
Transmission of Hypervirulence Traits via Sexual Reproduction within and between Lineages of the Human Fungal Pathogen
Since 1999 a lineage of the pathogen Cryptococcus gattii has been infecting humans and other animals in Canada and the Pacific Northwest of the USA. It is now the largest outbreak of a life-threatening fungal infection in a healthy population in recorded history. The high virulence of outbreak strains is closely linked to the ability of the pathogen to undergo rapid mitochondrial tubularisation and proliferation following engulfment by host phagocytes. Most outbreaks spread by geographic expansion across suitable niches, but it is known that genetic re-assortment and hybridisation can also lead to rapid range and host expansion. In the context of C. gattii, however, the likelihood of virulence traits associated with the outbreak lineages spreading to other lineages via genetic exchange is currently unknown. Here we address this question by conducting outgroup crosses between distantly related C. gattii lineages (VGII and VGIII) and ingroup crosses between isolates from the same molecular type (VGII). Systematic phenotypic characterisation shows that virulence traits are transmitted to outgroups infrequently, but readily inherited during ingroup crosses. In addition, we observed higher levels of biparental (as opposed to uniparental) mitochondrial inheritance during VGII ingroup sexual mating in this species and provide evidence for mitochondrial recombination following mating. Taken together, our data suggest that hypervirulence can spread among the C. gattii lineages VGII and VGIII, potentially creating novel hypervirulent genotypes, and that current models of uniparental mitochondrial inheritance in the Cryptococcus genus may not be universal.
Published in the journal:
. PLoS Genet 9(9): e32767. doi:10.1371/journal.pgen.1003771
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003771
Summary
Since 1999 a lineage of the pathogen Cryptococcus gattii has been infecting humans and other animals in Canada and the Pacific Northwest of the USA. It is now the largest outbreak of a life-threatening fungal infection in a healthy population in recorded history. The high virulence of outbreak strains is closely linked to the ability of the pathogen to undergo rapid mitochondrial tubularisation and proliferation following engulfment by host phagocytes. Most outbreaks spread by geographic expansion across suitable niches, but it is known that genetic re-assortment and hybridisation can also lead to rapid range and host expansion. In the context of C. gattii, however, the likelihood of virulence traits associated with the outbreak lineages spreading to other lineages via genetic exchange is currently unknown. Here we address this question by conducting outgroup crosses between distantly related C. gattii lineages (VGII and VGIII) and ingroup crosses between isolates from the same molecular type (VGII). Systematic phenotypic characterisation shows that virulence traits are transmitted to outgroups infrequently, but readily inherited during ingroup crosses. In addition, we observed higher levels of biparental (as opposed to uniparental) mitochondrial inheritance during VGII ingroup sexual mating in this species and provide evidence for mitochondrial recombination following mating. Taken together, our data suggest that hypervirulence can spread among the C. gattii lineages VGII and VGIII, potentially creating novel hypervirulent genotypes, and that current models of uniparental mitochondrial inheritance in the Cryptococcus genus may not be universal.
Introduction
Cryptococcus neoformans and C. gattii are the causative agents of cryptococcosis in humans. C. neoformans typically infects HIV-infected individuals and other patients with immunodeficiencies, but has also been found in apparently immunocompetent individuals in the Far East [1], [2]. C. gattii is a primary pathogen that causes disease in otherwise healthy people [3], [4], but has also been found in HIV patients in Malawi, Africa and California, USA [5], [6]. C. gattii accounts for less than 1% of all cryptococcosis cases, and until the late 1990s occurred mostly in subtropical regions of the world. However, in 1999, an outbreak of C. gattii was reported on Vancouver Island in domestic pets and people [7]–[9]. This outbreak spread to mainland Canada and then into the northwestern states of the United States [10]–[13] and currently numbers more than 400 cases [14]–[17].
C. gattii is divided into distinct clades (VGI-VGIV) [14], with the outbreak originating on Vancouver Island, and a more recent outbreak in Oregon [18], , being caused by three clonal groups within VGII (VGIIa, VGIIb and VGIIc) [20]. These hypervirulent outbreaks are characterized by an unusual ability of the pathogen to parasitise host phagocytic cells: upon engulfment by macrophages, outbreak strains initiate mitochondrial tubularisation and rapid intracellular proliferation of the fungus [21].
Cryptococcosis is not spread from infected animals or humans to susceptible hosts but rather infections are acquired from the environment. Hence, cryptococcal species likely experience strong selective pressure from factors encountered within environmental niches. Genetic recombination by meiotic sexual reproduction in eukaryotic pathogens is a widely-occurring mechanism that generates genetic diversity (and hence novel phenotypic diversity) but carries the risk of destroying beneficial gene combinations [22]. The genetic distance across which genetic recombination occurs yields very different outcomes. Outcrossing and hybridization can result in dramatic changes to genotype and resulting virulence phenotypes. For example, Grigg et al. [23] have demonstrated that outcrossing sexual recombination can be a major force in shaping eukaryotic pathogens, since recombinant Toxoplasma progeny from crosses between two distinct ancestral lines type II and type III are significantly more virulent than either parent. A similar hypothesis has been proposed for the origin of C. gattii outbreak strains [24]. However, outcrossing can also come at the cost of breaking up highly-fit coadapted gene-complexes, such as those that enable host adaptation [25], [26], and can result in lethal levels of genetic load resulting in widespread inviability.
Therefore, estimating how likely it is for hypervirulence traits to move between C. gattii lineages by recombination is critical both for predicting the likelihood of novel hypervirulent genotypes occurring and, more broadly, in understanding the origins of infectious outbreaks. In addition, given that the expression of mitochondrially-encoded genes correlates with virulence in C. gattii, but mitochondrial genes do not contribute to virulence in C. neoformans [21], [27], important questions remain about the relative role of mitochondrially-encoded, versus nuclear-encoded, genes in controlling virulence in this pathogen. Here we address both of these questions via a series of genetic crosses, followed by comprehensive phenotypic analyses. Our findings demonstrate that hypervirulence in C. gattii is a complex, multigenic trait. Surprisingly, however, this trait can be transmitted relatively easily to other lineages and is not strictly limited to one mitochondrial genotype. Finally, we show that, in contrast to existing paradigms, mitochondrial inheritance in C. gattii is not strictly uniparental and thus current models of genetic exchange in this pathogenic clade should be revisited.
Results
Experimental Design of Ingroup and Outgroup Crosses
This study addresses two questions:
-
How likely is the spread of hypervirulence traits between C. gattii lineages?
-
What is the contribution of the mitochondrial genome in controlling virulence in C. gattii?
Recent work has demonstrated that the ability to change mitochondrial morphology is closely linked to intracellular proliferation and thus hypervirulence in C. gattii [21]. In the related pathogen C. neoformans, as in most eukaryotes, mitochondria are inherited from only one parent (in this case the MATa parent) following mating [28], [29]. To exploit this uniparental inheritance and to test the likelihood of virulence traits spreading within the C. gattii population, we conducted a series of crosses in which progeny would inherit mitochondria either from a hypervirulent parent, or from a non-outbreak strain exhibiting wild type virulence. If phenotypes associated with hypervirulence (mitochondrial tubularisation in response to phagocytosis and rapid intracellular proliferation) were solely determined by mitochondrial genotypes then all progeny from each cross would have the same phenotype as the MATa parent (Figure 1A).

The ability to proliferate within macrophages is a proven predictor of virulence in C. neoformans and C. gattii [5], [21]. To assess virulence in a comprehensive progeny set in this study, we utilized intracellular proliferation as a proxy-measure of virulence and investigated its relationship to mitochondrial tubularisation.
Our experimental approach included both outgroup crosses, between strains from two different molecular groups (VGII and VGIII), and ingroup crosses, between strains from the same molecular group (VGII) (Figure 1B). Despite the fact that VGIII strains are more fertile than other C. gattii strains [24], [30], [31], experimental mating of C. gattii strains is extremely difficult in the laboratory setting. However, within the VGIII lineage, the VGIII pair B4546 (MATa) and NIH312 (MATα) had previously been identified as mating test strains in an extensive screening study [32]. These strains also exhibit low intracellular proliferation rates and hence were chosen for the outgroup crosses in this study. Disappointingly, after various attempts, we and others were unable to mate MATa-VGII with MATα–VGII strains that exhibit explicitly distinct intracellular proliferation values. We were, however, able to conduct mating between VGII strains with more similar intracellular proliferation rates allowing for dissection of individual spores.
