
Mutations in Global Regulators Lead to Metabolic Selection during Adaptation to Complex Environments
Changing environmental conditions are the norm in biology. However, understanding adaptation to complex environments presents many challenges. For example, adaptation to resource-rich environments can potentially have many successful evolutionary trajectories to increased fitness. Even in conditions of plenty, the utilization of numerous but novel resources can require multiple mutations before a benefit is accrued. We evolved two bacterial species isolated from the gut of healthy humans in two different, resource-rich media commonly used in the laboratory. We anticipated that under weak selection the population would evolve tremendous genetic diversity. Despite such a complex genetic background we were able to identify a strong degree of parallel evolution and using a combination of population proteomic and population genomic approaches we show that two global regulators, arcA and rpoS, are the principle targets of selection. Up-regulation of the different metabolic pathways that are controlled by these global regulators in combination with up-regulation of transporters that transport nutrients into the cell revealed increased use of the novel resources. Thus global regulators can provide a one-step model to shift metabolism efficiently and provide rapid a one-step reprogramming of the cell metabolic profile.
Published in the journal:
. PLoS Genet 10(12): e32767. doi:10.1371/journal.pgen.1004872
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004872
Summary
Changing environmental conditions are the norm in biology. However, understanding adaptation to complex environments presents many challenges. For example, adaptation to resource-rich environments can potentially have many successful evolutionary trajectories to increased fitness. Even in conditions of plenty, the utilization of numerous but novel resources can require multiple mutations before a benefit is accrued. We evolved two bacterial species isolated from the gut of healthy humans in two different, resource-rich media commonly used in the laboratory. We anticipated that under weak selection the population would evolve tremendous genetic diversity. Despite such a complex genetic background we were able to identify a strong degree of parallel evolution and using a combination of population proteomic and population genomic approaches we show that two global regulators, arcA and rpoS, are the principle targets of selection. Up-regulation of the different metabolic pathways that are controlled by these global regulators in combination with up-regulation of transporters that transport nutrients into the cell revealed increased use of the novel resources. Thus global regulators can provide a one-step model to shift metabolism efficiently and provide rapid a one-step reprogramming of the cell metabolic profile.
Introduction
Adaptation to novel environments can proceed either through many mutations with small effects or through few mutations with large effects [1]. Adaptation to complex environments is the norm in biology, but a clear understanding of the adaptive processes employed by organisms in ecologically diverse environments is challenging. Ecological complexity can arise from increased species diversity, spatial or temporal heterogeneity or different resources. The availability of countless resources in complex environments can lead to a rapid and substantial increase in genetic diversity that can obscure broader biochemical principles of adaptation. For example, if resources are varied and plentiful, the population can accumulate mutations in genes that are not essential for survival in the selective environment [2], [3]. Thus, in nutrient-rich environments, specialists with narrower niches can persist by using alternative resources without necessarily improving their fitness relative to the ancestor. Adaptation then occurs through specialization via fitness improvements or via metabolic erosion possibly without fitness improvements relative to the ancestor [3]–[5]. We hypothesized that, despite the tremendous opportunities for increased genetic diversity under conditions of plenty, consistent adaptive responses could be observed as parallel evolution across and within independently evolved populations. We further reasoned that such general and consistent adaptive responses could be driven by global metabolic regulators to provide an efficient reprogramming of metabolic networks with a minimal number of steps.
Experimental evolution of model organisms under novel conditions is a versatile approach for understanding the evolutionary dynamics of adaptation and the functional constraints that shape the physiological evolvability of an organism. Typically, microbial model organisms are selected for adaptation to a single or a few distinct resources [6]–[11], to antibiotics [12], [13], or to temperature [14], [15]. Experiments in such relatively simple selective environments have shown that during adaptation to a single resource, the evolving population typically climbs a single-peak fitness landscape in an incremental manner with diminishing returns epistasis [16]–[18]. Even in such simple environments, resource partitioning or spatial or temporal heterogeneity can lead to the evolution of different specialists and complex ecological interactions [10], [11], [19]–[26].
To better understand the evolutionary and adaptive dynamics in ecologically complex environments, we focused on resource availability and conducted selection experiments in very nutrient-rich conditions. Unlike adaptation to a single limiting resource that is often conceptualized as a single fitness peak, a wealth of resources will potentially present abundant peaks in the fitness landscape. Because resources may differ only slightly, the selection differences can be very small and are reflected as very modest fitness peaks. As a consequence, selection will be weak and lead to an increase in genetic variation through the accumulation of mutations, though the fixation of any specific mutation would be unlikely. Identifying adaptive mutations in such genetically diverse populations can be difficult. However, we reasoned that adaptive mutations should evolve repeatedly in independent populations, while neutral or deleterious mutations should not show any discernible degree of parallelism. Parallel evolution has been readily observed in nature regardless of the ecological complexity [11], [27], [28]. Such adaptive, phenotypic convergence can be based on different underlying genetic changes, such that adaptive, parallel changes can occur in the same gene, or in different genes of the same pathway or functional group [12], [15].
In an environment where selection is weak and selective differences are small, it is hard to imagine a scenario where an individual can quickly accumulate mutations along a multi-protein pathway that will lead to increased fitness. Instead, mutations in regulatory genes such as transcriptional or translational regulators that can simultaneously affect many operons or entire pathways could produce much larger benefits and circumvent potential complications from epistatic interactions among different mutations. One example of a gene with such large pleiotropic effects is the global stress response regulator rpoS, which is activated during late exponential and stationary phase [29]. Mutations in rpoS are often among the first mutations to evolve during experimental evolution of E. coli [30] and have been routinely observed in different selective conditions [31]–[33]. These mutations lead to changes in the stress response and nutrient acquisition [34], change the stress induced mutation rates [35], [36] and increase long-term viability [37]. Knocking out rpoS leads to a down-regulation of the starvation stress response and efflux pumps, and to increased nutrient efficiency via the up-regulation of proteins such as porins. The trade-off of stress resistance and nutritional competence was termed the SPANC balance (self preservation and nutritional competence) by Ferenci [34]. While the prevalence of rpoS knockout mutants is low in wild isolates [38], considerable variation in rpoS expression has been observed among wild strains [39]. We therefore hypothesized that mutations in global regulators could be especially beneficial in ecologically complex, nutrient-rich environments that induce weak selection.
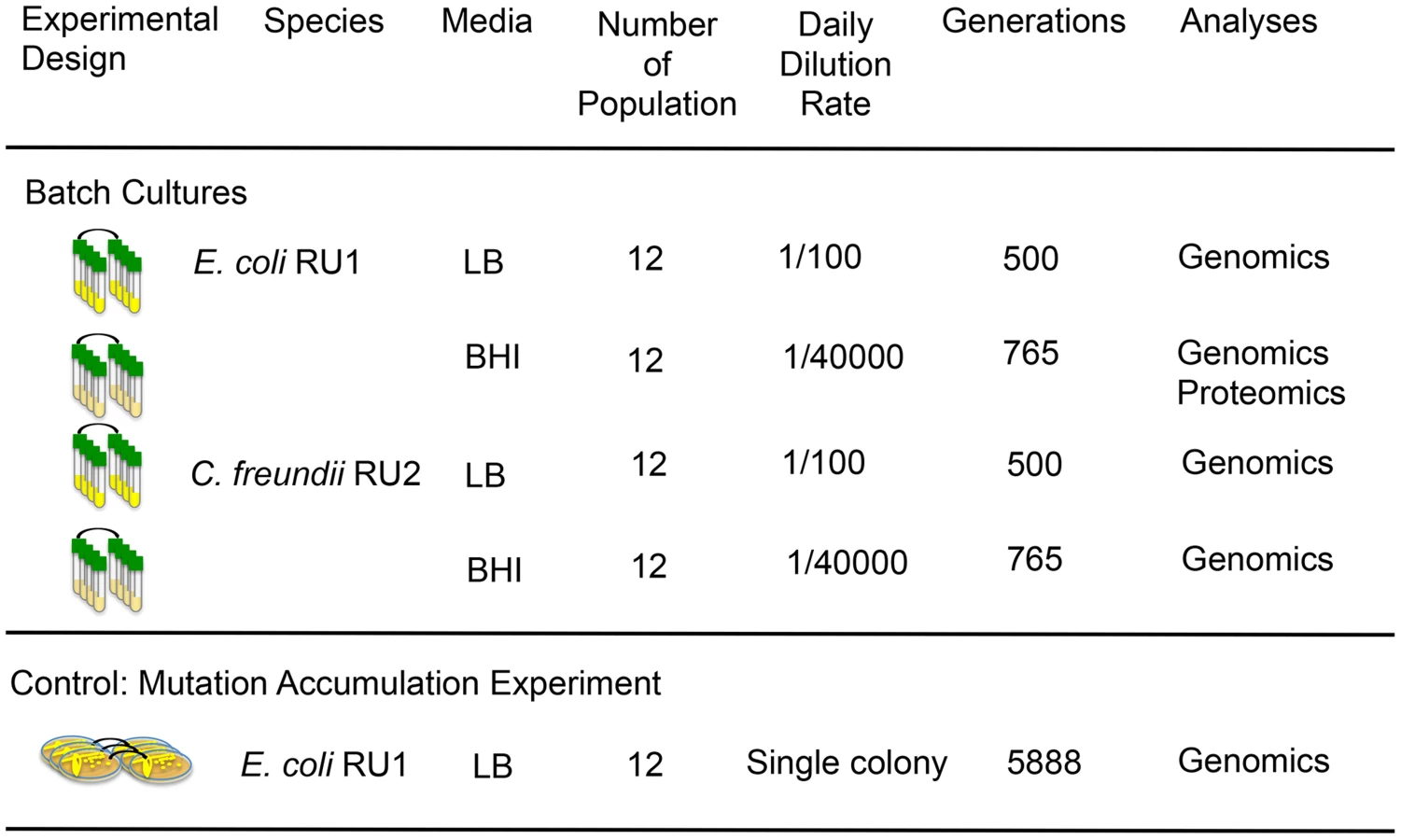
In contrast to previous experiments where laboratory adapted strains were evolved in rich media commonly used in the laboratory [40], we isolated naïve strains from their natural habitat, the gut of healthy humans, and used rich media as novel, selective environments. In the gut, E. coli and C. freundii interact with hundreds of other strains as well as their human host. More than 90% of these commensal gut bacteria are anaerobes, which convert non-digestible complex carbohydrates into short-chain fatty acids and produce the simple mono - and di-saccharides favored by E. coli [41], [42]. In return, facultative anaerobes like E. coli and C. freundii play an important role in maintaining a low-oxygen environment. We chose two complex media (BBL BHI and LB Miller) that differed primarily in the composition and amounts of amino acids, vitamins and carbohydrates (S1 Text). In addition to the populations selected in complex media (two genotypes in two environments resulting in four treatments, Fig. 1), we also performed a control experiment, where we reduced selection as much as possible by daily bottlenecking the population to a single cell, the approach commonly used for mutation accumulation (MA) experiments [43]–[45]. In mutation accumulation experiments, independently evolved lines are expected to accumulate a random set of mutations with little to no parallel evolution.
