
G×G×E for Lifespan in : Mitochondrial, Nuclear, and Dietary Interactions that Modify Longevity
It is widely recognized that mitochondrial function plays an important role in longevity and healthy aging. Considerable attention has been focused on the extension of longevity by caloric or dietary restriction and mutations that alter this process, and these interventions commonly are associated with shifts in mitochondrial function. While the genetic bases of these effects are the focus of much interest, relatively little effort has been directed at understanding the role that mitochondrial DNA (mtDNA) polymorphisms play in the diet restriction response. This work presents a comprehensive effort to quantify the effects of mtDNA variants, nuclear genetic variants and dietary manipulations on longevity in Drosophila, with a focus on testing for the importance of the interactions among these factors. We found that mitochondrial genotypes can have significant effects on longevity and the diet restriction response but these effects are highly dependent on nuclear genetic (G) background and the specific diet environment (E). For example, a mitochondrial haplotype that shortens lifespan in one nuclear background or diet regime shows no such effect when the genetic background or diet regime is changed. Our experiments indicate that identifying individual mitochondrial, nuclear or dietary effects on longevity is unlikely to provide general results without quantifying the prevalent mitochondrial × nuclear × diet (G×G×E) interactions.
Published in the journal:
. PLoS Genet 10(5): e32767. doi:10.1371/journal.pgen.1004354
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004354
Summary
It is widely recognized that mitochondrial function plays an important role in longevity and healthy aging. Considerable attention has been focused on the extension of longevity by caloric or dietary restriction and mutations that alter this process, and these interventions commonly are associated with shifts in mitochondrial function. While the genetic bases of these effects are the focus of much interest, relatively little effort has been directed at understanding the role that mitochondrial DNA (mtDNA) polymorphisms play in the diet restriction response. This work presents a comprehensive effort to quantify the effects of mtDNA variants, nuclear genetic variants and dietary manipulations on longevity in Drosophila, with a focus on testing for the importance of the interactions among these factors. We found that mitochondrial genotypes can have significant effects on longevity and the diet restriction response but these effects are highly dependent on nuclear genetic (G) background and the specific diet environment (E). For example, a mitochondrial haplotype that shortens lifespan in one nuclear background or diet regime shows no such effect when the genetic background or diet regime is changed. Our experiments indicate that identifying individual mitochondrial, nuclear or dietary effects on longevity is unlikely to provide general results without quantifying the prevalent mitochondrial × nuclear × diet (G×G×E) interactions.
Introduction
The extension of longevity by dietary or caloric restriction (DR or CR) has been demonstrated in a wide range of organisms, suggesting evolutionarily conserved pathways that regulate this response [1], [2], [3], [4], [5]. However, dissecting the genetic and cellular mechanisms of DR remains a great challenge. Several pathways have been identified that mediate the DR response such as the insulin [6], [7], mTOR [8], [9], [10], AMPK [11], [12], [13], [14] and sirtuin pathways [15], [16], [17], [18]. These pathways intersect through downstream targets and involve mechanisms of regulation and feedback. Thus, the control of DR involves a network of pathways rather than any one critical pathway, which underlies the challenge of identifying singular genetic and biochemical mechanisms for the DR response. While efforts to identify single genes, pathways or nutrients that extend longevity have been productive, studies that focus on the interaction among relevant factors have received relatively little attention [19]. Longevity, and its extension by genetic or dietary interventions, are complex phenotypes, and it is increasingly apparent that gene-by-gene (G×G) and gene-by-environment (G×E) interactions are fundamental components of these kinds of traits [20], [21]. Given the complexity of the genetic, nutritional and physical environments in which most organisms live, including humans, explicit studies of the role of these interaction effects are critically important to determine the generality of single factor analyses.
The mitochondrion has received increasing attention as a nexus for regulation of the longevity extending effects associated with DR [10], [22], [23], [24], [25]. Mitochondria generate critical energy stores in the form of ATP and NADH that can promote cellular maintenance and longevity, but also generate reactive oxygen species (ROS) that can cause cellular damage and senescence. As a hub for input from multiple pathways affecting longevity, mitochondria provide a compelling target for studies seeking to understand gene-by-gene and gene-by-environment effects in aging. Mitochondrial function and biogenesis are dependent on genes encoded in both the mtDNA and the nuclear chromosomes. Animal mtDNA encodes 13 protein coding subunits of oxidative phosphorylation complexes of the electron transport chain (ETC) with more than 70 remaining subunits encoded by nuclear genes and imported in to the mitochondrion [26], [27]. mtDNA also encodes a minimal set of RNAs (2 rRNAs and 22 tRNAs) that comprise the translation machinery inside the mitochondrion, with the remaining protein components, such as ribosomal proteins and tRNA synthetases, encoded by nuclear genes and imported into the organelle. Thus, mitochondrial function depends critically on the coordination of mitochondrial and nuclear encoded components and this co-dependence provides the basis of mitochondrial-nuclear interactions (hereafter mitonuclear interactions), which is one mode of gene-by-gene interaction, or epistasis, that may affect fitness, disease and aging [28], [29], [30].
Specific genes have been identified that play important roles in modifying mitochondrial function in response to DR. The TOR pathway regulates nutrient sensing and protein translation [31], [32], and 4E-BP regulates differential translation of mRNAs encoding proteins targeted for mitochondria vs. cytosolic function [10]. The PGC-1α family of transcriptional co-activators also plays a critical role in regulating mitochondrial biogenesis with systemic and tissue-specific effects on longevity [22], [33]. Altered expression of specific subunits of electron transport chain (ETC) complexes can reduce the efficacy of longevity extension by DR [10], [34], suggesting that the coordination of nuclear and mtDNA-encoded components of mitochondrial function are important in mediating a proper response to DR. Genetic analyses of longevity extension by DR are examples of the more general problem of genotype-by-environment interaction, as the question concerns the identification of genes or alleles that generate a novel phenotype (longer life) in a novel environment (reduced protein or calories). Thus, mitochondrial regulation of DR or CR not only requires the complex mitonuclear interactions, but also involves the impact of the dietary environment on this gene-by-gene interaction. Collectively, this network can be summarized as a three-way interaction between mitochondrial genotype, nuclear genotype and dietary environment denoted as G×G×E in quantitative and ecological genetics.
In this study we explicitly unite all three of these issues: mtDNA variation, nuclear gene variation and dietary variation to test hypotheses about the generality of single-factor effects on longevity under DR and CR. By employing a 5-diet design, we attempt to make the distinction between CR and DR. In caloric restriction (CR) only the caloric content of the diet is reduced while the relative composition of macro-nutrients is kept untouched. In dietary restriction (DR), one focal nutrient of the diet, such as the proportion of protein or sugar, is restricted with the caloric content kept constant (see Figure 1). We address the questions of main effects and interaction effects directly using Drosophila genetic tools to jointly manipulate mitochondrial and nuclear gene mutations, and examine these individual and combined effects on lifespan in response to dietary alterations. We demonstrate that mtDNA genotype alters the dietary effect on lifespan in each of two commonly used nuclear genetic backgrounds of D. melanogaster. We also find that mtDNA haplotypes can alter the effect of SIR2 overexpression on life span in a diet dependent manner. These results demonstrate the three-way mito-nuclear-diet interaction as an important factor in shaping longevity outcomes. These results imply that an accurate assessment of mitochondrial factors cannot be made without the context of nuclear genetic background and dietary regime, each of which has been implicated as a single important factor influencing longevity.

Results
Caloric and dietary restriction have genotype-specific effects on longevity
For each of two axes of dietary manipulation, both the concentration axis and the composition axis (see Figure 1), there was a robust lifespan response in each of two standard D. melanogaster strains, w1118 and OreR (Figure 2 and Table 1). The concentration axis is made of food type I, II and III (Figure 2A and C). There is a clear pattern that mean lifespan declines with food concentration. Strain w1118 is shorter-lived than OreR (comparing Figure 2A, B to Fig. 2C, D), which also agrees with earlier work where w1118 is generally found to be a highly fecund but short-lived strain [35], [36].