Overall, there was a low rate of spore germination in both the VGII a x VGIII α and VGII α x VGIII a mating pairs. This is consistent with population genetic evidence [24], [33] that these molecular types are genetically isolated with respect to nuclear DNA exchange and hence consistent with assignment as distinct species [34]. For each mating (B4546 x R265, CBS10090 x NIH312 and JF101 x AIg289), at least 50 individual spores (50, 50, 63) were dissected, and in all cases none germinated (0/163). After extensive attempts we were able to obtain six viable microdissected spores (6/140) from an outgroup cross (strains YL4 x 97/433). Crosses across species boundaries in Escherichia, Salmonella and Saccharomyces species are known to suffer from extensive DNA mismatches and cause serious problems during meiosis attributable to the mismatch repair system aborting homologous recombination [35]–[37]. To circumvent this substantial barrier and in an attempt to generate a more comprehensive working progeny set, a region of highly dense spores and hyphae was selected, plated, and colonies that arose were isolated and characterized. This type of analysis is therefore subject to possible isolation of parental yeast cells, blastospores (yeast cells derived from hyphae post-fusion but prior to nuclear fusion and meiosis), diploid fusion products, and true haploid meiotic progeny. For these reasons, and to better understand the dynamics of mating between these two distinct molecular types, the resulting progeny sets were subjected to molecular (MultiLocus Sequence Typing; MLST/Fluorescence-activated cell sorting; FACS) and phenotypic (self-filamentation) analyses.
As an additional approach, VGIII strains carrying the crg1::NEO mutation were crossed with a VGII strain carrying the bwc2::NAT mutation (JF101xAIg289 and JF109xAIg254) (Figure 1B). The parental strain AIg289 also carried a mutation within the FUR1 gene that confers resistant to 5-fluorouracil. Mutation of both Crg1 and Bwc2 enhance mating under conditions that normally repress it. Basidiospores were unviable (0/63 germinated). However, putative fusion or post-meiotic strains were isolated by plating onto YPD medium containing both nourseothricin and G418. Strains were examined for phenotypes and PCR-RFLP markers.
Molecular Characterisation of Progeny from Outgroup Cross R265 x B4546
In the mating pair between VGII α (R265) x VGIII a (B4546), we isolated a total of 18 progeny as described above. Amplification of the ATP6 gene (encoded by the mitochondrial genome) revealed that 100% of the progeny inherited the a mitochondrial genome (Figures 2A&B and Supplementary Information), consistent with previous studies showing uniparental mitochondrial inheritance during a-α mating [28], [38], [39].

Based on FACS analysis, we then determined that 18/18 isolates showed signs of diploidy (Figure 2 C&D) and, in line with this, MLST analysis showed signs of heterozygosity at 7/8 markers (Figure 2C). In all of the isolates, the remaining marker (PLB1) specifically amplified the VGII allele, most likely due to primer bias or loss of heterozygosity caused by mitotic gene conversion or partial chromosomal loss. Interestingly, while all 18 strains retained copies of both SXI1/SXI2, only 17% (3/18) were self-fertile (Figure 2D).
Six progeny that were restored towards haploidy following extensive passage on YPD medium, and these showed recombinant genotypes in MLST analysis with alleles contributed by both parental strains, and ploidy was assessed by FACS analysis (Figure 2E). None of the progeny showed signs of self-filamentation, although none were derived from one of the three self-filamentous parents (Figure 2E).
It is problematic to distinguish aneuploid progeny by FACS analysis alone. MLST analysis showed signs of heterozygosity for the markers MPD, GPD1 and LAC1 for all restored “haploid” progeny. We therefore sequenced the genomes of four of the diploid progeny as well as the respective “haploid restored” progeny and examined read mapping coverage and variant ratios to determine the ploidy in these strains. This analysis revealed that all progeny were broadly diploid/haploid, but most also carried aneuploid regions within at least one of their chromosomes (Supplementary Information and Figure S1). This analysis also provided further information about the progeny and restored “haploid” strains' genetic background: PLB1 was found to be monoallelic in MLST analysis for progeny and restored “haploid” strains but is heterozygous according to the genome data. IGS and TEF1 are also monoallelic in the MLST analysis but biallelic according to the genomic data whereas CAP10 is monoallelic in both MLST and genomic data, as is the MAT locus for which the entire chromosome is monoallelic in all strains.
Similar chromosomal abnormalities have previously been described as a common feature in C. gattii and C. neoformans and been suggested as an adaptive mechanism to stresses such as exposure to antifungals [4], [40]–[44].
Molecular Characterisation of Progeny from Outgroup Cross CBS10090 x NIH312
In the mating between VGII a (CBS10090) x VGIII α (NIH312), we isolated a total of 16 progeny. Mitochondrial amplification of the ATP6 gene (mitochondrial) revealed that 100% of the progeny exclusively inherited the a mitochondrial genome (Figure 3B). Based on FACS analysis, we found that ∼half (7/16) were haploid (1N) and the rest (9/16) were diploid (2N) although this level of analysis cannot distinguish aneuploid isolates (Figure 3A). When each of the progeny was analyzed at eight unlinked MLST loci, including one sex-specific marker (SXI1/SXI2), 5/16 showed no signs of nuclear exchange (all α/haploid) but all five carried an a mitochondrial genome (Figure 3B). This indicates that these five mitochondrial exchange strains are the product of blastospores (i.e., monokaryotic yeast budding off of dikaryotic hyphae). We also show that two isolates harbor alleles from both nuclear genomes, have uniparental mitochondrial inheritance from the a parent, and also are haploid by FACS analysis, indicating that these two isolates were produced via meiosis, although one of the two shows increased levels of inheritance from the VGIII parent (Progeny 2 has 7/8 MLST loci from the VGIII parent while P3 has 4/8 MLST loci from the VGIII parent). The 9/16 remaining isolates show signs of aneuploidy: they all retain markers with sequences from both parental nuclear genomes and are all 2N or greater than 1N based on FACS analysis (Figure 3). These nine isolates also show self-fertility and 7/9 have both sex determining alleles (Figure 3B). The remaining two progeny (1 and 13) show no amplification of the SXI2a allele, however, these are self-fertile α isolates likely exhibiting robust α-α unisexual reproduction. In all of the isolates, the CAP10 locus specifically amplified the VGIII allele, again suggesting either an amplification bias or the loss of this region of chromosome 11.

Molecular Characterisation of Progeny from Additional Outgroup Crosses
In the mating between VGIII α (97/433) x VGII a (YL4), we isolated a total of 7 progeny as described above. None of the progeny showed signs of self-fertility. Nuclear markers indicated that all progeny except SP130, which received all tested alleles from the VGIII α parent (97/433), were recombinant. Amplification of the ATP6 gene (encoded by the mitochondrial genome) revealed that 4/7 of the progeny inherited the a and 3/7 the α mitochondrial genome (Figure 4A) indicating non-uniparental inheritance in this outgroup cross.

For the crosses with marked strains VGII α (AIg254) x VGIII a (JF109) and VGIII α (JF101) x VGII a (AIG289) three and six viable progeny were isolated. (Figure 4B&C). All nine progeny were recombinant compared to the parental isolates. They also all had inherited their mitochondrial genotype from the a parent.
Collectively, our molecular and phenotypic findings indicate that the rate of successful meiosis is low during VGII x VGIII mating with only 2/16 viable progeny being haploid recombinants in a VGII a x VGIII α mating and all of the viable progeny from the VGII α x VGIII a cross being diploid (2N). Thus, both sets of crosses indicate the presence of a restrictive barrier in meiosis due to cryptic speciation between molecular types VGII and VGIII. Compared to the high germination rate observed between VGIII x VGIII F1 progeny [32] both crosses produced few viable progeny. Although more than 163 spores that could be individually manipulated were produced in VGII x VGIII crosses, the spores did not germinate, suggesting that most progeny were largely inviable, as has been previously reported for sexual crosses between the related species Saccharomyces cerevisiae and S. bayanus [45], although at lower frequency.
Inheritance of Macrophage Interaction Traits during Outgroup Crosses R265 x B4546
Given the involvement of mitochondria in cryptococcal hypervirulence [21] we considered whether mitochondrial genotype is the sole determinant of hypervirulence. If so, then we would anticipate the intracellular proliferation rate and mitochondrial tubularisation pattern of the progeny from these crosses to match that of the mitochondrial-donor parent.
Indeed in the outgroup cross R265 x B4546, all 18 hybrid diploid progeny showed intracellular proliferation rates similar to that of the low-virulence MATa (mitochondrial donor) parent B4546 (Figure 5A). In addition, none of these F1 strains were able to trigger extensive mitochondrial tubularisation in response to engulfment by a host macrophage (Figure 5B), suggesting that the replacement of the R265-type mitochondrion with that from B4546 eliminated the hypervirulence trait in these progeny.

Because the progeny from these cross were all diploid, we tested whether this hybrid nuclear genotype may be ‘masking’ virulence phenotypes. However, when we “restored six” of the strains to haploidy via repeated rounds of mitotic passage, both mitochondria tubularisation and intracellular proliferation rates remained low (Figures 5C&D and Figure S2A).