We used a powerful combination of population proteomics and population genomics to reveal phenotypic convergence to identify potential biochemical mechanisms of adaptation. Despite the complexities imposed by the tremendous amount of underlying genetic diversity accumulated during adaptation to complex nutrient rich environments, we identified clear genomic signatures of adaptation across and within independently evolved populations. Strikingly, changes in the global regulators arcA and rpoS evolved consistently, while changes in other global regulators were largely absent. Subsequent proteomic and carbohydrate analysis of populations adapting to BHI showed increased abundance of enzymes associated with the TCA cycle and amino acid metabolism to make use of abundant amino acids, resulting in the secretion of the polyamine putrescine as a nitrogen sink. Thus in complex media, where the adaptive landscape is relatively flat and has many potential modest peaks requiring many changes to produce a substantive increase in fitness, the “go to” strategy may be to use global regulators such as arcA and rpoS to overcome epistasis by changing the regulation of whole metabolic pathways in a coordinated manner. This allows populations to rapidly reprogram resource utilization and to adapt to complex fitness landscapes in a much smaller number of moves.
Results
Parallel adaptation to complex environments is readily observed across populations despite a large amount of genetic variation
We isolated E. coli RU1 and C. freundii RU2 (S1 Figure) de novo from the gut flora of healthy humans using only two overnight growths on agar to reduce any selection prior to our adaptation experiment. For each species, 12 populations were established and allowed to adapt over a minimum of 500 generations to media and conditions that were very rich in resources and substantially different than the environment of the human gut (Fig. 1). We chose LB and BHI as novel environments since they are likely to be very different from the gut resource base, but still support robust growth of both ancestral strains (S1 Table).
Over the course of the selection experiments, we observed modest but significant changes in various fitness components, consistent with adaptation under weak selection conditions. While lag time decreased in most treatments, maximum growth remained constant in LB and decreased in BHI (S1 Table). In all but one treatment, LB-evolved C. freundii, the populations significantly increased their stationary phase density (OD600) indicating enhanced abilities to utilize the resources efficiently. While we observed significant changes, the differences between ancestor and evolved populations were modest and consistent with permissive environments inducing weak selection pressures (S1 Table, S1 Text).
As a consequence of weak selection, considerable genetic variation evolved over the course of our experiment. This was evident both at the phenotypic as well as the genotypic level. We observed considerable phenotypic variation in colony size and in the ability to utilize arabinose (Fig. 2), in redox activity, in exopolysaccharide content and loss of motility (S2A-D Figure, respectively). Interestingly, evolved E. coli populations had at least one colony among the 8 colonies assessed per population that lost motility, but only one single C. freundii colony out of all the colonies assessed (two sets of 96 colonies in total) lost motility (S1 Text).

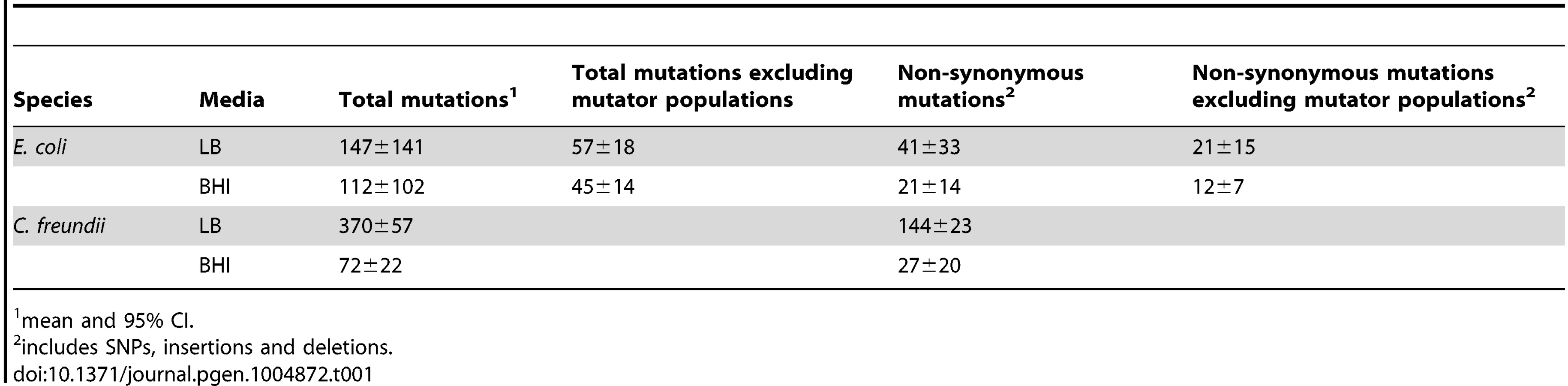
To assess the evolved genetic variation and identify adaptive mutations, we sequenced the evolved populations and identified mutations in coding regions that occurred at a minimum frequency of 0.05 in a population. Two BHI-evolved populations (E.coli BHI5 and C. freundii BHI20) could not be aligned properly and were omitted from further genomic analyses. The number of mutations ranged from 29 to 725 per population. The number of mutations per population did not differ significantly among the E. coli populations evolved in LB or BHI (Fig. 3; Table 1). Two populations evolved to become mutators in each environment (LB4, LB11, BHI6, and BHI10, S1 Text). If the mutator populations are excluded, the average number of mutations between the LB and BHI-evolved populations was reduced, although there was still no significant difference in the number of mutations across environments. In contrast, the LB-evolved C. freundii populations accumulated significantly more mutations than the BHI-evolved populations. Overall, the number of mutations differed significantly both between media and species (Full factor ANOVA with Media and Strain as fixed factors: Media F1,42 = 15.1, p = 0.0004, Species F1,42 = 4.5, p = 0.039, Media×Species F1,42 = 9.5, p = 0.0036). While synonymous mutations can have fitness effects [46], [47], we focused our analyses on non-synonymous mutations, which include SNPs, insertions, and deletions. The number of non-synonymous mutations ranged from 5 to 198 in a population, with more mutations arising in the LB than in BHI in the E. coli population (Fig. 3, Table 1). Excluding the mutator populations reduced the average non-synonymous mutations per population further (LB: 21±15, BHI: 12±7). Among the C. freundii populations, the average number of non-synonymous mutations was significantly higher in the LB-evolved populations than in the BHI-evolved populations. Again, we observed significant differences among media and species (Media = F1,42 = 23.8, p<0.0001, Species: F1,42 = 13.2, p = 0.0007, Media×Species: F1,42 = 9.4, p = 0.0037).
