
A Genetic Selection for Mutants Reveals an Interaction between DNA Polymerase IV and the Replicative Polymerase That Is Required for Translesion Synthesis
Bacterial DNA polymerase IV (Pol IV) is capable of replicating damaged DNA via a process termed translesion DNA synthesis (TLS). Pol IV-mediated TLS can be accurate or error-prone, depending on the type of DNA damage. Errors made by Pol IV contribute to antibiotic resistance and adaptation of bacterial pathogens. In addition to catalyzing TLS, overproduction of Escherichia coli Pol IV impedes growth. In the current work, we demonstrate that both of these functions rely on the ability of Pol IV to bind the β sliding processivity clamp and switch places on DNA with the replicative Pol, Pol III. This switch requires that Pol IV contact both Pol III as well as two discrete sites on the β clamp protein. Taken together, these results provide a deeper understanding of how E. coli manages the actions of Pol III and Pol IV to coordinate high fidelity replication with potentially error-prone TLS.
Published in the journal:
. PLoS Genet 11(9): e32767. doi:10.1371/journal.pgen.1005507
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005507
Summary
Bacterial DNA polymerase IV (Pol IV) is capable of replicating damaged DNA via a process termed translesion DNA synthesis (TLS). Pol IV-mediated TLS can be accurate or error-prone, depending on the type of DNA damage. Errors made by Pol IV contribute to antibiotic resistance and adaptation of bacterial pathogens. In addition to catalyzing TLS, overproduction of Escherichia coli Pol IV impedes growth. In the current work, we demonstrate that both of these functions rely on the ability of Pol IV to bind the β sliding processivity clamp and switch places on DNA with the replicative Pol, Pol III. This switch requires that Pol IV contact both Pol III as well as two discrete sites on the β clamp protein. Taken together, these results provide a deeper understanding of how E. coli manages the actions of Pol III and Pol IV to coordinate high fidelity replication with potentially error-prone TLS.
Introduction
Despite the actions of several DNA repair pathways, lesions capable of blocking progression of the replicative DNA polymerase (Pol) persist in the DNA template. Translesion DNA synthesis (TLS) represents one evolutionarily conserved mechanism by which organisms cope with these replication-blocking lesions [1–3]. In contrast to repair functions, which either reverse or excise the damage, TLS acts to bypass the damaged site using one or more specialized Pol, allowing the replication fork to proceed past the lesion [1]. Depending on the TLS Pol used, the DNA lesion and its sequence context, bypass may be either accurate or inaccurate [1–3]. Furthermore, due to the fact that most TLS Pols lack intrinsic proofreading activity and possess an open active site, these Pols display a significantly reduced fidelity when replicating undamaged DNA. Thus, TLS Pols may cause mutations by catalyzing inaccurate TLS, or by gaining inappropriate access to undamaged DNA during normal replication. A growing body of evidence supports the view that mutations introduced by TLS Pols contribute to antibiotic resistance and adaptation of microbial pathogens [4–7], as well as genome instability and cancer development in metazoans [8–11]. As a result, the actions of TLS Pols must be tightly regulated to limit unwanted mutations. Furthermore, TLS Pols replicate considerably slower than replicative Pols [12–14]. As a result, their unregulated access to the replication fork would significantly slow replication. The eukaryotic proliferating cell nuclear antigen (PCNA) and the bacterial β sliding clamp proteins play crucially important roles in managing the actions of TLS Pols, and in coordinating their activities with those of their respective replisomes via a process termed ‘Pol switching’ [12,15–20]. Pol-Pol interactions are also suggested to contribute to Pol switching [21–23]. However, several important questions regarding the mechanisms by which TLS Pols switch with replicative Pols, as well as the biological importance of the switching mechanism to regulation of TLS in vivo remain unanswered.
