
Loss of Function Mutation in the Palmitoyl-Transferase HHAT Leads to Syndromic 46,XY Disorder of Sex Development by Impeding Hedgehog Protein Palmitoylation and Signaling
Disorders of gonadal development represent a clinically and genetically heterogeneous class of DSD caused by defects in gonadal development and/or a failure of testis/ovarian differentiation. Unfortunately, in many cases the genetic aetiology of DSD is unknown, indicating that our knowledge of the factors mediating sex determination is limited. Using exome sequencing on a case of autosomal recessive syndromic 46,XY DSD with testicular dysgenesis and chondrodysplasia, we found a homozygous missense mutation (G287V) within the coding sequence of the O-acetyl-transferase HHAT gene. The HHAT gene encodes an enzyme required for the attachment of palmitoyl residues that are critical for multimerization and long range signaling potency of hedgehog secreted proteins. We found that HHAT is widely expressed in human organs during fetal development, including testes and ovaries around the time of sex determination. In vitro assays show that G287V mutation impairs HHAT palmitoyl-transferase activity and mice lacking functional Hhat exhibit testicular dysgenesis as well as other skeletal, neuronal and growth defects that recapitulate most aspects of the syndromic 46,XY DSD patient. These data provide the first clinical evidence of the essential role played by lipid modification of Hedgehog proteins in human testicular organogenesis and embryonic development.
Published in the journal:
. PLoS Genet 10(5): e32767. doi:10.1371/journal.pgen.1004340
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004340
Summary
Disorders of gonadal development represent a clinically and genetically heterogeneous class of DSD caused by defects in gonadal development and/or a failure of testis/ovarian differentiation. Unfortunately, in many cases the genetic aetiology of DSD is unknown, indicating that our knowledge of the factors mediating sex determination is limited. Using exome sequencing on a case of autosomal recessive syndromic 46,XY DSD with testicular dysgenesis and chondrodysplasia, we found a homozygous missense mutation (G287V) within the coding sequence of the O-acetyl-transferase HHAT gene. The HHAT gene encodes an enzyme required for the attachment of palmitoyl residues that are critical for multimerization and long range signaling potency of hedgehog secreted proteins. We found that HHAT is widely expressed in human organs during fetal development, including testes and ovaries around the time of sex determination. In vitro assays show that G287V mutation impairs HHAT palmitoyl-transferase activity and mice lacking functional Hhat exhibit testicular dysgenesis as well as other skeletal, neuronal and growth defects that recapitulate most aspects of the syndromic 46,XY DSD patient. These data provide the first clinical evidence of the essential role played by lipid modification of Hedgehog proteins in human testicular organogenesis and embryonic development.
Introduction
Disorders of sex development (DSD) are rare “congenital conditions in which development of the chromosomal, gonadal or anatomical sex is atypical” [1], and which display a wide spectrum of phenotypes. One clinically and genetically heterogeneous class of DSD is partial or complete 46,XY gonadal dysgenesis [2], caused by a defect in gonadal development and/or a failure of testis differentiation. Individuals with 46,XY complete gonadal dysgenesis (46,XY CGD) are characterized by a 46,XY karyotype, normal female external genitalia, undeveloped (“streak”) gonads, no sperm production, and the presence of Müllerian structures. Despite considerable progress in understanding the genetic factors involved in gonadal differentiation, the causative mutation for individuals with 46,XY CGD remains unknown in 80% of the cases [1], [3], [4]. The majority of resolved cases involve mutations or deletions in genes coding for SRY, desert hedgehog (DHH) , MAP3K1 [5] and NR5A1 (SF1) while the prevalence of duplications involving genes coding for NR0B1 (DAX1) and WNT4 represent ∼1% of the resolved cases [6]. One characteristic of DSD with gonadal dysgenesis is their frequent association with other congenital malformations such as growth or mental retardation, conditions that can be referred to as syndromic DSD [7]. The large variation in cases of syndromic 46,XY DSD involving gonadal dysgenesis suggests that among the network of genes essential for proper development of testes and ovaries, some genes may have pleiotropic actions. The study of syndromic DSD thus provides an opportunity to discover new genes involved in human sex determination and improve the diagnosis and clinical management of DSD patients.
The hedgehog (Hh) family of signaling molecules is composed of three members, namely sonic hedgehog (SHH), desert hedgehog (DHH) and indian hedgehog (IHH). Hh molecules function as morphogens that signal at both short and long range through the patched 1 receptor (PTCH1) in a concentration dependent manner. All Hh ligands are initially synthesized as precursor proteins that undergo auto-proteolytic cleavage and dual lipid post-translational modifications. A cholesterol molecule is attached to the C-terminus of the Hh signaling moiety [8], [9], while the N-terminus is palmitoylated by the membrane-bound O-acyl-transferase hedgehog acyl-transferase (HHAT) [10], [11]. These lipid modifications are required for efficient signaling at both long and short range [12], [13]. In particular, palmitate attachment to SHH is required for the formation of multimeric SHH complexes and increases its signaling potency [10], [12], [14], [15].
Hedgehog ligands play a major role in embryonic patterning, growth and differentiation for a variety of organs and tissues. For instance, SHH acts on neural, cranial and skeletal patterning as well as kidney, lung, tooth and eye development. In the case of bone growth and pancreas development it acts in a synergistic manner with IHH (for review, read [16]). Loss of SHH expression or function in humans results in pathologies as diverse as holoprosencephaly [17], preaxial polydactyly [18] or non-syndromic colobomatous microphthalmia [19]. Mutations in IHH, on the other hand, more specifically lead to bone growth defects (brachydactyly or acrocapitofemoral dysplasia [20]). Interestingly, several mutations of DHH have been described in patients with a non-syndromic form of 46,XY DSD with partial or complete gonadal dysgenesis [2], [21], [22], [23]. Similarly, ablation of Dhh in mice leads to severe testis dysgenesis including apolar Sertoli cells, anastomotic testis cords, a reduced number of fetal Leydig cells and insufficient production of androgens, resulting in male infertility, hypogonadism, underdeveloped male accessory glands and feminized external genitalia [24], [25], [26]. In the fetal mouse testis, Dhh is expressed specifically in the Sertoli cells from E11.5 onwards [27] and thus is required for testis cord formation, Leydig cell differentiation and possibly germ cell survival [24], [25], [26], [28].