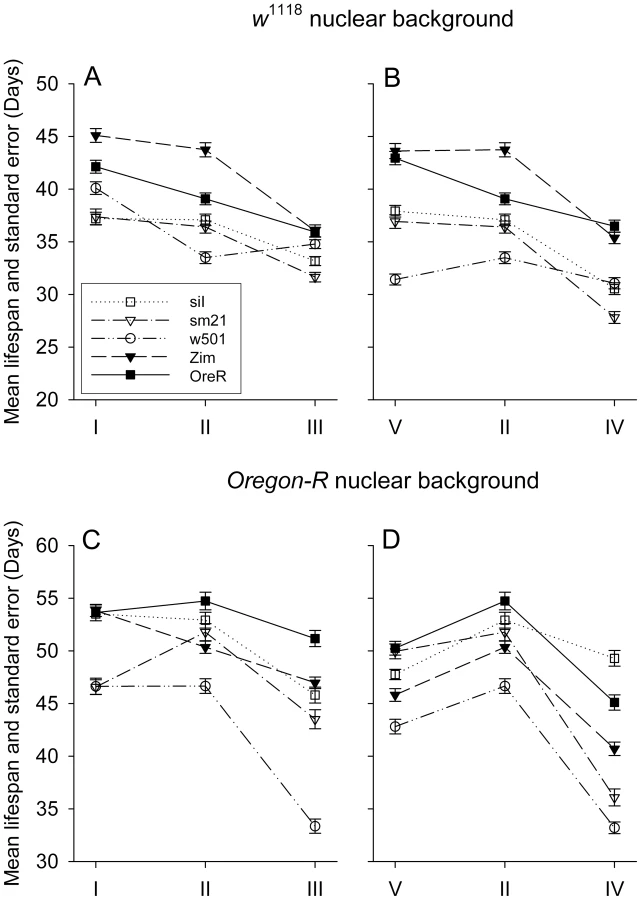

Food type V, II and IV make up a composition axis, where the sugar:yeast ratio is 3∶1, 1∶1 and 1∶3, respectively. In both nuclear genetic backgrounds, it is apparent that the low protein and the balanced diets results in long lifespan, with the high protein diet resulting in reduced lifespan (Figure 2B, D). Pooling data crosses all mitotypes and just focusing on the dietary affect, the estimates of hazard ratio of different diets are summarized in Table 1. It shows that compared to the median level diet (type II), the high yeast diet (type IV) and high caloric diet (type III) roughly double the chance of death (see Table 1, column ‘hazard ratio’: for w1118 the risks of death for diet types III and IV are 1.9442 and 2.0007 relative to type II; for OreR these values are 1.6809 and 2.1390).
In Drosophila studies, yeast is usually the only protein source, and increase in yeast concentration leads to high fecundity and short lifespan [36]. It has been further demonstrated that rather than yeast concentration alone, sugar/yeast ratio is what leads to the longevity and reproductive changes [37], [38]. Here we have a high sugar food, a high yeast food and a balanced food with their caloric content roughly being equal. While altering either the diet composition or concentration significantly affects lifespan (P<0.001 for both strains), different nuclear genotypes appear to have different optimal sugar/yeast ratios that result in maximum longevity (compare w1118 to OreR in Figure 2B and D; Note that this figure presents data for multiple mtDNA types, described in the following section).
mtDNA effects are restricted to individual haplotypes, not species-level divergence
To examine the effects of mtDNA genotype on longevity, we generated 18 ‘mito-nuclear’ genotypes by placing mtDNA from strains of Drosophila melanogaster and D. simulans onto controlled nuclear backgrounds of D. melanogaster (Oregon-R, w1118, SIR2 overexpression and control; see Methods). In each diet regime, on each nuclear background, the individual mitotypes show significant variation in longevity (P<2×10−16; Table 2, effect of mito; note: ‘nucleartype’ = nuclear genetic background; ‘mitotype’ = individual mtDNA genotype). Mitochondrial effects are largely due to variation among individual mitotypes while species-level mtDNA sequence divergence surprisingly contributes very little (compare D. simulans mitoypes siI, sm21, w501 to D. melanogaster mitotypes OreR and Zim; Figure 2 A–D). Although in some cases it appears D. simulans mitotypes are shorter lived than D. melanogaster mitotypes on specific diets (w1118 background under CR, Figure 2A), this species divergence effect is not significant across all diets given the magnitude of within-species variation observed. Comparing a model where individual mitotypes are treated separately to a model where mtDNA variation is treated as a species divergence effect with individual mitotypes nested within species divergence, the latter model is not significantly better in the OreR nuclear background (table 2, χ2 = 0.1113, n.s), indicating the lack of support for an mtDNA species-divergence effect. In the w1118 nuclear background, the species effect (χ2 = 3.7314, P = 0.053) is marginally significant. While the support for a species-level effect of mtDNA on longevity is again not strong, this effect does appear to be stronger in w1118 than in the OR nuclear background. The variation among mitotypes for longevity is, however, very significant in each nuclear background (table 2, χ2 = 156.07 for OreR and χ2 = 556.76 for w1118, d.f. = 2, P<0.001 for both).

Dietary effects are modulated by mitochondrial genotypes and this genotype-by-environment interaction (G×E; technically a mitotype-by-environment (M×E) interaction) is also a big contributor to survivorship. We found dietary effects to be variable in different mitotypes (Table 2, χ2 = 149.68 in the OreR nucleartype background and χ2 = 45.813 in the w1118 nucleartype background, d.f. = 1, both P<0.001). However, the same is not true for species mtDNA divergence: dietary effect is virtually not altered by the species-level mtDNA divergence effect on either nuclear background (table 2, χ2 = 0.0129 for OreR and χ2 = 0.0001 for w1118, d.f. = 1, both insignificant). The use of independent contrast approaches, or scaling for mtDNA branch length does not alter the outcome of these analyses (data not shown). Together these results indicate that mtDNA polymorphisms have a more pronounced effect on longevity than the species-level mtDNA divergence of ∼100 amino acids and a total of 571 fixed nucleotide substitutions between the mtDNAs of these two species.
A two-nucleotide mito-nuclear epistasis shortens lifespan
In the statistical analyses for the previous section, mitotype w501 was excluded from the results reported in Table 2 as it is known to have a strong negative epistatic interaction with the Oregon-R nuclear background [29]. The D. simulans w501 and sm21 mtDNAs differ from each other by six mutations, 5 of which are synonymous or indels in non-coding DNA, and the only putative functional mutation is in the anticodon stem of the mitochondrial tRNATyr gene. Further genetic mapping shows that the w501 mtDNA interacts negatively with a mutation in the nuclear-encoded gene for mitochondrial-tRNATyr synthetase [29]. This gene is polymorphic in D. melanogaster: the OreR genetic background carries the mutant allele and w1118 carries the wildtype allele for mitochondrial tRNATyr synthetase on chromosome 2. Transgenic analyses of this single nucleotide show that it is responsible for the negative interaction with the point mutation in the tRNATyr gene of w501 mtDNA [29].
Our longevity results show that the w501 mitotype is generally shorter-lived than sm21 in OreR but not in w1118, which carries the wild type allele of the nuclear-encoded tRNATyr synthetase (Figure 2 a–d). Therefore, the lifespan difference caused by the mt-tRNATyr mutation between w501 and sm21 appears to be strongly dependent on nuclear background, as is true for several other phenotypes [29], [39]. This empirical pattern has very strong statistical support. The w501 mtDNA -by-nuclear term is highly significant (χ2 = 132.98 on 1 degrees of freedom, P<0.001). It can be further determined that in the OreR genetic background, the sm21 mitotype has about 53.8% risk of death relative to the w501 mitotype, which is highly significantly lower (Table 3, P<0.001). In contrast, in the w1118 genetic background, the risk of death in sm21 and w501 mitotypes are not significantly different (Table 3, P = 0.952).