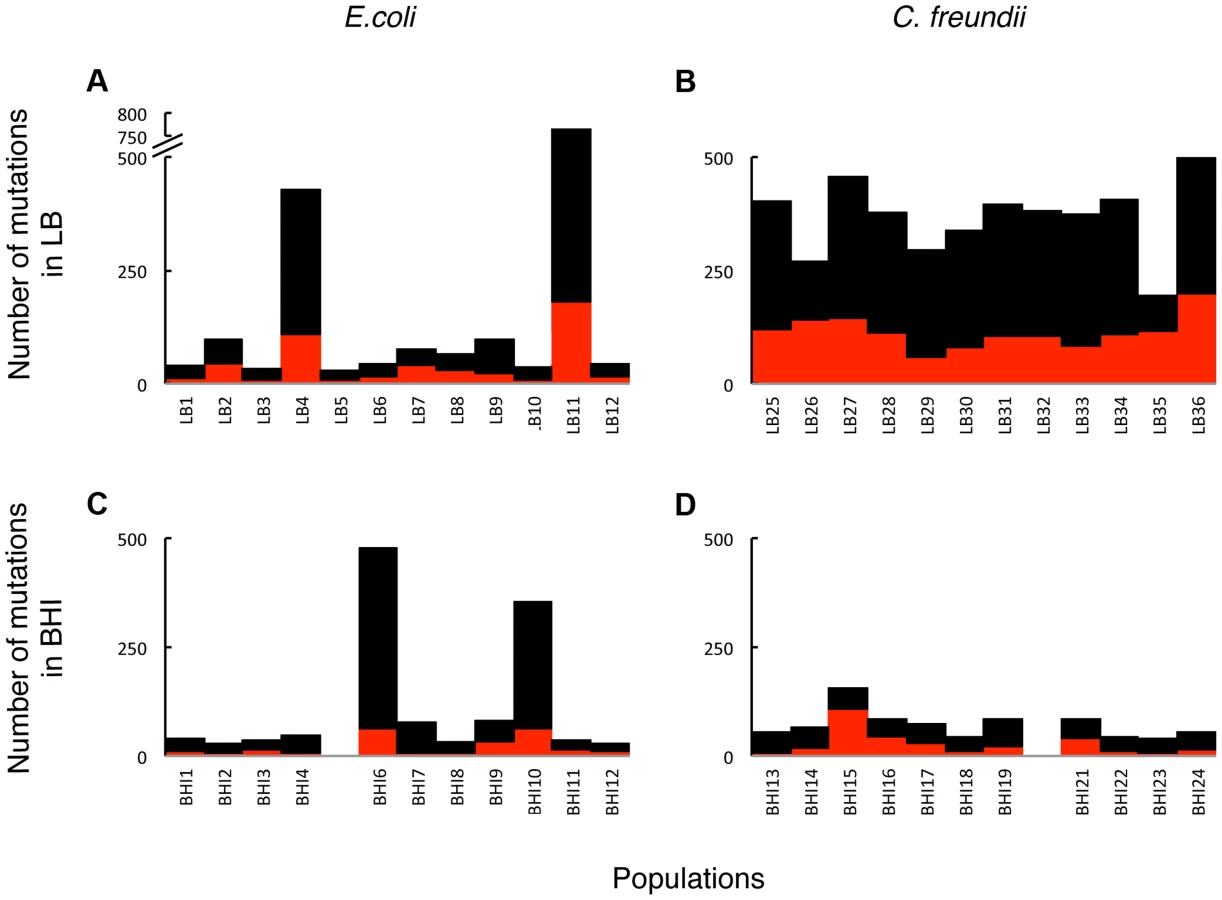
The accumulation of largely non-adaptive mutations complicates the identification of adaptive changes within a single, polymorphic population. However, we expected that important adaptive trajectories would exist across independently evolved populations. Therefore, we focused our analyses on parallel, non-synonymous mutations that evolved consistently across populations, both within and across species and media. Most mutations occurred in only one or a few populations, consistent with the presence of large non-adaptive genetic variation (Fig. 4). Strong parallel evolution across environments and species occurred in the global regulator arcA, which acquired mutations in all 24 LB-evolved populations and in nine of eleven BHI-evolved E. coli populations (Fig. 5). The probability that mutations evolved in the same gene in 24 independently evolved populations at random is very small considering that E. coli RU1 and C. freundii RU2 had 4565 and 5068 annotated genes, respectively (p = (1/4565)12 *(1/5068)12) and suggests that these mutations are adaptive. Surprisingly, C. freundii adaptation to BHI did not implicate arcA. The second most commonly mutated gene was the global stress response regulator rpoS [29], [48], which had mutations in nine of 23 E. coli and eight of 23 C. freundii populations. None of the mutation accumulation lines had mutations in arcA or rpoS.


Besides arcA and rpoS, only a few other genes acquired mutations in replicated populations, and unlike arcA and rpoS, these other mutations occurred only within a treatment and not across species and selective environment. No other mutation evolved with any degree of parallelism in the E. coli populations. Among the C. freundii populations, mutations in a gene encoding the Valine-Glycine Repeat Protein G, vgrG, a homolog to the tailspike of bacteriophage T4, and in a gene encoding adenosylmethionine-8-amino-7-oxononanoate aminotransferase, an enzyme involved in biotin biosynthesis, occurred in almost all evolved populations in both selective environments, while mutations in different mobile elements, in the peptide deformylase, the methionyl-tRNA formyltransferase and the sodium/glutamate symport protein were only common among the LB-evolved C. freundii populations (for further details see S1 Text).
Mutations in arcA and rpoS drive adaptation to the complex selective environments
The highly parallel evolution of mutations in arcA and rpoS combined with their global effects suggests that these mutations are driving adaptation in these complex selective environments. Mutations in these genes were very common with multiple different alleles co-occurring within the same population. The cumulative frequencies of arcA mutations in particular reached high frequencies in LB (average 0.75±0.08 (mean and 95%CI) across 24 populations (Fig. 6). In only one population did we observe the fixation of a single arcA mutation (LB5).

We observed 46 unique mutations in arcA, both within and among populations (Fig. 7). Strikingly, none of these mutations introduced a stop codon or a frame shift; 44 of these 46 unique mutations were non-synonymous substitutions, one mutation resulted in a C-terminal deletion of three amino acids, and one mutation was an insertion of one amino acid. To independently confirm some of the mutations identified from population genomics, we directly sequenced arcA from eight single colonies isolated from six of the LB-evolved E. coli populations. We were able to confirm eleven of the 46 mutations identified in the whole population samples (L2: I122M, Y137C, I22S, L4: N116T, L6: R16H, A76T; L8: E94K; L10: A25T, G59S, L50Q; and L12: L50Q). In addition, we identified two new mutations (L8: G62D and 218ΔTPE; and L12: G62D) suggesting that our cutoff of 5% in the deep sequencing population analysis still missed many arcA variants. Each clone had only one mutation in arcA, suggesting that the one mutation was sufficient to achieve a beneficial effect.

The response regulator arcA is part of the two-component arcAB signal transduction system. The membrane bound sensor kinase ArcB phosphorylates ArcA (ArcA-P) in response to a variety of environmental challenges to maintain redox and metabolic homeostasis [49]–[51]. Mutations in the sensor kinase arcB could also affect the regulation of arcA. We therefore examined the whole genome sequencing data for mutations in the sensor kinase arcB and found mutations in six of the BHI-evolved (BHI1, BHI2, BHI3, BHI4, BHI9 and BHI10) and in one of the LB-evolved E. coli populations (LB8). Among the C. freundii populations, mutations in arcB were less frequent, with only one population evolving mutations in arcB in each environment (LB26 and BHI24). As with arcA, all mutations were non-synonymous, though one mutation did result in a frame shift. Importantly, neither arcA nor arcB evolved mutations in the mutation accumulation lines suggesting that changes in arcAB are under selection and not random.
The amount of variation in rpoS mutations was not quite as dramatic as in arcA, but substantial nonetheless. In the LB-evolved E. coli populations we predominately observed SNPs, while in the BHI-evolved E. coli populations mutations in rpoS resulted in stop codons, large deletions or frame shift mutations, suggesting a loss of function (S3 Figure). Among the five LB-evolved and three BHI-evolved C. freundii populations with mutations in rpoS, only one population (LB31) acquired a mutation resulting in a stop codon while the rest acquired substitutions. To confirm some of these mutations independently, we sequenced rpoS from eight single colony isolates for three of the LB-evolved populations (LB6, LB8 and LB12) and confirmed the A199T mutation in population LB8 as well as a new rpoS mutation (Y283C) in LB6.