Inheritance of Macrophage Interaction Traits during Outgroup Cross CBS10090 x NIH312
In contrast to the R265 x B4546 cross, the cross between CBS10090 and NIH312, in which genotyping indicated all offspring carried mitochondria from the hypervirulent MATa strain CBS10090, yielded F1 strains showing a wide range of intracellular proliferation rates (Figure 6A). Notably, two haploid recombinant offspring (Progeny 2 and 3) carry mitochondria from the virulent (CBS10090) parent, and yet only one (Progeny 3) shows a high intracellular proliferation rate. Thus a VGII mitochondrial genome, at least alone, is not sufficient to confer hypervirulence in this context.

Interestingly, progeny derived from haploid blastospores are isolates in which the nuclear genome is identical to the α parent NIH312, but the mitochondrial genome has been inherited from the a parent CBS10090. Such isolates show variable intracellular proliferation rates and tubularisation behaviour, e.g. Progeny 14 presents with IPR similar to the hypervirulent a parent CBS10090, whereas Progeny 5, 6, 15, and 12 proliferate less well within macrophages. This indicates that additional mechanisms might contribute to hypervirulence. In particular the sex induced silencing pathway becomes activated in blastospore progeny produced during the sexual cycle [46]. Thus, epigenetic processes might also contribute to altering biological properties of blastospore progeny, in addition to the exchange of the mitochondrial genome, leading to modified virulence phenotypes in blastospore progeny that are identical in their nuclear and mitochondrial genomes, yet differ phenotypically.
Lastly, we note that recombinant progeny from this cross no longer showed concordance between intracellular proliferation and mitochondrial tubularisation rates upon engulfment, with many strains showing high levels of tubularisation even under control conditions (Figure 6B and Figure S2B).
Inheritance of Macrophage Interaction Traits in Additional Outgroup Crosses
To independently verify these observations, we undertook additional crosses using marked strains, as described above. Crosses between a hypervirulent MATa and a low virulence MATα parent resulted in range of intracellular proliferation rates (Figure 7A&B) whereas the reverse cross between a hypervirulent MATα and a low virulence MATa parent only produced progeny with low intracellular proliferative capacity (Figure 7C). Within these outgroup crosses, we also observed misregulated mitochondria (Figure 7D–F and Figure S2C–E).

Taken together, the data from these outgroup crosses thus strongly suggest that:
-
The inheritance of mitochondria from a hypervirulent parent is, alone, not sufficient to confer high intracellular proliferation rates and,
-
Multiple nuclear genomic regions are likely to interact with the mitochondrial genome to regulate hypervirulence in this group.
Molecular Characterisation of Progeny from Ingroup Crosses
For the two ingroup VGII x VGII crosses (CBS1930 x R265 and LA584 x R265), nuclear markers indicated that all progeny were recombinant, although one (#37) from the cross between CBS1930 and R265 is likely aneuploid, because for one chromosome both parental alleles were amplified. Remarkably, however, for both crosses significant numbers of progeny inherited their mitochondria from the unexpected (R265, MATα) parent: 9/36 for the cross with CBS1930 (Figure 8A) and 4/13 for the cross with LA584 (Figure 8B). Thus, it appears that, in contrast to outgroup crosses, crosses within the VGII clade produce a high proportion of viable recombinant progeny but that non-uniparental inheritance of mitochondria (i.e. in which either parent can donate mitochondria to daughter cells, but not at the same time) occurs more frequently than anticipated (25–30% compared to 5% in previous studies). This is analogous to the situation that occurs in atypical diploid-haploid crosses, in which mitochondria are inherited from the MATα parent at a high rate [47].

Inheritance of Macrophage Interaction Traits following Ingroup Crosses
In contrast to the situation with outgroup crosses, several ingroup crosses resulted in a significant number of progeny that exhibited intracellular proliferation rates that were as high or even higher than the hypervirulent MATα parent (R265), despite inheriting their mitochondrion from a lower-virulence MATa parent (Figures 9A&B). Conversely, several progeny inherited the mitochondria from the hypervirulent α parent R265 and yet displayed low intracellular proliferation rates. Thus, hypervirulent phenotypes can spread within VGII independently of the mitochondrial genotype. We had anticipated that in these crosses the progeny would inherit mitochondria from a lower virulence MATa parent (CBS1930 or LA584) but, as described above, in fact both crosses demonstrated a higher rate of mitochondrial inheritance from the MATα parent (25 to 30%), indicating that mitochondrial inheritance is not strictly uniparental in this group.

Interestingly, unlike the CBS10090 x NIH312 outgroup cross (Figure 7), both ingroup crosses produced progeny that remained able to correctly tubularise their mitochondria in response to phagocyte engulfment (Figures 9C&D and Figure S2F&G), which most likely explains the ability of these recombinant progeny to continue to proliferate rapidly within host cells.
Increased mitochondrial genome copy number or a higher number of mitochondria can affect mitochondrial inheritance. Similarly, the larger cell size of one parent might lead to an increased cytoplasmic and/or mitochondrial contribution to progeny. For instance, hyper-suppressive RHO mutants of S. cerevisiae exhibit deletions in the mitochondrial genome and are ‘petite’ variants. However, the S. cerevisiae mitochondrial genome replicates faster than the wild type and consequently, when crossed with wild type, all progeny inherit the mutant mitochondrion [48]–[50]. To test whether such a phenomenon may account for the non-uniparental mitochondrial inheritance we observed during in-group crosses, we measured the size of our parental strains under control conditions in vitro and after macrophage passage. However, cell size does not appear to be a contributing factor for changes in virulence (data not shown). In addition, previously published data on mitochondrial DNA copy number [21] showed no increase of mitochondrial genetic information and hence makes it unlikely that a higher copy number from R265 leads to a ‘leak’ from the α parent in those crosses.
Recombination of Mitochondrial Genomes in C. gattii
The surprising result of biparental mitochondrial inheritance in the ingroup crosses (mitochondria inherited from a parent 70–75% of the time and from the α parent 25–30% of the time), in contrast to uniparental inheritance from only the a parent in outgroup crosses, prompted us to further investigate the unexpected phenotypes of progeny from the outgroup cross between CBS10090 and NIH312. By utilising whole-genome sequence data for the two parental strains and Progeny 5, which had been made available as part of a larger sequencing project, we found 440 mitochondrial single nucleotide polymorphisms (SNPs) that differed between CBS10090 and NIH312. Progeny 5 shared 320 SNPs with CBS10090 and 120 with NIH312. Aligning these sites across the 34 kb mitochondrial genome showed that, with the exception of a single SNP, all sites from a single parent formed contiguous blocks (Figure 10A). Thus this pattern of polymorphisms represents very strong evidence for mitochondrial recombination in Progeny 5 from the VGII x VGIII outgroup cross CBS10090 x NIH312 (Figure 10B). In addition, by using three mitochondrial markers to assess the inheritance of mitochondrial DNA in the VGII x VGII crosses, we identified four examples of recombinant mitochondrial genotypes (Figure 10C). These findings support previous evidence for mitochondrial recombination both in C. neoformans [27], [51] and C. gattii [52].

Discussion
In this study we conducted a systematic analysis to test the potential spread of hypervirulence in C. gattii. In crosses between a hypervirulent VGII strain and strains from a different clade, VGIII, it appears that a mitochondrial genotype originating from within the outbreak is necessary, but not sufficient, to confer hypervirulence. Thus simple transmission of a mitochondrial lineage is unlikely to spread outbreak traits to a new population of C. gattii.
We generated multiple sets of VGII x VGIII F1 progeny, one from a VGII a x VGIII α mating, and the other from a VGII α x VGIII a mating. The first conclusion from the progeny sets was that germination was very infrequent (less than 1%, 0/163 spores), supporting the hypothesis that these molecular types are cryptic species manifesting strong reproductive barriers. To circumvent this, isolation of progeny had to be done without individual spore dissections (selecting probable progeny from spore-dense regions); resulting in a need to clearly type all progeny using a multilocus sequence typing (MLST) or PCR-RFLP based approaches.
Several independent studies have shown that, based on sequence analysis, there is no allelic exchange observed between the four molecular types of C. gattii, leading to a hypothesis that they are independent species within the pathogenic Cryptococcus species complex and adhere to a phylogenetic species concept [11], [24], [33], [34], [53]. The results presented in this manuscript demonstrate that there are clear reproductive barriers between isolates from two of the molecular types examined, VGII and VGIII. This reproductive barrier is post-zygotic, and supported with examples from both VGII α x VGIII a as well as VGII a x VGIII α crosses. Therefore, these two lineages appear to also adhere to a biological species concept. Specifically, analysis of progeny from such crosses showed predominantly hybrid diploids, followed by mitochondrial exchange strains (i.e., blastospores), and finally only 1–2/34 (3–6%) haploid recombinants. This post-zygotic barrier parallels studies examining progeny isolated from sexual crosses between the related species Saccharomyces cerevisiae and S. bayanus, whereby viable spores from tetrads were found at a frequency of about 1/10,000 [45]. Furthermore, similar post-zygotic barriers between closely related species from within both the Microbotryum and Neurospora genera and AD hybrids in C. neoformans result in low levels of viable progeny [54]–[56].