Dietary modification of a mitonuclear epistasis: G×G×E
The strong mitonuclear interaction for longevity with the w501 mtDNA is further modified by dietary environment. To quantify this, a series of contrasts were performed where the w501 and sm21 mtDNAs were evaluated as either two separate mitotypes or as one pooled class of mitotype, based on the fact that w501 is most closely related to the sm21 mtDNA (both are siII mtDNA haplotypes of D. simulans differing by only 6 nucleotides; see above). Two groups of models were evaluated (see Table 4). The first model evaluates main effects of mitotype and diet, and contrasts a model of five mitotypes (si1, sm21, w501, OreR, and Zim) vs. a four-mitotype model (si1, sm21 and w501 pooled, OreR and Zim), to test whether w501 has a significant main effect. The second set of contrasts was performed between nested models (mitotype effect nested within diet effect), and again we compared the models containing 5 mitotypes versus the reduced models of 4 mitotypes (w501 and sm21 pooled) to test whether the w501-by-diet effect is significant. These contrasts were run using data for all five diets, just the DR conditions (diets IV, II, V), or just the CR conditions (diets I, II, III).
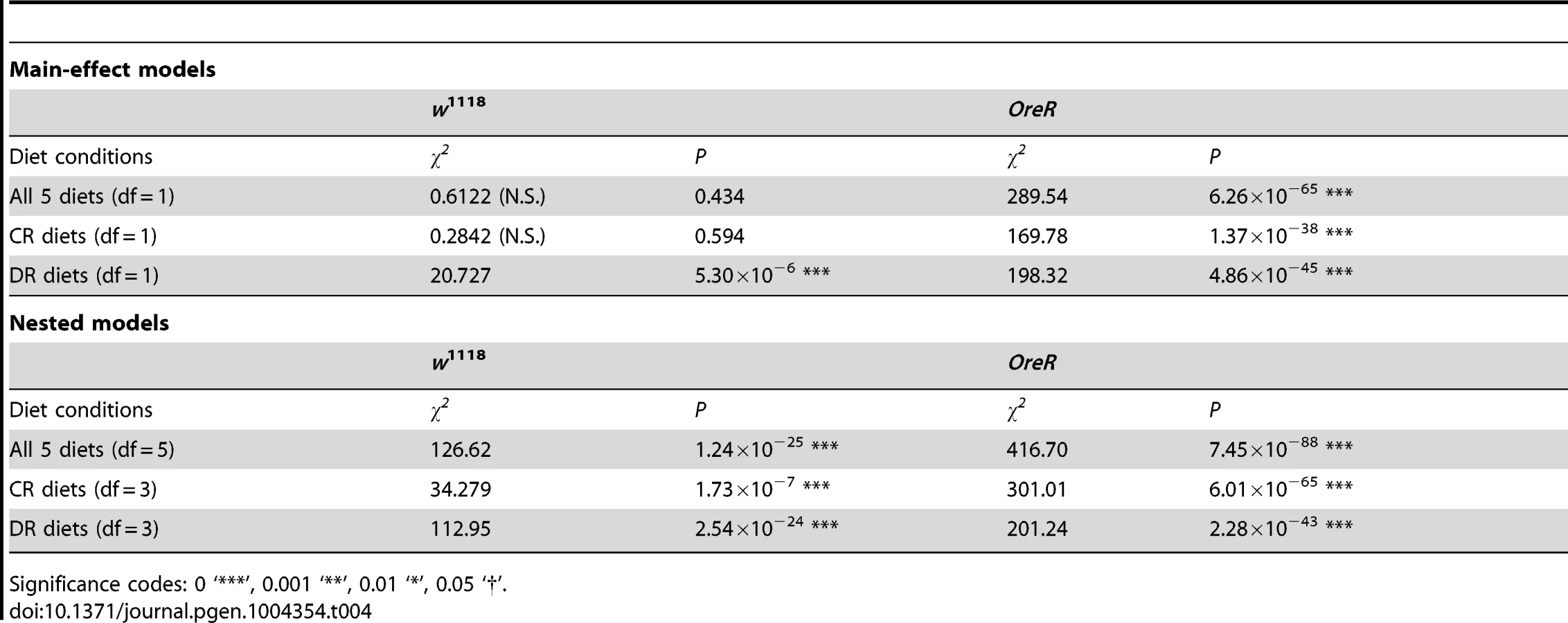
The top half of Table 4 shows the results of the main-effect models. In the w1118 nuclear background the sm21/w501 distinction is only significant under the DR conditions, whereas in the OreR nuclear background the sm21/w501 distinction significantly improves the models under all diet conditions (see Table 4, top half). These analyses confirm that the w501 effect on longevity is greater on the OreR background, as expected from the known epistatic interaction [29]. However, the nested models reveal that even though w501 does not have a significant main effect in the w1118 background, the diet-by-w501 effect improves the model greatly compared to the main effect model (compare entries from top half and bottom half of Table 4). In the OreR background where the w501 main effect is significant, the diet-by-w501 effect greatly improves the model when all diets are analyzed, and under CR diets. Notably, in the OreR nuclear background under DR diets, there is little improvement in the model fit between main-effect and nested models (χ2 = 198.32 vs. 201.24). These analyses provide a specific example of G×G×E interactions: a particular mtDNA with distinct effects on longevity in alternative nuclear backgrounds generates unique responses to the dietary environments that modify longevity.
Mitochondrial DNA copy number also shows a w501-by-nuclear epistatic effect. We observed that the OreR nucleartype carrying the w501 mtDNA has a significant increase in mtDNA/nDNA copy number ratio over the sm21 mitotype and such an increase is not present in w1118 nuclear background (Figure 3) (F2,30 = 9.79, P = 5.34×10−4, General Linear Model: mitotype effect nested within nuclear background effect). Given that mtDNA/nDNA ratio is a good indicator of mitochondrial biogenesis, this pattern may be due to the compensatory response in the presence of a w501 mito OreR nuclear incompatibility.

SIR2 overexpression, dampens the response to caloric restriction but not yeast:sugar ratio alteration
Two mitotypes of each species (OreR, Zim, sm21 siI) were placed on to SIR2 overexpression and control nuclear backgrounds to test the hypothesis of a SIR2-mediated mitonuclear interaction, and to examine the response of these genotypes to DR and CR dietary environments. The SIR2 overexpression genotypes show similar mean lifespans on each of the diets of the caloric restriction gradient (diet type I, II and III) which displays a lack of CR response, while the SIR2 control genotypes display a robust CR effect (compare the slopes of the lines in Figures 4A and 4C). The reduced response of the SIR2 overexpression genotypes compared to the controls is consistent across different mitotypes. Statistical analyses of these data reveal that increase of caloric content increases the risk of death, however, the magnitude of increase is modulated by SIR2 overexpression. A very strong dietary effect on survivorship is observed in the control genotypes, (Table 5, 60.1% increase in the risk of death with each diet level, P = 5.29×10−103). In the SIR2 overexpression genotypes, this increase in risk is small but remains significant (Table 5, Caloric manipulations, hazard ratio column, 5.51% increased risk with each diet level, P = 0.0132).