E. coli contains five distinct Pols, which are named Pols I-V. Pols II, IV and V act in TLS [1,3], while Pol I functions in DNA repair and Okazaki fragment maturation [24]. The 20-subunit Pol III holoenzyme serves as the bacterial replicase, and is composed of 2 homodimeric β sliding clamps and 3 core complexes (Pol IIIαεθ), which are tethered together via interaction of Pol IIIα with the heptameric DnaX ATPase (τ3δδ’ψχ) that acts to load the β clamp onto primed DNA [25,26]. The Pol IIIαεθ core complex performs DNA synthesis functions. Within this complex, Pol IIIα catalyzes DNA polymerization, Pol IIIε functions in proofreading and Pol IIIθ modestly stimulates Pol IIIε proofreading activity [25,26]. With the possible exception of Pol I, each of the 5 E. coli Pols contains either a pentameric (QLxLF) or hexameric (QLxLxL) clamp-binding motif (CBM) that is required for biological activity [27–31]. The CBM interacts with a hydrophobic cleft located near the C-terminus of each β clamp protomer. The different Pols also contact non-cleft surfaces of the β clamp, and these interactions likewise contribute to Pol function and/or Pol switching [17,32–37]. To date, structural information regarding these non-cleft contacts is restricted to Pol IV. In the co-crystal structure of the complex consisting of the Pol IV little finger domain (Pol IVLF; residues 243–351) bound to the β clamp, residues 303VWP305 of Pol IV interacted with positions E93 and L98 on the rim of the β clamp, while the C-terminal hexameric CBM (346QLVLGL351) of Pol IV extended over the β clamp dimer interface to interact with the β clamp cleft of the adjacent β clamp protomer [32]. Using a primer extension assay, we previously demonstrated that while only the Pol IV-β clamp cleft interaction was required for processive replication, both the β clamp rim and cleft interactions contributed to Pol III-Pol IV switching in vitro [12,17,38]. In contrast to our findings, Gabbai and colleagues, utilizing a different assay that may more accurately represent the structure and composition of the replisome, concluded that the Pol IV-β clamp rim contact stimulated but was not required for a Pol III-Pol IV switch in vitro [18]. In light of this finding, they suggested direct competition between Pol III and Pol IV for the β clamp cleft as an alternative mechanism for their switching. In addition to switching, Pol IV can be recruited directly to single strand (ss) DNA gaps generated by Pol III skipping over lesions in the template strand to continue replication downstream of the block [1,39]. In summary, recruitment of TLS Pols to lesions is suggested to occur by two different mechanisms: (i) β clamp may recruit TLS Pols post-replicatively to lesions within ssDNA DNA gaps generated by Pol III skipping, or (ii) TLS Pols may be recruited to the replication fork and access lesions after undergoing a switch with Pol III. However, the extent to which these proposed mechanisms are utilized in vivo has not yet been determined. Furthermore, the biological relevance of the Pol IV-β clamp rim interaction to the TLS function of Pol IV is also unknown.
E. coli Pol IV catalyzes accurate bypass of N2-dG lesions induced by nitrofurazone (NFZ), as well as alkylated adducts such as N3-methyladenine (N3-mdA) caused by methyl methanesulfonate (MMS). As a result, E. coli strains lacking Pol IV function (i.e., ΔdinB) display sensitivity to these agents [17,40–43]. In contrast to this protective role, overproduction of Pol IV is lethal to E. coli [34,44,45]; similarly, aberrant expression of Pol κ, the eukaryotic ortholog of dinB, promotes genome instability in human cells [46]. Lethality in aerobically cultured E. coli cells was suggested to result from toxic levels of double strand (ds) DNA breaks resulting from efforts to repair closely spaced 8-oxo-7,8-deoxyguanosine (8-oxo-dG) adducts incorporated during replication of undamaged DNA by Pol IV [47]. However, sensitivity of a dnaN159 strain to ~4-fold higher than SOS-induced levels of Pol IV [34,44], and of a dnaN+ E. coli strain to significantly higher than SOS-induced levels (i.e., ~70-fold; [45]), were both independent of Pol IV catalytic activity, suggesting that at least in these cases, lethality relied on one or more alternative mechanisms. The dnaN159 allele encodes a mutant form of the β sliding clamp that is deficient in regulation of proper access of the different E. coli Pols to DNA [34,35,38,44,48,49]. Thus, sensitivity of the dnaN159 strain to elevated levels of Pol IV was suggested to result from its enhanced ability to replace the bacterial Pol III replicase at the replication fork, thereby disrupting DNA replication [34,44,48]. Consistent with a Pol III-Pol IV switch underlying this lethal phenotype, mutations in Pol IV that disrupt its ability to interact with either the cleft (Pol IVC; see Table 1) or the rim (Pol IVR) of the β clamp abrogated its ability to kill the dnaN159 strain [38]. Finally, SOS-induced levels of Pol IV modestly slowed the rate of DNA replication in vitro [13,14,45], while overproduction of Pol IV severely impeded it, possibly by replacing Pol III [13,14,45]. The finding that Pol IVLF-β clamp interactions were dispensable when Pol IV was expressed at ~70-fold higher than SOS-induced levels suggested that Pol IV may interact with Pol III to effect its displacement from the β clamp [45]. Based on these results, Pol IV was suggested to act as a DNA damage checkpoint effector that acts to slow replication fork progression in response to DNA damage [13,45].

