
, a Direct Transcriptional Target, Modulates T-Box Factor Activity in Orofacial Clefting
Among the most common human congenital anomalies, cleft lip and palate (CL/P) affects up to 1 in 700 live births. MicroRNA (miR)s are small, non-coding RNAs that repress gene expression post-transcriptionally. The miR-17-92 cluster encodes six miRs that have been implicated in human cancers and heart development. We discovered that miR-17-92 mutant embryos had severe craniofacial phenotypes, including incompletely penetrant CL/P and mandibular hypoplasia. Embryos that were compound mutant for miR-17-92 and the related miR-106b-25 cluster had completely penetrant CL/P. Expression of Tbx1 and Tbx3, the DiGeorge/velo-cardio-facial (DGS) and Ulnar-mammary syndrome (UMS) disease genes, was expanded in miR-17-92 mutant craniofacial structures. Both Tbx1 and Tbx3 had functional miR seed sequences that mediated gene repression. Analysis of miR-17-92 regulatory regions uncovered conserved and functional AP-2α recognition elements that directed miR-17-92 expression. Together, our data indicate that miR-17-92 modulates expression of critical T-box transcriptional regulators during midface development and is itself a target of Bmp-signaling and the craniofacial pioneer factor AP-2α. Our data are the first genetic evidence that an individual miR or miR cluster is functionally important in mammalian CL/P.
Published in the journal:
. PLoS Genet 9(9): e32767. doi:10.1371/journal.pgen.1003785
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003785
Summary
Among the most common human congenital anomalies, cleft lip and palate (CL/P) affects up to 1 in 700 live births. MicroRNA (miR)s are small, non-coding RNAs that repress gene expression post-transcriptionally. The miR-17-92 cluster encodes six miRs that have been implicated in human cancers and heart development. We discovered that miR-17-92 mutant embryos had severe craniofacial phenotypes, including incompletely penetrant CL/P and mandibular hypoplasia. Embryos that were compound mutant for miR-17-92 and the related miR-106b-25 cluster had completely penetrant CL/P. Expression of Tbx1 and Tbx3, the DiGeorge/velo-cardio-facial (DGS) and Ulnar-mammary syndrome (UMS) disease genes, was expanded in miR-17-92 mutant craniofacial structures. Both Tbx1 and Tbx3 had functional miR seed sequences that mediated gene repression. Analysis of miR-17-92 regulatory regions uncovered conserved and functional AP-2α recognition elements that directed miR-17-92 expression. Together, our data indicate that miR-17-92 modulates expression of critical T-box transcriptional regulators during midface development and is itself a target of Bmp-signaling and the craniofacial pioneer factor AP-2α. Our data are the first genetic evidence that an individual miR or miR cluster is functionally important in mammalian CL/P.
Introduction
The evidence that there is a genetic component underlying CL/P is compelling. Analysis of a Danish cohort of CL/P cases revealed that relatives of patients with CL/P have a higher relative risk for CL/P compared to background risk levels. This notion of CL/P heritability is also supported by twin studies [1], [2]. Genome wide association studies (GWAS) and mouse genetics studies have also pointed to genes and genomic regions that are associated with CL/P [3], [4].
MiRs repress gene expression post-transcriptionally by Watson-Crick base pairing to the seed sites in the 3′UTR of target genes. The miR-17-92 cluster, encoding miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92a-1, is within a region on chromosome 13q that when deletion is associated with CL/P, lung hypoplasia, microphthalmia, microcephaly, and small stature in human patients and has phenotypic similarities to Feingold syndrome [5], [6]. Moreover, miR-17-92 is found in an amplified region associated with small cell lung cancer, as well as in B-cell lymphomas, and is over-expressed in several solid tumor types, including breast, colon, lung, pancreas, and prostate cancers [7].
The mouse embryos with miR-17-92 loss-of-function have smaller body size, microphthalmia, heart defects, and lung hypoplasia [6], [8], [9]. Moreover, the miR-17-92 gain-of-function mutants develop lymphoma, indicating that the mouse is an accurate model for the human syndrome [10]. Unlike the miR-17-92 loss-of-function mice, its two homologous clusters, miR-106a-363 and miR-106b-25 loss-of-function embryos do not exhibit any gross abnormalities.
Our previous findings indicated that miR-17-92 is directly regulated by Bmp-signaling in heart development [9]. Bmp-signaling deficiency in mice and humans has been shown to cause CL/P and other craniofacial anomalies [11], [12]. Interestingly, miR-17-92 has also been shown to be directly regulated by Myc family transcription factors [7]. Here, we show that miR-17-92 deficiency results in orofacial clefting and that the human disease genes Tbx1 and Tbx3 are direct targets for miR-17-92. Our findings also reveal that miR-17-92 is a direct target for the master regulator of cranial neural crest development AP-2α.
Results
miR-17-92 mutant embryos have orofacial clefting
We found that miR-17-92 (miR17-92null/null) mutant embryos had severe craniofacial defects including CL/P and mandibular hypoplasia with notching, revealing that miR-17-92 is a critical regulator of craniofacial development (Figure 1A–H, Figure S1). Moreover, by genetically reducing miR-106b-25 dose on the miR-17-92null background, the clefting phenotype was both more severe and completely penetrant, indicating that there is genetic redundancy between these two miR complexes (Figure 1C–F, Figure S1E–H and Table S1).