Unlike arcA, we observed many loss of function mutations in rpoS, which is consistent with previous selection experiments where knock-out mutations evolved relatively rapidly under different selective conditions [31]–[33]. RpoS levels could also be attenuated through changes in the regulation of its expression. The expression of rpoS is repressed during exponential growth and activated upon starvation during late exponential and stationary phase using different transcriptional and translational mechanisms. Transcription of rpoS is up-regulated through spoT/(p)ppGpp and BarA/UvrY and repressed by arcAB, while translation is up-regulated by two small RNAs, DsrA and RprA and repressed by a third small RNA ArcZ [29]. Presumably, mutations in any of these genes could also affect the up-regulation of rpoS and lead to reduced expression, resulting in similar phenotypes as the knockout mutants. We did not observe any mutations in spoT, though mutations in this gene evolve readily in minimal media [6], [52], [53]. Four of the LB-evolved C. freundii populations (LB27, LB32, LB35, LB36), however, had a frame shift mutation in barA/uvrY. Three of these populations did not have a mutation in rpoS, suggesting that the effect of loss of function mutations in barA/uvrY could lead to reduced transcription of rpoS and result in a similar phenotype to rpoS knockout mutations. Mutations in the small RNAs DsrA and RprA could also lead to reduced translation of rpoS and result in a similar phenotype as rpoS knockout mutants. We looked for mutations in DsrA and RprA in E. coli and did not detect any mutations in these small RNAs. While we also searched for the small RNAs in C. freundii using sequences retrieved from Citrobacter, we were unable to locate the two small RNAs in our reference genome.
Linking mutations to functional changes across regulatory sequences is more difficult as it requires excellent annotation and understanding of the transcriptional regulators. Nonetheless, it is certainly likely that changes in regulatory regions could provide adaptive changes. To test whether arcA or rpoS expression were altered by mutations within their regulatory regions, we examined the 500 nucleotides preceding the start codons of these two genes and found no mutation in any of the evolved populations or the MA lines.
Evolved populations showed improved utilization of abundant amino acids through up-regulation of enzymes in the TCA cycle
Population-level proteomic analysis of the BHI-evolved E. coli populations showed significant changes in protein abundance between the ancestor and evolved populations as well as a remarkable degree of parallelism among the evolved populations. We observed significant and highly parallel decreases of ArcA abundance in the evolved populations and increases of proteins of the TCA cycle, amino acid metabolism and transporters (Fig. 8). We identified 4469 unique peptides in 39 samples (ancestor and twelve evolved populations with three replicates each), corresponding to 488 proteins (see S1 Text for more details). Quantitative analysis of the 488 proteins revealed 166 proteins that were significantly different between the ancestor and the evolved populations (p<0.01; log2-fold change>±0.7). Of those, 58 proteins decreased and 108 proteins increased significantly over the course of the selection experiment (S2 Table). All observed proteins associated with the TCA cycle (aconitate hydratase, isocitrate dehydrogenase, 2-oxoglutarate dehydrogenase, succinyl-CoA ligase, succinate dehydrogenase, fumarate hydratase, and malate dehydrogenase) and glyoxylate shunt (isocitrate lyase) significantly increased (Fig. 8). The up-regulation of the TCA cycle and the decrease in ArcA is consistent with previous studies that observed increased flux through the TCA cycles in arcA knock-out mutants [50], [54]. RpoS was not detected in our proteomic analyses in any of the samples and therefore we cannot draw any conclusions about its abundance.

Mutations in rpoS reduce starvation stress response and increase nutrient acquisition and utilization
Reduced starvation stress is associated with increased nutrient acquisition and metabolism and reduced stress responses [34] – conditions we expected in our resource rich environments. Altered resource utilization can be developed by increasing C/N acquisition via the up-regulation of porins, which allow nutrients to flow through the outer membrane, and a concomitant decrease of the effluxers that provide protection from toxins during starvation stress [34], [55]. Consistent with increased C/N acquisition from amino acids and small peptides, we observed significant increases of peptidases (alpha-aspartyl dipeptidase peptidase E, peptidase B, and methionine aminopeptidase) and of proteins associated with ABC transporter systems responsible for the transport of amino acids or peptides (glutamate aspartate (GltI), lysine-arginine-ornithine (ArgT), glutamine (GlnH), histidine (HisJ) and oligopeptide sytems (OppA)), and carbohydrates (galactose/methyl galactoside (MglB), ribose (RbsB), maltose/maltodextrin (MalE)).
Genomic analyses suggested some loss of function among specific efflux pumps consistent with low stress conditions. We identified mutations in several RND efflux pumps including cmeA and cmeB that are found in multiple copies within the E. coli and C. freundii genomes. Mutations in cmeA and cmeB ranged in frequency from 0.05 to 0.41 and occurred in 15 of 24 populations across both environments and organisms, suggesting that decreases in CmeA and CmeB function are under selection during adaptation. Eight out of twelve LB-evolved E. coli populations acquired mutations in either cmeA or cmeB. Mutations in cmeA that resulted in likely loss of function (all either insertions, deletions or SNPs to stop codons) evolved in four populations (LB5, LB9, LB11 and LB12), while five different populations had mutations in one of the cmeB copies (LB1, LB4, LB5, LB7 and LB8). Mutations in cmeA and cmeB were not as prevalent among the LB-evolved and completely absent among the BHI-evolved C. freundii populations. One LB-evolved C. freundii population acquired a substitution in cmeB (LB25), one had an insertion (LB34) and a third population had an insertion in the RND efflux transporter (LB2). One MA line acquired an insertion in both cmeA and cmeB. The cmeA and B mutations resulting in loss of function mutations support the SPANC balance conditions of low starvation stress and increased nutrient uptake and decreased efflux.
While rpoS mutants are predicted to have a decreased stress response, our proteomic data (Fig. 8, S2 Table) suggested that changes in the stress response, were more nuanced and that some stress pathways, such as the starvation and acid stress responses were up-regulated while others such as protein unfolding stress were diminished. Across the twelve BHI-evolved E. coli populations, we observed decreases in chaperones associated with protein folding stress (DnaK) and heat shock proteins (GroES), and in proteins involved in the oxidative stress response through glutathione (glutaredoxin 2 and 3, and glutathione peroxidase). Conversely, proteins involved in the oxidative stress response through thioredoxin (thioredoxin reductase, universal stress proteins AEFG, superoxide dismutase, glutathione S-transferase), acid stress (HdeAB) and another heat shock protein (HchA/Hsp31) increased.
E. coli populations adapted to BHI show increased putrescine secretion consistent with altered nitrogen homeostasis
The up-regulation of the TCA cycle and the increased amino acid acquisition and metabolism could lead to increased production of ammonia or polyamines to maintain nitrogen homeostasis. To test this hypothesis, we determined whether the evolved populations produced and secreted more polyamines or ammonia. We began by testing the pH of spent media after 24 hours of growth, and observed a significant increase in pH from 8.1 to 8.4 in the BHI-evolved E. coli populations (S1 Table) compared to the ancestor. Similarly, the pH also increased significantly in the LB-evolved C. freundii populations, but not in the other two treatments. Increased pH is consistent with proteomic data that suggested significantly increased TCA cycle activity and amino acid metabolism. The breakdown of amino acids by the decarboxylation of ornithine or of arginine to agmatine can result in the production of the polyamine putrescine [56]. Indeed, putrescine was significantly higher in the spent media of the BHI-evolved E. coli population compared to the ancestor (t-test: t = 6.08, df = 22, p<0.0001), but not in the cell extract (t-test: t = 0.3, df = 20, p = 0.76)(S4 Figure). While we only have quantitative data for the BHI-evolved E. coli populations, the odor of the C. freundii populations at stationary phase suggested that they, too, all produced and secreted increased amounts of putrescine.
Discussion
The natural world presents organisms with complex and variable environments. Resources often range from rich and varied, to poor and limiting. An abundance of new, but usable, resources may induce very weak selection pressures and result in a complex multi-peaked adaptive landscape, where most single nucleotide changes or mutations to specific components of a metabolic pathway would not generate enough fitness gains to facilitate rapid success. One path to adaptation would be to change the global regulators that control the management of metabolic flux to provide a simple “one-step” adaptation for the entire physiology of the organism. Adaptation by such a one-step mechanism constitutes a higher order ‘metabolic selection’ that allows the organism to capture larger gains in fitness and circumvent the complications of multi-gene epistasis. To test this idea, we used wild isolates of E. coli and C. freundii and investigated their adaptive responses under weak selection as they were moved from their natural habitat, the human gut, to a rich and markedly different resource base. We found that, as expected, weak selection induced by rich complex environments resulted in large genetic variation and likely allowed even deleterious mutations to persist. We observed a striking diversity of phenotypes across all populations. Underlying genetic diversity could be observed readily as a tremendous variation in colony sizes and physical appearance on different indicator agar plates, as well as loss of motility (Fig. 2 and S1 Text). Whole population sequencing of the evolved populations identified arcA and rpoS as the targets of selection. Whole population proteomics of the BHI-evolved E. coli populations showed that these populations up-regulated several amino acid and carbohydrate transporters to move abundant nutrients into the cell and up-regulated the proteins of the TCA cycle needed to use them efficiently (Fig. 9). We also observed significantly increasing putrescine production consistent with increased utilization of amino acids as C/N sources. The combination of whole genome sequencing and whole population proteomics proved to be a powerful approach for the mapping of genotypic changes to biochemical mechanisms that, in turn, produce altered phenotypes.