Compared to the distinct responses of SIR2 overexpression and control genotypes to the CR gradient, these same genotypes have clearly parallel responses to the DR gradient, i.e. when the animals receive dietary manipulation in a form of food composition change (sugar:yeast ratio axis, diet type IV, II and V; compare the slopes of the lines in Figures 4B to 4D). Overall, a diet rich in yeast (S/Y = 1∶3) reduces longevity whereas a diet with relatively low yeast (S/Y = 3∶1) extends longevity, relative to the balanced diet. This dietary gradient has similar effects in both SIR2 overexpression and the control genetic background: the higher yeast diet causes a 54.9% increased risk of death in the control and 67.8% in the overexpression genotypes, P<0.001 for both; see Table 5, S/Y manipulations, column hazard ratio).
The observation that SIR2 overexpression reduces the response to caloric restriction agrees well with the original report in D. melanogaster in which SIR2 overexpression extended life span on high caloric diet but gains no additional extension on CR diet [40]. The new finding that SIR2 overexpression has relatively little impact on longevity changes across a diet composition gradient, while diet V of this gradient does result in longevity extension (Figures 4B and D) suggests that the mechanisms of nutrient sensing of S/Y ratio are either independent of, or abrogate, the main effects of SIR2. Our results provide a clear example that CR and DR are very different processes and reveal considerable complexity underlying diet-genotype interactions that modify longevity.
SIR2 overexpression effects on longevity are mitotype-dependent
The plots of mean lifespan indicate that the variance among mitotypes is lower in the SIR2 overexpression background than in the control background (Figure 4). The random effect Cox model confirms this observation. Across the CR axes (diet I II and III), the variance of the mitochondrial effect is 0.4216 in the control genotype and drops to 0.1506 with SIR2 overexpression (Table 6). Across the DR axes (diet V, II and IV), our results show the mitotype effect to be larger than across the CR axes. Moreover, SIR2 overexpression reduces the mitotype effect from 0.8138 to 0.3858 (Table 6). Figure 4 shows that the impact of SIR2 overexpression is evident for mitotypes OreR, Zim and sm21, with mitotype si1 showing less response. With the si1 mitotype removed, the variance component among mitotypes diminishes (Table 6). The different responses of the two D. simulans mtDNAs (sm21 and si1) result in little or no impact of species-level mtDNA divergence on longevity. It is apparent that the SIR2 overexpression reduces longevity of each mitotype on the CR and DR axes (Table 5), but the si1 mitotype is less sensitive to this effect than the other three (OreR, Zim, sm21). The distinct response of the si1 mitotype to these SIR2 and diet experiments, and the minor role for species-level divergence, indicates that nucleotide polymorphisms between sm21 and si1 mtDNAs play a role in mediating the combined effects of SIR2 overexpression and diet on longevity.

It is possible that the strong si1-SIR2 effect is due to a very different expression level of the UAS-SIR2 construct in the si1 mitotype background, rather than a specific mito-SIR2 interaction. We quantified the overexpression level of UAS-SIR2 in the siI and OreR mitotype backgrounds to test this possibility. The expression level of SIR2 is not significantly different between siI and the most-divergent OreR (Figure 5). We note that only females were included in our demography experiment and the overexpression level is about 5× in females (Figure 5). While the 40× overexpression shown in males would likely be deleterious, we did not use males for longevity assays. Based on these results we conclude that mitotype itself does not affect the level of overexpression, indicating that the SIR2-siI interaction is not likely due to a spurious differential over expression mediated by mtDNA-effects on the GAL4-UAS system.

Overall, we found strong support for the effects of nuclear genotypes (SIR2 overexpression), diet, and mitochondrial genotype on lifespan. As summarized in Table 7, all three of these terms are highly significant (P<0.001). We found the diet effect to be significantly altered by SIR2 overexpression (Table 7 χ2 = 233.94 P<0.001) on the CR axis and not significantly modified by SIR2 on the DR axis (χ2 = 3.09 P = 0.0793). An epistatic interaction between mitotype and SIR2 is well supported (χ2 = 77.272 and 64.074 for CR and DR respectively, P<0.001 for both). A mitotype-by-diet interaction is also found to be significant (χ2 = 53.423 and 305.95 for CR and DR respectively, P<0.001 for both). As shown in the w1118 and OreR experiments, we again see little support for the effect due to species level mtDNA divergence (Table 7). In summary, the results indicate that in addition to diet, mitochondrial and nuclear genotypes are important factors modulating lifespan, and that the epistatic interaction between them also has substantial effects.

Discussion
In this study we have quantified the effects of mitochondrial genotypes, nuclear genotypes and dietary modifications on the lifespan of Drosophila. Our goal was to provide a comprehensive survey of the range of effects across each of these interacting genetic and dietary factors. No previous study has examined the complexity of interactions among these factors in an effort to sort out main effects, epistatic effects and genotype-by-environment interactions.
Our results show that the main effects of variation in mtDNA, nuclear genetic background and diet can be significantly modified by the interaction between each factor, using controlled orthogonal designs capable of quantify interaction effects. Among mtDNAs, there are mitotypes that show parallel shifts in lifespan across diets or nuclear backgrounds, but there are others that show very different responses with crossing reactions norms among diets (Figures 2 and 4). For nuclear backgrounds, there are distinct responses to diet for each of the four genotypes studied (w1118 and OreR, SIR2 overexpression and control). And for caloric or dietary restriction, the efficacy of this manipulation is highly dependent on genotype, both nuclear and mitochondrial. Overall, our results reveal that the interactions are arguably more important than the main effects alone for Drosophila life span. The interactions are not simply pair-wise, as mito-nuclear interactions are modified by diet. These genotype-by-genotype-by-environment, or G×G×E (technically Mito × Nuclear × Diet) effects raise the question of how best to proceed with the identification of general effects that modify longevity.
MtDNA mutations, mitonuclear epistasis and purifying selection
A general result regarding mtDNA effects on longevity is the different impacts of point mutations versus deeper mtDNA divergence. The high mutation rate of mtDNA results in high levels of mtDNA variation in most animals, including humans. These new mutants are actively tested for function with nuclear genetic variation across each generation, and chance combinations of nuclear and mitochondrial alleles can generate novel phenotypes (epistatic interactions) that are not predicted from the main effects of either genome. The large number of nuclear-encoded subunits of the mitochondrial electron transport chain and mitochondrial translation machinery make a particularly large mutational target for novel variants that must interact with the mtDNA encoded subunits that contribute to these critical cellular processes.
One result of particular interest is the effect of the tRNATyr mutation in w501 mtDNA, which reduces lifespan on the OreR nuclear background but can show average longevity on the w1118 background. This demonstrates a particular case of 2-nucleotide mitonuclear incompatibility due to mis-match between mitochondrial - and nuclear-encoded components. In this particular case of w501 mtDNA, the OreR and w1118 nuclear backgrounds carry different tRNA-synthetase alleles that functionally interact with the w501 tRNATyr mutation. Although, admittedly, a D. simulans mitochondrial-D. melanogaster nuclear genome combination is unlikely in nature, mitochondrial dysfunction due to tRNA mutation is not at all rare. Notably, exercise intolerance in humans results from a mutation in this same mt-tRNATyr gene [41], [42], [43]. Indeed, mitochondrial tRNA diseases involving similar mitochondrial-nuclear dysfunction in human are common, such as MERRF syndrome (Myoclonic Epilepsy with Ragged Red Fibers) [44], which is caused by mutation in mitochondrial tRNALys. Therefore, the principle of mitonuclear epistatic interactions is a general problem that has received little attention until recently [28], [29], [30].
One rather unexpected finding is that the w501 - OreR interaction appears to result in small, yet significant, upregulation of mitochondrial copy number. The regulatory mechanism of steady level of mtDNA copy number is thought to be a complex network consisting of both mitochondrial and nuclear encoded components, with many details yet to be elucidated. The major nuclear encoded components includes mitochondrial transcription factor A (TFAM), mitochondrial DNA polymerase γ (PLOG), mitochondrial single strand binding protein (mtSSB) [45]. The sequence of the mtDNA in the origin of replication region of the D-loop, presumably also plays an important role affecting mtDNA level [46]. Our result suggests that mitochondrial translational deficiency, such as the one leading to the w501 - OreR interaction, may also be a part in the mtDNA level regulatory network, possibly acting though a compensatory mechanism.