In addition to cleft lip and mandible defects, both miR-17-92 mutants and miR-17-92null; miR-106b-25null compound mutants had cleft secondary palate (Figure 1 B, D, H). Expression of mitotic cell marker phospho-Histone H3 (pHH3) was greatly reduced in miR-17-92 mutants, indicating that miR-17-92 is required for normal progenitor cell proliferation during orofacial development (Figure 1I–L, Figure S2). Taken together, these data provide the first genetic evidence that miRs are important regulators of mammalian orofacial development and are involved in CL/P.
miR-17-92 is expressed in craniofacial structures
We generated a miR-17-92 bacterial artificial chromosome (BAC) transgenic LacZ reporter line to follow the expression of primary (pri)-miR-17-92 (Figure S3A). Three individual transgenic lines showed similar LacZ expression pattern, revealing that pri-miR-17-92 was expressed in branchial arches and frontonasal process (Figure 2A). LacZ was also detected in the nasal structures, calvarial bones, auricle, periocular mesenchyme, and limb mesenchyme (Figure 2K). Sagittal sections on E11.5 embryos revealed LacZ activity in epithelium and mesenchyme of first branchial arch and frontonasal process (Figure 2I). Coronal sections through E12.5 and E13.5 embryos demonstrated LacZ staining in distal tips of the palatal shelves, the mandibular mesenchyme and mesenchyme of forming frontal bones (Figure 2J, L).

In situ with a pri-miR-17-92 probe revealed similar expression pattern as the transgenic LacZ data (Figure 2E–F). Furthermore, in situ analysis with locked nuclei acid (LNA) probes to detect mature miR-17 and miR-92a showed that miR-17 and miR-92a were highly expressed in branchial arch and frontonasal process (Figure 2 B–D, G–H). Unlike miR-17-92, expression of miR-106b-25 was relatively low (Figure S3B–E).
Tbx genes are repressed by miR-17-92
Tbx1 gain-of-function causes cleft lip and is a miR-17-92 target in the heart [9], [13]. We evaluated the expression of candidate craniofacial miR-17-92 target genes based on bioinformatics analysis, including Tbx3, Fgf10, Pax9, Shox2 and Osr1, in both miR-17-92 null and conditional knock out mutants using AP-2α cre driver [14]. In situ hybridization in miR-17-92 mutants demonstrated up-regulated Tbx3 expression in mandible, frontonasal-derived structures, tongue, and secondary palate at E13.5 (Figure S4A–F, Figure S5E–F) and in paired maxillary processes and nasal process at E10.0 and E10.5 (Figure 3A–B, Figure S4 G–N). Changes in Tbx1 and Fgf10 expression were not detected at E10.0 likely because the expression changes were not dramatic enough to be detected by in situ hybridization (data not shown). Tbx1 was expanded primarily in the secondary palate, tongue, and oral ectoderm at E13.5 (Figure 3C–D, Figure S5A–D). In addition, Fgf10 was expanded in distal mandible and tongue (Figure S5I–L), while ectopic Shox2 expression was observed in distal mandible at E13.5 (Figure S5G–H). Expression of Osr1 was upregulated in the distal mandible and frontonasal structures at E13.5 (Figure S5M–P). In contrast, the expression pattern of Pax9 in miR-17-92 null mutant embryos was unchanged compared to control embryos (data not shown). qRT-PCR experiments also showed up-regulation of Tbx1, Tbx3, Fgf10, Shox2 and Osr1 in miR-17-92; miR106b-25 compound mutants at E13.5 (Figure 3E).
