
Null Mutation in PGAP1 Impairing Gpi-Anchor Maturation in Patients with Intellectual Disability and Encephalopathy
Glycosylphosphatidylinositols (GPI) are glycolipid anchors that anchor various proteins to the cell surface. At least 26 genes are involved in biosynthesis and modification of the GPI anchors. Recently, mutations in eight of those genes have been described. Although those mutations do not fully abolish the functions of encoded enzymes, they lead to a decreased expression of surface GPI-anchored proteins and to different forms of intellectual disability. Here we report a mutation in PGAP1 that encodes a protein that modifies the GPI anchor. We found that the mutation leads to a full loss of PGAP1 enzyme activity, but that the patient cells still express normal levels of surface GPI-anchored proteins. However, the GPI anchors have an abnormal lipid structure that is resistant to cleavage by phosphatidylinositol-specific phospholipase C. Our results add PGAP1 to the growing list of GPI abnormalities that cause intellectual disability and indicate that the fine structure of GPI-anchors is also important for a normal neurological development.
Published in the journal:
. PLoS Genet 10(5): e32767. doi:10.1371/journal.pgen.1004320
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004320
Summary
Glycosylphosphatidylinositols (GPI) are glycolipid anchors that anchor various proteins to the cell surface. At least 26 genes are involved in biosynthesis and modification of the GPI anchors. Recently, mutations in eight of those genes have been described. Although those mutations do not fully abolish the functions of encoded enzymes, they lead to a decreased expression of surface GPI-anchored proteins and to different forms of intellectual disability. Here we report a mutation in PGAP1 that encodes a protein that modifies the GPI anchor. We found that the mutation leads to a full loss of PGAP1 enzyme activity, but that the patient cells still express normal levels of surface GPI-anchored proteins. However, the GPI anchors have an abnormal lipid structure that is resistant to cleavage by phosphatidylinositol-specific phospholipase C. Our results add PGAP1 to the growing list of GPI abnormalities that cause intellectual disability and indicate that the fine structure of GPI-anchors is also important for a normal neurological development.
Introduction
Many eukaryotic cell-surface proteins with various functions are anchored to the membrane via glycosylphosphatidylinositol (GPI) [1]–[3]. After biosynthesis in the endoplasmic reticulum (ER), GPI-anchors are transferred to the proteins by the GPI transamidase and the structure of the GPI-anchor is then remodeled, which is critical for sorting, regulating and trafficking of the GPI anchored proteins (GPI-APs) [3]. This remodeling starts in the ER by eliminating the acyl-chain linked to the inositol in the GPI-anchor by PGAP1 [4], then a side-chain of ethanolamine-phosphate on the second mannose of the GPI-anchor is removed by MPPE1 (PGAP5) [5]. GPI-APs are then transported from the ER to the plasma membrane through the Golgi apparatus, where further remodeling by PGAP3 and PGAP2 takes place [6], [7]. Germline mutations in eight genes that are involved in the GPI-anchor biosynthesis and remodeling have been described (Table 1) [8]–[22]. The mutations in all of those, PIGA, PIGL, PIGM, PIGV, PIGN, PIGO, PIGT and PGAP2, are hypomorphic and lead to partially decreased cell surface expression of various GPI-APs, thus causing a wide phenotypic spectrum ranging from syndromic disorders with various malformations to non-specific forms of intellectual disability. The reported mutations in genes of early steps of the GPI-anchor synthesis such as PIGA (MIM 311770), PIGL (MIM 605947), and PIGM (MIM *610273), or in a gene involved in GPI transfer to proteins such as PIGT (MIM *610272) are supposed to result in a degradation of precursor non-GPI-anchored proteins by ER associated degradation, whereas mutations in genes that are involved in later steps of the pathway, such as PIGV (MIM *610274), PIGO (MIM *614730), and PGAP2 (MIM *615187) result in partial secretion of non-GPI-anchored proteins such as alkaline phosphatase (in case of PIGV or PIGO deficiency) [23] or of proteins bearing cleaved GPI-anchor (in case of PGAP2 deficiency), and are therefore characterized by hyperphosphatasia. Here we report on the identification of a mutation in PGAP1 that encodes the GPI inositol-deacylase [4]. This leads to a new type of GPI-anchor deficiency manifesting non-specific autosomal recessive intellectual disability (ARID), in which cell surface levels of GPI-APs are not affected whereas the structure of GPI moiety is abnormal.