In both the wild type nuclear backgrounds, w1118 and OreR, we demonstrated that dietary restriction effect is highly dependent on mitotype (Table 2). These genome-wide patterns provide strong motivation to test the hypothesis that epistasis also exists between mitotypes and genetic pathways known to influence dietary restriction. We tested this hypothesis using a mito-SIR2 interaction. The up-regulation of SIR2 leads to lifespan extension in many model organisms [15], [18], [40], [47], [48], [49] and there are sound reasons why this process may involve a mito-SIR2 interaction. First, SIR2 is an NAD+ consuming deacetylase [50], [51] and mitochondria oxidize NADH to NAD+ during oxidative phosphorylation by NADH dehydrogenase (Complex I). Interaction between SIR2 and mitochondria can stem from this sharing of the same cofactor pool. Second, SIR2-mito interactions may also arise through the upregulation of mitochondrial function by mimicking DR, which also involves other members of the sirtuin family [27], [52]. This pattern of epistatic interaction is consistent with a number of studies showing that mitochondrial function is enhanced, not only by sirtuins, but also other pathways that mimic DR [10], [14], [22], [23], [34], [53].
Due to the sequence divergence of the mitotypes included in this study, different mitotypes are expected to cause different levels of mitonuclear incompatibility, some mild while others may be severe. To the extent that overexpressing SIR2 mimics DR and enhances mitochondrial function, the distinct set of mtDNA haplotypes used in our study has the potential to identify novel functional interactions between SIR2 and mitochondria. Our initial expectation was that the divergent D. simulans mtDNA would generate higher levels of incompatibility and show greater differences in longevity. However, it was a particular mitotype, not the species-level divergence that is most informative (Figure 4, siI mitotype). Further genetic mapping and biochemical analyses are required to understand how siI mitotype mediates a unique response to SIR2, but the mutations that are unique to the siI mitotype provide a list of candidates that could guide these studies (see Table S1).
Our results have consistently falsified the hypothesis that divergent mtDNAs from a different species represent ‘strongly incompatible’ mtDNA genotypes. In most nuclear backgrounds, and across most diet treatments, our data show that the species-level divergence of mtDNAs (∼100 amino acids, see supplementary Table S1) has little phenotypic effect, while individual mtDNAs can have pronounced effects (e.g., w501 and siI mtDNAs; Figures 2 and 4, Tables 2 and 7). If purifying selection has been a general force during the divergence of mtDNAs, the large sequence divergence between species may be accompanied by little accumulated functional divergence. Likewise, the nature of purifying selection will determine the mutation-selection balance that permits individual mutant mitotypes carrying deleterious mutations to persist in populations, with measurable effects on longevity and other performance traits. These results suggests that additional screens of very closely related mtDNA may uncover informative SNPs showing strong epistatic interactions with nuclear factors affecting longevity.
Distinct mitonuclear mechanisms for dietary and caloric restriction
Our results also imply that the responses to food concentration or composition changes involve different mechanisms. Altering caloric content and changing yeast concentration are currently two main approaches of caloric vs. dietary restriction (CR and DR, respectively) in D. melanogaster. The former changes diet concentration while keeping sugar/yeast ratio constant [37], [54]. The latter approach can alter the sugar/yeast ratio while keeping total calories constant [37], or restrict the amount of yeast, resulting in both sugar/yeast ratio and total caloric changes [55]. We found that the SIR2 control genotype shows a strong response to CR, but SIR2 overexpression does not respond to CR, and lifespan changes little on different diets as long as yeast:sugar ratio is unchanged (compare Figure 4A to 4C). In contrast, the lifespan of SIR2 overexpression still displays response to isocaloric DR involving sugar/yeast ratio changes (compare Figure 4B to 4D). These observations collectively suggest that SIR2 is involved in sensing food concentration changes but has less of a role in sensing composition changes or yeast:sugar ratio. Thus, our results are consistent with the hypothesis that SIR2 regulates lifespan, but clearly show that SIR2's manner of longevity regulation is dependent on diet, which is indeed a case of genotype × diet interaction. A possible model to explain this result is that SIR2 is only involved in CR but not DR. When SIR2 is already overexpressed, additional CR can't result in further extension lifespan. However under DR, which may be SIR2 independent, this dietary modification remains effective regardless of SIR2 level. Therefore in both control and SIR2 overexpression condition, DR results in similar extension of lifespan.
In contrast to other reports, we found overexpression of SIR2 overall decreases life span. SIR2 is an important gene in aging research as it has suggested a link between metabolism and longevity [16], [56]. A polyphenol resveratrol, found in red wine, was suggested to be a SIR2 activator and has been demonstrated to lead to variety of benefits that are associated with DR [18], [47]. However, the roles of both SIR2 and resveratrol have been challenged [57], [58], [59]. Absence of life span extension by SIR2 overexpression has also been reported before [60], [61], [62], [63]. In our case, we suspect the lifespan decrease we observe is due to the expression level of SIR2 that is not optimal for longevity extension or related to developmental effects in larval life. Here we found the expression of SIR2 driven by a strong ubiquitous da-GAL4 driver to be about 5 times that of controls in females and 40 times that of controls in males (we used females for all longevity experiments). Previously, the similar ∼5× over-expression of SIR2 has been shown, in two studies, to increase or have no effect on longevity in Drosophila [61], [64]. It is known that SIR2 expression is high in the embryo stage but moderate in adult with high expression in brain, salivary gland, female ovary and spermatheca [65], [66], displaying a highly tissue specific pattern. Therefore, the absence of lifespan extension in our experiments is also possibly due to SIR2 expression in tissues and stages where overexpression can become detrimental. We note that one goal of this study was to identify genetic interactions between SIR2 and mtDNA variants as they affect the response to DR and CR, and the si1 mtDNA-SIR2 interaction is a promising result. However, we acknowledge further studies using stage - and tissue-specific SIR2 overexpression with moderate dosage are needed to resolve the details of SIR2-mtDNA genetic interactions.
Aging is a complex process and extending life span requires the coordination of a large number of genes, pathways, and environments, among which dietary regime is probably specially important. The interaction between them is an intrinsic nature of the aging process. These interactions may often appear as annoying sources of experimental complication that reduces the repeatability in particular experiments. For example, different mouse genotypes respond very differently to the same dietary environment [36], [21], which is essentially a form of G×E interaction. Moreover, alternative combinations of alleles from different loci can lead to different phenotypic response [40], [61], which is in fact an epistatic or G×G interaction. Both G×E and G×G interactions have been demonstrated and are beginning to draw attention in aging research. However, the greater challenge, as we have elucidated in this work, is a more general G×G×E interaction. That is, the effects of mutations that affect lifespan are heavily dependent on the interaction between genetic background and dietary regime simultaneously. If a goal in aging research is to understand the complexity of factors that may help ensure increased healthspan in humans, we must confront the complexity of interactions presented by genetically variable humans living in the variety of dietary environments so common today. Here we have sought to provide an example of such an effort in a model organism where we can begin to manipulate these factors and eventually elucidate the mechanisms of the interactions that have undoubtedly been important throughout evolution.