Taking advantage of next generation sequencing technologies, we identified by whole exome sequencing a missense mutation in the palmitoyl transferase gene HHAT in a unique case of autosomal recessive chondrodysplasia-46,XY DSD with gonadal dysgenesis (Nivelon-Nivelon-Mabille syndrome). These findings provide the first clinical evidence of the essential role that post-translational lipid modifications have on the functional performance of Hh proteins and hence on human testicular organogenesis and embryonic development.
Results
A familial case of syndromic 46,XY complete gonadal dysgenesis with chondrodysplasia
We studied a family with two children affected by a rare autosomal recessive syndrome associated with DSD and chondrodysplasia (OMIM 600092) [29], [30] (Fig. 1A). The first sibling displayed severe dwarfism with generalized chondrodysplasia, a narrow, bell-shaped thorax, micromelia, brachydactyly, severe microcephaly with cerebellar vermis hypoplasia, facial anomalies, hypoplastic irides, and coloboma of both optic discs. Follow up diagnostic tests at 16 years old confirmed previous clinical observations and identified mild mental retardation, muscular hypertrophy, myopia and other facial anomalies such as upslanting palpebral fissures, puffy eyelids, large mouth, and mild prognathism [30]. The karyotype was 46,XY but the patient exhibited clinical features of a 46,XY DSD with complete gonadal dysgenesis (CGD), including normal external female genitalia, lack of pubertal development, primary amenorrhea, and hypergonadotrophic hypogonadism. Histology confirmed the testicular dysgenesis and identified persistent Müllerian and Wolffian duct structures, in particular epididymal remnants. The left gonad contained large inclusions of immature testicular tissue mainly composed of dysplastic immature seminiferous tubules (Fig. 1C - arrowhead). These structures were delimited with a dense interstitial compartment containing a greatly reduced proportion of cells expressing the cholesterol side-chain cleavage enzyme CYP11A1 compared to normal adult testis (Fig. S1). The right gonad was hypoplastic and largely composed of fibroblastic and endothelial tissues. It contained some structures that resembled immature seminiferous tubules surrounded by myoid cells (Fig. 1D - arrowheads). No CYP11A1-expressing cells were detected around these structures (data not shown). It is possible that a small group of functional steroidogenic cells may have developed and persisted in the dysgenic gonads of the patient, in a number sufficient to explain the presence of vestigial Wolffian duct derivatives, however insufficient to achieve complete and normal development of the male genitalia.

The second sibling had a 46,XX karyotype with histologically normal ovaries, but otherwise exhibited similar phenotypic abnormalities including severe dwarfism and generalized chondrodysplasia [29]. Pregnancy was interrupted after ultrasound examination at 17 weeks of gestation. To date no other similar case has been reported.
Homozygous G287V missense mutation in the HHAT gene as a cause for Nivelon-Nivelon-Mabille syndrome
A 244K aCGH analysis performed on genomic DNA extracted from blood cells of the patient did not reveal any chromosomal rearrangements that could account for the pathology. All the copy number variations (CNV) identified during the analysis were annotated in the database of genomic variants (DGV) as benign (data not shown). We then performed targeted exon capture and next-generation sequencing on DNA from the 46,XY DSD proband and her parents (Table 1). Variants were evaluated using VariantMaster [31], for de novo, X-linked and recessive models (Table 2).


One de novo variant was identified in CELSR2 (NM_001408.2:c.50T>C:p.(Leu17Pro), OMIM: 604265) which is predicted as non-pathogenic by PolyPhen2 and Mutation Taster, and pathogenic by SIFT. RNAi-based knockouts of Celsr2 in mice resulted in prominent simplification of the dendritic arbors of cortical pyramidal neurons and Purkinje neurons [32], which in combination with the low pathogenicity score led us to not consider this variant further. Among the X-linked variants the NM_000132.3:c.1272-5T>C in F8 (OMIM: 300841) was not considered, as both programs (i.e. NNSplice and ESEfinder) predicted a slight decrease of the acceptor splice site, which we did not consider as potentially pathogenic. No data were found in the literature that allowed us to evaluate the potential role of WWC3 in the patients' phenotype. XPNPEP2 (OMIM: 300145) has been implicated in ACE inhibitor-induced angioedema [33]. The rather high allelic frequency for both WWC3 and XPNPEP2 variants (∼1/2000) coupled with the absence of a clear functional link between these genes and the patient phenotype led us to consider them as non-pathogenic or at least non-related to the patient's phenotype.
The NM_001170564.1:c.449G>T:p.(Gly150Val) in the HHAT short isoform, equivalent to NM_001122834:c.860G>T:p.(Gly287Val) in the HHAT long isoform (OMIM: 605743), was the only variant remaining after filtering variants for the recessive model. The variant is homozygous in the patient and heterozygous in the parents. Inheritance of the newly identified mutation was confirmed by Sanger sequencing (Fig. 1D). All pathogenicity prediction algorithms, i.e. SIFT, PolyPhen and Mutation Taster, consider this mutation as deleterious, damaging or pathogenic (Table 2). G287V is located just next to the 6th predicted transmembrane domain and in the membrane bound O-acyltransferase (MBOAT) domain, two highly conserved regions of HHAT (Fig. 1E). The G287 residue is also well conserved (GERP++: 4.98) across species from zebrafish to mice and humans (Fig. 1E), suggesting an important role for this amino-acid in either HHAT protein folding, stability or the acyltransferase activity required for palmitoylation of Hh proteins. In addition, the pleiotropic phenotype including dwarfism, chondrodysplasia, brachydactyly and gonadal dysgenesis is consistent with previously described defects in SHH, DHH and IHH signaling [34].
HHAT is widely expressed in mouse and fetal human organs including ovaries and Sertoli cells in developing testes
In order to obtain insight into the potential role of HHAT during fetal development, we characterized its expression in several human tissues, including fetal testes and ovaries, just after the time of sex determination (gestation week (GW) 9). Expression analysis revealed that HHAT is expressed at various levels in most of the fetal human organs investigated, including testes and ovaries (Fig. 2A). Interestingly, the different hedgehog genes showed more specific expression profiles. As expected, DHH is specifically expressed in the embryonic testis, whereas strong SHH expression is detected in the fetal lung and eye and is absent from the fetal heart at this developmental stage (Fig. 2A). We observed a strong expression of IHH in the small intestine and digits, consistent with its well-established role in the development of these structures [35], [36]. In the adult testis, qRT-PCR from purified human adult Sertoli cells, expressing SOX9, and Leydig cells, expressing INSL3, indicated that HHAT is expressed at high levels in Sertoli cells, the unique gonadal source of Hh ligands [27], [37] (Fig. 2B). In mice, transcriptional analysis of XX and XY SF1-positive gonadal cells during sex determination confirmed that Dhh is strongly and specifically expressed in XY cells starting at E11.5 (Fig. 2C), a profile consistent with its reported expression in Sertoli cells [27]. In contrast, Hhat transcripts are already present in the somatic progenitors of both XX and XY gonads at E10.5 prior to sex determination. Following testicular differentiation, there is a 2.5-fold increase in Hhat transcripts levels in XY SF1+ cells by E13.5 (Fig. 2C). Overall, the broad expression of HHAT during development is consistent with the wide-ranging functions of Hh in mediating organogenesis and embryonic patterning. In gonads, our expression analysis suggests that HHAT is expressed in the supporting cell lineage at the time of sex determination and later in Sertoli cells.