Results
Clinical manifestations
We undertook clinical characterization, mapping [24] and exome sequencing in a large cohort of families with non-specific ARID. We identified the PGAP1 mutation in the Syrian family MR079. The parents in family MR079 are the first-degree cousins and the family has one healthy girl and two affected children that carry the mutation in a homozygous status. The affected girl (III-2) was 4 years and 5 months old and the affected boy (III-3) was 2 years and 9 months old at the time of examination (Figure 1). Pregnancy, delivery, and birth parameters of both children were unremarkable. In the neonatal period, III-2 was hypotonic and III-3 was a floppy baby. Motor development was delayed; III-2 could sit at age of 18 months and at age of 45/12 years first tried to walk independently. At age of 29/12, III-3 could only roll from back to stomach and back. Both children did not finish potty training and were still partially fed with milk bottles. Both children have a developmental delay and severe intellectual disability with an estimated IQ below 35. III-2 could only babble a few syllables. While III-2 had major and absence epilepsy, III-3 did not yet have seizures. Sleeping patterns of both children were normal. They showed some stereotypic movements such as hitting on their own mouth and some washing movements of the hands. Both children seemed to see and hear properly, but specific tests could not be done. Brain CT scan of III-2 at age of one year revealed pronounced brain atrophy. At the time of examination, III-2 was 96 cm tall (25th percentile) with a head circumference of 46 cm (2 cm below the 5th percentile). III-3 was also of normal height and had a head circumference of 47 cm (1.5 cm below the 5th percentile). Their parents had head circumferences of 52 and 53 cm, also in the lower percentiles. Both children have large ears and a flattened nasal root. G-banding, cytogenetic examination and genome wide copy number variants analyses were unremarkable. We did not have information on the levels of alkaline phosphatase and it was not possible to obtain blood probes retrospectively.

Exome sequencing revealed a homozygous mutation in PGAP1
Autozygosity mapping [24] in family MR079 led to the identification of six candidate regions of a total length of 64 Mb. Subsequently, exome sequencing using DNA from individual III-3 was performed as described in former studies [21], [25] resulting in an average coverage of 53.28. 66% of the target sequences were covered with a depth of at least 20×, and 80.51% were covered with a depth of at least 5×. A total of 42,352 SNVs and 2,529 indels were identified. 342 SNVs and 64 indels were neither annotated, nor reported in 1000Genomes and Exome Variant Server, nor in in-house controls, and may affect the protein sequence (non-synonymous, splicing, or UTR). Of those, only two, in PGAP1 and SLC40A, were located in a candidate region, conserved, and predicted to be pathogenic by in silico programs. To exclude further candidate mutations, we repeated the exome sequencing using DNAs of both affected siblings. We enriched the exome using a PCR based targeting method (Ion AmpliSeq Exome Kit) and sequenced on the Ion Proton. The average coverage of III-3 and III-2 was 149.6× and 94.6×, respectively. 91.1% and 85.0% of the target sequences were covered with a depth of at least 20×, 96.3% and 93.4% with a depth of at least 5×, respectively. A total of 49,455 and 47,693 SNVs as well as 3,343 and 3,167 indels were identified. When applying the above mentioned filtering steps, we were by both affected children once again left with the variants in PGAP1 and SLC40A. Since mutations in SLC40A cause hemochromatosis of type 4 and have no effect on cognition (MIM 606069) [26], [27], we focused on the variant in PGAP1, NM_024989.3:c.589_591delCTT, NP_079265.2:p.Leu197del. Genotyping the variant in PGAP1 in 372 healthy Syrian adults using Sanger sequencing revealed no further carriers. Taking the minor allele frequency of 0 in the Exome Sequencing Project (ESP) data set and in our control sample of 372 healthy Syrian individuals, it seems that the mutation has prevalence far less than 0.001.
Molecular modeling using the GeneSilico fold recognition metaserver [28] and Modeler9.9 [29] using the closest related hydrolase (PDB code: 3LP5) as template highlighted the detrimental effect of the deletion of leucine 197 on the structure of PGAP1. Leucine 197 is located in the central strand of a β-sheet and is oriented towards the hydrophobic core of the enzyme where it forms multiple stabilizing interactions with the adjacent helices (Figure 2A, B). Deletion of this amino acid would place Ile198 at the position originally occupied by Leu197 (Figure 2C). The Cβ-branched side-chain of isoleucine cannot be accommodated at this sequence position resulting in several clashes with adjacent amino acids (Leu184, Ile194) of the hydrophobic core (Figure 2C). This will disrupt the packing of the hydrophobic core and consequently of the entire β-sheet topology, thus leading to a loss of tertiary structure and enzymatic activity.