Materials and Methods
Strains and mitonuclear genotypes
Longevity analyses were carried out on lines of D. melanogaster carrying alternative mtDNAs from D. melanogaster and its sister species D. simulans. We generated three sets of lines on each of three different nuclear genetic backgrounds, Oregon R (OreR), w1118 (Bloomington stock number 6326) and daughterless-GAL4 (da-GAL4, Bloomington stock number 5460), each carrying alternative mtDNAs. The D. melanogaster mitochondria are from the Oregon R (OreR) strain and Zim53 (Zim), from Zimbabwe [67], [68]. Also included are Drosophila simulans mitochondrial haplotypes si1 from Hawaii, sm21 from strain C167.4 (Drosophila Genetic Resource Center stock number 107850), and w501, from strain white501 (w501, Drosophila Species Stock Center stock number 14021-0251.195). Introgression of D. simulans mitochondria into flies carrying D. melanogaster nuclear genome is made possible by a rescue cross involving D. simulans strain C167.4 as females and D. melanogaster strain In(1A)B as males [28], [39], followed by balancer chromosome replacement to ensure the mito-replaced strains have the same set of D. melanogaster nuclear chromosomes of the original strain (Supplementary Figure S1). For simplicity, we will refer to different mitochondrial haplotypes as “mitotypes” (cf. mitochondrial genotypes) and nuclear chromosomal composition as “nucleartype”. When contrasting mitotypes in different nucleartypes, the following terminology will be used: ‘the OreR mitotype has different longevity on alternative nuclear backgrounds’; or the w1118 nucleartype shows different longevities on different mtDNA backgrounds'. We use the term “genotype” or “mitonuclear genotype” to refer to the combination of mtDNA and nuclear chromosomes that are constructed in a particular genetic line, using the notation: mtDNA; nuclearDNA. For example, the mitonuclear genotype w501; w1118 carries the D. simulans w501 mtDNA and the D. melanogaster w1118 nuclear chromosomes.
The mitotype panel covers a wide range of mitochondrial mutations: the OreR mitotype differs from the D. melanogaster Zim mitotype by 18 amino acid substitutions, and by 103 amino acid substitutions from the most diverged mitotype, D. simulans si1. The mitotypes are all fully sequenced and details of the molecular evolutionary divergence and the phylogenetic relationships can be found in Ballard [68], and Montooth et al.[39]. The Genebank sequence accession numbers for the sequences of mitotypes are: OreR AF200828, Zim AF200829, siI AF200835, sm21 KC244283 and w501 KC244284.
Overexpression of SIR2 was achieved by driving the expression of UAS-SIR2 via a ubiquitous daughterless-GAL4 driver. UAS-SIR2 construct was kindly provided by Drs. Jason Wood and Stephen Helfand. Each mitotype described above was introduced into the da-GAL4 construct using balancer chromosome substitution. SIR2 overexpression in different mitotype backgrounds is made possible by the mito-replaced da-GAL4 strains. Overexpression genotypes were obtained by crossing UAS-SIR2 males to mitotype-replaced da-GAL4 females. The control genotypes are generated by crossing the w- strain, in which UAS-SIR2 is constructed, to da-GAL4 females. To keep UAS-SIR2 and its control w- strain (+) has the same genetic background, they were maintained as UAS-SIR2/+ heterozygote and sorted by eye color to get UAS-SIR2/UAS-SIR2 and +/+ males to minimizing the chance of possible fixed genetic divergence that may arise between the SIR2 and the + genotypes if both were to be maintained as individual homozygote strains. Additional control genotypes, such as a control for the GAL4 line, would have been informative. However, the primary goal is to determine the reality (and magnitude) of interaction effects. Therefore, the alternative design is not employed. In our approach the over-expression genotype (da-GAL4/UAS-SIR2) and the non-overexpression genotype (da-GAL/+) both carry the same whole genome heterozygous background (da-GAL4 background/UAS-SIR2 background). Therefore the whole genome heterozygous background will minimize the effect of any fixed mutations on either the da-GAL4 background or the UAS-SIR2 background, and will reveal a more general picture how SIR2 and mtDNA will interact in any randomly chosen genetic background from a wild population.
Dietary manipulations
A five-diet design was used to study the effects of both food composition and food concentration on life span, termed diet I, II, II, IV, V (see Figure 1). The ingredients of the five diets (all in grams per liter); type I: 50 sucrose + 50 yeast, type II: 100 sucrose + 100 yeast, type III: 150 sucrose + 150 yeast, type IV: 50 sucrose + 150 yeast and type V: 150 sucrose + 50 yeast. All of these are prepared with 15 g/dl agar and supplemented with 0.2 g/dl Tegosept as fungal suppressor. The food making procedure follows the protocol described in [35]. Briefly, ingredients are well mixed in hot water and then autoclaved at liquid cycle (120°C for 25 min) to achieve sterility and uniform cooking conditions. The food mix is then allowed to cool down before addition of Tegosept using 20 g/dl stock solution. Note that type I, II and III are exactly the same as 0.5 N, 1.0 N and 1.5 N food used by other groups [35], [36], [40]. Type I, II and III form a concentration gradient where the yeast sugar ratio is constant and the total concentration and caloric content varies. Mair et al. reported very similar caloric contents for carbohydrate and autolysed yeast powder, 4.0 and 4.02 kcal/g respectively [37]. Therefore, type V, II and IV form a composition gradient where the caloric content is approximately constant but the food composition (sugar/yeast ratio) varies. “Standard food” is the common cornmeal food for general stock keeping and is made of 10 g/l agar, 100 g/l sucrose, 50 g/l cornmeal and 50 g/l cornmeal yeast.
Demography
Fly lifespan was measured by counting individual deaths in demography cages. Briefly, newly eclosed flies were collected over a 24 hour period, held on regular food for 4 days as mixed sex adults, and then sorted by sex. Demography experiments were only conducted on females. 100 females were placed in 1 liter cages in triplicate and were supplied with different types of food. On Mondays, Wednesdays and Fridays, food was renewed and death recorded. Cages were kept in climate controlled chambers maintaining a 12/12 hours light/dark cycle at 25°C.
Molecular assays
Quantification of the ratio of mitochondrial to nuclear DNA (mtDNA/nDNA) and SIR2 expression level were carried out by a quantitative PCR (qPCR) based method. To avoid amplification bias due to relative high A/T richness of mtDNA compared to genomic DNA we chose a G/C rich region in mtDNA to make mtDNA and nDNA primer sets comparable. Both mtDNA primers and nDNA primers were aligned against available sequence data in the NCBI database to ensure they map to highly conserved regions and are not likely to be affected by mismatch due to sequence polymorphism. We followed the DNA extraction method suggested by Guo et.al [69] to avoid underestimating the abundance of mitochondrial DNA. The primer sequences are as follows: mtDNA: 5′-GATTAGCTACTTTACATGGAACTC-3′ and 5′-CTGCTATAATAGCAAATACAGCTC-3′ adjacent to the mitochondrial genomic region of cytochrome c oxidase subunit I gene; nDNA: 5′-AACTCTGCTGCTACTTATCG-3′ and 5′-CAGGATCAGGATGGAATAGTATC-3′ adjacent to the nuclear region of NADH dehydrogenase 51 kDa subunit gene.
SIR2 expression level is quantified using ddCT method with a pair of primers within SIR2: 5′-TCATGAAGCCGGATATCGT-3′ and 5′-GGTATGCTGCTGGGAATG-3′ together with the reference primers in house keeping gene GAPDH: 5′-CCACTGCCGAGGAGGTCAACTAC-3′ and 5′-CATGCTCAGGGTGATTGCGTATGC-3′. Both qPCR experiments are done with 10-day-old whole flies reared on regular food. mtDNA/nDNA was measured in males only while SIR2 expression levels were assayed in both males and females. For both experiments, 3 biological replicates were assayed with 3 technical replicates.