The finding that a majority of progeny are diploid hybrids is intriguing, as it suggests that, both environmentally and clinically, there may be a potential for inter-molecular type hybrids, such as VGII x VGIII hybrids, to occur. This would parallel several seminal studies examining both C. neoformans var. neoformans/C. neoformans var. grubii (AD) and C. neoformans/C. gattii (BD and AB) hybrids [41], [54], [57]–[62]. These studies show that the hybrids are able to infect hosts, as several are clinical in origin, and our IPR assays, at a minimum, suggest that VGIII x VGII diploid hybrids can be virulent.
Mitochondrial inheritance in C. neoformans, as in most eukaryotes, is uniparental [63], [64], with progeny inheriting mitochondria from the MATa parent [29]. However, environmental factors such as high temperature and UV irradiation can lead to biparental inheritance and recombination of mitochondrial DNA [65]. Our data indicate that, at least within VGII, inheritance of mitochondria from either parent (rather than only one) can occur relatively frequently (25–30%), even in the absence of such stresses, and that recombination can occur for the mitochondrial genome. Previous studies have shown that while a-α mating leads to uniparental inheritance, regulated by the mating type-specific homeodomain genes SXI1α and SXI2a, α-α mating has biparental inheritance, which enables increased mitochondrial genome recombination [66]. Other recent studies have demonstrated roles for genes not within the mating type locus in the control of uniparental inheritance [67]. Furthermore, in a congenic VGII MATa/MATα strain pair in the R265 background, uniparental inheritance was observed [68]. This supports an interesting hypothesis whereby α-α mating may contribute to the formation of recombinant mitochondrial genomes with higher predispositions for virulence, possibly explaining the hypervirulence among specific VGII genotypes.
In addition to demonstrating biparental organelle inheritance in C. gattii, we provide clear genomic evidence for mitochondrial recombination following mating, in support of previous MLST/AFLP data that suggested the occurrence of mitochondrial recombination at the population level [38], [52]. Together, these data suggest that there may be less stringent control over uniparental mitochondrial inheritance by the mating type locus in the C. gattii VGII lineage as compared to much stricter uniparental mitochondrial inheritance in C. neoformans.
Whilst the overall populations of strains still showed a significant correlation between intracellular proliferation and mitochondrial tubularisation (Figure S2E), one striking observation from these crosses was the high frequency of ‘misregulated’ mitochondria, in which mitochondrial tubularisation (which, in outbreak strains, is a response to the stressful environment of the host cell involving mitochondrial fusion) occurs even under control growth conditions (Figure 5). Mitochondrial morphology is regulated by proteins encoded within the nuclear genome and, in S. cerevisiae, fusion events are controlled by a protein complex consisting of Fzo1p, Mgm1p, and Ugo1p [69]–[72]. Thus effective interaction of this machinery might be interrupted by non-compatible mitochondrial and nuclear genomes, reducing the ability of recombinant progeny to regulate virulence traits.
In contrast, for crosses between parents within the VGII clade, hypervirulence traits appear to spread easily and are no longer strictly dependent on the presence of a mitochondrial genotype originating from within the outbreak. Thus many, perhaps all, VGII mitochondria are capable of tubularising under host conditions and driving rapid intracellular proliferation, and therefore virulence. This presumably reflects the compatibility of nuclear and mitochondrial ‘cross-talk’ across VGII genotypes, allowing mitochondrial morphology to be correctly regulated in recombinant progeny. This may explain why multiple, distinct outbreaks of disease have all been caused by VGII isolates that differ in their genotype [11].
Our experimental model indicates that hypervirulence in C. gattii is a complex, multigenic trait, requiring regions of the mitochondrial genome and regions of the nuclear genome to confer hypervirulence, which can be attained by a variety of genetic combinations after sexual mating. Thus, there are potentially multiple routes by which such traits could disperse through the C. gattii population, suggesting that surveillance efforts should consider the possibility of independent outbreaks caused by distinct lineages of this pathogen. Our studies also provide phenotypic and molecular evidence that the VGII and VGIII molecular types of C. gattii are distinct species, separated by post-zygotic reproductive barriers. Finally, we provide evidence that mitochondrial inheritance in this species is more complex than currently appreciated, with both biparental mitochondrial inheritance and mitochondrial recombination being observed.
Materials and Methods
Yeast Strains, Mammalian Cells and Growth Conditions
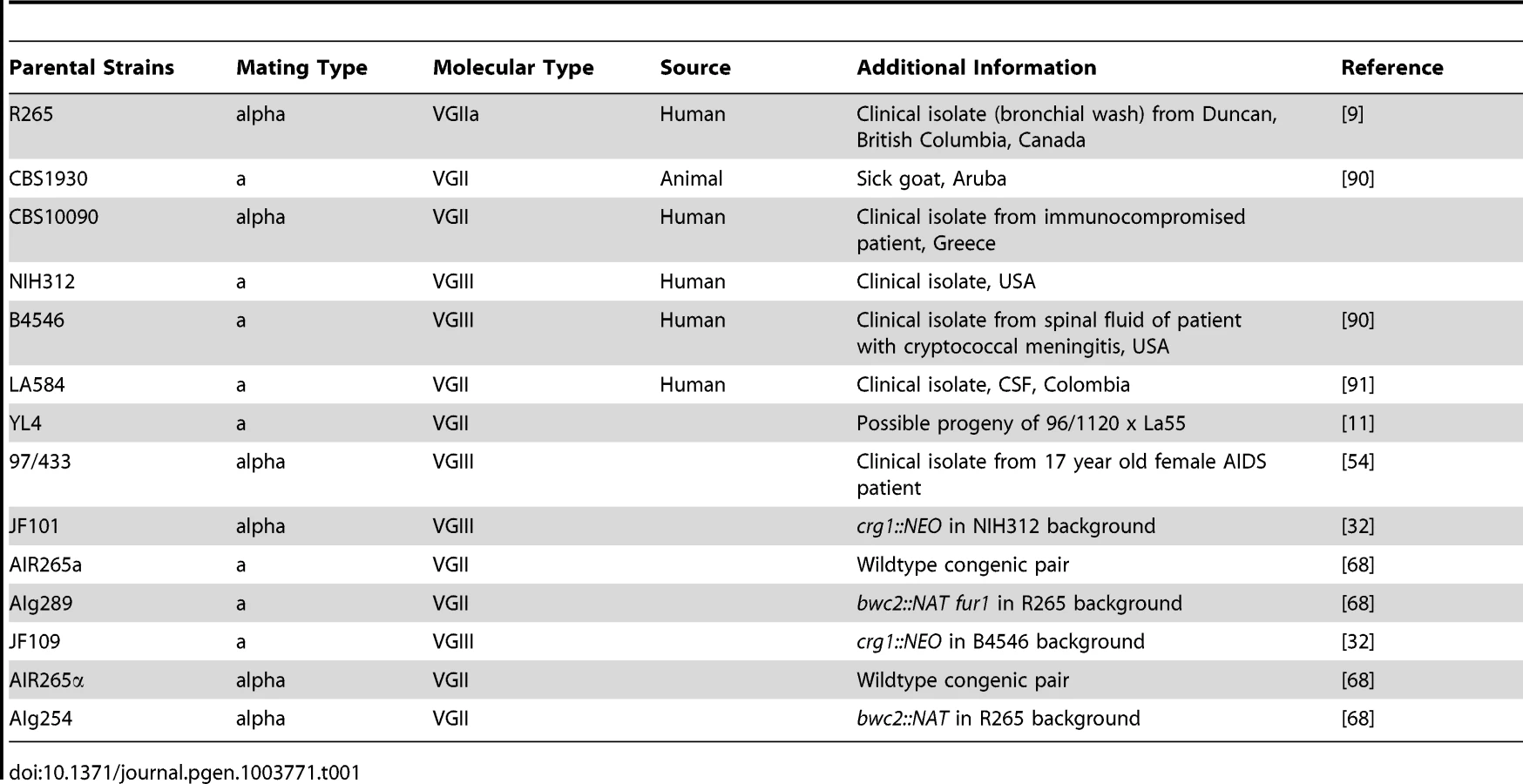
Cryptococcus gattii strains (Table 1 and Table 2) used in this study were cultured in liquid or agar YPD media (1% peptone, 1% yeast extract, 2% D-(+)-glucose) for 24 h at 25°C rotating at 20 rpm prior to experiments [73]. Mammalian J774 cells were grown as described previously [73].