The goal of this study was to define the relationship between the physiological function of Pol IV in TLS and its ability to impede E. coli growth when overproduced. With this goal in mind, the hypersensitivity of the dnaN159 strain was exploited to identify 13 novel mutant Pol IV proteins that failed to confer lethality. Genetic and biochemical characterization of these Pol IV mutants strongly suggest that the ability of overproduced levels of Pol IV to inhibit E. coli growth is a consequence of its ability to gain inappropriate access to the replication fork via a switch that is mechanistically similar to that used under physiological conditions to coordinate the actions of Pol IV with Pol III. Importantly, further analysis of one of the mutants, Pol IV-T120P, revealed novel insights into the mechanism by which Pol IV gains access to DNA lesions in vivo. Specifically, Pol IV-T120P retained complete catalytic activity in vitro but, like Pol IVR and Pol IVC, failed to support Pol IV TLS function in vivo. Using a single molecule primer extension assay, we demonstrated that the T120P mutation abrogated a biochemical interaction of Pol IV with Pol III that was required for Pol III-Pol IV switching. Taken together, these results suggest that Pol III-Pol IV switching involves interaction of Pol IV with both Pol III and the β clamp rim and cleft regions, and provide strong support for the view that Pol III-Pol IV switching represents a vitally important mechanism for regulating TLS in vivo by managing access of Pol IV to the DNA.
Results
Identification of novel Pol IV mutations that fail to impede growth of the dnaN159 E. coli strain
Based on results of a quantitative transformation assay, the hypersensitivity of the dnaN159 lexA51(Def) strain (MS105) to ~4-fold higher than SOS-induced levels of Pol IV expressed from its native LexA-regulated promoter present in pRM102 was independent of Pol IV catalytic activity ([38]; see Pol IV-D103N in S1 Table). Moreover, the ability of Pol IV to impede growth of MS105 was independent of dsDNA breaks stemming from the incorporation into nascent DNA of oxidized precursors, since lethality was observed regardless of whether MS105 was grown aerobically or anaerobically (S2 Table). In contrast, lethality of the dnaN159 strain required the ability of Pol IV to interact with both the rim and cleft of the β clamp ([38]; see Pol IVR and Pol IVC in S1 Table). Taken together, these results suggest that lethality was caused by inappropriate access of Pol IV to the replication fork, rather than its ability to incorporate oxidized precursors into nascent DNA [47]. With the goal of gaining insight into the mechanism by which elevated levels of Pol IV impeded growth, a genetic assay was used to select for novel Pol IV mutants that are unable to kill the dnaN159 strain. From a total of ~2 x 107 independent clones, 16 plasmid-encoded dinB mutants expressing a full length Pol IV protein were identified (S1 Fig). These mutants corresponded to 13 unique dinB alleles: 12 contained a single missense mutation, while one contained two missense mutations (Table 2). Using our quantitative transformation assay referred to above [38], we confirmed that each of these mutant Pol IV-expressing plasmids was unable to impede growth of the dnaN159 strain (S1 Table). For comparison, the wild type Pol IV-expressing plasmid pRM102 was more than 1,000-fold less efficient at transforming MS105 relative to the pWSK29 control (S1 Table).
In addition to impeding E. coli growth, overproduced levels of Pol IV also promote –1 frameshift mutations within homopolymeric runs of dG or dA [50], while SOS-induced levels confer UV sensitivity upon the dnaN159 strain [38,48]. Since these phenotypes appear to result from the ability of Pol IV to gain inappropriate access to the replication fork [34,44,48], we hypothesized that the Pol IV mutations described above would likewise be impaired for these functions. Based on results of a lacZ–→LacZ+ reversion assay [51,52], all 13 mutant dinB alleles were impaired for promoting –1 frameshift mutations (S2 Fig). Likewise, these mutants were also impaired for conferring UV sensitivity upon the dnaN159 strain (S3 Fig). Taken together, these results suggest that the mutations identified in Pol IV that prevented it from killing the dnaN159 strain served to impair its catalytic activity and/or its ability to gain access to the DNA template in vivo.