Statistical analyses
To test the effect of mitotype, nuclear genotype and diet on lifespan, we analyzed the demography data using a mixed-effect Cox proportional hazard model. The mixed-effect Cox model is an extension to the popular Cox proportional hazard model [70] to include random effects. The Cox model estimates the log(hazard ratios) from survivorship of different groups. Two groups having a hazard ratio equal to 1 (or equivalently log(hazard) equal to 0) suggests the groups are equally likely to die. Similarly, negative log(hazard) or positive log(hazard) suggests one group has higher or lower probability to die than the other group and subsequently survives shorter or longer, respectively. In a mixed-effect Cox model, random effects are included. Random effects are modeled assuming their log(hazard ratio) are drawn from Gaussian distributions with zero mean and unknown variances. The unknown Gaussian variances are numerically solved by finding the maximum of integrated partial likelihood using an iterative method [71]. The model was originally developed for clinical application. For example, a type of drug is tested in multiple hospitals in order to determine its ability to extend patient survival (by comparing treatment group survival to control group in each hospital). However, it is also important to know the amount of variation among hospitals especially how big the variation is in relation to the treatment effect. The among-hospital variation is considered as a random effect as it estimates the variation among all hospitals represented by the hospitals sampled. The treatment effect is, on the contrary, considered as a fixed effect. The mixed-effect Cox model enabled us to examine both fixed terms (such as diet type analogous to drug effect in this example) and random terms (such as genetic background effect and replicate noise, analogous to variation among hospitals) simultaneously. It also made examining variance components possible such that we can determine the relative magnitude of different random effects. Therefore their relative importance can be evaluated. In more practical terms, variance components estimated by the mixed-effect Cox proportional hazard model is a metric of the magnitude of effects, with a larger value indicating an effect is substantial and a small value indicating an effect is negligible. These tests are not possible with a classic Cox proportional hazard model with support for only fixed effects. The mixed effect Cox model is implemented using R package coxme() (http://cran.r-project.org/web/packages/coxme/). Additionally, the significance tests of overall contributions of each term are conducted by likelihood ratio test by comparing full models versus reduced models. Log-likelihood ratio tests are implemented using R Analysis of Deviance package anova() (http://stat.ethz.ch/R-manual/R-patched/library/stats/html/anova.glm.html).
Supporting Information
Zdroje
1. McCayCM, MaynardLA, SperlingG, BarnesLL (1939) Retarded growth, life span, ultitimate body size and age changes in the albino rat after feeding diets restricted in calories. Journal of Nutrition 18 : 1–13.
2. WeindruchR, WalfordRL, FligielS, GuthrieD (1986) The retardation of aging in mice by dietary restriction: longevity, cancer, immunity and lifetime energy intake. Journal of Nutrition 116 : 641–654.
3. BrossTG, RoginaB, HelfandSL (2005) Behavioral, physical, and demographic changes in Drosophila populations through dietary restriction. Aging Cell 4 : 309–317.
4. PartridgeL, GemsD (2002) Mechanisms of ageing: Public or private? Nature Reviews Genetics 3 : 165–175.
5. WalkerG, HouthoofdK, VanfleterenJR, GemsD (2005) Dietary restriction in C. elegans: From rate-of-living effects to nutrient sensing pathways. Mechanisms of Ageing and Development 126 : 929–937.
6. KenyonC (2005) The plasticity of aging: Insights from long-lived mutants. Cell 120 : 449–460.
7. TatarM, BartkeA, AntebiA (2003) The endocrine regulation of aging by insulin-like signals. Science 299 : 1346–1351.
8. SarbassovDD, AliSM, SabatiniDM (2005) Growing roles for the mTOR pathway. Current Opinion in Cell Biology 17 : 596–603.
9. TokunagaC, YoshinoK, YonezawaK (2004) mTOR integrates amino acid - and energy-sensing pathways. Biochemical and Biophysical Research Communications 313 : 443–446.
10. ZidBM, RogersAN, KatewaSD, VargasMA, KolipinskiMC, et al. (2009) 4E-BP Extends Lifespan upon Dietary Restriction by Enhancing Mitochondrial Activity in Drosophila. Cell 139 : 149–160.
11. CantoC, AuwerxJ (2009) PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Current Opinion in Lipidology 20 : 98–105.
12. CantoC, JiangLQ, DeshmukhAS, MatakiC, CosteA, et al. (2010) Interdependence of AMPK and SIRT1 for Metabolic Adaptation to Fasting and Exercise in Skeletal Muscle. Cell Metabolism 11 : 213–219.
13. KempBE, MitchelhillKI, StapletonD, MichellBJ, ChenZP, et al. (1999) Dealing with energy demand: the AMP activated protein kinase. Trends in Biochemical Sciences 24 : 22–25.
14. FinleyLWS, HaigisMC (2009) The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Research Reviews 8 : 173–188.
15. CohenHY, MillerC, BittermanKJ, WallNR, HekkingB, et al. (2004) Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305 : 390–392.
16. GuarenteL, PicardF (2005) Calorie restriction - the SIR2 connection. Cell 120 : 473–482.
17. NemotoS, FergussonMM, FinkelT (2004) Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 306 : 2105–2108.
18. WoodJG, RoginaB, LavuS, HowitzK, HelfandSL, et al. (2004) Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 430 : 686–689.
19. SchleitJ, JohnsonSC, BennettCF, SimkoM, TrongthamN, et al. (2013) Molecular mechanisms underlying genotype-dependent responses to dietary restriction. Aging Cell 12 : 1050–61.
20. HuangW, RichardsS, CarboneMA, ZhuD, AnholtRR, et al. (2012) Epistasis dominates the genetic architecture of Drosophila quantitative traits. Proceedings of the National Academy of Sciences of the United States of America 109 : 15553–15559.
21. LiaoCY, RikkeBA, JohnsonTE, DiazV, NelsonJF (2010) Genetic variation in the murine lifespan response to dietary restriction: from life extension to life shortening. Aging Cell 9 : 92–95.
22. FinleyLWS, LeeJ, SouzaA, Desquiret-DumasV, BullockK, et al. (2012) Skeletal muscle transcriptional coactivator PGC-1α mediates mitochondrial, but not metabolic, changes during calorie restriction. Proceedings of the National Academy of Sciences of the United States of America 109 : 2931–2936.
23. Lopez-LluchG, HuntN, JonesB, ZhuM, JamiesonH, et al. (2006) Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proceedings of the National Academy of Sciences of the United States of America 103 : 1768–1773.
24. HirscheyMD, ShimazuT, GoetzmanE, JingE, SchwerB, et al. (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464 : 121–U137.
25. QiuXL, BrownK, HirscheyMD, VerdinE, ChenD (2010) Calorie Restriction Reduces Oxidative Stress by SIRT3-Mediated SOD2 Activation. Cell Metabolism 12 : 662–667.
26. TaanmanJW (1999) The mitochondrial genome: structure, transcription, translation and replication. Biochimica Et Biophysica Acta-Bioenergetics 1410 : 103–123.
27. GuarenteL (2008) Mitochondria - A nexus for aging, calorie restriction, and sirtuins? Cell 132 : 171–176.
28. RandDM, FryA, SheldahlL (2006) Nuclear-mitochondrial epistasis and Drosophila aging: Introgression of Drosophila simulans mtDNA modifies longevity in D.melanogaster nuclear backgrounds. Genetics 172 : 329–341.
29. MeiklejohnCD, HolmbeckMA, SiddiqMA, AbtDN, RandDM, et al. (2013) An Incompatibility between a Mitochondrial tRNA and Its Nuclear-Encoded tRNA Synthetase Compromises Development and Fitness in Drosophila. PLoS Genetics 9(1): e1003238.
30. HoutkooperRH, MouchiroudL, RyuD, MoullanN, KatsyubaE, et al. (2013) Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497 : 451–457.
31. KapahiP, ZidBM, HarperT, KosloverD, SapinV, et al. (2004) Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Current Biology 14 : 885–890.
32. SarbassovDD, GuertinDA, AliSM, SabatiniDM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307 : 1098–1101.
33. ReraM, BahadoraniS, ChoJ, KoehlerCL, UlgheraitM, et al. (2010) Modulation of Longevity and Tissue Homeostasis by the Drosophila PGC-1 Homolog. Cell Metabolism 14 : 623–634.