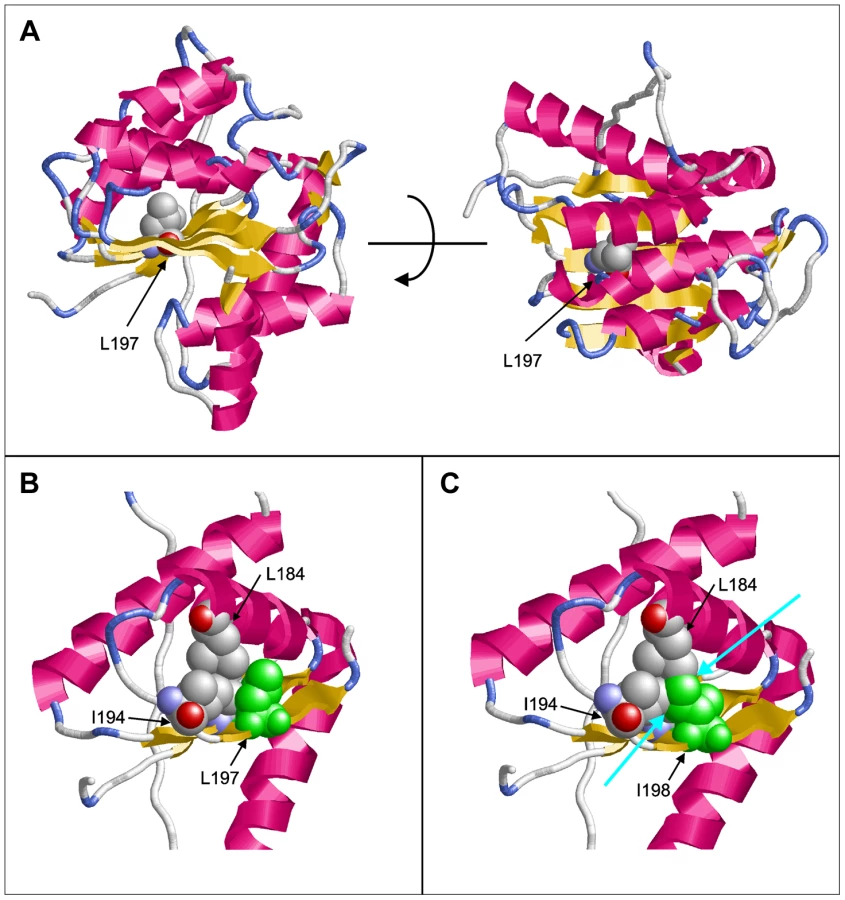
We then ran large scale homozygosity mapping using PLINK in our sample of over 100 consanguineous families [24] and over 600 sporadic cases of ID [30] and identified 7 index patients, 2 from consanguineous families with multiple affected children and 5 from outbreed families with single affected patients, that are homozygous at the PGAP1. Sequencing all seven individuals using Sanger did not reveal any mutations in PGAP1.
We then screened the exome variant server for functional variants in PGAP1. 149 variants are reported in this gene, of those 44 were coding or at splice sites. All of those are extremely rare (0.0077%–0.569%, i. e. 1–74 alleles out of ca. 13000 alleles). Based on the conservation of the variants and the prediction of in silico programs (Table S1), we roughly estimate that a maximum of 48 individuals may carry a mutation in PGAP1 (carrier rate of 48/6500 = 0.0073) and that the prevalence of the disease would be about 13 per million. If we take more conservative in silico prediction numbers, the prevalence of the disease would be 7 per million inhabitants (Table S1). The two most frequent variants in the ESP data were p.Lys111Glu and p.Gln585Glu and were observed in a heterozygous form 15 and 74 times out of 12992 and 12932 alleles, respectively. Both sites are well conserved in the mammalian. Molecular modeling showed that the most common variant Gln585Glu is located outside of catalytic active domains and it was not possible to make a prediction for this variant. Lys111Glu is at the C terminus of a helix of the deacylase domain. The charging pattern of the helix is highly conserved so that we expect that the change from Lys to Glu would change the charge of the protein and destabilize the helix.
Flow cytometry of B-lymphoblastoid cell lines
To determine effects of p.Leu197del alteration on cellular GPI-APs, we investigated the surface expression of GPI-APs on B-lymphoblastoid cell lines (LCLs) derived from the homozygous individual III-3 (−/−), 2 heterozygous parents (+/−), and the healthy sister (+/+) (Figure 3), as well as 6 healthy volunteers with a confirmed wild type genotype (data not shown). Using flow cytometry analysis, the respective surface expressions of CD59, CD55/DAF, and CD48 were quantified. Surface expression of these GPI-APs on LCLs from an affected person, other family members or healthy volunteers showed no significant difference, indicating that the PGAP1 mutation did not affect the surface expression levels of various GPI-APs (Figure 3A, dotted lines). The surface expression of the GPI anchor itself was quantified using fluorochrome conjugated aerolysin (FLAER, Pinewood Scientific), a bacterial toxin that specifically binds GPI anchors, and did not show significant differences between the affected individual, the heterozygous individuals, and the controls (data not shown).