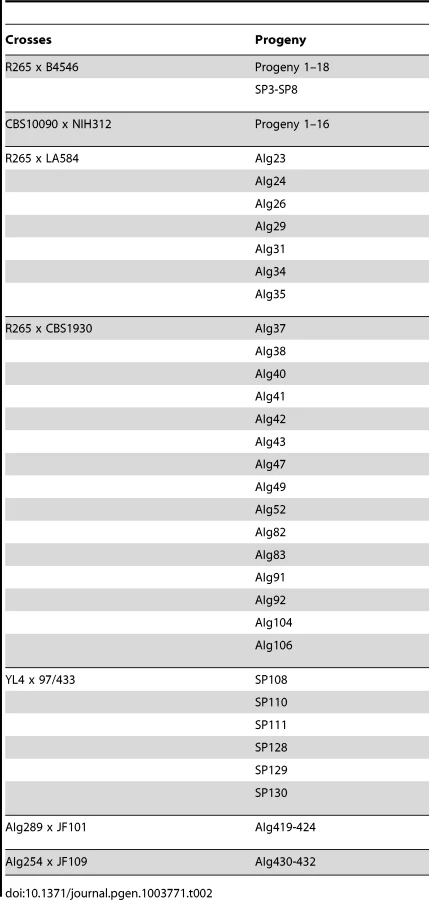
Crosses
For outgroup crosses between VGII a CBS10090, VGII α R265, VGIII a B4546, and VGIII α NIH312, mating assays were conducted on V8 media (5% V8 juice, 0.5 g/L KH2PO4, 4% agar; pH = 5) to generate spores for progeny analyses. Isolates were incubated at room temperature in the dark for 2–4 weeks in dry conditions. Fertility was assessed by light microscopy to identify basidiospore formation at the periphery and surface of the mating patch. To collect progeny from the crosses, basidiospores were isolated with a micromanipulator as described previously with the slight modification that due to low germination rate, other suspected resultant colonies were collected from areas where groups of basidiospores were collected [74], [75]. To summarize, spore-dense regions were collected using a glass Pasteur pipette and spread on a YPD agar plate. In total, the progeny set from the CBS10090 x NIH312 cross yielded 16 collected progeny and the R265 x B4546 cross yielded 18 collected progeny. Progeny from crosses YL4x97/433, AIg289xJF101 and AIg254xJF109 were established as described above. For AIg289 x JF101 and AIg254 x JF109, the cell mixture was plated onto YPD supplemented with nourseothricin and G-418 (both at 100 µg/ml).
To derive haploid progeny (SP3-SP8), the diploid progeny were passaged on YPD agar every 48 hours for 24 days (total 12 passages). A single colony from the previous passage was used to initiate the next passage. FACS analysis was performed on five colonies from every passage to determine the ploidy. Serial passaging was stopped when cells from at least one of the five colonies were found to be haploid. Cells from these haploid colonies were used to grow overnight cultures in liquid YPD, which were stored frozen at −80°C in glycerol and subsequently used for MLST and virulence analyses.
Crosses between R265 and CBS1930 or LA584 were established on V8 juice agar medium (pH unadjusted). Yeasts were mixed on the plate and examined 2–4 weeks later for the presence of basidia and basidiospore chains. Spores and surrounding parental yeasts were transferred using a gel-loading tip to YPD agar medium. Individual basidiospores were micromanipulated with a dissecting microscope. Genomic DNA from progeny was prepared by disrupting cells in buffer (10 mM Tris-HCl [pH 7.5], 10 mM EDTA, 0.5 M NaCl, 1% SDS)+½ volume chloroform with 425–600 µm glass beads, aided by two rounds of vortexing and freezing at −20°C. After centrifugation, the DNA in the supernatant was precipitated with an equal volume of isopropanol. For preparing larger quantities of DNA, a CTAB-based extraction buffer was used on lyophilized cells harvested from 50 ml cultures [76].
Identification and Detection of Polymorphic Regions between Strains
For the outgroup crosses sequence data for the MLST alleles of the parental isolates were previously published (Table S1) [11], [24]. For each F1 progeny isolate, DNA was isolated (Epicentre), and genomic regions were PCR amplified (Table S2), purified (ExoSAP-IT, Qiagen), and sequenced. Sequences from both forward and reverse strands were assembled and manually edited using Sequencher version 4.8 (Gene Codes Corporation). Based on the sequences of the parental strains, alleles of the progeny were assigned to three distinct categories: exclusively from the α parent, exclusively from the a parent, or heterozygous. Heterozygosity at each allele was based on alignments with parental alleles and clear observations of multiple positive nucleotide traces in regions that differ between the two parental sequences, which are indicative of two unique sequences (i.e., one from each parent) being analyzed. Additionally, mitochondrial inheritance was assayed using ATP6 mitochondrial specific forward (ACTTGCGGCTGAATGATAAAATCTAA) and reverse (GTGGAGATGTAATAAAGTGTGTCATG) primers, whereby the product (including the 5′ UTR and part of the ORF) from the VGIII (a and α) mitochondrial genome is larger than the product from the VGII (a and α) mitochondrial genome [5], [11].
For progeny from crosses R265 x CBS1930 and R265 x LA584, polymorphic regions were identified in multilocus sequence typing (MLST) data in GenBank and by sequencing fragments of the CBS1930 genome. MLST differences between the strains were used for three PCR-RFLP markers [24]. To identify other polymorphic regions, a small genomic DNA library was constructed from DNA of strain CBS1930. HindIII restriction fragments were cloned into the HindIII site of plasmid pBluescript. The ends of the inserts were sequenced, and the sequence compared by BLASTn to the R265 strain genome database at the Broad Institute [77]. Either single nucleotide polymorphisms (SNPs) affecting restriction enzymes sites or with multiple differences between the two strains were used for the design of oligonucleotides primers for PCR amplification of alleles from either parent. PCR reactions, digested with restriction enzyme where necessary, were resolved on 1× TAE agarose gels. Primer sequences and details about the polymorphisms are in Table 3. Sections of the mitochondrial genome were amplified and sequenced from R265, CBS1930, and LA584 to identify a polymorphism.

Analysis of nuclear and mitochondrial markers was conducted to test if progeny were recombinant was conducted. The strains from the cross between R265 and CBS1930 were assessed for 16 genetic markers. One was the mating type phenotype and 14 were PCR markers that amplified polymorphic parts of the nuclear genome. The markers are located on eight of the 14 chromosomes of C. gattii [77]. For the mitochondrial genome, the COX1 and COB1 genes were amplified and sequenced. A single nucleotide polymorphism was identified in intron 2 of COB1 (submitted as GenBank accessions JX486912 and JX486913). Subsequently, the CBS1930 strain was subject to Illumina genome sequencing, and the mitochondrial genome analyzed for other differences between this strain and R265. Two other regions were used to track the inheritance of the mitochondrial genome in the VGII x VGII progeny.
Ploidy Determination by Fluorescence Flow Cytometry
Cells were processed for flow cytometry as described previously, with slight modifications [54], [78]. Briefly, cells were harvested from YPD medium, washed once in phosphate-buffered saline (PBS) buffer, and fixed in 1 ml of 70% ethanol overnight at 4°C. Fixed cells were washed once with 1 ml of NS buffer (10 mM Tris-HCl (pH 7.5), 250 mM sucrose, 1 mM EDTA (pH 8.0), 1 mM MgCl2, 0.1 mM CaCl2, 0.1 mM ZnCl2) and then stained with propidium iodide (12.5 mg/ml) in 0.2 ml of NS buffer containing RNaseA (1 mg/ml) at 4°C for 16 h. Next, 0.5 ml of stained cells were diluted into 0.5 ml of 50 mM Tris-HCl (pH 8.0). Flow cytometry was performed on 10,000 cells and analyzed on the FL1 channel with a Becton-Dickinson FACScan (Duke University Medical Center Flow Cytometry Core Facility).
Self-Filamentation Assays
Filamentation assays were conducted on V8 media (pH = 5) and filamentation agar [79]. Isolates were incubated at room temperature in the dark for 2–4 weeks in dry conditions. Filamentation was assessed by light microscopic examination for hyphae formation at the periphery and surface of the incubated patches. All assays were conducted on both media types. If there were no signs of filamentation after a four-week period, isolates were scored as having no self-filamentation phenotype.