To identify common adaptive strategies, we focused on mutations that arose repeatedly in independently evolved populations. The two most common targets of selection were arcA and rpoS, both global regulators with large pleiotropic effects. Our overall picture for adaptation is one in which the adaptation through mutations in the global regulators arcA and rpoS drive the large metabolic changes essential for adaptation to nutrient rich environments under these selection conditions. Both of these global regulators affect up to 10% of the genes within their host genome [29], [49], [50]. Mutations in arcA evolved consistently in the majority of the populations. ArcA consists of two domains, the receiver domain (residues 1–123) that includes the site of phosphorylation (Asp54) [57]-[59] and a DNA binding domain (124–238) [59]. Phosphorylation stimulates formation of an ArcA-P dimer that binds to a variety of specific DNA sequence motifs with high affinity to repress or activate transcription of up to 229 operons directly or indirectly in response to the environment [49], [50], [54]. The majority of the diverse mutations in arcA were found in the receiver domain (Fig. 7). Mapping these mutations onto the three-dimensional crystal structure (1XHE) of the receiver domain revealed that the vast majority of mutations are in surface positions and solvent accessible loops (S5 Figure), with only a few mutations mapping to the hydrophobic core. While it is likely that some of these mutations could result in a complete loss of function, the likelihood that all 46 mutations do so is slim. It is interesting that all but two mutations in arcA were SNPs and the two exceptions were an insertion and a deletion at the C-terminus and likely resulting in a largely functional protein. This suggests that a complete loss-of-function that eliminates arcA function is not as beneficial as modifying its activity and is consistent with our proteomic data that showed a decrease in ArcA levels rather than a complete absence in ArcA. Mutations within the receiver domain could decrease ArcA signaling by a number of mechanisms including: 1) reducing ArcA stability; 2) decreasing the extent of ArcA phosphorylation during signaling; 3) increasing the rate of dephosphorylation; or 4) decreasing the extent of phosphorylation-dependent oligomerization. Only a few mutations mapped to the DNA binding domain but these could also alter ArcA function by decreasing DNA binding or any of the aforementioned mechanisms for altered receiver domain function. Mutations to arcA were also observed in previous selection experiments, notably during adaptation to glucose limited media [60], [61] and LB [40], [62]. Interestingly, in those studies mutations in arcA only evolved in the aerobic cultures, suggesting that oxygen deprivation and anaerobic stress were not the driver for arcA mutations.
Unlike arcA, it was striking that all mutations to rpoS in the BHI-evolved E. coli populations and only one mutation in the other three treatments appeared to produce a complete loss of function. One explanation could be the differences in the composition of the media, mainly the presence of glucose in BHI. Carbohydrates and glucose in particular are utilized first before switching to amino acids [63]–[65]. This is reflected in a diauxic growth pattern with a second lag and exponential growth phase. The depletion of carbohydrates could induce the RpoS regulated starvation stress response, which might delay the transition to amino acid metabolism. Losing RpoS function might therefore be beneficial to a fast switching response to other nutrient sources. In LB we never observed diauxic growth patterns, which is consistent with the low concentration of carbohydrates in the media. Mutations in rpoS have also been shown to improve longevity during stationary phase [37]. The populations reached stationary phase within eight hours in LB and as a consequence, these populations remained in stationary phase much longer. SNPs that improve persistence in stationary phase could therefore be selected in LB.
Proteomic analyses of BHI-evolved E. coli populations showed increased abundance of enzymes of the TCA cycle, which is consistent with the known phenotypes of arcA and rpoS knockout mutants based on flux analyses [50], [54], [66]. Knocking out either rpoS or arcA resulted in two-fold increases in metabolic flux through the TCA cycle, while knocking out arcB did not have an effect on the flux through the TCA cycle, consistent with our observation that mutations in arcB were rare [54], [66]. While proteomics and flux analyses use different measurements, the effect of mutations in arcA and rpoS seem very similar. This is even more remarkable considering that our proteomic analyses are based on polymorphic populations and the small changes in proteomics are population averages.
In addition, we see strong up-regulation of peptidases, amino acid metabolism, amino acid transporters and oxidative stress responses, indicating that these populations are metabolizing the media at an increased rate (Fig. 8 and 9, S2 Table). Again, this pattern is consistent with previous metabolic studies [54], [66]. ArcA has been shown to either directly or indirectly regulate many operons such as amino acid and polyamine production, beta-oxidation of fatty acid and operons encoding pathways for the utilization of aromatic compounds and peptides [49]. Knock-out mutants of rpoS not only had increased TCA cycle activity but also increased amino acid metabolism [66].
The arginine, asparagine and glutamine metabolism pathways and the TCA cycle feed into the urea cycle. While we did not observe changes in expression of enzymes of the urea cycle, we observed a significant increase in the production and secretion of putrescine in BHI-adapted population. The polyamine putrescine can be produced during the breakdown of amino acids by the decarboxylation of ornithine or by the decarboxylation of arginine via agmatine [56]. Arginine decarboxylase has been proposed to localize in the cell envelope, where it converts exogenous arginine to putrescine via agmatine [67]. Because we observed a significant decrease of agmatinase in the evolved populations, it seems more likely that putrescine is produced from ornithine, a component of the urea cycle. In the adapted populations, putrescine might be acting as a nitrogen sink for catabolism of amino acids via the urea cycle or, alternatively, may help with the increased oxidative stress resulting from increased metabolism. There are many roles for polyamines in metabolism including as C/N sources, oxidative stress response, and signaling [68]–[73], so an understanding of increased secretion of putrescine by our adaptive populations will require further biochemical studies.
We expected that parallel evolution could also evolve along pathways and lead to phenotypic convergence by mutating different genes along the pathways [12], [15]. Indeed, we did see some convergence in the regulation of rpoS, where not all populations had mutations in rpoS and instead had mutations in genes that regulate the expression of rpoS. We interrogated the genomic data for such phenotypic convergence by analyzing parallel evolution for different functional categories, but did not observe any evidence for phenotypic convergence at different levels of increasing complexity (see S1 Text, S6–S8 Figure). Nonetheless, we did see a high degree of parallel evolution among the populations when we grouped the proteins with significant changes to pathways. This is even more remarkable considering that we performed our proteomic analyses on whole, polymorphic populations and as such only measure the average change of a populations compared to the ancestor. This parallelism shows a clear response to selection. The global up-regulation of metabolism and the lack of clear phenotypic convergence of mutations along the pathway further support our assertion that mutations in arcA and rpoS drive adaptation to the rich selective conditions and lead to the observed metabolic changes. These relatively small changes observed in the proteomic data also further support our previous observations that very small biochemical changes can have large fitness effects [74] and are likely very relevant to adaptation in nature.
The consistent evolution of mutations in global regulators arcA and rpoS that each affect expression of about 10% of the genome supports the model in which adaptation evolved through the evolution of a few mutations with large beneficial effects. Instead of acquiring beneficial mutations in every gene involved in the TCA cycle and the various amino acid metabolism pathways, acquiring mutations in regulators affecting all these genes simultaneous is undoubtedly more efficient. Selection studies in other, less complex environments also implicated mutations in global regulators that lead to stable coexistence and polymorphic populations suggesting that mutations in global regulators are beneficial in different environments [60], [61]. In contrast to those earlier studies under glucose-limiting conditions, mutations to arcA and rpoS arose very rapidly and repeatedly across our populations. Diverse phenotypes and genotypes suggest that our populations are polymorphic as well. For example, the sequential utilization of carbohydrates and amino acids [64], [65] could select for different mutations specialized to either carbohydrate or amino acid utilizations, similar to the stable coexistence of different temporal specialist observed in previous studies [10], [19], [20], [25]. It is possible that arcA and rpoS mutations provide selective benefits in different phases of the growth cycle and the coexistence of these mutations in the populations could be an indication of such temporal and potentially nutritional specialization.