34. BahadoraniS, HurJH, LoT, VuK, WalkerDW (2010) Perturbation of mitochondrial complex V alters the response to dietary restriction in Drosophila. Aging Cell 9 : 100–103.
35. SkorupaDA, DervisefendicA, ZwienerJ, PletcherSD (2008) Dietary composition specifies consumption, obesity, and lifespan in Drosophila melanogaster. Aging Cell 7 : 478–490.
36. GrandisonRC, WongR, BassTM, PartridgeL, PiperMDW (2009) Effect of a Standardised Dietary Restriction Protocol on Multiple Laboratory Strains of Drosophila melanogaster. PLoS One 4(1): e4067.
37. MairW, PiperMDW, PartridgeL (2005) Calories do not explain extension of life span by dietary restriction in Drosophila. PLoS Biology 3(7): e223.
38. LeeKP, SimpsonSJ, ClissoldFJ, BrooksR, BallardJWO, et al. (2008) Lifespan and reproduction in Drosophila: New insights from nutritional geometry. Proceedings of the National Academy of Sciences of the United States of America 105 : 2498–2503.
39. MontoothKL, MeiklejohnCD, AbtDN, RandDM (2010) Mitochondrial-nuclear epistasis affects fitness within species but does not contribute to fixed incompatibilities between species of Drosophila. Evolution 64 : 3364–3379.
40. RoginaB, HelfandSL (2004) Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proceedings of the National Academy of Sciences of the United States of America 101 : 15998–16003.
41. PulkesT, SiddiquiA, Morgan-HughesJA, HannaMG (2000) A novel mutation in the mitochondrial tRNATyr gene associated with exercise intolerance. Neurology 55 : 1210–1212.
42. RileyLG, CooperS, HickeyP, Rudinger-ThirionJ, McKenzieM, et al. (2010) Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. American Journal of Human Genetics 87 : 52–59.
43. RaffelsbergerT, RossmanithW, Thaller-AntlangerH, BittnerRE (2001) CPEO associated with a single nucleotide deletion in the mitochondrial tRNATyr gene. Neurology 57 : 2298–2301.
44. SeibelP, DegoulF, RomeroN, MarsacC, KadenbachB (1990) Identification of point mutations by mispairing PCR as exemplified in MERRF disease. Biochemical and Biophysical Research Communications 173 : 561–565.
45. MoraesCT (2001) What regulates mitochondrial DNA copy number in animal cells? Trends in Genetics 17 : 199–205.
46. LeeHC, LiSH, LinJC, WuCC, YehDC, et al. (2004) Somatic mutations in the D-loop and decrease in the copy number of mitochondrial DNA in human hepatocellular carcinoma. Mutation Reseach 547 : 71–78.
47. HowitzKT, BittermanKJ, CohenHY, LammingDW, LavuS, et al. (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425 : 191–196.
48. KaeberleinM, McVeyM, GuarenteL (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes & Development 13 : 2570–2580.
49. TissenbaumHA, GuarenteL (2001) Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410 : 227–230.
50. LandryJ, SlamaJT, SternglanzR (2000) Role of NAD+ in the deacetylase activity of the SIR2-like proteins. Biochemical and Biophysical Research Communications 278 : 685–690.
51. LandryJ, SuttonA, TafrovST, HellerRC, StebbinsJ, et al. (2000) The silencing protein SIR2 and its homologs are MAD-dependent protein deacetylases. Proceedings of the National Academy of Sciences of the United States of America 97 : 5807–5811.
52. HebertAS, Dittenhafer-ReedKE, YuW, BaileyDJ, SelenES, et al. (2012) Calorie Restriction and SIRT3 Trigger Global Reprogramming of the Mitochondrial Protein Acetylome. Molecular Cell 49 : 186–199.
53. VerdinE, HirscheyMD, FinleyLWS, HaigisMC (2010) Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends in Biochemical Sciences 35 : 669–675.
54. ChapmanT, PartridgeL (1996) Female fitness in Drosophila melanogaster: An interaction between the effect of nutrition and of encounter rate with males. Proceedings of the Royal Society B-Biological Sciences 263 : 755–759.
55. MinKJ, TatarM (2006) Restriction of amino acids extends lifespan in Drosophila melanogaster. Mechanisms of Ageing and Development 127 : 643–646.
56. GuarenteL (2000) Sir2 links chromatin silencing, metabolism, and aging. Genes & Development 14 : 1021–1026.
57. BassTM, WeinkoveD, HouthoofdK, GemsD, PartridgeL (2007) Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans. Mechanisms of Ageing and Development 128 : 546–552.
58. PacholecM, BleasdaleJE, ChrunykB, CunninghamD, FlynnD, et al. (2010) SRT1720, SRT2183, SRT1460, and Resveratrol Are Not Direct Activators of SIRT1. Journal of Biological Chemistry 285 : 8340–8351.
59. WalleT, HsiehF, DeLeggeMH, OatisJE, WalleUK (2004) High absorption but very low bioavailability of oral resveratrol in humans. Drug Metabolism and Disposition 32 : 1377–1382.
60. AstromSU, ClineTW, RineJ (2003) The Drosophila melanogaster sir2+ gene is nonessential and has only minor effects on position-effect variegation. Genetics 163 : 931–937.
61. BurnettC, ValentiniS, CabreiroF, GossM, SomogyvariM, et al. (2011) Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 477 : 482–458.
62. GriswoldtAJ, ChangKT, RunkoAP, KnighttMA, MinKT (2008) Sir2 mediates apoptosis through JNK-dependent pathways in Drosophila. Proceedings of the National Academy of Sciences of the United States of America 105 : 8673–8678.
63. NewmanBL, LundbladJR, ChenY, SmolikSM (2002) A Drosophila homologue of Sir2 modifies position-effect variegation but does not affect life span. Genetics 162 : 1675–1685.
64. WhitakerR, FaulknerS, MiyokawaR, BurhennL, Henriksen, etal (2013) Increase expression of Drosophila Sir2 extends lifespan in a dose-dependent manner. Aging (Albany NY) 5 : 682–691.
65. ChintapalliVR, WangJ, DowJAT (2007) Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nature Genetics 39 : 715–720.
66. RoyS, ErnstJ, KharchenkoPV, KheradpourP, NegreN, et al. (2010) Identification of Functional Elements and Regulatory Circuits by Drosophila modENCODE. Science 330 : 1787–1797.
67. BallardJWO (2000) Comparative genomics of mitochondrial DNA in members of the Drosophila melanogaster subgroup. Journal of Molecular Evolution 51 : 48–63.
68. BallardJWO (2000) Comparative genomics of mitochondrial DNA in Drosophila simulans. Journal of Molecular Evolution 51 : 64–75.
69. GuoW, JiangL, BhasinS, KhanSM, SwerdlowRH (2009) DNA extraction procedures meaningfully influence qPCR-based mtDNA copy number determination. Mitochondrion 9 : 261–265.
70. Therneau TM, Grambsch PM (2001) Modeling Survival Data: Extending the Cox Model; Dietz K, Gail M, Krickeberg K, Samet J, Tsiatis A, editors. New York: Springer-Verlag. 350 p.
71. RipattiS, PalmgrenJ (2000) Estimation of multivariate frailty models using penalized partial likelihood. Biometrics 56 : 1016–1022.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 5
Nejčtenější v tomto čísle
- PINK1-Parkin Pathway Activity Is Regulated by Degradation of PINK1 in the Mitochondrial Matrix
- Null Mutation in PGAP1 Impairing Gpi-Anchor Maturation in Patients with Intellectual Disability and Encephalopathy
- Phosphorylation of a WRKY Transcription Factor by MAPKs Is Required for Pollen Development and Function in
- p53 Requires the Stress Sensor USF1 to Direct Appropriate Cell Fate Decision