Cryptococcal Intracellular Proliferation Assay and Mitochondrial Staining
Macrophages were infected with yeast cells and intracellular proliferation monitored as previously described [80]. Cryptococcal mitochondrial morphology was determined as described previously [11]. In brief, to determine the intracellular proliferation rate (IPR) of individual strains following phagocytosis, J774 macrophage cells were exposed to cryptococcal cells that were opsonized with 18B7 antibody (a kind gift from Arturo Casadevall) for 2 hr as described previously [73]. Each well was washed with PBS in quadruplicate to remove as many extracellular yeast cells as possible and 1 ml of fresh serum-free DMEM was then added. For time point T = 0, the 1 ml of DMEM was discarded and 200 µl of sterile dH2O was added into wells to lyse macrophage cells. After 30 minutes, the intracellular yeast were released and collected. Another 200 µl dH2O was added to each well to collect the remaining yeast cells. The intracellular yeast were then counted with a haemocytometer. For the subsequent five time points (T = 18 hrs, T = 24 hrs, T = 48 hrs, T = 72 hrs), intracellular cryptococcal cells were collected and counted. For each strain tested, the time course was repeated at least three independent times, using different batches of macrophages. The IPR value was calculated by dividing the maximum intracellular yeast number by the initial intracellular yeast number at T = 0. We confirmed that Trypan Blue stains 100% of the cryptococcal cells in a heat-killed culture, but only approximately 5% of cells from a standard overnight culture. The mitochondrial morphology assays were conducted in a similar way to those in previous studies, with modifications [21]. C. gattii cells were grown overnight at 37°C in DMEM untreated or isolated from macrophages 24 hr after infection. The cells were harvested, washed with PBS twice and re-suspended in PBS containing the Mito-Tracker Red CMXRos (Invitrogen) at a final concentration of 20 nM. Cells were incubated for 15 min at 37°C. After staining, cells were washed three times and re-suspended in PBS. For each condition, more than 100 yeast cells per replicate for each of the tested strains were chosen randomly and analysed. To quantify different mitochondrial morphologies, images were collected using a Zeiss Axiovert 135 TV microscope with a 100× oil immersion Plan-Neofluar objective or a Nikon Eclipse Ti Plan Apo VC 60× oil immersion objective. Both fluorescence images and phase contrast images were collected simultaneously. Images were captured with identical settings on a QIcam Fast 1394 camera using the QCapture Pro51 version 5.1.1 software. All images were processed identically in ImageJ and mitochondrial morphologies were analysed and counted blindly [11]. IPR and tubularisation data were analysed for statistically significant differences using one-way ANOVA analysis with multiple comparisons by Tukey Honestly Significant Difference (HSD) posthoc test. A p-value of <0.05 after controlling for multiplicity was considered to be statistically significant.
Illumina Sequencing
Genomic DNA from C. gattii strains NIH312, CBS10090, and progeny 5 from the cross between NIH312 and CBS10090 was isolated with the EpiCentre MasterPure Yeast DNA Purification Kit according to a modified version of the instruction manual. Briefly, the strains were grown in liquid YPD media for 24 h at 25°C rotating at 20 rpm. Cells from 3 ml of culture were harvested by centrifugation at 17,000× g for 5 minutes. Cells were lysed in 300 µl of Yeast Cell Lysis solution by mechanical disruption with 0.1 mm silica spheres (FastPrep Lysing Matrix, MP Biomedicals) twice for 30 seconds at 6,800 rpm in a Precellys24 and incubation at 65°C for 15 minutes. Samples were cooled down on ice for 5 minutes and proteins removed by vortexing with 150 µl of MPC Protein Precipitation Reagent and following centrifugation for 10 minutes at 17,000× g. DNA was recovered with 500 µl isopropanol and centrifugation at 17,000× g for 10 minutes. DNA was purified by RNase A treatment for 60 minutes at 37°C followed by phenol:chloroform extraction and ethanol precipitation. DNA yield and quality was determined by spectrophotometry. 2 µg of genomic DNA were used for library preparation: DNA was fragmented to 150–500 bp using Covaris shearing and processed with the TruSeq DNA Sample Prep Kit (Illumina) according to instructions, purification steps were performed with Agencourt AMPureXP magnetic particles (Beckham Coulter) on a magnetic stand (AmBio). Whole genomes were sequenced on an Illumina HiSeq2000 at the MRC Clinical Science Centre, Imperial College London (UK).
Illumina Sequence Analysis
Alignment and SNP calling parameters were initially optimized. The nuclear and mitochondrial genome sequences and feature files for C. gattii isolate R265 (VGII) were downloaded from http://www.broadinstitute.org/ (GenBank project accession number AAFP01000000). Illumina reads were aligned to the genome sequence using Burrows-Wheeler Aligner (BWA) v0.5.9 [81] with default parameters and converted to pileup format using Samtools v.0.1.18 [82]. To act as a control for sequencing, alignment and SNP calling, we resequenced the reference strain R265. We used a False Discovery (FDR) approach [83] to test our SNP-calling method, which we set at a minimum required depth of four reads, with 90% disagreeing from the reference base and agreeing with each other. First, we randomly modified 63,193 and 698,535 nucleotides within the reference sequence, corresponding with the maximum number of SNPs identified within the VGII group and within any of our C. gattii isolates, respectively. We aligned the reads of R265 to these two altered genome sequences and called SNPs using our chosen parameters. We identified 99.32% and 99.26% of true positives, whilst only calling 3553 (5.6%) and 3363 (0.48%) false positives or genuine discrepancies with the reference sequence respectively. For analysis we used all SNPs that were covered by ≥4 reads in all isolates, leaving a total 740 mitochondrial sites. To ensure that these sites could not have been heterogeneous in the progeny sequence we examined the allele frequencies at each variant site and found that each site had greater than 95% agreement with the called SNP. We simulated 100 bp reads with 0.01% uniform error for 200× coverage from each parental mitochondrial genotype and used them in a single combined BWA alignment and SNPs calling protocol as before. We detected only seven sites that show shared differences from the reference sequence of R265. The other SNPs were not detected because they did not reach the 90% agreement threshold.
De Novo Assemblies of Mitochondrial Genomes
Two to four million 100 bp Illumina paired end reads were assembled with velvet (version 1.2.08) [84]. Redundant assembly runs with varying k-mer lengths were performed for each strain, and each time resulted in identical circular contigs, which only differed in lengths of overlapping ends.
Chromosomal Ploidy Analysis of VGII x VGIII Progeny
Supercontigs of C. gattii R265 were obtained from Broad Institute (http://www.broadinstitute.org), and grouped in chromosomal context by alignment to C. gattii WM276 chromosomes [77] with MUMmer (version 3.23) [85], [86]. The resulting tiled R265 chromosomes served as reference in downstream analyses.
Ploidy of EJB and SP strains was assessed by read coverage and allele frequencies at variant sites. After mapping with Bowtie 2 (version 2.1.0) [87], read coverages were calculated and plotted with CNV-seq [88]. C. gattii WM276 was used as mapping genome, a theoretical VGII x VGIII diploid of equally pooled R265, B4546 reads as reference, and R265 x B4546 progeny (Progeny and SP strains) reads as testing samples, respectively.
Allele frequencies of variant sites in Progeny and SP progeny were calculated after Bowtie 2 read mapping to R265 chromosomes, and variant calling with samtools/bcftools from the samtools package (version 0.1.18 (r982 : 295)) [82] Allele frequencies were extracted from DP4 fields of VCF output, and nucleotide variant ratios calculated for each position by division of reads depth/number of variant bases. A separate mapping of R265 reads was performed as control, and observed positions removed as background noise from Progeny and SP variant calls. Additional details on the ploidy analyses are available in the supplementary information (Text S1).
Alignment of Mitochondrial Genomes
To assess mitochondrial recombination, genomes of CBS10090 and NIH312 were compared to CBS10090 x NIH312 progeny strain Progeny 5. Sequences were aligned, and corresponding regions visualized with Progressive Mauve (version 2.3.1) [89]. Additional details on the analysis of mitochondrial genomes are available in the supplementary information (Text S1).
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. ChengPY, ShamA, KronstadJW (2009) Cryptococcus gattii isolates from the British Columbia cryptococcosis outbreak induce less protective inflammation in a murine model of infection than Cryptococcus neoformans. Infect Immun 77 : 4284–4294.
2. LuiG, LeeN, IpM, ChoiKW, TsoYK, et al. (2006) Cryptococcosis in apparently immunocompetent patients. QJM 99 : 143–151.
3. IdnurmA, BahnYS, NielsenK, LinX, FraserJA, et al. (2005) Deciphering the model pathogenic fungus Cryptococcus neoformans. Nat Rev Microbiol 3 : 753–764.
4. KronstadJW, AttarianR, CadieuxB, ChoiJ, D'SouzaCA, et al. (2011) Expanding fungal pathogenesis: Cryptococcus breaks out of the opportunistic box. Nat Rev Microbiol 9 : 193–203.
5. ByrnesEJ (2011) A diverse population of Cryptococcus gattii molecular type VGIII in southern Californian HIV/AIDS patients. PLoS Pathog 7: e1002205.
