of high mobility group AT-hook 2 (HMGA2) as a positional candidate gene. Genetics Selection Evolution 44 : 6.
41. HellemansJ, PreobrazhenskaO, WillaertA, DebeerP, VerdonkPCM, et al. (2004) Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat Genet 36 : 1213–1218.
42. BarshGS (1996) The genetics of pigmentation: From fancy genes to complex traits. Trends in Genetics 12 : 299–305.
43. JacksonIJ (1997) Homologous pigmentation mutations in human, mouse and other model organisms. Human Molecular Genetics 6 : 1613–1624.
44. BaxterLL, HouL, LoftusSK, PavanWJ (2004) Spotlight on spotted mice: A review of white spotting mouse mutants and associated human pigmentation disorders. Pigment Cell Research 17 : 215–224.
45. YamadaT, OhtaniS, SakuraiT, TsujiT, KuniedaT, et al. (2006) Reduced expression of the endothelin receptor type B gene in piebald mice caused by insertion of a retroposon-like element in intron 1. Journal of Biological Chemistry 281 : 10799–10807.
46. SantschiEM, PurdyAK, ValbergSJ, VrotsosPD, KaeseH, et al. (1998) Endothelin receptor B polymorphism associated with lethal white foal syndrome in horses. Mammalian Genome 9 : 306–309.
47. FuchsS, AmielJ, ClaudelS, LyonnetS, CorvolP, et al. (2001) Functional characterization of three mutations of the endothelin B receptor gene in patients with Hirschsprung's disease: Evidence for selective loss of G(i) coupling. Molecular Medicine 7 : 115–124.
48. SaravananK, HariprasadG, JiteshO, DasU, DeyS, et al. (2007) Endothelin and its receptor interactions: Role of extracellular receptor domain and length of peptide Ligands. Protein and Peptide Letters 14 : 779–783.
49. TakasukaT, SakuraiT, GotoK, FuruichiY, WatanabeT (1994) Human endothelin receptor ET(B) - amino-acid-sequence requirements for super stable complex-formation with its ligand. Journal of Biological Chemistry 269 : 7509–7513.
50. GrantcharovaE, FurkertJ, ReuschHP, KrellHW, PapsdorfG, et al. (2002) The extracellular N terminus of the endothelin B (ETB) receptor is cleaved by a metalloprotease in an agonist-dependent process. Journal of Biological Chemistry 277 : 43933–43941.
51. VerspurtenJ, GevaertK, DeclercqW, VandenabeeleP (2009) SitePredicting the cleavage of proteinase substrates. Trends in Biochemical Sciences 34 : 319–323.
52. KaelinCB, XuX, HongLZ, DavidVA, McGowanKA, et al. (2012) Specifying and sustaining pigmentation patterns in domestic and wild cats. Science 337 : 1536–1541.
53. OkumuraN, HayashiT, SekikawaH, MatsumotoT, MikawaA, et al. (2006) Sequencing, mapping and nucleotide variation of porcine coat colour genes EDNRB, MYO5A, KITLG, SLC45A2, RAB27A, SILV and MITF. Animal Genetics 37 : 80–82.
54. FangM, LarsonG, Soares RibeiroH, LiN, AnderssonL (2009) Contrasting mode of evolution at a coat color locus in wild and domestic pigs. PLoS Genet 5: e1000341 doi:10.1371/journal.pgen.1000341.
55. LiJ, YangH, LiJr, LiHp, NingT, et al. (2010) Artificial selection of the melanocortin receptor 1 gene in Chinese domestic pigs during domestication. Heredity 105 : 274–281.
56. Silvers W (1979) The coat colors of mice. New York: Springer Verlag. Online at http://www.informatics.jax.org/wksilvers. Chapter 11.
57. PicardoM, CardinaliG (2011) The genetic determination of skin pigmentation: KITLG and the KITLG/c-Kit pathway as key players in the onset of human familial pigmentary diseases. Journal of Investigative Dermatology 131 : 1182–1185.
58. WangZ-Q, SiL, TangQ, LinD, FuZ, et al. (2009) Gain-of-function mutation of KIT ligand on melanin synthesis causes familial progressive hyperpigmentation. American Journal of Human Genetics 84 : 672–677.
59. HadjiconstantourasC, SargentCA, SkinnerTM, ArchibaldAL, HaleyCS, et al. (2008) Characterization of the porcine KIT ligand gene: expression analysis, genomic structure, polymorphism detection and association with coat colour traits. Animal Genetics 39 : 217–224.
60. OkumuraN, HayashiT, UenishiH, FukudomeN, KomatsudaA, et al. (2010) Sequence polymorphisms in porcine homologs of murine coat colour-related genes. Animal Genetics 41 : 113–121.
61. MillerCT, BelezaS, PollenAA, SchluterD, KittlesRA, et al. (2007) cis-regulatory changes in kit ligand expression and parallel evolution of pigmentation in sticklebacks and humans. Cell 131 : 1179–1189.
62. SulemP, GudbjartssonDF, StaceySN, HelgasonA, RafnarT, et al. (2007) Genetic determinants of hair, eye and skin pigmentation in Europeans. Nature Genetics 39 : 1443–1452.