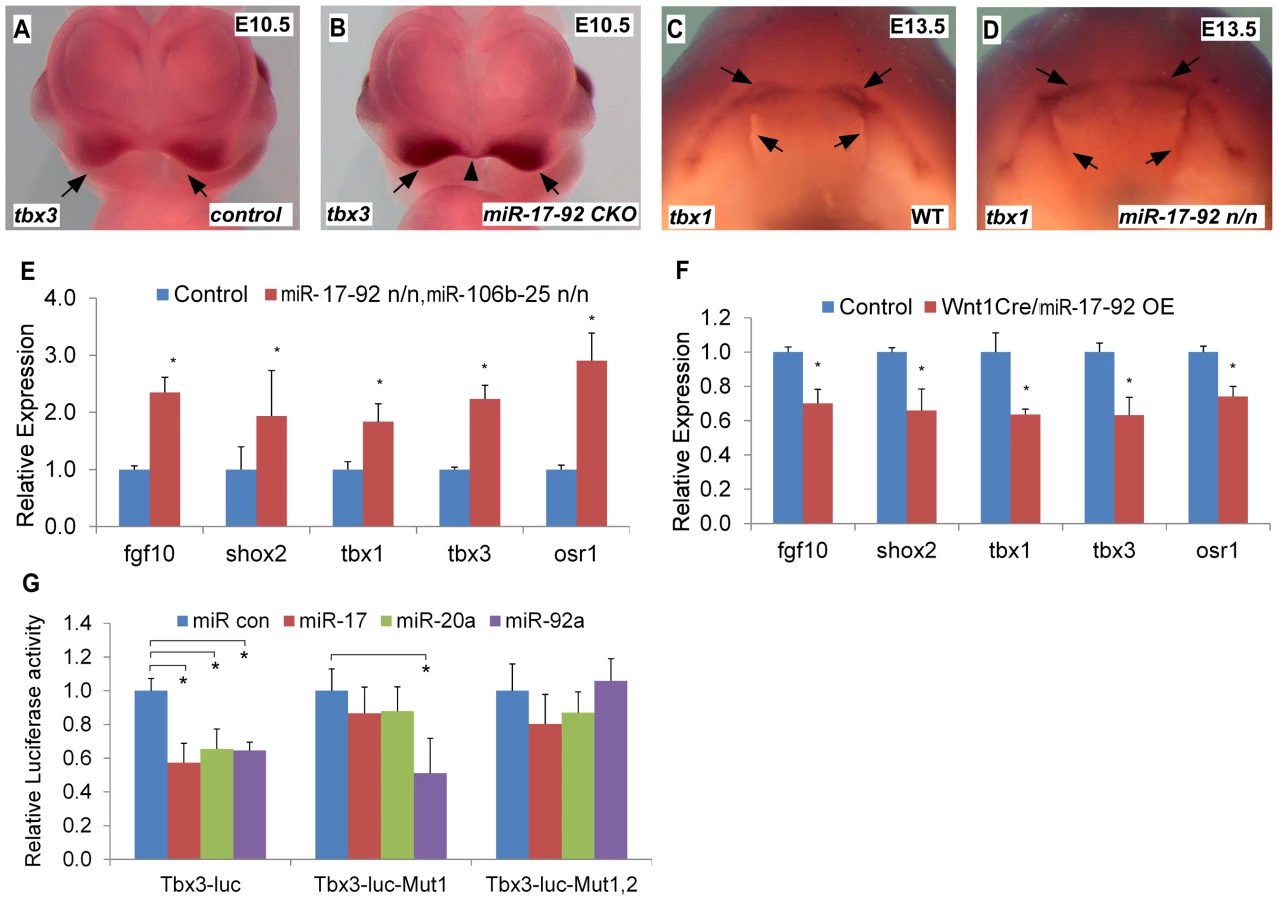
To evaluate the expression changes of the above genes, we used a miR-17-92 conditional, cre-activated gain-of-function line (miR-17-92OE) and the Wnt1cre driver to activate miR-17-92 in cranial neural crest (CNC) [10], [15]. qRT-PCR analysis from Wnt1cre; miR-17-92OE orofacial tissue revealed that Fgf10, Tbx1, Tbx3, Osr1 and Shox2 were significantly repressed (Figure 3F), while there was no obvious morphological defect detected in miR-17-92 overexpression mutants potentially due to moderaterepression of the miR-17-92 target genes.
Direct regulation of craniofacial development genes by miR-17-92
Target genes that are repressed by miR-17-92 have a mixture of miR-17/20a/106b and miR-92a/25 family seed sites in their 3′UTRs (Figure S6). Bioinformatics analysis revealed conserved miR-17/20a/106b family seed sequence in the 3′ UTR of Fgf10, Shox2 and Osr1 (Figure S6A–C, F). The Tbx3 3′ UTR contained both a miR-17/20a/106b family seed site and a miR-92a/25 family seed site (Figure S6D–E). We cloned the 3′ UTRs of Fgf10, Shox2, Tbx3 and Osr1 into luc reporter plasmids to test miR seed sequence function in vitro. Transfections with miR mimics of miR-17-92 resulted in drastic reduction in luciferase activity for all of the reporter plasmids (Figure 3G, Figure S7). Mutation of the respective miR seed sequences within 3′ UTRs of those genes ablated the inhibition by the corresponding miR (Figure 3G, Figure S7). These data suggest that miR-17-92 directly inhibits Fgf10, Shox2, Tbx3 and Osr1.
Bmps regulate miR-17-92 complex in craniofacial development and miR-17-92 overexpression suppresses orofacial clefting in Bmp mutant mice
Previous work showed that conditional inactivation of Bmpr1a, Bmp4, and Bmp2;Bmp4 in developing facial processes using the Nestincre transgenic driver result in orofacial clefting ([11] and Figure S8). This cre driver directs cre activity in facial prominences [11]. Moreover, miR-17-92 is a direct target for Bmp-signaling in cardiac progenitors [9]. We crossed the miR-17-92OE line into Nestincre, Bmp4, Bmp7 conditional mutant background to test whether miR-17-92 gain-of-function could genetically rescue the defects in Bmp mutants. All NestinCre, Bmp4 flox/+, Bmp7 flox/+ embryos (23 out of 23) and embryos without NestinCre (29 out of 29) had normal morphology (Figure 4A, Figure S9A, D, table S2), while all NestinCre, Bmp4 flox/flox, Bmp7 flox/+ mutant embryos (6 out of 6) had bi-lateral cleft lip and heart defects with incompletely penetrant embryonic lethality at E12.0 likely due to heart defects (Figure 4B, Figure S9B, E, table S2). Most (5 out of 6) NestinCre, Bmp4 flox/flox, Bmp7 flox/+, miR-17-92OE embryos were rescued by miR-17-92 overexpression (significant different compared to NestinCre, Bmp4 flox/flox, Bmp7 flox/+ mutants, CHI-TEST, p<0.01), with full suppression of cleft lip and heart defect caused by Bmp loss-of-function, but not eye defect (Figure 4C, Figure S9C, F, table S2).