G287V mutation impairs HHAT palmitoyl-transferase activity in vitro
To further investigate the effect of the G287V mutation on HHAT function, plasmids encoding wild type and G287V HA-tagged HHAT were generated and expressed in COS-1 cells. Both wild type and G287V HHAT were expressed at equivalent levels in the presence or absence of co-transfected SHH, which in turn was expressed at the same level in the presence of either wild type or mutant HHAT (Fig. 3A). Immunofluorescence staining revealed that the subcellular localization of wild type and G287V HHAT was similar; both proteins localized in the endoplasmic reticulum (ER) and the Golgi (Fig. 3B). To test whether the G287V mutation altered protein stability, cells expressing wild type or mutant HHAT were treated with cycloheximide and chloramphenicol, and the amount of HHAT protein remaining at various time points was measured. The stability of the G287V mutant was comparable to that of wild type HHAT (Fig. 3C).
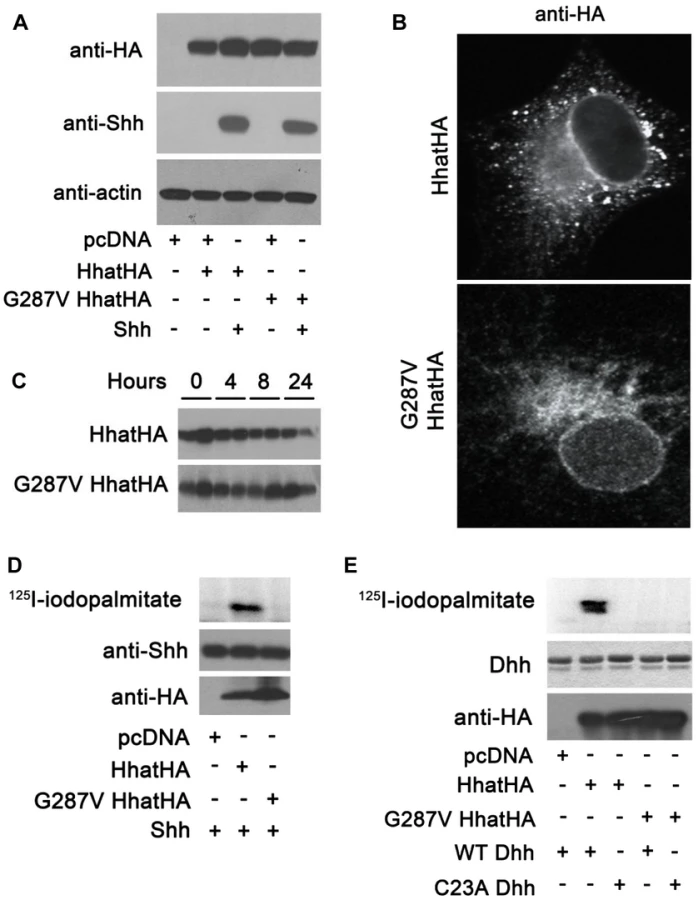
We next used an in vitro assay for HHAT activity, which quantifies the incorporation of 125I-iodopalmitate into Hh proteins, to compare the activity of G287V HHAT to that of wild type. Membranes from cells expressing either wild type or G287V HHAT were incubated with 125I-iodo palmitoyl CoA and purified, recombinant SHH protein. In the presence of wild type HHAT, robust incorporation of 125I-iodopalmitate into SHH was detected (Fig. 3D). By contrast, no incorporation of 125I-iodopalmitate into SHH was detected with G287V HHAT. DHH is the only hedgehog protein expressed in gonads and is likely the more relevant Hh family substrate in this system. To date, no study has tested the ability of HHAT to palmitoylate DHH. We purified a recombinant DHH protein encompassing the N-terminal signaling fragment, along with a C23A DHH mutant, which lacks the N-terminal Cys residue required for palmitoylation. Using the in vitro HHAT activity assay, we provide the first evidence that HHAT directly palmitoylates DHH and that the G287V mutation results in loss of activity (Fig. 3E). Taken together, these results clearly indicate that G287V HHAT lacks the ability to palmitoylate hedgehog proteins.
Palmitoylation of hedgehog ligands is required for testis organogenesis in mice
In mice, Hhat loss of function recapitulates many of the skeletal and growth defects observed in the patients described above, as well as in human individuals bearing mutations in genes encoding Hh proteins [34]. Mice lacking functional HHAT developed to term but died soon after birth. Mutant mice displayed severe dwarfism, short-limbs, diminished chondrogenesis and osteogenesis, facial defects, and holoprosencephaly together with acrania and agnathia [12], [38]. Since defects in testicular differentiation and development have never been investigated in these mutant mice, we analyzed by histological means and immunofluorescence the pattern of expression of key somatic and germ cell markers in the developing gonads of wild type (WT) and HhatCreface/Creface mutant (Creface) mice [38]. The developing XX mutant gonads (E11.5–E15.5) were histologically indistinguishable from XX control gonads (Fig. S2A–H). As expected, none of the testis-specific markers including SOX9 and CYP11A1 were expressed (data not shown), but cells positive for the key ovarian-promoting factors FOXL2 were normally present in XX mutant gonads at E13.5 (Fig. S2J). As expected, female germ cells were normal in numbers and entered meiosis at around E13.5, as shown by the expression of MVH and γH2AX, which were similar in both control and mutant XX gonads (Fig. S2M,N & O,P). Thus, we conclude that ovarian differentiation occured normally in XX mutant gonads.