6. ChenJ, VarmaA, DiazMR, LitvintsevaAP, WollenbergKK, et al. (2008) Cryptococcus neoformans strains and infection in apparently immunocompetent patients, China. Emerg Infect Dis 14 : 755–762.
7. BartlettKH, KiddSE, KronstadJW (2008) The emergence of Cryptococcus gattii in British Columbia and the Pacific Northwest. Curr Infect Dis Rep 10 : 58–65.
8. KiddSE, BachPJ, HingstonAO, MakS, ChowY, et al. (2007) Cryptococcus gattii dispersal mechanisms, British Columbia, Canada. Emerg Infect Dis 13 : 51–57.
9. KiddSE, HagenF, TscharkeRL, HuynhM, BartlettKH, et al. (2004) A rare genotype of Cryptococcus gattii caused the cryptococcosis outbreak on Vancouver Island (British Columbia, Canada). Proc Natl Acad Sci U S A 101 : 17258–17263.
10. ByrnesEJ, HeitmanJ (2009) Cryptococcus gattii outbreak expands into the Northwestern United States with fatal consequences. F1000 Biol Reports 1 : 62.
11. ByrnesEJ (2010) Emergence and pathogenicity of highly virulent Cryptococcus gattii genotypes in the northwest United States. PLoS Pathog 6: e1000850.
12. ByrnesEJ (2009) First reported case of Cryptococcus gattii in the Southeastern USA: implications for travel-associated acquisition of an emerging pathogen. PLoS ONE 4: e5851.
13. DattaK, BartlettKH, BaerR, ByrnesE, GalanisE, et al. (2009) Spread of Cryptococcus gattii into Pacific Northwest region of the United States. Emerg Infect Dis 15 : 1185–1191.
14. ByrnesEJ (2011) Cryptococcus gattii: an emerging fungal pathogen infecting humans and animals. Microbes Infect 13 : 895–907.
15. GalanisE, MacdougallL, KiddS, MorshedM (2010) Epidemiology of Cryptococcus gattii, British Columbia, Canada, 1999–2007. Emerg Infect Dis 16 : 251–257.
16. PatrickS, TurabelidzeG, YatesK, MyersA, NasciR, et al. (2010) Emergence of Cryptococcus gattii - Pacific Northwest, 2004–2010. MMWR Morb Mortal Wkly Rep 59 : 865–868.
17. MarrKA (2012) Cryptococcus gattii as an important fungal pathogen of western North America. Expert Rev Anti Infect Ther 10 : 637–643.
18. ByrnesEJ (2009) Molecular evidence that the range of the Vancouver Island outbreak of Cryptococcus gattii infection has expanded into the Pacific Northwest in the United States. J Infect Dis 199 : 1081–1086.
19. MoreraN, Juan-SallésC, TorresJM, AndreuM, SánchezM, et al. (2011) Cryptococcus gattii infection in a Spanish pet ferret (Mustela putorius furo) and asymptomatic carriage in ferrets and humans from its environment. Medical Mycology 49 : 779–784.
20. KiddSE, GuoH, BartlettKH, XuJ, KronstadJW (2005) Comparative gene genealogies indicate that two clonal lineages of Cryptococcus gattii in British Columbia resemble strains from other geographical areas. Eukaryot Cell 4 : 1629–1638.
21. MaH, HagenF, StekelDJ, JohnstonSA, SionovE, et al. (2009) The fatal fungal outbreak on Vancouver Island is characterized by enhanced intracellular parasitism driven by mitochondrial regulation. Proc Natl Acad Sci U S A 106 : 12980–12985.
22. HeitmanJ (2010) Evolution of eukaryotic microbial pathogens via covert sexual reproduction. Cell Host Microbe 8 : 86–99.
23. GriggME, BonnefoyS, HehlAB, SuzukiY, BoothroydJC (2001) Success and virulence in Toxoplasma as the result of sexual recombination between two distinct ancestries. Science 294 : 161–165.
24. FraserJA, GilesSS, WeninkEC, Geunes-BoyerSG, WrightJR, et al. (2005) Same-sex mating and the origin of the Vancouver Island Cryptococcus gattii outbreak. Nature 437 : 1360–1364.
25. HeitmanJ (2006) Sexual reproduction and the evolution of microbial pathogens. Curr Biol 16: R711–725.
26. WendteJM, MillerMA, LambournDM, MagargalSL, JessupDA, et al. (2011) Self-mating in the definitive host potentiates clonal outbreaks of the apicomplexan parasites Sarcocystis neurona and Toxoplasma gondii. PLoS Genet 6: e1001261.
27. ToffalettiDL, NielsenK, DietrichF, HeitmanJ, PerfectJR (2004) Cryptococcus neoformans mitochondrial genomes from serotype A and D strains do not influence virulence. Curr Genet 46 : 193–204.
28. XuJ, AliRY, GregoryDA, AmickD, LambertSE, et al. (2000) Uniparental mitochondrial transmission in sexual crosses in Cryptococcus neoformans. Curr Microbiol 40 : 269–273.
29. YanZ, XuJ (2003) Mitochondria are inherited from the MATa parent in crosses of the basidiomycete fungus Cryptococcus neoformans. Genetics 163 : 1315–1325.
30. NgamskulrungrojP, SorrellTC, ChindampornA, ChaiprasertA, PoonwanN, et al. (2008) Association between fertility and molecular sub-type of global isolates of Cryptococcus gattii molecular type VGII. Med Mycol 46 : 665–673.
31. HallidayCL, CarterDA (2003) Clonal reproduction and limited dispersal in an environmental population of Cryptococcus neoformans var. gattii isolates from Australia. J Clin Microbiol 41 : 703–711.
32. FraserJA, SubaranRL, NicholsCB, HeitmanJ (2003) Recapitulation of the sexual cycle of the primary fungal pathogen Cryptococcus neoformans var. gattii: implications for an outbreak on Vancouver Island, Canada. Eukaryot Cell 2 : 1036–1045.
33. BoversM, HagenF, KuramaeEE, BoekhoutT (2008) Six monophyletic lineages identified within Cryptococcus neoformans and Cryptococcus gattii by multi-locus sequence typing. Fungal Genet Biol 45 : 400–421.
34. NgamskulrungrojP, GilgadoF, FaganelloJ, LitvintsevaAP, LealAL, et al. (2009) Genetic diversity of the Cryptococcus species complex suggests that Cryptococcus gattii deserves to have varieties. PLoS One 4: e5862.
35. ChambersSR, HunterN, LouisEJ, BortsRH (1996) The mismatch repair system reduces meiotic homeologous recombination and stimulates recombination-dependent chromosome loss. Mol Cell Biol 16 : 6110–6120.
36. HunterN, ChambersSR, LouisEJ, BortsRH (1996) The mismatch repair system contributes to meiotic sterility in an interspecific yeast hybrid. EMBO J 15 : 1726–1733.
37. RayssiguierC, ThalerDS, RadmanM (1989) The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature 342 : 396–401.
38. XuJ, YanZ, GuoH (2009) Divergence, hybridization, and recombination in the mitochondrial genome of the human pathogenic yeast Cryptococcus gattii. Mol Ecol 18 : 2628–2642.
39. YanZ, HullCM, HeitmanJ, SunS, XuJ (2004) SXI1alpha controls uniparental mitochondrial inheritance in Cryptococcus neoformans. Curr Biol 14: R743–744.
40. Heitman J, Kozel TR, Kwon-Chung J, Perfect JR, Casadevall A (2011) Cryptococcus: from human pathogen to model yeast. Washington, DC: ASM Press.
41. LengelerKB, CoxGM, HeitmanJ (2001) Serotype AD strains of Cryptococcus neoformans are diploid or aneuploid and are heterozygous at the mating-type locus. Infect Immun 69 : 115–122.
42. Kwon-ChungKJ, ChangYC (2012) Aneuploidy and drug resistance in pathogenic fungi. PLoS Pathog 8: e1003022.
43. FraserJA, HuangJC, Pukkila-WorleyR, AlspaughJA, MitchellTG, et al. (2005) Chromosomal translocation and segmental duplication in Cryptococcus neoformans. Eukaryot Cell 4 : 401–406.
44. SionovE, LeeH, ChangYC, Kwon-ChungKJ (2010) Cryptococcus neoformans overcomes stress of azole drugs by formation of disomy in specific multiple chromosomes. PLoS Pathog 6: e1000848.
45. SebastianiF, BarberioC, CasaloneE, CavalieriD, PolsinelliM (2002) Crosses between Saccharomyces cerevisiae and Saccharomyces bayanus generate fertile hybrids. Res Microbiol 153 : 53–58.
46. WangX, HsuehYP, LiW, FloydA, SkalskyR, et al. (2010) Sex-induced silencing defends the genome of Cryptococcus neoformans via RNAi. Genes Dev 24 : 2566–2582.