Consistently, qRT-PCR data indicated that pri-miR-17-92, miR-17, and miR-20a were reduced in Bmp2/4 mutant midface (Figure 4D). In situ analysis using miR-17 LNA probe also indicated that miR-17 was dramatically reduced in Bmp2/4 mutants (Figure S10C, D) compared to controls (Figure S10A, B). In addition, qRT-PCR indicated that Fgf10, Tbx1, Tbx3, Osr1 and Shox2 were up-regulated in the midface of Bmp2; Bmp4 conditional mutants (Figure S10E), further suggesting that these genes are regulated by a BMP-miR-17-92 genetic pathway in craniofacial structures.
Moreover, in vivo chromatin immunoprecipitation (ChIP) data using embryonic midface extracts showed enrichment in the anti-Smad1/5/8 immunoprecipitated chromatin, indicating that Smad1/5/8 directly binds miR-17-92 chromatin (Figure 4E). Co-transfection of a constitutively active Bmpr1a construct with miR-17-92 luc reporter resulted in approximately 3-fold induction supporting the hypothesis that Bmp signaling directly regulates miR-17-92 in developing craniofacial structures (Figure 4F). Together, a conserved Bmp-miR-17-92 genetic pathway plays a critical role in the orofacial development.
AP-2α regulates miR-17-92 in craniofacial development
Mouse mutants for AP-2α have CL/P and human patients have branchio-oculo-facial syndrome that has CL/P as a cardinal feature (BOFS MIM 113620). ChIP-sequencing (ChIP-seq) indicated that AP-2α bound to miR-17-92 chromatin in cultured human neural crest [16] (Figure 4H, S11A). To determine if AP-2α directly regulates miR-17-92, we evaluated pri-miR-17-92, mature miR-17, and mature miR-20a levels in the AP-2α mutant midface. qRT-PCR experiments indicated that pri-miR-17-92 and mature miRs were down-regulated in AP-2α mutants (Figure 4G). We used ChIP-PCR to determine whether AP-2α binds to miR-17-92 chromatin in developing midface tissue. Because there are multiple predicted AP-2α binding sites in miR-17-92, we subdivided miR-17-92 into four regions based on ChIP-seq (Figure 4 H–J and S11). ChIP-PCR experiments using midface extracts indicated that AP-2α bound to miR-17-92 regions 1, region 2, and region 4 (Figure 4 K). Transfection experiments with a miR-17-92 reporter containing AP-2α binding sites revealed that AP-2α transcriptionally activated miR-17-92 although synergism with Smad1 was not detected using this miR-17-92 reporter (Figure 4 L). Moreover, AP-2α may also directly regulate miR-106b-25 as suggested by the analysis of ChIP-seq data [16] (Figure S12).
Discussion
We report the first miR underlying mammalian CL/P. miR-17-92 is located on human chromosome 13q31.3 in a critical region for CL/P associated with 13q deletion syndrome highlighting the importance of our findings to human disease. Our data indicated that miR-17-92 promotes proliferation in developing midface by regulating a group of progenitor genes including Tbx1 and Tbx3 that are known human disease genes (Figure S13). Our findings reveal that timely down-regulation of progenitor genes in developing midface by miR-17-92 is critical for normal midface development.
Mir-17-92 regulates Tbx1 and Tbx3 genes in craniofacial development
Tbx1 loss - and gain-of-function result in cleft palate in human DGS patients and mouse models [13], [17]–[19]. Consistent with our findings, Tbx1 gain-of-function results in cell cycle arrest [17]. DGS is characterized by highly variable phenotypes indicating that there are strong modifiers in the human genome [18], [20]. Our findings suggest miR-17-92 as a candidate genetic modifier for Tbx1 since it fine-tunes Tbx1 expression levels.
Mouse mutants for Tbx3 and the related Tbx2 have cleft palate [21]. Furthermore, human patients with UMS have abnormal and distinct facial appearance indicating a requirement for Tbx3 in human craniofacial development [22]. While our findings suggest that elevated Tbx3 inhibits proliferation, there is other evidence suggesting that Tbx3 promotes proliferation [23]. However, an in vivo study reveals that Tbx3 overexpression results in reduced cardiomyocyte proliferation in the zebrafish heart [24]. More work will be required to evaluate Tbx3 function and target genes in vivo in the context of the miR-17-92 mutant midface to better understand contextual Tbx3 function.