As summarized in Fig 1A, the identified Pol IV mutations are distributed throughout all 4 structural domains of Pol IV. To gain insight into the possible defect(s) associated with each mutant Pol IV, the positions of the mutations identified in each of the 13 dinB alleles were represented on in silico models for the structure of the Pol IV-β clamp complex assembled on primed DNA in either a non-replicative (meaning Pol IV is bound to both the β clamp rim and cleft, but not the DNA; see Fig 1B) or a replicative mode (meaning Pol IV is bound only to the β clamp cleft, as well as the DNA; see Fig 1C). Based upon these structural models, combined with our current understanding of E. coli Pol IV structure-function [32,53], as well as published studies of the homologous Sulfolobus solfataricus P2 Dpo4 [54,55], we inferred likely functions for several of the mutated residues in Pol IV (Table 2). Residues A15, G52, C66 and A143 likely contribute to either structural integrity or the hydrophobic core (Fig 1A–1C), suggesting that mutations of these residues most likely alter overall Pol IV structure. Position D10 likely contributes to structural integrity and is adjacent to D8, D103 and E104 (Fig 1D), which together act to coordinate 2 Mg+2 ions to constitute the catalytic core of Pol IV, suggesting that substitution of D10 with glycine might affect the structure of the Pol IV catalytic center. Residue R75 resides in the fingers domain and appears to form a hydrogen bond with residue D20 in the palm (Fig 1E), possibly contributing to the tertiary structure of Pol IV. Residues G183, G219, and R323 are all in close proximity to the DNA template and may be involved in Pol IV-DNA interactions (Fig 1F–1H). Finally, H302 and Q342 of Pol IV are immediately adjacent to residues previously demonstrated by Bunting et al. to directly contact the β clamp rim (303VWP305) or cleft (346QLVLGL351), respectively, suggesting that the H302Q and Q342K substitutions disrupt these interactions ([32]; Fig 1B). While presumed functions could be assigned for many of the mutants based upon previous studies, possible defects of the Pol IV mutants bearing substitutions of residues A44, T120 or A149 remain unclear (Table 2).

Overexpression of Pol IV catalytic domain mutants fails to impede growth of dnaN+ E. coli
In order to gain insight into the relationship between the abilities of elevated levels of Pol IV to impede growth of the dnaN159 strain [34,38,44] and overproduced levels of Pol IV to kill the dnaN+ strain [45], a quantitative transformation assay was used to analyze the phenotypes of pBAD derivatives bearing the relevant Pol IV mutations (see Table 1). The ability of overproduced levels of full-length Pol IV (pDB10) or the catalytic domain of Pol IV (Pol IVCD; residues 1–230 expressed from pDB12) to impede growth of E. coli was independent of both its catalytic activity [45] and aerobic growth (S4 Fig), similar to the situation discussed above for the dnaN159 strain (S1 Table and S2 Table). Since overexpression of Pol IVCD was necessary and sufficient to impede growth ([45]; see Fig 2), we focused on Pol IV mutations mapping within the first 230 residues of Pol IV. Despite the fact that the mutant Pol IV proteins appeared stable when expressed from their native dinB promoter contained within a low copy number plasmid (S1 Fig), 6 of the 11 mutants containing substitutions within the first 230 residues of Pol IV displayed either poor solubility or signs of extensive proteolysis following their overproduction from the T7 promoter while one mutant (R75L) was specifically unstable when cloned into Pol IVCD-expressing pBAD plasmid (see Table 2). These Pol IV mutants were not pursued further. Results for the other 4 Pol IV mutants are summarized in Fig 2. The plasmids overproducing Pol IVCD-D10G (pDB20), Pol IVCD-C66S (pDB21), Pol IVCD-T120P (pDB23), or Pol IVCD-G183V (pDB25) each transformed the dnaN+ strain with an efficiency comparable to that of the pBAD control, both in the presence or absence of arabinose (albeit most exhibited tiny to small colonies), while the plasmid overproducing wild type Pol IV (pDB10) or Pol IVCD (pDB12) failed to transform in the presence of arabinose (Fig 2). However, in contrast to the other mutants, which formed tiny to small colonies in the presence of arabinose (Fig 2), the strain overproducing Pol IVCD-T120P displayed robust colonies that were indistinguishable from the strain bearing either the empty pBAD plasmid or overproducing Pol IVLF (pDB14). In contrast to the robust growth observed for the strain overproducing Pol IVCD-T120P, the strain overproducing the full length Pol IV-T120P (pDB33) failed to grow in the presence of arabinose (Fig 2). We confirmed that the Pol IVCD-T120P mutant was expressed in a soluble form and at a level comparable to wild type Pol IVCD (see legend to Fig 2). Based on this observation and results discussed later in this report, the difference between the growth phenotype of the strain expressing Pol IVCD-T120P and that expressing full length Pol IV-T120P appears to be a result of the Pol IVLF domain, which contributes to the ability of Pol IV to impede E. coli growth [16,45]. Taken together, these findings suggest that a common mechanism underlies the ability of overproduced levels of Pol IVCD to impede growth of the dnaN+ strain and near-physiological levels of Pol IV to impede growth of the dnaN159 strain. Furthermore, the fact that Pol IVCD lacks the β clamp-binding Pol IVLF domain, yet is nevertheless able to displace Pol III from the β clamp in vitro [16,45], suggests that Pol IVCD interacts physically with one or more subunit of Pol III holoenzyme. Thus, the ability of T120P to alleviate the lethal phenotype may be indicative of this mutant being impaired for a Pol III-Pol IV interaction.