63. HirookaH, de KoningDJ, HarliziusB, van ArendonkJAM, RattinkAP, et al. (2001) A whole-genome scan for quantitative trait loci affecting teat number in pigs. Journal of Animal Science 79 : 2320–2326.
64. RodriguezC, TomasA, AlvesE, RamirezO, ArqueM, et al. (2005) QTL mapping for teat number in an Iberian-by-Meishan pig intercross. Animal Genetics 36 : 490–496.
65. Arnaud-DabernatS, BourbonPM, DierichA, Le MeurM, DanielJY (2003) Knockout mice as model systems for studying nm23/NDP kinase gene functions. Application to the nm23-M1 gene. Journal of Bioenergetics and Biomembranes 35 : 19–30.
66. DeplagneC, PeuchantE, MoranvillierI, DubusP, DabernatS (2011) The anti-metastatic nm23-1 gene is needed for the final step of mammary duct maturation of the mouse nipple. PLoS ONE 6: e18645 doi:10.1371/journal.pone.0018645.
67. WarrissPD, KestinSC, BrownSN, NuteGR (1996) The quality of pork from traditional pig breeds. Meat Focus International 5 : 179–182.
68. CameronND, EnserMB (1991) Fatty acid composition of lipid in Longissimus dorsi muscle of Duroc and British Landrace pigs and its relationship with eating quality. Meat Science 29 : 295–307.
69. BrobergerC (2005) Brain regulation of food intake and appetite: molecules and networks. Journal of Internal Medicine 258 : 301–327.
70. ReynoldsCB, EliasAN, WhisnantCS (2010) Effects of feeding pattern on ghrelin and insulin secretion in pigs. Domestic Animal Endocrinology 39 : 90–96.
71. GondretF, RiquetJ, TacherS, DemarsJ, SanchezMP, et al. (2011) Towards candidate genes affecting body fatness at the SSC7 QTL by expression analyses. Journal of Animal Breeding and Genetics 129 : 316–324.
72. Epstein H, Bichard M (1984) Pig. In: Mason I, editor. Evolution of Domesticated Animals. London: Longman. pp.145–162.
73. MegensHJ, CrooijmansR, CristobalMS, HuiX, LiN, et al. (2008) Biodiversity of pig breeds from China and Europe estimated from pooled DNA samples: differences in microsatellite variation between two areas of domestication. Genetics Selection Evolution 40 : 103–128.
74. RamosAM, CrooijmansR, AffaraNA, AmaralAJ, ArchibaldAL, et al. (2009) Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS ONE 4: e6524 doi:10.1371/journal.pone.0006524.
75. SanCristobalM, ChevaletC, HaleyCS, JoostenR, RattinkAP, et al. (2006) Genetic diversity within and between European pig breeds using microsatellite markers. Animal Genetics 37 : 189–198.
76. R Core Team (2011) R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing.
77. WiggintonJE, CutlerDJ, AbecasisGR (2005) A note on exact tests of Hardy-Weinberg equilibrium. American Journal of Human Genetics 76 : 887–893.
78. WrightS (1931) Evolution in Mendelian populations. Genetics 16 : 97–159.
79. WilkinsonS, WienerP, ArchibaldAL, LawA, SchnabelRD, et al. (2011) Evaluation of approaches for identifying population informative markers from high density SNP Chips. BMC Genetics 12 : 45.
80. WeirBS, CockerhamCC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38 : 1358–1370.
81. AkeyJM, ZhangG, ZhangK, JinL, ShriverMD (2002) Interrogating a high-density SNP map for signatures of natural selection. Genome Research 12 : 1805–1814.
82. MoradiMH, Nejati-JavaremiA, Moradi-ShahrbabakM, DoddsKG, McEwanJC (2012) Genomic scan of selective sweeps in thin and fat tail sheep breeds for identifying of candidate regions associated with fat deposition. BMC Genetics 13 : 10.
83. BadkeYM, BatesRO, ErnstCW, SchwabC, SteibelJP (2012) Estimation of linkage disequilibrium in four US pig breeds. BMC Genomics 13 : 24.
84. FalushD, StephensM, PritchardJK (2003) Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 164 : 1567–1587.
85. ZhaoK, WrightM, KimballJ, EizengaG, McClungA, et al. (2010) Genomic diversity and introgression in O. sativa reveal the impact of domestication and breeding on the rice genome. PLoS ONE 5: e10780 doi:10.1371/journal.pone.0010780.
86. LiH, HandsakerB, WysokerA, FennellT, RuanJ, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 : 2078–2079.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 4
Nejčtenější v tomto čísle
- The G4 Genome
- Neutral Genomic Microevolution of a Recently Emerged Pathogen, Serovar Agona
- The Histone Demethylase Jarid1b Ensures Faithful Mouse Development by Protecting Developmental Genes from Aberrant H3K4me3
- The Tissue-Specific RNA Binding Protein T-STAR Controls Regional Splicing Patterns of Pre-mRNAs in the Brain