47. SkosirevaI, JamesTY, SunS, XuJ (2010) Mitochondrial inheritance in haploid x non-haploid crosses in Cryptococcus neoformans. Curr Genet 56 : 163–176.
48. EphrussiB, de Margerie-HottinguerH, RomanH (1955) Suppressiveness: A new factor in the genetic determinism of the synthesis of respiratory enzymes in Yeast. Proc Natl Acad Sci U S A 41 : 1065–1071.
49. de ZamaroczyM, MarottaR, Faugeron-FontyG, GoursotR, ManginM, et al. (1981) The origins of replication of the yeast mitochondrial genome and the phenomenon of suppressivity. Nature 292 : 75–78.
50. BlancH, DujonB (1980) Replicator regions of the yeast mitochondrial DNA responsible for suppressiveness. Proc Natl Acad Sci U S A 77 : 3942–3946.
51. NielsenK, CoxGM, WangP, ToffalettiDL, PerfectJR, et al. (2003) Sexual cycle of Cryptococcus neoformans var. grubii and virulence of congenic a and α isolates. Infect Immun 71 : 4831–4841.
52. BoversM, HagenF, KuramaeEE, BoekhoutT (2009) Promiscuous mitochondria in Cryptococcus gattii. FEMS Yeast Res 9 : 489–503.
53. MeyerW, AanensenDM, BoekhoutT, CogliatiM, DiazMR, et al. (2009) Consensus multi-locus sequence typing scheme for Cryptococcus neoformans and Cryptococcus gattii. Med Mycol 1–14.
54. LinX, LitvintsevaAP, NielsenK, PatelS, FloydA, et al. (2007) Alpha AD alpha hybrids of Cryptococcus neoformans: evidence of same-sex mating in nature and hybrid fitness. PLoS Genet 3 : 1975–1990.
55. DettmanJR, JacobsonDJ, TurnerE, PringleA, TaylorJW (2003) Reproductive isolation and phylogenetic divergence in Neurospora: comparing methods of species recognition in a model eukaryote. Evolution 57 : 2721–2741.
56. Le GacM, HoodME, GiraudT (2007) Evolution of reproductive isolation within a parasitic fungal species complex. Evolution 61 : 1781–1787.
57. BoekhoutT, TheelenB, DiazM, FellJW, HopWC, et al. (2001) Hybrid genotypes in the pathogenic yeast Cryptococcus neoformans. Microbiology 147 : 891–907.
58. LinX, PatelS, LitvintsevaAP, FloydA, MitchellTG, et al. (2009) Diploids in the Cryptococcus neoformans serotype A population homozygous for the alpha mating type originate via unisexual mating. PLoS Pathog 5: e1000283.
59. BoversM, HagenF, KuramaeEE, DiazMR, SpanjaardL, et al. (2006) Unique hybrids between the fungal pathogens Cryptococcus neoformans and Cryptococcus gattii. FEMS Yeast Res 6 : 599–607.
60. LitvintsevaAP, KestenbaumL, VilgalysR, MitchellTG (2005) Comparative analysis of environmental and clinical populations of Cryptococcus neoformans. J Clin Microbiol 43 : 556–564.
61. AminnejadM, DiazM, ArabatzisM, CastanedaE, LazeraM, et al. (2012) Identification of novel hybrids between Cryptococcus neoformans var. grubii VNI and Cryptococcus gattii VGII. Mycopathologia 173 : 337–346.
62. BoversM, HagenF, KuramaeEE, HoogveldHL, DromerF, et al. (2008) AIDS patient death caused by novel Cryptococcus neoformans x C. gattii hybrid. Emerg Infect Dis 14 : 1105–1108.
63. GyawaliR, LinX (2011) Mechanisms of uniparental mitochondrial DNA inheritance in Cryptococcus neoformans. Mycobiology 39 : 235–242.
64. WangL, LinX (2011) Mechanisms of unisexual mating in Cryptococcus neoformans. Fungal Genet Biol 48 : 651–660.
65. YanZ, SunS, ShahidM, XuJ (2007) Environment factors can influence mitochondrial inheritance in the fungus Cryptococcus neoformans. Fungal Genet Biol 44 : 315–322.
66. YanZ, HullCM, SunS, HeitmanJ, XuJ (2007) The mating type-specific homeodomain genes SXI1alpha and SXI2a coordinately control uniparental mitochondrial inheritance in Cryptococcus neoformans. Curr Genet 51 : 187–195.
67. GyawaliR, LinX (2013) Prezygotic and postzygotic control of uniparental mitochondrial DNA inheritance in Cryptococcus neoformans. mBio 4: e00112–00113.
68. ZhuP, ZhaiB, LinX, IdnurmA (2013) Congenic strains for the genetic analysis of virulence traits in Cryptococcus gattii. Infect Immun 81 : 2616–2625.
69. MeeusenS, NunnariJ (2003) Evidence for a two membrane-spanning autonomous mitochondrial DNA replisome. J Cell Biol 163 : 503–510.
70. WongED, WagnerJA, GorsichSW, McCafferyJM, ShawJM, et al. (2000) The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion competent mitochondria. J Cell Biol 151 : 341–352.
71. SesakiH, SouthardSM, YaffeMP, JensenRE (2003) Mgm1p, a dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol Biol Cell 14 : 2342–2356.
72. SesakiH, JensenRE (1999) Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol 147 : 699–706.
73. MaH, CroudaceJE, LammasDA, MayRC (2006) Expulsion of live pathogenic yeast by macrophages. Curr Biol 16 : 2156–2160.
74. HsuehYP, IdnurmA, HeitmanJ (2006) Recombination hotspots flank the Cryptococcus mating-type locus: implications for the evolution of a fungal sex chromosome. PLoS Genet 2: e184.
75. HsuehYP, FraserJA, HeitmanJ (2008) Transitions in sexuality: recapitulation of an ancestral tri - and tetrapolar mating system in Cryptococcus neoformans. Eukaryot Cell 7 : 1847–1855.
76. PitkinJW, PanaccioneDG, WaltonJD (1996) A putative cyclic peptide efflux pump encoded by the TOXA gene of the plant-pathogenic fungus Cochliobolus carbonum. Microbiology 142 : 1557–1565.
77. D'SouzaCA, KronstadJW, TaylorG, WarrenR, YuenM, et al. (2011) Genome variation in Cryptococcus gattii, an emerging pathogen of immunocompetent hosts. mBio 2: e00342–00310.
78. LinX, HullCM, HeitmanJ (2005) Sexual reproduction between partners of the same mating type in Cryptococcus neoformans. Nature 434 : 1017–1021.
79. WickesBL, MayorgaME, EdmanU, EdmanJC (1996) Dimorphism and haploid fruiting in Cryptococcus neoformans: association with the alpha-mating type. Proc Natl Acad Sci U S A 93 : 7327–7331.
80. VoelzK, LammasDA, MayRC (2009) Cytokine signaling regulates the outcome of intracellular macrophage parasitism by Cryptococcus neoformans. Infect Immun 77 : 3450–3457.
81. LiH, DurbinR (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 : 1754–1760.
82. LiH, HandsakerB, WysokerA, FennellT, RuanJ, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 : 2078–2079.
83. FarrerRA, HenkDA, MacLeanD, StudholmeDJ, FisherMC (2013) Using false discovery rates to benchmark SNP-callers in next-generation sequencing projects. Sci Rep 3 : 1512.
84. ZerbinoDR, BirneyE (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18 : 821–829.
85. KurtzS, PhillippyA, DelcherAL, SmootM, ShumwayM, et al. (2004) Versatile and open software for comparing large genomes. Genome Biol 5: R12.
86. DelcherAL, KasifS, FleischmannRD, PetersonJ, WhiteO, et al. (1999) Alignment of whole genomes. Nucleic Acids Res 27 : 2369–2376.
87. LangmeadB, SalzbergSL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9 : 357–359.
88. XieC, TammiMT (2009) CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics 10 : 80.
89. DarlingAE, MauB, PernaNT (2010) progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5: e11147.
90. BoekhoutT, van BelkumA, LeendersAC, VerbrughHA, MukamurangwaP, et al. (1997) Molecular typing of Cryptococcus neoformans: taxonomic and epidemiological aspects. Int J Syst Bacteriol 47 : 432–442.
91. ChuckSL, SandeMA (1989) Infections with Cryptococcus neoformans in the acquired immunodeficiency syndrome. N Engl J Med 321 : 794–799.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 9
Nejčtenější v tomto čísle
- A Genome-Wide Systematic Analysis Reveals Different and Predictive Proliferation Expression Signatures of Cancerous vs. Non-Cancerous Cells
- Recent Acquisition of by Baka Pygmies
- The Condition-Dependent Transcriptional Landscape of
- Histone Chaperone NAP1 Mediates Sister Chromatid Resolution by Counteracting Protein Phosphatase 2A