Altered GPI anchors are resistant to PI-PLC cleavage
We then investigated the expected structural abnormality of GPI-anchors by testing sensitivity of GPI-APs to phosphatidylinositol-specific phospholipase C (PI-PLC) [31]. The LCLs were incubated with 10 unit/ml of PI-PLC for 1.5 h at 37°C and the remaining surface GPI-APs were determined by flow cytometry. Of GPI-APs, 61% to 90% were removed from the surface of LCLs of the healthy sister with a homozygous wildtype (Figure 3A, solid line) and healthy control individuals (data not shown). In contrast, no significant or only slight reduction of the surface GPI-APs was seen with LCLs from the affected person (Figure 3A), indicating that almost all GPI-APs on the affected LCLs had abnormal GPI anchors resistant to PI-PLC [4]. This is a strong indication that the p.Leu197del mutation causes null or almost null activity of the PGAP1 enzyme. GPI-APs on LCLs from heterozygous parents were only partially sensitive to PI-PLC (Figure 3A), indicating that the p.Leu197del mutation causes haplo-insufficiency. These defective sensitivities of affected the person's and parents' GPI-APs to PI-PLC were fully restored by transfection of wild-type PGAP1 cDNA (Figure 3B, solid lines).
Finally, the functional effect of the p.Leu197del mutation was tested in the PGAP1 deficient Chinese hamster ovary (CHO) cell system [4]. GPI-APs expressed on the PGAP1 deficient CHO cells are resistant to PI-PLC and the activity of PGAP1 cDNA can be assessed by its ability to make PI-PLC-sensitive GPI-APs after transfection. CHO cells defective for PGAP1 were transiently transfected with N-terminally-FLAG-tagged wild-type and p.Leu197del mutant human PGAP1 cDNA in an expression vector with a strong SRα promoter, or an empty vector. Four days after transfection, each transfectant was treated with or without PI-PLC, and the surface expression of CD59, DAF and urokinase plasminogen activator receptor (uPAR) were assessed by flow cytometry. The wild-type PGAP1 cDNA rescued PI-PLC sensitivity (Figure 4A, left panels). In contrast, the transfection of the mutant p.Leu197del cDNA did not increase the sensitivity to PI-PLC, thus indicating functional loss of the mutant PGAP1 cDNA (Figure 4A, center panels). To determine PGAP1 protein levels, lysates were prepared two days after transfection, immunoprecipitated with anti-FLAG beads and analyzed by SDS-PAGE/Western blotting. The p.Leu197del mutant protein was not detected at all, indicating that the deletion of Leu197 caused an unstable protein (Figure 4B).
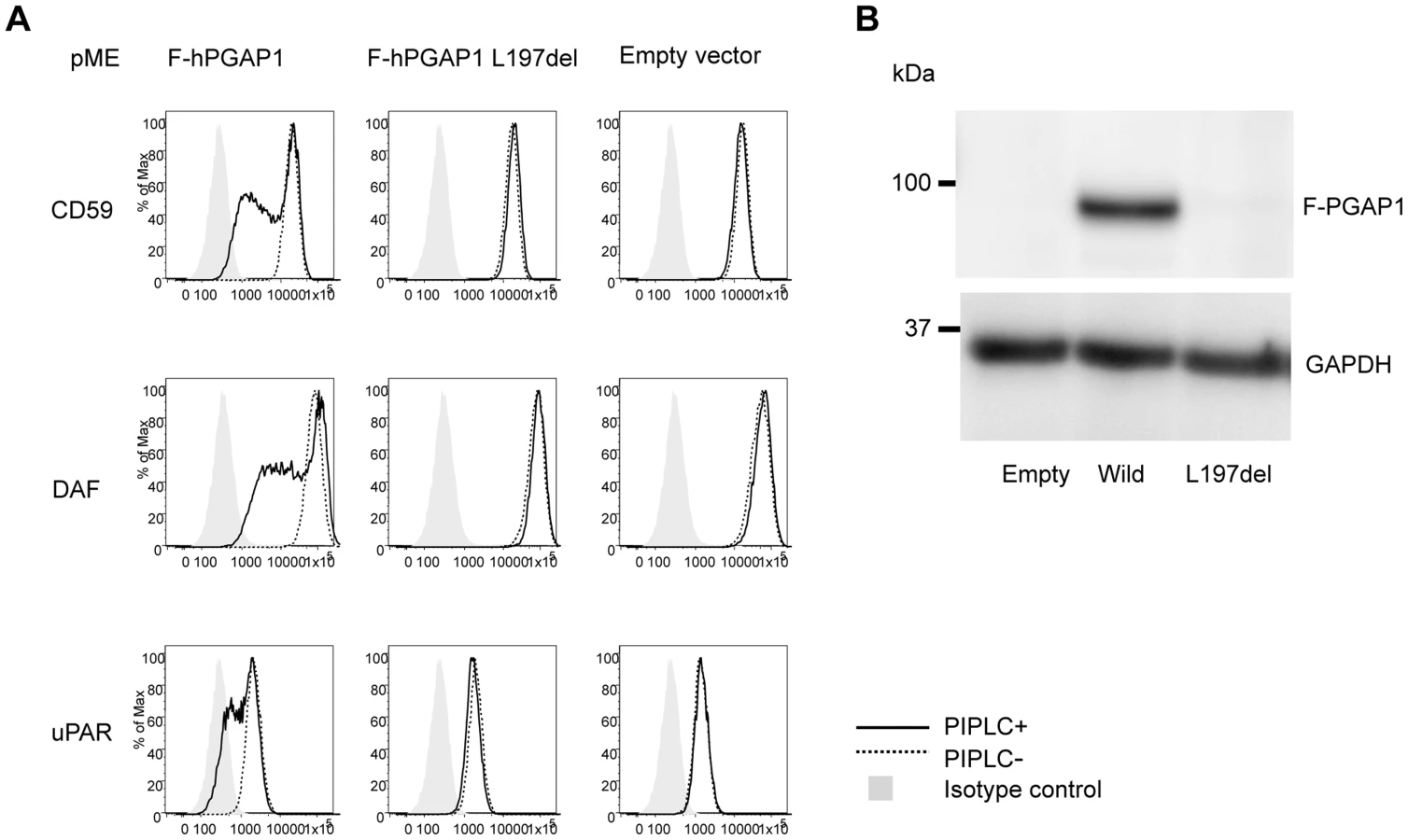
In order to evaluate other known variants in PGAP1, we screened the public database of ESP (see above). Of listed variants, we chose the two most frequent variants: rs142320636: c.331A>G (p.Lys111Glu) and rs62185645: c.1753C>G (p.Gln585Glu), and tested the functional effect of these mutations in the PGAP1 deficient Chinese hamster ovary (CHO) cell system. Transfection of the mutant p.Lys111Glu cDNA did not increase the sensitivity to PI-PLC, indicating functional loss of the mutant PGAP1 cDNA. Mutant p.Gln585Glu showed an activity comparable to the wild type PGAP1 (Figure S1). Thus, it is possible that homozygosity of p.Lys111Glu leads to ARID.