MiR-17-92 regulates Fgf signaling
Both Fgf10 and Fgfr null mice have cleft secondary palate [25], [26]. Mutations in Fgf10 and Fgf receptors cause lacrimo-auricular-dento-digital (LADD) syndrome in human patients indicating a requirement for Fgf-signaling in human craniofacial development [27]. Fgf10 mRNA is enriched in anterior and middle regions of the secondary palate. Moreover, Fgf10 deficiency results in abnormal fusion of the palatal to oral cavity epithelium, suggesting that Fgf10 is required for maturation of palate epithelium. Importantly, elevated Fgf signaling is pathologic in human patients as shown by the extensive investigations into Fgf receptor mediated craniosynostosis [28]. Homozygosity for the Fgfr2 gain-of-function Crouzon mutation in mice results in cleft palate, as well as, craniosynostosis [29] indicating that elevated Fgf signaling also causes cleft palate. Our data demonstrate that miR-17-92 directly represses Fgf10 as a mechanism to maintain correct levels of Fgf10 during palate closure.
Micro RNAs in human orofacial development and disease
Currently, there are no other genetic loss-of-function data indicating that single miRs or miR clusters are important in mammalian orofacial clefting. Data from zebrafish indicate that miR-140 targets pdgfra to regulate primary palate development [30]. GWAS in human patients reveal important genome regions that are associated with CL/P, including 8q24 [3], [31]. Within the 8q24 region is the c-myc gene, a known miR-17-92 regulator [32], [33]. Chromosomal deletions that include miR-17-92 cause a variant of Feingold syndrome in human patients with small stature and skeletal abnormalities [6]. Human patients with hemizygous miR-17-92 deletion do not have CL/P likely reflecting phenotypic heterogeneity in miR-17-92 loss of function families. These data are consistent with our findings indicating that there is incomplete penetrance of the CL/P phenotype in miR-17-92 mutant mouse embryos (table S1).
Mir-17-92 regulation in midface development by Bmp and AP-2α
Consistent with previously finding that Bmp-deficiency results in CL/P in mice and humans [11], [12], our data indicate that Bmp signaling activates miR-17-92 in craniofacial development. Moreover, we show that AP-2α also regulates miR-17-92 expression although our transfection assays failed to uncover synergistic miR-17-92 activation by AP-2α and Bmp-signaling (not shown). One possibility is that Bmp-signaling and AP-2α activate miR-17-92 sequentially during craniofacial progenitor cell development. The assays we employed here cannot easily distinguish molecular events that occur in neighboring or closely apposed cells rather than in the same cell. We also failed to detect up-regulated miR-17-92 target genes in AP-2α mutants perhaps due to functional redundancy with other AP-2 family members [34]–[36]. Nonetheless our findings have important implications since AP-2α has been shown to regulate Irf6, a common genetic defect in syndromic and non-syndromic CL/P in human patients [37]. AP-2α regulation potentially connects miR-17-92 to a gene regulatory network that may be involved in a large portion of human CL/P. In summary, we identified a miR-mediated genetic pathway that plays critical roles during orofacial development (Figure S13).
Materials and Methods
Ethics statement
All animal experiments detailed within the manuscript were approved by the Baylor College of Medicine review board.
Mouse alleles and transgenic lines
The miR-17-92 and miR-106b-25 alleles, Bmp2, Bmp4 and Bmp7 conditional null, AP-2α cre, Nestin cre and Wnt1cre alleles were previously described [8], [10], [11], [14], [15], [38]. To generate miR-17-92-lacZ reporter transgenic lines, we obtained the BAC from BACPAC Resources Center, Children's hospital Oakland Research Institute (BAC number: RP23-89P9) and replaced miR-17-92 sequence with lacZ coding sequence by recombineering, followed by pro-nuclear injection. Constructs were generated using PCR, cloning and recombineering. A LoxP site flanked neo cassette was isolated from PL452 plasmid using BamHI and EcoRI. lacZ coding sequence was isolated from hsp68-lacZ plasmid using BamHI and NcoI. All fragments were cloned into pBluescript SK+ to generate miR-17-92-lacZ construct, followed by recombineering and subsequently Cre mediated recombination for removal of the neo cassette (Figure S3A).
Scanning Electron Microscopy (SEM)
Mouse embryos were harvested in ice-cold Phosphate Buffered Saline (PBS), then fixed overnight (O/N) in 4% paraformaldehyde (PFA) and 2% glutaraldehyde in PBS at 4°C. The samples were then dehydrated in ethanol series to a final 100% ethanol, followed by transferring to graded series of increasing concentrations of hexamethyldisilazane (HMDS) for 5 min each and air dried O/N. Samples were mounted on to double-stick carbon tabs (Ted Pella. Inc.), which have been pre-mounted on to aluminum specimen mounts (Electron Microscopy Sciences). The samples were then coated with a thickness of 25 nm platinum alloy under vacuum using a Balzer MED 010 evaporator (Technotrade International), then immediately flash carbon coated under the same vacuum. The samples were transferred to a desiccator for later examination. JSM-5910 scanning electron microscope (JEOL, USA, Inc.) was used at an accelerating voltage of 5 kV.
Immunofluorescence
Embryos were fixed in 4% PFA, embedded in paraffin and cut to 5 µm sections mounted on Superfrost/Plus slides (Fisher Scientific). The antigens were retrieved by incubating in the citrate buffer (10 mM) for 2 minutes in microwave oven. The primary antibody was anti-Phospho-Histone H3 with 1∶200 dilution (Cell Signaling). Broad HRP conjugated secondary antibody (Invitrogen) was used and visualized using TSA Plus Fluorescence Systems from PerkinElmer on a Zeiss LSM 510 Confocal Microscope. Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI).