In XY embryos, no obvious differences were observed between mutant and control XY gonads at E11.5 (Fig. S3A, B). SRY-positive pre-Sertoli cells were present (Fig. S3C, D), indicating that the testicular program was normally initiated, despite the absence of functional HHAT. However, our analysis revealed that testis development was severely affected at subsequent stages, leading ultimately to testicular dysgenesis. We observed a drastic reduction in testis size, which was apparent from E12.5 to E15.5 (Fig. 4A–F). In addition, H&E staining (Fig. 4A–F) as well as immunofluorescence for marker proteins of germ cells (ECADH and MVH) and Sertoli cells (SOX9 and WT1) (Fig. 4G–V), demonstrated not only a decrease in testis cord numbers between wild-type and mutant testes, but also an alteration of the size and shape of testis cords at all stages investigate, which appeared irregular and anastomotic (Fig. 4). Interestingly, while testis cord formation was affected, the differentiation of Sertoli cells appeared to be normal. Both, the expression of SOX9 and WT1, two Sertoli cells markers, were present in XY mutant gonads at all stages investigated (Fig. 4G–L & S–V). These data collectively suggest that HHAT is not required for Sertoli cell commitment but plays a role in proper testis cord formation.

The interstitial compartment of the developing mutant testes at E13.5 and E15.5 exhibited a high cellular density typical of irregular and dense connective tissues, in sharp contrast with the loose structure generally observed in control testes at these developmental ages (compare arrows in Fig. 4 D&F with C&E). In fact, we found that the differentiation of fetal Leydig cells and steroidogenesis were not initiated in mutant gonads, indicated by the absence of the Leydig cell-specific marker CYP11A1 at E12.5 and E13.5 (Fig. 4 T&V). Nevertheless, few rare CYP11A1-positive cells were observed at E15.5 (arrows in Fig. 4X), an expression pattern reminiscent to what was found in the patient gonads (Fig. S1D). Overall, the analysis of the HhatCreface/Creface mutant XY gonads confirmed that the loss of functional HHAT affects the development of fetal Leydig cells as well as the proper formation of testis cords, leading ultimately to testicular dysgenesis.
Discussion
We used whole-exome sequencing (WES) for the identification of the underlying genetic cause of a unique case of autosomal recessive syndromic 46,XY DSD with testicular dysgenesis and chondrodysplasia. Analysis of WES results using an autosomal recessive model revealed a homozygous G287V missense mutation in the hedgehog acyl-transferase (HHAT) gene. This gene encodes an endoplasmic reticulum protein, HHAT, which catalyzes the transfer of palmitate onto hedgehog ligands. The Gly287 residue is a highly conserved amino acid which, when mutated, leads to a non-functional HHAT protein that lacks the ability to palmitoylate hedgehog proteins. Consistent with the patient phenotype, HHAT is expressed in a wide variety of human fetal organs as well as in Sertoli cells, the unique source of DHH in the testis. In addition, Hhat loss-of-function in mice recapitulates most of the testicular, skeletal, neuronal and growth defects observed in the patient. Overall, these data emphasize the essential role played by post-translational lipid modification of Hh ligands in mediating patterning of many aspects of the body, including the testis, limbs, axial skeleton and the central nervous system.
Role of Hedgehog palmitoylation in embryonic development
The patient familial history indicates that the patient and sibling were conceived by a healthy, non-consanguineous caucasian couple [29]. Despite an extensive search through large cohorts of patients suffering from skeletal disorders and/or DSD, we have not been able to identify a second familial case with a pathogenic mutation in the HHAT locus. This suggests that the frequency of pathogenic variants of HHAT is extremely low within the general population, which would explain the absence of other listed cases of Nivelon-Nivelon-Mabille syndrome. It is possible that other mutations in the HHAT locus may lead to severe defects in HHAT cellular localization, expression and/or activity that might cause lethality at the embryonic or fetal stages. Alternatively, additional variants may be found in the non-coding regulatory regions of the gene, as is the case for SHH and other genes [39], [40].
The wide range of developmental and testicular anomalies observed in both the patient and mice lacking the capacity to palmitoylate Hh proteins is consistent with a perturbation of SHH, DHH and IHH signaling. When tested in vivo or with in vitro cell-based assays, palmitoylation of SHH has been shown to be required for the formation of soluble multimeric complexes which enhances its signaling potency [10], [12], [14], [15]. In agreement, mice with Hhat loss-of-function fail to properly establish a long-range Hh signaling gradient [38] which results in testicular, neuronal, skeletal and limb patterning defects [12], [38] similar to those observed in Shh [41], [42], Dhh [24], [25], [26] and Ihh [35], [43] knockout mice. Interestingly, mouse embryos expressing a non-palmitoylated form of SHH (ShhC25S) exhibit similar defects to that of Hhat mutant embryos due to defective SHH signaling [12]. These similar phenotypes indicate that Hh proteins are indeed the major targets of HHAT.
Surprisingly, the clinical characteristics and defects observed in the patient bearing the G287V mutation are not as severe as the phenotypes found in ShhC25S mutant or Hhat knockout mice [12], [38]. While homozygous Hhat mutant mice die around birth, the patient exhibits normal viability despite severe skeletal, craniofacial and neuronal defects. This could be explained by species differences and/or a small residual transferase activity in the G287V-HHAT mutant protein that is not detected in our in vitro assays.
The role of Hh proteins in regulating embryonic pattern formation and adult tissue maintenance in mammals and the deleterious effects that accompany their ablation has been extensively studied (for review see [16]). In the present study, we face a novel and peculiar situation where Hh primary sequences are not affected, but rather the establishment of morphogen gradients and subsequent long range signaling actions of all three Hh ligands. This is reflected by the variable severity of the multiple Hh gene-related symptoms described in this patient. For instance, the impaired testicular development suffered by the patient reaches a severity that is close, if not identical, to what was observed in previous cases of DHH mutation [2], [21], with the generation of streak gonads that do not contain functional reproductive structures. Similarly, the patient suffered a severe form of type A1 brachydactyly, which affects each of the five fingers – a situation typically observed in patients with IHH mutations [44]. In these particular cases, the severity of the symptoms might reflect the necessity to establish a proper long-range Hh gradient for gonadal organogenesis and finger development. On the other hand, the relatively mild abnormalities observed in optical disc and the absence of holoprosencephaly in the patient are in stark contrast with the severe craniofacial defect and abnormal forebrain development found in patients with SHH loss of function [17], [45]. These differences in severity might account for the fact that forebrain development into two separated hemispheres does not rely on the establishment of a long range SHH gradient as much as other patterning SHH-related processes.