Discussion
Eight GPI deficiencies caused by hypomorphic mutations in the coding regions of GPI biosynthesis genes PIGM, PIGA, PIGL, PIGV, PIGN, PIGO, PIGT, and PGAP2 have been reported. Except PIGM, all lead to a decreased surface expression of GPI-APs and result in intellectual disability, often associated with epilepsy, distinct facial characteristics, and further organ malformations [9]–[22]. We showed here that complete PGAP1 deficiency did not affect the surface expression of GPI-APs but expressed structurally abnormal GPI-APs with the acylated inositol.
In previous works, we have reported that Pgap1 knock-out mice had otocephaly, male infertility, growth retardation, and often died right after birth [32]. Also further two mutant mouse strains, otoxray (oto for otocephaly) [33], [34] and beaker [35] were reported to have disrupted Pgap1. Both mice strains showed developmental abnormalities of the forebrain; the recessive lethal otoxray showed a truncation of the forbrain and the breaker mutant displayed a holoprosencephaly-like phenotype. Both Wnt signaling and Nodal signaling were reported to be affected in these mutant mice. These data emphasize the importance of PGAP1 for vital functions and for brain development. It was also indicated that the Pgap1 mutant mice phenotypes are dependent upon the genetic background since otocephaly and holoprosencephaly are not seen in some mouse strains [34], [35].
Based on our mapping results, exome sequencing data and functional experiments that proved pathogenicity of the mutation, the previous reports on intellectual disability caused by mutations in the GPI synthesis pathway, and the mouse models that clearly show an association between the disruption of Pgap1 and abnormalities of brain, we consider the deletion of leucine197 to be causative for the severe non-specific autosomal recessive intellectual disability in our examined patients of family MR079. PGAP1 is the ninth gene of the GPI synthesis pathway that is now associated to a human phenotype (Table 1). Further mutations in PGPA1 are needed to confirm our findings. Also, describing further patients with different mutations is necessary to delineate the phenotypes of the GPI deficiencies. For example, considering the defect in the modification of the GPI anchors, the alkaline phosphatase would not be elevated in patients with PGAP1 mutations, but this needs to be confirmed.
In conclusion, null mutations in PGAP1 lead to severe intellectual disability and encephalopathy with no obvious malformations; we add PGAP1 to the growing number of genes involved in GPI-anchor deficiencies with human phenotypes. PGAP1 deficiency causes a defect in the ER part of the GPI-AP biosynthesis that involves the remodeling of the anchors after attachment to proteins, and it leads to normal protein expression on the cell surface but to abnormal anchor structure.
Materials and Methods
The study was approved by the Ethic Committees of the Universities of Bonn and of Erlangen-Nürnberg in Germany, and Osaka University in Japan. Informed consent of all examined persons or of their guardians was obtained.