By investigating adaptation of wild organisms to resource rich environments we have shown that adaptation occurs within five hundred generations through mutations in global regulators, leading to increased rates of metabolism. Mutations to global regulators might be more common during selection in permissive environments. At niche boundaries such as a thermal limits, single mutations could greatly increase the fitness of an organism [75], mostly because fitness at the niche boundaries can be dramatically reduced compared to the niche optimum [76], [77] and thus a small number of mutations or even a single mutation can result in substantial fitness gains.
Both rpoS and arcA have been linked to virulence [78], [79]. Our findings suggest that it is important to appreciate the role of laboratory adaptation when evaluating strains for pathogenicity, especially in light of the fact that rpoS loss-of-function mutations evolve readily in the laboratory but are not found in natural populations [38]. The transition from the natural habitat to laboratory conditions suggests how we might improve experimental evolution studies that require handling and adaptation of pathogens as well as provide a starting point for forensic attribution of strains during outbreaks of novel pathogens.
Materials and Methods
Strain isolation and identification
E.coli RU1 (hence forth referred to as E. coli) and C. freundii RU2 (hence forth referred to as C. freundii) were isolated from the stool of healthy humans (S1 Text, S5 Figure). To minimize any potential for adaptation during the isolation process, we plated stool samples on MacConkey agar plates. After a single overnight growth, half of a single colony was flash frozen at −80°C in Trypticase soy broth with 20% Glycerol (BD, USA) and the other half was used for phenotypic strain characterization. Strain identification by 16S sequencing was done from the frozen sample. All experimental evolution studies started from the frozen primary isolates by using a single clonalized colony derived from the initial snap frozen isolate. The identity of the wild "un-adapted" strains was based on the results of API 20 E (Biomerieux, USA) test strips and species-specific PCR (using forward primer: AGAGTTTGATCMTGGCTCAG, reverse primer: GWATTACCGCGGCKGCTG).
Selection experiments
Single colony transfers (mutation accumulation control experiments)
Twelve clones of E. coli RU1 were evolved independently for 200 days by daily transfers of a single, randomly chosen colony to a fresh LB plate (Fig. 1). Every 15 days, a sample of the transferred colony was frozen at −80°C. To calculate the generations per transfer, we assessed the number of cells in a colony after 24 hours of growth on LB agar and calculated the number of doublings as log2(number of cells/colony) at generation 0, 105 and 200 for two randomly chosen lines. The average number of generations per transfer was calculated as 29.4, which results in 5888 generations over 200 single cell bottlenecks.
Population flask transfers (experimental evolution of populations)
A single colony from the frozen cultures of E. coli RU1 and C. freundii RU2 were each used to inoculate twelve independent cultures in Luria Broth Miller (LB, BD) and brain-heart infusion (BBL BHI, BD) (Fig. 1, S3 Table). Populations evolved in liquid media were transferred as described previously [10], [11]. LB-evolved populations were grown in 10 ml liquid LB Miller broth in 25mm test tubes at 250 rpm and 37°C, and transferred daily by 100-fold dilutions into fresh media (∼6.6 generations/day) for a total of 75 days or 500 generations. Every 15 days, we froze a sample of the populations at −80°C for further analyses. The BHI-evolved populations were cultured in 10 ml of BHI broth under the same conditions as the LB cultures, but transferred daily by 40,000-fold dilutions into fresh media (∼15.2 generation/day) for a total of 50 days or 765 generations. This dilution was chosen to achieve the same effective population size at transfer of 105 cells/ml as in a parallel experiment performed with Yersinia pestis to allow direct comparisons. The BHI-evolved populations were frozen at −80°C after 25 and 50 days.
Phenotypic assays
Assays were performed either at the population level or the single colony level. Cells or populations were grown in their selective media (LB or BHI) and grown in liquid media or plated on agar plates made with their selective media, unless otherwise stated. Adaptation to the selective environments was assessed as changes in lag time and growth rate by measuring OD600 over 24 hours of growth in liquid media following Walkiewicz et al. [74]. To test for changes in the pH of spent media, we grew the populations to stationary phase and measured the pH of the media after removing the cells. To test for genetic variation within the populations, we plated the populations at low density on tetrazolium arabinose plates and observed considerable variation in both colony size and in the ability to utilize arabinose. We plated the populations on the selective media supplemented with agar and isolated eight randomly chosen colonies from each of the 12 populations per treatment. These test sets of 96 individual isolates per species and environment were used for three phenotypic assays: 1) the redox state by plating on methylene blue (0.065 g/liter); 2) differences in exopolysaccharides by plating on Congo Red (0.15 g/liter); and 3) loss of motility by plating cells on soft agar (0.25% DIFCO). For more information see S1 Text.
Sequencing, alignment and mutation identification
Ancestral genomes
Genomic DNA of the two ancestral strains was isolated using the Ultra Clean Microbial DNA isolation Kit (Mo Bio Laboratories, Inc.) and sequenced on the Roche 454 platform according to standard sequencing methods. The reference sequences of E. coli RU1 and C. freundii RU2 were assembled de novo using Newbler v2.6 and annotated using RAST [80]. The annotated genes were grouped by function and assigned to subsystems using SEED [81] (S6 Figure).
Evolved lines and whole populations sequencing
Genomic DNA of single clones (MA lines: MA1–MA12) and whole populations (LB1–LB12, LB25–LB36, and BHI1–BHI24) was isolated as described for the ancestors and sequenced on Illumina HiSeq in 100 bp paired-end reads. Reads were aligned to the reference sequence using the breseq-0.24.rc6 pipeline (with options –j2 –c –p)[82]. Mutations that occurred at a minimum frequency of 0.05, which is an order of magnitude above the overall error rate as determined following Saxer et al. [44] were considered. Because we used such a low acceptance threshold for mutations in polymorphic populations, we used additional measures to reduce the false positive rate, that can be due to errors in the reference sequence, duplications that were incorrectly resolved in the reference sequence, repeat regions or sequence properties, or methylation patterns [83]. Mutations arise at random, and under a model of selective neutrality, the frequency of a mutation should be proportional to the time it arose, while the frequency of a beneficial mutation should be determined by the time it arose and by its selection coefficient. Therefore, mutations that arise at random cannot be expected to occur at almost the same frequency in replicated polymorphic populations. In clonal samples, polymorphic sites could arise when a single colony is grown to high density for DNA isolation. However, such mutations should be rare within a single sample and very unlikely to occur in multiple independent clonal samples. In addition, the relaxed selection experience by the MA lines makes it unlikely that the same mutation would arise in multiple independently evolved lines. While beneficial mutations can evolve more than once in mutation accumulation experiments, it is unlikely that such mutations would occur at the exact same site (nucleotide) at approximately the same frequencies in independently evolved mutation accumulation lines, and that these same mutations would be polymorphic in a clonal sample (i.e. at lower frequency than 0.8–0.9 among reads aligning to that site).
We aligned the reads of the clonal MA lines as if they were polymorphic populations and identified mutations that occurred in 4 or more lines. These sites were removed from BHI - and LB-evolved populations. We performed the same removal procedure for mutations that occurred in one to twelve MA lines and observed a significant change in the number of mutations removed based on their presence in three or four MA lines. This resulted in the removal of 233 mutations that occurred in three treatments (LB, BHI and MA), 72 mutations that occurred in two treatments (MA and LB, or MA and BHI) and 3 mutations that occurred in the MA lines. The majority of these mutations were in intergenic regions, in transposable elements, hypothetical genes and in the large and small ribosomal subunits. In addition, we removed mutations in genes encoding for genes annotated as core proteins, the Magnesium and cobalt transport protein, CorA and tRNA dihydrouridine synthase A.
Based on our experience with E. coli and the work of Lang et al. [84], we used a different approach to filter out false positive mutations for C. freundii. Adaptive mutations that arise in multiple populations evolve at different times and in populations with a different assortment of genotypes, which is reflected in different frequencies in the populations. The frequencies of false positive mutations that are a result of systematic errors on the other hand will be very similar among populations. Therefore, we filtered out any site for which the frequencies in BHI and LB did not significantly differ based on Levene's test [85].
Proteomic and putrescine analyses of evolved populations
Cell extracts and spent media samples were prepared by growing the evolved populations and ancestral populations to stationary phase, separating the cells from the supernatant by centrifugation and inactivating remaining cells with 70% (v/v) ethyl alcohol.
LC-MS analysis of proteins
Protein was extracted by re-suspending cell pellets in 50 µL lysis buffer [6 M urea (Sigma U-0631) and 14.3 mM 2-mercaptoethanol (Sigma M6240) in 100 mM triethylammonium bicarbonate (TEAB), pH 9 (Sigma T7408)]. The extracted protein was digested with trypsin and the tryptic peptides analyzed by LC-MS using the accurate mass and time (AMT) tag proteomics approach [86]. See S1 Text for details. The software program DAnTE [87] was employed to perform an abundance roll-up procedure to convert peptide abundance information to protein information, thereby inferring protein abundances. ANOVA analyses were applied to protein abundance data sets (p-value ≤0.01) to identify statistically significant differences in protein expression levels (S2 Table, S9 Figure).