Lipid modifications of Hh ligands are conserved during evolution
The core components required for Hh production, movement and signal transduction events are conserved between Drosophila and humans [46], [47]. This is particularly true for the mechanisms of post-translational dual lipid modification of Hh ligands [9], [10]. This is required to generate soluble, multimeric forms of Hh, create the Hh-signaling gradient and to enhance Hh-signaling potency [13]. Palmitoylation is one of the two critical lipid modifications [48] and is mediated by the hedgehog HHAT. This protein is highly conserved, particularly in transmembrane regions and MBOAT domains. It is noteworthy that the mutated G287 residue, one of the highly conserved residues in vertebrate evolution, is positioned adjacent to the predicted 6th transmembrane region and 2nd MBOAT domain (Fig. 1E). Importantly, the G287V mutation does not affect the subcellular localisation and stability of mutant HHAT, both of which are comparable to that of wild type HHAT. It does, however, impair its ability to palmitoylate hedgehog ligands. Further work will be required to define the precise structural role of G287 in mediating HHAT protein folding and its palmitoyl-transferase activity.
HHAT is essential for testicular development and fetal Leydig cell differentiation
Our expression studies using human and mouse embryonic tissues as well as purified cells indicate that HHAT is expressed in developing gonads around the time of sex determination, probably in the supporting cell lineage, and later in Sertoli cells, which are the sole source of hedgehog ligand in the gonad [27], [37]. The absence of functional HHAT leads to testicular dysgenesis both in humans and in the knockout mouse model. Careful analysis of testis development during the sex determination period revealed that early stages of gonad development occur normally up to E11.5 in HhatCreface/Creface mice, and that the initiation of testicular program is not affected. However, by E12.5 testicular development was significantly impaired in XY embryos with a massive reduction in testis size, a reduction in the number of testis cords that also appeared irregular in shape and diameter, and a almost complete absence of fetal Leydig cells. Thus HHAT is not required for Sertoli cell commitment but plays a role in proper testis cord formation and the differentiation of Leydig cells. In this context, it is interesting to note that HhatCreface/Creface testicular phenotype was similar to the one observed in Dhh knockout mice. Knockout mice for the Dhh gene display testis dysgenesis including apolar Sertoli cells, anastomotic testis cords, a decreased number of fetal Leydig cells and an insufficient production of androgens resulting in male infertility, hypogonadism and feminized external genitalia [24], [25], [26]. Consistent with its role in the mouse, several mutations of DHH have also been described in patients with a non-syndromic form of 46,XY DSD with partial or complete gonadal dysgenesis [2], [21], [22], [23]. Although it is now clear that HHAT is essential for testis formation in both humans and mice, further work will be required to define the precise role of HHAT and Hh palmitoylation in mediating testis development and how it influences testis cord formation and the specification of fetal Leydig and peritubular myoid cells.
Materials and Methods
Patient management, sample collection and DNA extraction
All clinical investigations have been performed according to Declaration of Helsinki principles. The study was approved by the local French ethics committee (#DC2011-1332). The proposita was described in [29], [30] and referred at 16 years of age for follow-up. The patient was seen at the Medical Genetics Center of Dijon in September 2004. She presented mild mental retardation, a dysmorphic face, an anteriorly bent thorax with a rigid spine, brachydactyly, muscular hypertrophy and myopia. Endocrine studies performed because of lack of pubertal development showed hypergonadotrophic hypogonadism. Gonadectomy was performed in order to prevent malignant transformation. The proband's parents had a normal phenotype. Genomic DNA from the proband and her parents were isolated from blood samples using the Qiagen DNA mini kit (Qiagen, Valencia, California). An array-CGH 244K (Agilent) has excluded pathogenic microdeletions and microduplications.
Exome capture and sequencing
Exome capture was performed using the SureSelect Human All Exon v3 kit (Agilent Inc). Sequencing was carried out on an Illumina HiSeq 2000 instrument. Fastq files were obtained using the Illumina CASAVA v1.8.1 software and processed using our “in house” bioinformatic pipeline running on the Vital-IT Center for high-performance computing of the Swiss Institute of Bioinformatics (SIB; http://www.vital-it.ch). This pipeline utilizes published algorithms in a sequential manner (BWA [49] for map reads, SAMtools [50] for detection of variants, Pindel [51] for the detection of indels, ANNOVAR [52] for the annotation). The entire coding sequence corresponding to the RefSeq [53] coding genes was used as the reference for the calculation of coverage and reads on target. All experiments were performed using the manufacturer's recommended protocols without modifications.
Results were analyzed using the VariantMaster software [31] in order to identify de novo variants as well as variants respecting different Mendelian inheritance models (dominant with reduced penetrance, recessive, X-linked). Variant validation was performed by targeted Sanger sequencing.
Human tissue collection and Sertoli and Leydig cell purification
Human fetal gonads (9 gestational weeks) were obtained from pregnant women referred to the Rennes University Hospital (France) for legal abortion in the first trimester as previously described [54]. All women received information and gave verbal consent in accordance with national guidelines (Agence de la Biomédecine, authorization #PFS09-011) and protocols were approved by the local ethics committee of Rennes (#11-48). The termination of pregnancy was induced by treatment with Mifegyne (mifepristone) followed 48 h later by Cytotec treatment, and finalized by aspiration. None of the terminations were motivated by fetal abnormality. After recovery from the aspiration product under a binocular microscope (Olympus SZX7, Lille, France), gonads and other somatic organs were snap frozen and stored at −80°C until RNA extraction.
Adult human testes were collected either from prostate cancer patients undergoing orchiectomy who had not received any hormone therapy or from brain dead men with the authorization from the Agence de la Biomédecine (authorization # PFS09-015, “Etude de la spermatogenèse humaine normale et pathologique”). Leydig cells were isolated using a combination of mechanical dissociation, enzymatic digestion, filtration through nylon meshes and density Percoll gradient centrifugation, as previously described [55]. Human primary Sertoli cells were purchased from Lonza (Basel, Switzerland) following isolation procedures described elsewhere [56], [57].
Isolation of mouse Sf1:GFP positive cells and RNA extraction
Urogenital ridges from individual embryos bearing a Sf1-BAC-eGFP transgene-positive urogenital ridges [58], [59] were dissected at relevant stages (E10.5, E11.5, E12.5 and E13.5), digested with trypsin/EDTA, filtered through a 40-mm cell strainer to generate single-cell suspensions and then sorted using a FACS Vantage SE with a purity of above 97% as previously described [58].