Mapping and exome sequencing
Genomic DNA was extracted from EDTA blood probes by standard methods and genotyped with the Affymetrix Mapping array 6.0 (Affymetrix, Santa Clara, CA, USA). Analysis did not reveal pathogenic deletions or duplications. Mendelian segregation was calculated using PedCheck software and was confirmed in all instances. Autozygosity mapping was performed using HomozygosityMapper [36]. DNA from individual III-3 was enriched using the SureSelect Human All Exon Kit, which targets approximately 50 Mb of human genome (Agilent, Santa Clara, Ca, USA) and paired-end sequenced on a SOLiD 5500 xl instrument (Life Sciences, Carlsbad, CA, U.S.A.). Image analysis and base calling was performed using the SOLiD instrument control software with default parameters. Read alignment was performed with LifeScope 2.5 using the default parameters with human genome assembly hg19 (GRCh37) as reference. Single-nucleotide variants and small insertions and deletions (indels) were detected using LifeScope, GATK 2 and samtools/bcftools [37], [38]. To replicate the results, DNA from individuals III-2 and III-3 was amplified using the Ion AmpliSeq Exome Kit (Life Technologies, Carlsbad, CA, U.S.A.) which targets approximately 58 Mb of the human genome. After quality control on the Bioanalyzer High Sensitivity Chip (Agilent, Santa Clara, Ca, USA) and emulsion PCR (Ion PI Template OT2 200 Kit v3, Life Technologies, Carlsbad, CA, U.S.A.) the samples were sequenced on a Proton PI chip Version 2 (Life Technologies, Carlsbad, CA, U.S.A.). Base calling, pre-processing of the reads, short read alignment and variant calling was performed using the Torrent Suite including the Torrent Variant Caller (TVC, Version 4.0) with default parameters recommended for the Ampliseq Exome panel (low stringency calling of germline variants, Version September 2013). Variant annotation was performed using Annovar, integrating data from a variety of public databases [39], [40]. Additionally, variants were compared to an in-house database containing more than 350 sequenced exomes to identify further common variants which are not present in public databases. Finally, the variants were validated by PCR and Sanger sequencing according to the standard protocols to exclude technical artifacts and to test for segregation.
PI-PLC treatment and FACS analysis
Heparin blood samples were collected from one affected and from all unaffected siblings and parents. Lymphoblastoid Cell lines (LCLs) were generated and cultured in RPMI 1640 (Gibco, Life technologies, Darmstadt, Germany) that is supplemented with 10% FCS (PAA Biotech, Cölbe, Germany) and different other supplements. LCLs from one of the affected siblings (III-3) and the parents were transfected with empty pMEoriP vector or pMEoriP-FLAG-humanPGAP1. Cells from healthy sister were used without transfection. Cells (5×106) were suspended in 0.8 ml of Opti-MEM and electroporated with 20 µg each of the plasmids at 260 V and 960 µF using a Gene Pulser (Bio Rad, Hercules, CA). Four days after transfection, cells were treated with or without 10 unit/ml of PI-PLC (Molecular probes, Eugene, OR) for 1.5 h at 37°C. Surface expression of GPI-APs was determined by staining cells with mouse anti-human CD59 (5H8), -human DAF (IA10), -human CD48 (BJ40) antibodies and each isotype IgG followed by a PE-conjugated anti-mouse IgG antibody (BJ40, mouse IgG1 and IgG2a, and secondary antibody were purchased from BD Biosciences, Franklin Lakes, NJ) and analyzed by flow cytometer (Cant II; BD Biosciences) using Flowjo software (Tommy Digital Inc., Tokyo, Japan).
Functional analysis using CHO cells
pMEFLAG-hPGAP1 mutant (L197del) bearing patient's mutation was generated by site directed mutagenesis. PGAP1 deficient CHO cell (C10) [4] were transiently transfected with wild type or mutant pMEFLAG-hPGAP1 by electroporation. Cells (107) were suspended in 0.4 ml of Opti-MEM and electroporated with 20 µg each of the plasmids at 260 V and 960 µF using a Gene Pulser. Four days after transfection, cells were treated with or without 10 unit/ml of PI-PLC for 1.5 h at 37°C. Surface expression of GPI-APs was determined by staining cells with mouse anti-human CD59 (5H8), -human DAF (IA10), -hamster uPAR (5D6) antibodies and each isotype IgG, followed by a PE-conjugated anti-mouse IgG antibody and analyzed by flow cytometer using Flowjo software. Two days after transfection of each PGAP1 construct, lysates were immunoprecipitated with anti-FLAG beads and analyzed by SDS-PAGE/Western blotting.
Web resources
1000Genomes, http://www.1000genomes.org/
ABI, L.T. (2012). LifeScope.: http://www.lifetechnologies.com/lifescope.