In situ hybridization
Tissue preparation and in situ hybridization were as previously described [39], [40]. The gene probes were synthesized using DIG RNA Labeling Kit (Roche) following manufacturer's guidelines. The enzymes used for digestion and transcription of in situ constructs are SacII and T7 for Fgf10, XhoI and T7 for Shox2 (gift from Dr. Yiping Chen's lab), EcoRI and T7 for Osr1(gift from Dr. Rulang Jiang's lab), EcoRI and T3 for Tbx1(gift from Dr. Antonio Baldini's lab), PstI and T3 for Tbx3 (gift from Dr. Robert Kelly's lab). miRCURY LNA probes were purchased from Exiqon and used per manufacturer's guidelines.
Generation of constructs
To generate 3′ UTR luciferase reporter plasmids, 3′ UTR genomic sequence of genes including Fgf10, Osr1, Shox2 and Tbx3 were amplified using a high-fidelity PCR system (Roche) with designed oligonucleotides and subcloned into the pMIR-REPORT Luciferase miRNA Expression Reporter Vector (Ambion).
Oligonucleotides used to amplify 3′ UTR genomic sequence of Fgf10 are sense, 5′-CGACTAGTAAGAAAACACTGTTGGTGGATGCAG -3′, and antisense, 5′-GCACGCGTTTTTATTCTCTTTTCCCAGC-3′. Oligonucleotides used to amplify 3′ UTR genomic sequence of Osr1 are sense, 5′ - GACTAGTATAAACAGAGCCTGCGGG -3′, and antisense, 5′ - CGACGCGTGCCTGTAAAATAACCGTTTATTT -3′. Oligonucleotides used to amplify 3′ UTR genomic sequence of Shox2 are sense, 5′-ACTAGTCGCCGGCGCCAGCGCCACGGT-3′, and antisense, 5′-AAGCTTCTTTTTTGTATGAACGTCC-3′. Oligonucleotides used to amplify 3′ UTR genomic sequence of Tbx3 are sense, 5′ - GACTAGTAAACAAGAAAAACAAAATCGCC -3′, and antisense, 5′ - CCCAAGCTTTCATTTCAATAAAAATTTATTG -3′. Oligonucleotides used to amplify 3′ UTR genomic sequence of Tbx3 without mir17/mir-20a seed site are sense, 5′ - GACTAGTGTGTAACCAGGCTGCTGTTGCTTT -3′, and antisense, 5′ - CCCAAGCTTTGGTCGTTTGAACCAAGTCCCTCT -3′. Underlined letters represent enzyme restriction sites for subcloning. All PCR products were sequenced to make sure no mutations were introduced.
Site-directed mutagenesis
All site-directed mutagenesis of the miR seed sites in the 3′ UTR reporter constructs were achieved by using the QuikChange II site-directed mutagenesis kit (Stratagene). The sense-strand sequences of the oligonucleotides used for mutagenesis (underlined letters indicate the mutation of miR seed sites) were: 5′-TAAGACACGCAAGCATTTACTGGAAAGACACTGGGTCATATCATATGCACAACCAAAG - 3′ (Fgf10-mut1,), 5′-CCCCATGCGCTCTCAGTTGACTTAATTTGACACTCTGCAATAAAAAACACCAGCAAT - 3′ (Fgf10-mut2), 5′-ACAGCAAATAGTGCAGACGTTGGATTCTTATTTCAACCCGCCATTTAGATTACTAAAGAGA - 3′ (Fgf10-mut3); 5′-GCTGACCTTTTTCTGCGAAGTTGAATTCAATAGGAGACATTTGATAAGAG - 3′ (Shox2-mut); 5′ - GCCGGGCGTTGTATTGCGACTGGGAATTCATGCTGACCATCGGTAACGGAC - 3′ (Osr1-mut); 5′ - GGACCATTAGTTCTTTTAACTGTATAGAATTCAACAAGGTTTTAAAAGATAATAATA - 3′ (Tbx3-mut). All PCR products were sequenced to make sure no unexpected mutations were introduced.
Chromatin immunoprecipitation
Wild type mouse embryonic orofaces were dissected at E12.5 (for Smad1/5/8 ChIP) or E10.5 (for AP-2α ChIP) and followed by ChIP analysis as previously described [9]. As control, normal rabbit immunoglobulin G was used as a replacement for the anti-Smad1/5/8 (sc-6031 X, Santa Cruz) and 3B5 mouse monoclonal AP-2α antibody [41] to reveal nonspecific immunoprecipitation of the chromatin. The PCR products were evaluated for appropriate size on a 2% agarose gel and were confirmed by sequencing. The primers for amplifying the regulatory element in the 5′ upstream of mir-17-92 genomic sequence were: sense, 5′ - CTGGCGGGAAGCCTGAGC -3′, antisense, 5′-CACGGCGGCTCGTTCTTG -3′ (for AP-2α region1); sense, 5′ - CCTTCATTCACCCACATGGTCCTT -3′, antisense, 5′ - AGCAGCCGCCACCATCTT -3′ (for AP-2α region2); sense, 5′ - GCACACAATGGCCCTCGG -3′, antisense, 5′ - GCGCGCACAAAGTTTCGG -3′ (for AP-2α region3); sense, 5′ - CGCAGCCGCCCAGAAAC -3′, and antisense, 5′-TCCGCGCCAGCTTATCAAGAGAAA -3′ (for AP-2α region4 and Bmp/Smad regulatory element).