Quantitative Reverse Transcriptase PCR (qRT-PCR)
Total RNA from human samples was isolated using Macherey Nagel RNA isolation kit, and 250 ng of total RNA were DNase-treated and converted to 1st strand cDNA using SuperScript II Reverse Transcriptase following the manufacturer's instructions (Invitrogen Life Technologies). RNA from Sf1+ cells was extracted using RNeasy microkit from Qiagen according to the manufacturer's protocol and amplified RNAs were obtained and converted into double-stranded cDNA as previously described [58]. qPCR was carried out in optical 100-well plates and labeled by using the SYBR green master mix (Applied Biosystems). The fluorescence was quantified with a Prism 7900 HT sequence detection system (Applied Biosystems). The expression of each gene was assayed in triplicate as previously described [60], with mouse Gapdh and human RPL19 genes used as reference genes. Primers used for qRT-PCR are listed in Table S1 and were designed using the software PRIMER EXPRESS (Applied Biosystems). The statistical significance of fold-changes was determined by a paired Student's t-test.
Reagents and antibodies
Coenzyme A, CoA synthetase, and anti-HA antibodies were purchased from Sigma (St. Louis, MO). Anti-SHH and anti-CYP11A1 antibodies were purchased from Santa Cruz Biotechnology. Anti-γH2AX, anti-MVH and anti-ECADH antibodies were purchased from Cell Signaling Technology, R&D Systems and Becton Dickinson (USA), respectively. Anti-WT1 was purchased from Dakocytomation (CA, USA). Anti-SRY, anti-SOX9, anti-FOXL2 antibody were previously described [61]. Alexa-conjugated secondary antibodies were purchased from Invitrogen. [125I] NaI was obtained from Perkin Elmer.
Mammalian expression plasmids
The full length SHH and the wild type HHAT cDNA plasmids, in a pcDNA3.1 backbone with a C-terminal HA-tag, have been previously described [11]. The G287V HHAT construct was generated from wild-type HHAT by site directed mutagenesis using the QuikChange mutagenesis kit (Stratagene). A plasmid encoding full length DHH was purchased from Origene. To generate the DHH (23–198) construct for purification, an NdeI restriction site was introduced preceding basepair 69 of DHH. A BamHI site followed by a stop codon was introduced preceding basepair 597. The digested fragment was ligated into NdeI and BamHI digested pET19b (Novagen). The CATATG sequence at the beginning of DHH was deleted using QuikChange to generate the N-terminally His6-tagged human DHH (23–198), with an enterokinase cleavage site immediately preceding residue 23 of DHH. The C23A DHH construct was generated by site directed mutagenesis using QuikChange. All constructs and mutations were confirmed by DNA sequencing.
Cell culture and transfection
COS-1 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 50 units/ml penicillin, and 50 µg/ml streptomycin. 293FT cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 50 units/ml penicillin, 50 µg/ml streptomycin, 500 µg/ml Geneticin, 1 mM GlutaMAX (Invitrogen), 1 mM sodium pyruvate, and 0.1 mM nonessential amino acids. Transfections were carried out using Lipofectamine 2000 (Invitrogen).
Expression of purified recombinant DHH
His-tagged SHH, DHH (23–198) and C23A DHH (23–198) were expressed in Escherichia coli BL21(DE3)pLysS cells, purified on Ni-NTA-agarose resin, and dialyzed (20 mM Tris-HCL pH8.0, 350 mM NaCl, and 1 mM β-mercaptoethanol) in the presence of 6 ng/mL enterokinase. The protein concentration was measured using the DC protein assay (Bio-Rad). The N termini of both wild-type and mutant proteins were confirmed by Edman degradation.
In vitro palmitoylation assay
293FT cells were transfected with plasmids encoding wild type or G287V HHAT. Cells were lysed in hypotonic lysis buffer (0.2 mM MgCl2 and 10 mM HEPES, pH 7.3) 48 hours post transfection. After Dounce homogenization, sucrose was added to a final concentration of 0.25 M and the membrane fraction was pelleted by ultracentrifugation at 100,000 g for 45 min in a Beckman Ti-70.1 fixed angle rotor. The pellet was resuspended in the hypotonic lysis buffer containing 0.25 M sucrose and flash frozen. 10 µg of membranes were incubated with 10 µl of recombinant SHH or DHH (0.2 mg/ml in 20 mM MES (pH 6.5), 1 mM EDTA, and 1 mM DTT), followed by the addition of 30 µl of reaction buffer (167 mM MES (pH 6.5), 1.7 mM DTT, 0.083% Triton X-100, and 167 µM 125I-iodopalmitoylCoA (synthesized using CoA synthetase as previously described [62]). After one hour, the reaction was stopped by the addition of 50 µl of 2× sample buffer with 40 mM DTT. Samples were electrophoresed on SDS-PAGE gels, dried, and exposed to phosphorimaging for 12–18 h.
Protein stability assay
COS-1 cells were transfected with wild type or G287V HHAT. 48 hours post-transfection, cells were incubated in media supplemented with 100 µg/ml cyclohexamide and 40 µg/ml chloramphenicol for 0, 4, 8, 10, or 24 hours. Cells were lysed in 500 µl radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris (pH 7.4), 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, and 1 mM EDTA), electrophoresed on SDS-PAGE gels, transferred to PVDF membranes and probed with anti-HA antibody to determine protein levels.
Hhat knockout mouse line
The Hhat+/Creface mouse line was established and maintained by Paul A. Trainor at the Stowers Institute for Medical Research. These mice were housed in the Laboratory Animal Services Facility at the Stowers Institute for Medical Research according to IACUC animal welfare guidelines. HhatCreface/Creface embryos and control littermates were generated and genotyped by classic PCR as described previously [38]. Embryos were collected from timed matings and staged by designating noon of the day on which the mating plug was detected as E0.5. Embryos were fixed overnight in 4% paraformaldehyde (PFA) and embedded in paraffin. Sections (5-µm) were stained with hematoxylin and eosin (H&E) or processed for immunofluorescence (IF).
Histology and immunofluorescence
Cells were transfected with wild type or G827V HHAT and grown on coverslips for 24 hours. Cells on coverslips were fixed in 4% paraformaldehyde, permeabilized with 0.2% TritonX-100, and stained with indicated antibodies and Hoechst dye. Coverslips were mounted on slides using ProLong Gold mounting solution (Invitrogen). Images were collected on a Zeiss LSM 510 microscope using a 63× water immersion objective.