ANNOVAR: http://www.openbioinformatics.org/annovar/
GeneTalk: http://www.gene-talk.de
BWA, Burrows-Wheeler Aligner; http://bio-bwa.sourceforge.net/
dbSNP, NCBI: http://www.ncbi.nlm.nih.gov/snp/
GATK 2, Genome Analysis Toolkit: http://www.broadinstitute.org/gatk/index.php
Kyoto Encyclopedia of Genes and Genomes, KEGG, http://www.genome.jp/kegg/
MutationTaster: http://www.mutationtaster.org/ELAND, alignment algorithm, Illumina.com
NHLBI Exome Sequencing Project (ESP): http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM): http://www.omim.org
PolyPhen2: http://genetics.bwh.harvard.edu/pph2/
SIFT: http://sift.jcvi.org/
UCSC Genome Browser: www.genome.ucsc.edu
Supporting Information
{kind=link}
Zdroje
1. TiedeA, BastischI, SchubertJ, OrleanP, SchmidtRE (1999) Biosynthesis of glycosylphosphatidylinositols in mammalian and unicellular microbes. Biol Chem 380 : 503–523.
2. McConvilleMJ, MenonAK (2000) Recent developments in the cell biology and biochemistry of glycosylphosphatidylinositol lipids. Mol Membr Biol 17 : 1–16.
3. FujitaM, KinoshitaT (2012) GPI-anchor remodeling: potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim Biophys Acta 1821 : 1050–1058.
4. TanakaS, MaedaY, TashimaY, KinoshitaT (2004) Inositol deacylation of glycosylphosphatidylinositol-anchored proteins is mediated by mammalian PGAP1 and yeast Bst1p. J Biol Chem 279 : 14256–14263.
5. FujitaM, MaedaY, RaM, YamaguchiY, TaguchiR, et al. (2009) GPI glycan remodeling by PGAP5 regulates transport of GPI-anchored proteins from the ER to the Golgi. Cell 139 : 352–365.
6. TashimaY, TaguchiR, MurataC, AshidaH, KinoshitaT, et al. (2006) PGAP2 is essential for correct processing and stable expression of GPI-anchored proteins. Mol Biol Cell 17 : 1410–1420.
7. MaedaY, TashimaY, HoujouT, FujitaM, Yoko-oT, et al. (2007) Fatty acid remodeling of GPI-anchored proteins is required for their raft association. Mol Biol Cell 18 : 1497–1506.
8. AlmeidaAM, MurakamiY, BakerA, MaedaY, RobertsIA, et al. (2007) Targeted therapy for inherited GPI deficiency. N Engl J Med 356 : 1641–1647.
9. JohnstonJJ, GropmanAL, SappJC, TeerJK, MartinJM, et al. (2012) The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet 90 : 295–300.
10. van der CrabbenSN, HarakalovaM, BrilstraEH, van BerkestijnFM, HofstedeFC, et al. (2013) Expanding the spectrum of phenotypes associated with germline PIGA mutations: A child with developmental delay, accelerated linear growth, facial dysmorphisms, elevated alkaline phosphatase, and progressive CNS abnormalities. Am J Med Genet A 164 : 29–35.
11. SwobodaKJ, MargrafRL, CareyJC, ZhouH, NewcombTM, et al. (2013) A novel germline PIGA mutation in Ferro-Cerebro-Cutaneous syndrome: A neurodegenerative X-linked epileptic encephalopathy with systemic iron-overload. Am J Med Genet A 164 : 17–28.
12. KrawitzPM, MurakamiY, HechtJ, KrugerU, HolderSE, et al. (2012) Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am J Hum Genet 91 : 146–151.
13. KrawitzPM, SchweigerMR, RodelspergerC, MarcelisC, KolschU, et al. (2010) Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet 42 : 827–829.
14. HornD, WieczorekD, MetcalfeK, BaricI, PalezacL, et al. (2013) Delineation of PIGV mutation spectrum and associated phenotypes in hyperphosphatasia with mental retardation syndrome. Eur J Hum Genet doi:10.1038/ejhg.2013.241
15. KukiI, TakahashiYukitoshi, OkazakiShin, KawawakiHisashi, EharaEiji, InoueNorimitsu, KinoshitaTaroh, MurakamiYoshiko (2013) Case report on vitamin B6 responsive epilepsy due to inherited GPI deficiency. Neurology 81 : 1467–1469.
16. NakamuraK, OsakaH, MurakamiY, AnzaiR, NishiyamaK, et al. (2014) PIGO mutations in intractable epilepsy and severe developmental delay with mild elevation of alkaline phosphatase levels. Epilepsia 55(2): e13–e17.
17. MaydanG, NoymanI, Har-ZahavA, NeriahZB, Pasmanik-ChorM, et al. (2011) Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet 48 : 383–389.