Real time RT-PCR
Total RNA was isolated using RNeasy Micro Kit (QIAGEN) and real-time thermal cycling was performed using StepOne Real-Time PCR Systems (Applied Biosystems). Super Script II Reverse Transcriptase (Invitrogen) was used for RT-PCR and SYBR Green JumpStart Taq ReadyMix (SIGMA) was used for real-time thermal cycling. All error bars represent SEM.
Luciferase activity assay
Plasmids used for transfection were generated as described above or previously reported [9]. LS8 cells were transfected using Lipofectamine 2000 (Invitrogen). Luciferase activity assays were measured using the luciferase Assay System (Promega).
ChIP-seq analysis
hNCC AP-2α ChIP-seq and histone modification markers ChIP-seq datasets were accessed from GEO under accession number GSE28876 [16], [42]. Raw fastq reads were mapped to hg19 genome using Bowtie2 [43]. The total number of tags of each ChIP-seq run was normalized to 10 million. ChIP-seq tracks were visualized and compared in UCSC Genome Browser.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. GrosenD, BilleC, PedersenJK, SkyttheA, MurrayJC, et al. (2010) Recurrence risk for offspring of twins discordant for oral cleft: a population-based cohort study of the Danish 1936–2004 cleft twin cohort. Am J Med Genet A 152A: 2468–2474.
2. GrosenD, ChevrierC, SkyttheA, BilleC, MolstedK, et al. (2010) A cohort study of recurrence patterns among more than 54,000 relatives of oral cleft cases in Denmark: support for the multifactorial threshold model of inheritance. J Med Genet 47 : 162–168.
3. BeatyTH, MurrayJC, MarazitaML, MungerRG, RuczinskiI, et al. (2010) A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat Genet 42 : 525–529.
4. DixonMJ, MarazitaML, BeatyTH, MurrayJC (2011) Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet 12 : 167–178.
5. KirchhoffM, BisgaardAM, StoevaR, DimitrovB, Gillessen-KaesbachG, et al. (2009) Phenotype and 244k array-CGH characterization of chromosome 13q deletions: an update of the phenotypic map of 13q21.1-qter. Am J Med Genet A 149A: 894–905.
6. de PontualL, YaoE, CallierP, FaivreL, DrouinV, et al. (2011) Germline deletion of the miR-17 - 92 cluster causes skeletal and growth defects in humans. Nat Genet 43 : 1026–1030.
7. MendellJT (2008) miRiad roles for the miR-17-92 cluster in development and disease. Cell 133 : 217–222.
8. VenturaA, YoungAG, WinslowMM, LintaultL, MeissnerA, et al. (2008) Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132 : 875–886.
9. WangJ, GreeneSB, Bonilla-ClaudioM, TaoY, ZhangJ, et al. (2010) Bmp signaling regulates myocardial differentiation from cardiac progenitors through a MicroRNA-mediated mechanism. Dev Cell 19 : 903–912.
10. XiaoC, SrinivasanL, CaladoDP, PattersonHC, ZhangB, et al. (2008) Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol 9 : 405–414.
11. LiuW, SunX, BrautA, MishinaY, BehringerRR, et al. (2005) Distinct functions for Bmp signaling in lip and palate fusion in mice. Development 132 : 1453–1461.
12. SuzukiS, MarazitaML, CooperME, MiwaN, HingA, et al. (2009) Mutations in BMP4 are associated with subepithelial, microform, and overt cleft lip. Am J Hum Genet 84 : 406–411.
13. FulcoliFG, HuynhT, ScamblerPJ, BaldiniA (2009) Tbx1 regulates the BMP-Smad1 pathway in a transcription independent manner. PLoS One 4: e6049.
14. MacateeTL, HammondBP, ArenkielBR, FrancisL, FrankDU, et al. (2003) Ablation of specific expression domains reveals discrete functions of ectoderm - and endoderm-derived FGF8 during cardiovascular and pharyngeal development. Development 130 : 6361–6374.
15. ChaiY, JiangX, ItoY, BringasP, HanJ, et al. (2000) Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development 127 : 1671–1679.
16. Rada-IglesiasA, BajpaiR, PrescottS, BrugmannSA, SwigutT, et al. (2012) Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell 11 : 633–648.
17. FunatoN, NakamuraM, RichardsonJA, SrivastavaD, YanagisawaH (2012) Tbx1 regulates oral epithelial adhesion and palatal development. Hum Mol Genet 21 : 2524–2537.
18. WiddershovenJC, BowserM, SheridanMB, McDonald-McGinnDM, ZackaiEH, et al. (2013) A candidate gene approach to identify modifiers of the palatal phenotype in 22q11.2 deletion syndrome patients. Int J Pediatr Otorhinolaryngol 77 : 123–127.