The patient gonads and healthy human adult testes were fixed in formol whereas mouse samples used for immunofluorescence were fixed in paraformaldehyde. Five µm-sections were cut from paraffin embedded samples and stained with hematoxylin and eosin (H&E), or processed for IF and stained with indicated antibodies and DAPI. All images were obtained with a Zeiss Axioscop or a Nikon C1 Upright microscope.
Supporting Information
Zdroje
1. HughesIA, HoukC, AhmedSF, LeePA (2006) Consensus statement on management of intersex disorders. J Pediatr Urol 2 : 148–162.
2. DasDK, SanghaviD, GawdeH, Idicula-ThomasS, VasudevanL (2011) Novel homozygous mutations in Desert hedgehog gene in patients with 46,XY complete gonadal dysgenesis and prediction of its structural and functional implications by computational methods. Eur J Med Genet 54: e529–534.
3. EggersS, SinclairA (2012) Mammalian sex determination-insights from humans and mice. Chromosome Res 20 : 215–238.
4. HughesIA (2008) Disorders of sex development: a new definition and classification. Best Pract Res Clin Endocrinol Metab 22 : 119–134.
5. PearlmanA, LokeJ, Le CaignecC, WhiteS, ChinL, et al. (2010) Mutations in MAP3K1 cause 46,XY disorders of sex development and implicate a common signal transduction pathway in human testis determination. Am J Hum Genet 87 : 898–904.
6. DomeniceS, CorreaRV, CostaEM, NishiMY, VilainE, et al. (2004) Mutations in the SRY, DAX1, SF1 and WNT4 genes in Brazilian sex-reversed patients. Braz J Med Biol Res 37 : 145–150.
7. HiortaO, Gillessen-KaesbachbG (2009) Disorders of sex development in developmental syndromes. Endocr Dev 14 : 174–180.
8. PorterJA, YoungKE, BeachyPA (1996) Cholesterol modification of hedgehog signaling proteins in animal development. Science 274 : 255–259.
9. PorterJA, EkkerSC, ParkWJ, von KesslerDP, YoungKE, et al. (1996) Hedgehog patterning activity: role of a lipophilic modification mediated by the carboxy-terminal autoprocessing domain. Cell 86 : 21–34.
10. PepinskyRB, ZengC, WenD, RayhornP, BakerDP, et al. (1998) Identification of a palmitic acid-modified form of human Sonic hedgehog. J Biol Chem 273 : 14037–14045.
11. BuglinoJA, ReshMD (2008) Hhat is a palmitoylacyltransferase with specificity for N-palmitoylation of Sonic Hedgehog. J Biol Chem 283 : 22076–22088.
12. ChenMH, LiYJ, KawakamiT, XuSM, ChuangPT (2004) Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev 18 : 641–659.
13. BuglinoJA, ReshMD (2012) Palmitoylation of Hedgehog proteins. Vitam Horm 88 : 229–252.
14. GoetzJA, SinghS, SuberLM, KullFJ, RobbinsDJ (2006) A highly conserved amino-terminal region of sonic hedgehog is required for the formation of its freely diffusible multimeric form. J Biol Chem 281 : 4087–4093.
15. TaylorFR, WenD, GarberEA, CarmilloAN, BakerDP, et al. (2001) Enhanced potency of human Sonic hedgehog by hydrophobic modification. Biochemistry 40 : 4359–4371.
16. VarjosaloM, TaipaleJ (2008) Hedgehog: functions and mechanisms. Genes Dev 22 : 2454–2472.
17. BelloniE, MuenkeM, RoesslerE, TraversoG, Siegel-BarteltJ, et al. (1996) Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet 14 : 353–356.
18. LetticeLA, HeaneySJ, PurdieLA, LiL, de BeerP, et al. (2003) A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet 12 : 1725–1735.
19. SchimmentiLA, de la CruzJ, LewisRA, KarkeraJD, ManligasGS, et al. (2003) Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. Am J Med Genet A 116A: 215–221.
20. ByrnesAM, RacachoL, GrimseyA, HudginsL, KwanAC, et al. (2009) Brachydactyly A-1 mutations restricted to the central region of the N-terminal active fragment of Indian Hedgehog. Eur J Hum Genet 17 : 1112–1120.
21. CantoP, SoderlundD, ReyesE, MendezJP (2004) Mutations in the desert hedgehog (DHH) gene in patients with 46,XY complete pure gonadal dysgenesis. J Clin Endocrinol Metab 89 : 4480–4483.
22. CantoP, VilchisF, SoderlundD, ReyesE, MendezJP (2005) A heterozygous mutation in the desert hedgehog gene in patients with mixed gonadal dysgenesis. Mol Hum Reprod 11 : 833–836.
23. UmeharaF, TateG, ItohK, YamaguchiN, DouchiT, et al. (2000) A novel mutation of desert hedgehog in a patient with 46,XY partial gonadal dysgenesis accompanied by minifascicular neuropathy. Am J Hum Genet 67 : 1302–1305.
24. ClarkAM, GarlandKK, RussellLD (2000) Desert hedgehog (Dhh) gene is required in the mouse testis for formation of adult-type Leydig cells and normal development of peritubular cells and seminiferous tubules. Biol Reprod 63 : 1825–1838.
25. Pierucci-AlvesF, ClarkAM, RussellLD (2001) A developmental study of the Desert hedgehog-null mouse testis. Biol Reprod 65 : 1392–1402.
26. YaoHH, CapelB (2002) Disruption of testis cords by cyclopamine or forskolin reveals independent cellular pathways in testis organogenesis. Dev Biol 246 : 356–365.
27. BitgoodMJ, McMahonAP (1995) Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev Biol 172 : 126–138.
28. MakelaJA, SaarioV, Bourguiba-HachemiS, NurmioM, JahnukainenK, et al. (2011) Hedgehog signalling promotes germ cell survival in the rat testis. Reproduction 142 : 711–721.
29. NivelonA, NivelonJL, MabilleJP, MaroteauxP, FeldmanJP, et al. (1992) New autosomal recessive chondrodysplasia–pseudohermaphrodism syndrome. Clin Dysmorphol 1 : 221–227.
30. Thauvin-RobinetC, MugneretF, CallierP, ChouchaneM, GarronE, et al. (2005) Unique survival in chrondrodysplasia-hermaphrodism syndrome. Am J Med Genet A 132A: 335–337.