18. OhbaC, OkamotoN, MurakamiY, SuzukiY, TsurusakiY, et al. (2013) PIGN mutations cause congenital anomalies, developmental delay, hypotonia, epilepsy, and progressive cerebellar atrophy. Neurogenetics doi:10.1007/s10048-013-0384-7
19. NgBG, HackmannK, JonesMA, EroshkinAM, HeP, et al. (2012) Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am J Hum Genet 90 : 685–688.
20. KvarnungM, NilssonD, LindstrandA, KorenkeGC, ChiangSC, et al. (2013) A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet 50 521–528.
21. HansenL, TawamieH, MurakamiY, MangY, Ur RehmanS, et al. (2013) Hypomorphic Mutations in PGAP2, Encoding a GPI-Anchor-Remodeling Protein, Cause Autosomal-Recessive Intellectual Disability. Am J Hum Genet 92 : 575–583.
22. KrawitzPM, MurakamiY, RiessA, HietalaM, KrugerU, et al. (2013) PGAP2 Mutations, Affecting the GPI-Anchor-Synthesis Pathway, Cause Hyperphosphatasia with Mental Retardation Syndrome. Am J Hum Genet 92 : 584–589.
23. MurakamiY, KanzawaN, SaitoK, KrawitzPM, MundlosS, et al. (2012) Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. J Biol Chem 287 : 6318–6325.
24. Abou JamraR, WohlfartS, ZweierM, UebeS, PriebeL, et al. (2011) Homozygosity mapping in 64 Syrian consanguineous families with non-specific intellectual disability reveals 11 novel loci and high heterogeneity. Eur J Hum Genet 19 : 1161–1166.
25. Abou JamraR, PhilippeO, Raas-RothschildA, EckSH, GrafE, et al. (2011) Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am J Hum Genet 88 : 788–795.
26. MontosiG, DonovanA, TotaroA, GarutiC, PignattiE, et al. (2001) Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest 108 : 619–623.
27. NjajouOT, VaessenN, JoosseM, BerghuisB, van DongenJW, et al. (2001) A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat Genet 28 : 213–214.
28. KurowskiMA, BujnickiJM (2003) GeneSilico protein structure prediction meta-server. Nucleic Acids Res 31 : 3305–3307.
29. SanchezR, SaliA (2000) Comparative protein structure modeling. Introduction and practical examples with modeller. Methods Mol Biol 143 : 97–129.
30. RauchA, WieczorekD, GrafE, WielandT, EndeleS, et al. (2012) Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380 : 1674–1682.
31. VolwerkJJ, ShashidharMS, KuppeA, GriffithOH (1990) Phosphatidylinositol-specific phospholipase C from Bacillus cereus combines intrinsic phosphotransferase and cyclic phosphodiesterase activities: a 31P NMR study. Biochemistry 29 : 8056–8062.
32. UedaY, YamaguchiR, IkawaM, OkabeM, MoriiE, et al. (2007) PGAP1 knock-out mice show otocephaly and male infertility. J Biol Chem 282 : 30373–30380.
33. ZoltewiczJS, PlummerNW, LinMI, PetersonAS (1999) oto is a homeotic locus with a role in anteroposterior development that is partially redundant with Lim1. Development 126 : 5085–5095.
34. ZoltewiczJS, AshiqueAM, ChoeY, LeeG, TaylorS, et al. (2009) Wnt signaling is regulated by endoplasmic reticulum retention. PLoS One 4: e6191.
35. McKeanDM, NiswanderL (2012) Defects in GPI biosynthesis perturb Cripto signaling during forebrain development in two new mouse models of holoprosencephaly. Biol Open 1 : 874–883.
36. SeelowD, SchuelkeM, HildebrandtF, NurnbergP (2009) HomozygosityMapper–an interactive approach to homozygosity mapping. Nucleic Acids Res 37: W593–599.
37. McKennaA, HannaM, BanksE, SivachenkoA, CibulskisK, et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20 : 1297–1303.
38. LiH, HandsakerB, WysokerA, FennellT, RuanJ, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 : 2078–2079.
39. WangK, LiM, HakonarsonH (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164.
40. AbecasisGR, AltshulerD, AutonA, BrooksLD, DurbinRM, et al. (2010) A map of human genome variation from population-scale sequencing. Nature 467 : 1061–1073.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 5
Nejčtenější v tomto čísle
- PINK1-Parkin Pathway Activity Is Regulated by Degradation of PINK1 in the Mitochondrial Matrix
- Null Mutation in PGAP1 Impairing Gpi-Anchor Maturation in Patients with Intellectual Disability and Encephalopathy
- Phosphorylation of a WRKY Transcription Factor by MAPKs Is Required for Pollen Development and Function in
- p53 Requires the Stress Sensor USF1 to Direct Appropriate Cell Fate Decision