19. PortnoiMF (2009) Microduplication 22q11.2: a new chromosomal syndrome. Eur J Med Genet 52 : 88–93.
20. AggarwalVS, MorrowBE (2008) Genetic modifiers of the physical malformations in velo-cardio-facial syndrome/DiGeorge syndrome. Dev Disabil Res Rev 14 : 19–25.
21. ZirzowS, LudtkeTH, BronsJF, PetryM, ChristoffelsVM, et al. (2009) Expression and requirement of T-box transcription factors Tbx2 and Tbx3 during secondary palate development in the mouse. Dev Biol 336 : 145–155.
22. JossS, KiniU, FisherR, MundlosS, PrescottK, et al. (2011) The face of Ulnar Mammary syndrome? Eur J Med Genet 54 : 301–305.
23. LeeJM, KimJY, ChoKW, LeeMJ, ChoSW, et al. (2007) Modulation of cell proliferation during palatogenesis by the interplay between Tbx3 and Bmp4. Cell Tissue Res 327 : 285–292.
24. RibeiroI, KawakamiY, BuscherD, RayaA, Rodriguez-LeonJ, et al. (2007) Tbx2 and Tbx3 regulate the dynamics of cell proliferation during heart remodeling. PLoS One 2: e398.
25. AlappatSR, ZhangZ, SuzukiK, ZhangX, LiuH, et al. (2005) The cellular and molecular etiology of the cleft secondary palate in Fgf10 mutant mice. Dev Biol 277 : 102–113.
26. RiceR, Spencer-DeneB, ConnorEC, Gritli-LindeA, McMahonAP, et al. (2004) Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest 113 : 1692–1700.
27. RohmannE, BrunnerHG, KayseriliH, UygunerO, NurnbergG, et al. (2006) Mutations in different components of FGF signaling in LADD syndrome. Nat Genet 38 : 414–417.
28. ChaiY, MaxsonREJr (2006) Recent advances in craniofacial morphogenesis. Dev Dyn 235 : 2353–2375.
29. EswarakumarVP, OzcanF, LewED, BaeJH, TomeF, et al. (2006) Attenuation of signaling pathways stimulated by pathologically activated FGF-receptor 2 mutants prevents craniosynostosis. Proc Natl Acad Sci U S A 103 : 18603–18608.
30. EberhartJK, HeX, SwartzME, YanYL, SongH, et al. (2008) MicroRNA Mirn140 modulates Pdgf signaling during palatogenesis. Nat Genet 40 : 290–298.
31. BirnbaumS, LudwigKU, ReutterH, HermsS, SteffensM, et al. (2009) Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat Genet 41 : 473–477.
32. O'DonnellKA, WentzelEA, ZellerKI, DangCV, MendellJT (2005) c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435 : 839–843.
33. DewsM, HomayouniA, YuD, MurphyD, SevignaniC, et al. (2006) Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet 38 : 1060–1065.
34. WingerQ, HuangJ, AumanHJ, LewandoskiM, WilliamsT (2006) Analysis of transcription factor AP-2 expression and function during mouse preimplantation development. Biol Reprod 75 : 324–333.
35. SatodaM, ZhaoF, DiazGA, BurnJ, GoodshipJ, et al. (2000) Mutations in TFAP2B cause Char syndrome, a familial form of patent ductus arteriosus. Nat Genet 25 : 42–46.
36. LiW, CornellRA (2007) Redundant activities of Tfap2a and Tfap2c are required for neural crest induction and development of other non-neural ectoderm derivatives in zebrafish embryos. Dev Biol 304 : 338–354.
37. RahimovF, MarazitaML, ViselA, CooperME, HitchlerMJ, et al. (2008) Disruption of an AP-2alpha binding site in an IRF6 enhancer is associated with cleft lip. Nat Genet 40 : 1341–1347.
38. BaiY, WangJ, MorikawaY, Bonilla-ClaudioM, KlysikE, et al. (2013) Bmp signaling represses Vegfa to promote outflow tract cushion development. Development 140 : 3395–3402.
39. YaylaogluMB, TitmusA, ViselA, Alvarez-BoladoG, ThallerC, et al. (2005) Comprehensive expression atlas of fibroblast growth factors and their receptors generated by a novel robotic in situ hybridization platform. Dev Dyn 234 : 371–386.
40. LuMF, PressmanC, DyerR, JohnsonRL, MartinJF (1999) Function of Rieger syndrome gene in left-right asymmetry and craniofacial development. Nature 401 : 276–278.
41. TurnerBC, ZhangJ, GumbsAA, MaherMG, KaplanL, et al. (1998) Expression of AP-2 transcription factors in human breast cancer correlates with the regulation of multiple growth factor signalling pathways. Cancer Res 58 : 5466–5472.
42. Rada-IglesiasA, BajpaiR, SwigutT, BrugmannSA, FlynnRA, et al. (2011) A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470 : 279–283.
43. LangmeadB, TrapnellC, PopM, SalzbergSL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 9
Nejčtenější v tomto čísle
- A Genome-Wide Systematic Analysis Reveals Different and Predictive Proliferation Expression Signatures of Cancerous vs. Non-Cancerous Cells
- Recent Acquisition of by Baka Pygmies
- The Condition-Dependent Transcriptional Landscape of
- Histone Chaperone NAP1 Mediates Sister Chromatid Resolution by Counteracting Protein Phosphatase 2A