31. SantoniFA, MakrythanasisP, NikolaevS, GuipponiM, RobyrD, et al. (2014) Simultaneous identification and prioritization of variants in familial, de novo, and somatic genetic disorders with VariantMaster. Genome Res 24 : 349–55.
32. ShimaY, KengakuM, HiranoT, TakeichiM, UemuraT (2004) Regulation of dendritic maintenance and growth by a mammalian 7-pass transmembrane cadherin. Dev Cell 7 : 205–216.
33. Cilia La CorteAL, CarterAM, RiceGI, DuanQL, RouleauGA, et al. (2011) A functional XPNPEP2 promoter haplotype leads to reduced plasma aminopeptidase P and increased risk of ACE inhibitor-induced angioedema. Hum Mutat 32 : 1326–1331.
34. NieuwenhuisE, HuiCC (2005) Hedgehog signaling and congenital malformations. Clin Genet 67 : 193–208.
35. GaoB, HuJ, StrickerS, CheungM, MaG, et al. (2009) A mutation in Ihh that causes digit abnormalities alters its signalling capacity and range. Nature 458 : 1196–1200.
36. van den BrinkGR, BleumingSA, HardwickJC, SchepmanBL, OfferhausGJ, et al. (2004) Indian Hedgehog is an antagonist of Wnt signaling in colonic epithelial cell differentiation. Nat Genet 36 : 277–282.
37. FrancoHL, YaoHH (2012) Sex and hedgehog: roles of genes in the hedgehog signaling pathway in mammalian sexual differentiation. Chromosome Res 20 : 247–258.
38. DennisJF, KurosakaH, IulianellaA, PaceJ, ThomasN, et al. (2012) Mutations in Hedgehog acyltransferase (Hhat) perturb Hedgehog signaling, resulting in severe acrania-holoprosencephaly-agnathia craniofacial defects. PLoS Genet 8: e1002927.
39. MakrythanasisP, AntonarakisS (2013) Pathogenic variants in non-protein-coding sequences. Clin Genet 84 : 422–428.
40. JeongY, LeskowFC, El-JaickK, RoesslerE, MuenkeM, et al. (2008) Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat Genet 40 : 1348–1353.
41. JeongY, DolsonDK, WaclawRR, MatiseMP, SusselL, et al. (2011) Spatial and temporal requirements for sonic hedgehog in the regulation of thalamic interneuron identity. Development 138 : 531–541.
42. ChiangC, LitingtungY, LeeE, YoungKE, CordenJL, et al. (1996) Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383 : 407–413.
43. St-JacquesB, HammerschmidtM, McMahonAP (1999) Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev 13 : 2072–2086.
44. GaoB, GuoJ, SheC, ShuA, YangM, et al. (2001) Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. Nat Genet 28 : 386–388.
45. TraiffortE, DubourgC, FaureH, RognanD, OdentS, et al. (2004) Functional characterization of sonic hedgehog mutations associated with holoprosencephaly. J Biol Chem 279 : 42889–42897.
46. InghamPW, NakanoY, SegerC (2011) Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet 12 : 393–406.
47. WilsonCW, ChuangPT (2010) Mechanism and evolution of cytosolic Hedgehog signal transduction. Development 137 : 2079–2094.
48. LeeJD, KrausP, GaianoN, NeryS, KohtzJ, et al. (2001) An acylatable residue of Hedgehog is differentially required in Drosophila and mouse limb development. Dev Biol 233 : 122–136.
49. LiH, DurbinR (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 : 1754–1760.
50. LiH, HandsakerB, WysokerA, FennellT, RuanJ, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 : 2078–2079.
51. WangK, LiM, HakonarsonH (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164.
52. YeK, SchulzMH, LongQ, ApweilerR, NingZ (2009) Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25 : 2865–2871.
53. PruittKD, TatusovaT, MaglottDR (2007) NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35: D61–65.
54. Mazaud-GuittotS, NicolazCN, Desdoits-LethimonierC, CoiffecI, MaamarMB, et al. (2013) Paracetamol, aspirin and indomethacin induce endocrine disturbances in the human fetal testis capable of interfering with testicular descent. J Clin Endocrinol Metab 98: E1757–67 doi:10.1210/jc.2013-2531
55. WilleyS, RouletV, ReevesJD, KergadallanML, ThomasE, et al. (2003) Human Leydig cells are productively infected by some HIV-2 and SIV strains but not by HIV-1. AIDS 17 : 183–188.
56. EganJJ, SaltisJ, WekSA, SimpsonIA, LondosC (1990) Insulin, oxytocin, and vasopressin stimulate protein kinase C activity in adipocyte plasma membranes. Proc Natl Acad Sci U S A 87 : 1052–1056.
57. LipshultzLI, MurthyL, TindallDJ (1982) Characterization of human Sertoli cells in vitro. J Clin Endocrinol Metab 55 : 228–237.
58. NefS, SchaadO, StallingsNR, CederrothCR, PitettiJL, et al. (2005) Gene expression during sex determination reveals a robust female genetic program at the onset of ovarian development. Dev Biol 287 : 361–377.
59. StallingsNR, HanleyNA, MajdicG, ZhaoL, BakkeM, et al. (2002) Development of a transgenic green fluorescent protein lineage marker for steroidogenic factor 1. Endocr Res 28 : 497–504.
60. CederrothCR, VinciguerraM, KuhneF, MadaniR, DoergeDR, et al. (2007) A phytoestrogen-rich diet increases energy expenditure and decreases adiposity in mice. Environ Health Perspect 115 : 1467–1473.
61. PitettiJL, CalvelP, RomeroY, ConneB, TruongV, et al. (2013) Insulin and IGF1 receptors are essential for XX and XY gonadal differentiation and adrenal development in mice. PLoS Genet 9: e1003160.
62. BuglinoJA, ReshMD (2008) Hhat is a palmitoylacyl transferase with specificity for N-palmitoylation of sonic hedgehog. J Biol Chem 283 : 22076–22088.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 5
Nejčtenější v tomto čísle
- PINK1-Parkin Pathway Activity Is Regulated by Degradation of PINK1 in the Mitochondrial Matrix
- Null Mutation in PGAP1 Impairing Gpi-Anchor Maturation in Patients with Intellectual Disability and Encephalopathy
- Phosphorylation of a WRKY Transcription Factor by MAPKs Is Required for Pollen Development and Function in
- p53 Requires the Stress Sensor USF1 to Direct Appropriate Cell Fate Decision