Putrescine secretion
Putrescine concentration in the cell extract and in spent media was measured during GC-MS analyses of carbohydrates. For specific details see S1 Text.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LandeR (1983) The Response to Selection on Major and Minor Mutations Affecting a Metrical Trait. Heredity 50 : 47–65.
2. CooperVS, LenskiRE (2000) The population genetics of ecological specialization in evolving Escherichia coli populations. Nature 407 : 736–739.
3. LeibyN, MarxCJ (2014) Metabolic erosion primarily through mutation accumulation, and not tradeoffs, drives limited evolution of substrate specificity in Escherichia coli. PLoS Biol 12: e1001789.
4. PengL, ShimizuK (2003) Global metabolic regulation analysis for Escherichia coli K12 based on protein expression by 2-dimensional electrophoresis and enzyme activity measurement. Appl Microbiol Biotechnol 61 : 163–178.
5. FutuymaDJ, MorenoG (1988) The Evolution of Ecological Specialization. Annual Review of Ecology and Systematics 19 : 207–233.
6. BarrickJE, LenskiRE (2009) Genome-wide mutational diversity in an evolving population of Escherichia coli. Cold Spring Harbor symposia on quantitative biology 74 : 119–129.
7. ElenaSF, LenskiRE (2003) Evolution experiments with microorganisms: The dynamics and genetic bases of adaptation. Nature Reviews Genetics 4 : 457–469.
8. LenskiRE, RoseMR, SimpsonSC, TadlerSC (1991) Long-term experimental evolution in Escherichia coli I: Adaptation and divergence during 2,000 generations. American Naturalist 138 : 1315–1341.
9. LenskiRE, TravisanoM (1994) Dynamics of adaptation and diversification: A 10,000-generation experiment with bacterial populations. Proceedings of the National Academy of Sciences of the United States of America 91 : 6808–6814.
10. SaxerG, DoebeliM, TravisanoM (2009) Spatial structure leads to ecological breakdown and loss of diversity. Proceedings of the Royal Society B-Biological Sciences 276 : 2065–2070.
11. SaxerG, DoebeliM, TravisanoM (2010) The Repeatability of Adaptive Radiation During Long - Term Experimental Evolution of Escherichia coli in a Multiple Nutrient Environment Plos One. 5: e14184.
12. MillerC, KongJY, TranTT, AriasCA, SaxerG, et al. (2013) Adaptation of Enterococcus faecalis to Daptomycin Reveals an Ordered Progression to Resistance. Antimicrobial Agents and Chemotherapy 57 : 5373–5383.
13. ToprakE, VeresA, MichelJB, ChaitR, HartlDL, et al. (2012) Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nature Genetics 44 : 101–U140.
14. CounagoR, WilsonCJ, PenaMI, Wittung-StafshedeP, ShamooY (2008) An adaptive mutation in adenylate kinase that increases organismal fitness is linked to stability-activity trade-offs. Protein Engineering Design & Selection 21 : 19–27.
15. TenaillonO, Rodriguez-VerdugoA, GautRL, McDonaldP, BennettAF, et al. (2012) The Molecular Diversity of Adaptive Convergence. Science 335 : 457–461.
16. ChouHH, ChiuHC, DelaneyNF, SegreD, MarxCJ (2011) Diminishing Returns Epistasis Among Beneficial Mutations Decelerates Adaptation. Science 332 : 1190–1192.
17. KhanAI, DinhDM, SchneiderD, LenskiRE, CooperTF (2011) Negative Epistasis Between Beneficial Mutations in an Evolving Bacterial Population. Science 332 : 1193–1196.
18. WiserMJ, RibeckN, LenskiRE (2013) Long-Term Dynamics of Adaptation in Asexual Populations. Science 342 : 1364–1367.
19. FriesenML, SaxerG, TravisanoM, DoebeliM (2004) Experimental evidence for sympatric ecological diversification due to frequency-dependent competition in Escherichia coli. Evolution 58 : 245–260.
20. SpencerCC, SaxerG, TravisanoM, DoebeliM (2007) Seasonal resource oscillations maintain diversity in bacterial microcosms. Evolutionary Ecology Research 9 : 775–787.
21. HellingRB, VargasCN, AdamsJ (1987) Evolution of Escherichia coli during growth in a constant environment. Genetics 116 : 349–358.
22. RosenzweigRF, SharpRR, TrevesDS, AdamsJ (1994) Microbial evolution in a simple unstructured environment: Genetic differentiation in Escherichia coli. Genetics 137 : 903–917.
23. StewartFM, LevinBR (1973) Partitioning of resources and outcome of interspecific competition: Model and some general considerations. American Naturalist 107 : 171–198.
24. HabetsMGJL, RozenDE, HoekstraRF, de VisserJAGM (2006) The effect of population structure on the adaptive radiation of microbial populations evolving in spatially structured environments. Ecology Letters 9 : 1041–1048.
25. RozenDE, LenskiRE (2000) Long-term experimental evolution in Escherichia coli VIII: Dynamics of a balanced polymorphism. American Naturalist 155 : 24–35.
26. RaineyPB, TravisanoM (1998) Adaptive radiation in a heterogeneous environment. Nature 394 : 69–72.
27. RundleHD, NagelL, BoughmanJW, SchluterD (2000) Natural selection and parallel speciation in sympatric sticklebacks. Science 287 : 306–308.
28. LososJB, JackmanTR, LarsonA, de QueirozK, Rodriguez-SchettinoL (1998) Contingency and determinism in replicated adaptive radiations of island lizards. Science 279 : 2115–2118.
29. BattestiA, MajdalaniN, GottesmanS (2011) The RpoS-Mediated General Stress Response in Escherichia coli. Annual Review of Microbiology Vol 65 65 : 189–213.
30. Notley-McRobbL, SeetoS, FerenciT (2003) The influence of cellular physiology on the initiation of mutational pathways in Escherichia coli populations. Proceedings of the Royal Society B-Biological Sciences 270 : 843–848.
31. BlankD, WolfL, AckermannM, SilanderOK (2014) The predictability of molecular evolution during functional innovation. Proc Natl Acad Sci U S A 111 : 3044–3049.
32. MaharjanR, SeetoS, Notley-McRobbL, FerenciT (2006) Clonal adaptive radiation in a constant environment. Science 313 : 514–517.
33. EydallinG, RyallB, MaharjanR, FerenciT (2013) The nature of laboratory domestication chagnes in freshly isolated Escherichia coli strains. Environmental Microbiology 16 : 611–905.
34. FerenciT (2005) Maintaining a healthy SPANC balance through regulatory and mutational adaptation. Molecular Microbiology 57 : 1–8.
35. BjedovI, TenaillonO, GerardB, SouzaV, DenamurE, et al. (2003) Stress-induced mutagenesis in bacteria. Science 300 : 1404–1409.
36. Al MamunAM, LombardoMJ, SheeC, LisewskiAM, GonzalezC, et al. (2012) Identity and Function of a Large Gene Network Underlying Mutagenic Repair of DNA Breaks. Science 338 : 1344–1348.
37. FinkelSE (2006) Long-term survival during stationary phase: evolution and the GASP phenotype. Nature Reviews Microbiology 4 : 113–120.
38. SnyderE, GordonDM, StoebelDM (2012) Escherichia coli Lacking RpoS Are Rare in Natural Populations of Non-Pathogens. G3-Genes Genomes Genetics 2 : 1341–1344.
39. KingT, IshihamaA, KoriA, FerenciT (2004) A regulatory trade-off as a source of strain variation in the species Escherichia coli. Journal of Bacteriology 186 : 5614–5620.
40. Puentes-TellezPE, HansenMA, SorensenSJ, van ElsasJD (2013) Adaptation and heterogeneity of Escherichia coli MC1000 growing in complex environments. Applied and environmental microbiology 79 : 1008–1017.
41. Maltby R, Leatham-Jensen MP, Gibson T, Cohen PS, Conway T (2013) Nutritional Basis for Colonization Resistance by Human Commensal Escherichia coli Strains HS and Nissle 1917 against E. coli O157:H7 in the Mouse Intestine. Plos One 8.
42. ToppingDL, CliftonPM (2001) Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiological Reviews 81 : 1031–1064.
43. LynchM (2010) Evolution of the mutation rate. Trends in Genetics 26 : 345–352.
44. Saxer G, Havlak P, Fox SA, Quance MA, Gupta S, et al. (2012) Whole Genome Sequencing of Mutation Accumulation Lines Reveals a Low Mutation Rate in the Social Amoeba Dictyostelium discoideum. Plos One 7.
45. Halligan DL, Keightley PD (2009) Spontaneous mutation accumulation studies in evolutionary genetics. Annual Review of Ecology Evolution and Systematics 40.
46. PlotkinJB, KudlaG (2011) Synonymous but not the same: the causes and consequences of codon bias. Nature Reviews Genetics 12 : 32–42.
47. AnderssonSGE, KurlandCG (1990) Codon Preferences in Free-Living Microorganisms. Microbiological Reviews 54 : 198–210.
48. Hengge-Aronis R (2002) Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol Mol Biol Rev 66: : 373–395, table of contents.
49. ParkDM, AkhtarMS, AnsariAZ, LandickR, KileyPJ (2013) The bacterial response regulator ArcA uses a diverse binding site architecture to regulate carbon oxidation globally. PLoS Genet 9: e1003839.
50. Shimizu K (2013) Metabolic Regulation of a Bacterial Cell System with Emphasis on Escherichia coli Metabolism. ISRN Biochemistry 2013.
51. LevanonSS, SanKY, BennettGN (2005) Effect of oxygen on the Escherichia coli ArcA and FNR regulation systems and metabolic responses. Biotechnology and Bioengineering 89 : 556–564.
52. Herron MD, Doebeli M (2013) Parallel Evolutionary Dynamics of Adaptive Diversification in Escherichia coli. PloS Biology 11.
53. CooperTF, RozenDE, LenskiRE (2003) Parallel changes in qene expression after 20,000 generations of evolution in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America 100 : 1072–1077.
54. PerrenoudA, SauerU (2005) Impact of global transcriptional regulation by ArcA, ArcB, Cra, Crp, Cya, Fnr, and Mlc on glucose catabolism in Escherichia coli. Journal of Bacteriology 187 : 3171–3179.
55. AmaralL, MartinsA, SpenglerG, MolnarJ (2014) Efflux pumps of Gram-negative bacteria: what they do, how they do it, with what and how to deal with them. Front Pharmacol 4 : 168.
56. TaborCW, TaborH (1985) Polyamines in microorganisms. Microbiol Rev 49 : 81–99.
57. IuchiS, LinEC (1992) Purification and phosphorylation of the Arc regulatory components of Escherichia coli. J Bacteriol 174 : 5617–5623.
58. GeorgellisD, KwonO, De WulfP, LinEC (1998) Signal decay through a reverse phosphorelay in the Arc two-component signal transduction system. J Biol Chem 273 : 32864–32869.
59. Toro-RomanA, MackTR, StockAM (2005) Structural analysis and solution studies of the activated regulatory domain of the response regulator ArcA: A symmetric dimer mediated by the alpha 4-beta 5-alpha 5 face. Journal of Molecular Biology 349 : 11–26.
60. PhilippeN, CrozatE, LenskiRE, SchneiderD (2007) Evolution of global regulatory networks during a long-term experiment with Escherichia coli. Bioessays 29 : 846–860.
61. PlucainJ, HindreT, Le GacM, TenaillonO, CruveillerS, et al. (2014) Epistasis and allele specificity in the emergence of a stable polymorphism in Escherichia coli. Science 343 : 1366–1369.
62. Puentes-TellezPE, KovacsAT, KuipersOP, van ElsasJD (2014) Comparative genomics and transcriptomics analysis of experimentally evolved Escherichia coli MC1000 in complex environments. Environmental Microbiology 16 : 856–870.
63. Neidhardt FC, Curtiss III R, Ingraham JL, Lin ECC, Low KB, et al. (1996) Escherichia coli and Salmonella: Cellular and Molecular Biology. Washington, D.C.: ASM Press.
64. BaevMV, BaevD, RadekAJ, CampbellJW (2006) Growth of Escherichia coli MG1655 on LB medium: monitoring utilization of sugars, alcohols, and organic acids with transcriptional microarrays. Applied microbiology and biotechnology 71 : 310–316.
65. BaevMV, BaevD, RadekAJ, CampbellJW (2006) Growth of Escherichia coli MG1655 on LB medium: monitoring utilization of amino acids, peptides, and nucleotides with transcriptional microarrays. Applied microbiology and biotechnology 71 : 317–322.
66. RahmanM, HasanMR, ObaT, ShimizuK (2006) Effect of rpoS gene knockout on the metabolism of Escherichia coli during exponential growth phase and early stationary phase based on gene expressions, enzyme activities and intracellular metabolite concentrations. Biotechnol Bioeng 94 : 585–595.
67. BuchJK, BoyleSM (1985) Biosynthetic arginine decarboxylase in Escherichia coli is synthesized as a precursor and located in the cell envelope. J Bacteriol 163 : 522–527.
68. TkachenkoAG, NesterovaL (2001) The role of putrescine in of oxidative stress defense genes expression regulation in Escherichia coli. Mikrobiologiia 70 : 168–173.
69. ChattopadhyayMK, TaborCW, TaborH (2009) Polyamines Are Not Required for Aerobic Growth of Escherichia coli: Preparation of a Strain with Deletions in All of the Genes for Polyamine Biosynthesis. Journal of Bacteriology 191 : 5549–5552.
70. SchneiderBL, HernandezVJ, ReitzerL (2013) Putrescine catabolism is a metabolic response to several stresses in Escherichia coli. Mol Microbiol 88 : 537–550.
71. SturgillG, RatherPN (2004) Evidence that putrescine acts as an extracellular signal required for swarming in Proteus mirabilis. Mol Microbiol 51 : 437–446.
72. BernierSP, LetoffeS, DelepierreM, GhigoJM (2011) Biogenic ammonia modifies antibiotic resistance at a distance in physically separated bacteria. Mol Microbiol 81 : 705–716.
73. ReitzerL (2003) Nitrogen assimilation and global regulation in Escherichia coli. Annual Review of Microbiology 57 : 155–176.
74. WalkiewiczK, CardenasASB, SunC, BacornC, SaxerG, et al. (2012) Small changes in enzyme function can lead to surprisingly large fitness effects during adaptive evolution of antibiotic resistance. Proceedings of the National Academy of Sciences of the United States of America 109 : 21408–21413.
75. Pena MI, Davlieva M, Bennett MR, Olson JS, Shamoo Y (2010) Evolutionary fates within a microbial population highlight an essential role for protein folding during natural selection. Molecular Systems Biology 6.
76. Quance MA, Travisano M (2009) Effects of temperature on the fitness cost of resistance to bacteriophage T4 in Escherichia coli. Evolution.
77. KniesJL, IzemR, SuplerKL, KingsolverJG, BurchCL (2006) The genetic basis of thermal reaction norm evolution in lab and natural phage populations. Plos Biology 4 : 1257–1264.
78. DongT, SchellhornHE (2010) Role of RpoS in Virulence of Pathogens. Infection and Immunity 78 : 887–897.
79. SenguptaN, PaulK, ChowdhuryR (2003) The global regulator arcA modulates expression of virulence factors in Vibrio cholerae. Infection and Immunity 71 : 5583–5589.
80. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, et al. (2008) The RAST server: Rapid annotations using subsystems technology. Bmc Genomics 9.
81. OverbeekR, DiszT, StevensR (2004) The SEED: A peer-to-peer environment for genome annotation. Communications of the Acm 47 : 46–51.
82. DeatherageDE, BarrickJE (2014) Identification of Mutations in Laboratory-Evolved Microbes from Next-Generation Sequencing Data Using breseq. Methods Mol Biol 1151 : 165–188.
83. RossMG, RussC, CostelloM, HollingerA, LennonNJ, et al. (2013) Characterizing and measuring bias in sequence data. Genome Biol 14: R51.
84. LangGI, RiceDP, HickmanMJ, SodergrenE, WeinstockGM, et al. (2013) Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature 500 : 571–574.
85. Whitlock MC, Schluter D (2009) The analysis of biological data. Greenwood Village, CO: Roberts and Company Publishers.
86. ZimmerJS, MonroeME, QianWJ, SmithRD (2006) Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom Rev 25 : 450–482.
87. PolpitiyaAD, QianWJ, JaitlyN, PetyukVA, AdkinsJN, et al. (2008) DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics 24 : 1556–1558.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 12
Nejčtenější v tomto čísle
- Tetraspanin (TSP-17) Protects Dopaminergic Neurons against 6-OHDA-Induced Neurodegeneration in
- Maf1 Is a Novel Target of PTEN and PI3K Signaling That Negatively Regulates Oncogenesis and Lipid Metabolism
- The IKAROS Interaction with a Complex Including Chromatin Remodeling and Transcription Elongation Activities Is Required for Hematopoiesis
- Echoes of the Past: Hereditarianism and