Pol IV-T120P catalyzes DNA replication similar to wild type Pol IV in vitro
In order to gain insight into the mechanistic basis for the phenotypes of the mutant Pol IV proteins, recombinant forms of each were purified for biochemical analyses. As noted above, we were able to overproduce all 13 mutant proteins. However, 6 of the 13 were either partially proteolyzed or poorly soluble following their overproduction from the T7 promoter (see Table 2), suggesting that their substitutions may affect the tertiary structure of Pol IV. The ability of the other 7 mutant Pol IV proteins to catalyze DNA replication in vitro was analyzed using a primer extension assay [17,34,38,56]. The DNA template consisted of a 32P labeled 30-mer annealed near the middle of a 100-mer (see depiction in Fig 3). Using this template, replication activity of each mutant Pol IV alone, as well as in the presence of single stranded DNA binding protein (SSB), β clamp and the DnaX (γ3δδ’) clamp loader accessory proteins was analyzed. As controls, we examined Pol IV-D103N, which lacks catalytic activity [17,38,57], as well as Pol IVR and Pol IVC, which are impaired for interaction with the β clamp rim or cleft, respectively [38]. Based on published in vitro studies, the Pol IV-β clamp rim interaction is required for Pol III-Pol IV switching, but is dispensable for Pol IV replication. In contrast, the β clamp cleft interaction is required for both Pol IV replication and the Pol III-Pol IV switch [17].

In the absence of accessory proteins, Pol IVR, Pol IVC, Pol IV-T120P and Pol IV-H302Q/Q342K exhibited replication activity roughly comparable to wild type Pol IV (Fig 3A). In contrast, Pol IV-R323S was only modestly active, while Pol IV-D10G, Pol IV-C66S, Pol IV-R75L and Pol IV-G183V lacked detectable activity, similar to Pol IV-D103N (Fig 3A). In the presence of SSB, β clamp and the DnaX complex, Pol IVR and Pol IV-T120P were again indistinguishable from wild type Pol IV (Fig 3B). Pol IV-C66S, Pol IV-R75L, Pol IV-G183V and Pol IV-R323S each displayed modest replication activity (Fig 3B), suggesting that the presence of accessory factors compensated in part for their intrinsic biochemical defects, possibly by helping to recruit the mutant Pol IV proteins to the primer/template junction, and/or by stabilizing an active conformation of the mutant Pol IV protein. Whereas Pol IV-H302Q/Q342K was indistinguishable from wild type Pol IV in the absence of accessory proteins (Fig 3A), it was impaired for processive replication in their presence compared to wild type (Fig 3B), suggesting that the Q342K mutation, which is adjacent to the Pol IV CBM (see Fig 1), interferes with the Pol IV-β clamp cleft interaction. Finally, Pol IV-D10G lacked detectable activity, similar to the D103N mutation, suggesting that D10 either participates directly in catalysis, or its substitution with glycine perturbs the structure of the catalytic center (see Fig 1D). Taken together, these results demonstrate that with the exception of Pol IV-D10G, each of the mutant proteins retained at least partial catalytic activity in vitro. Remarkably, Pol IV-T120P supported replication activity and processivity that were each comparable to that of wild type Pol IV.
In order to quantify the replication activity of Pol IV-T120P more rigorously, we measured its kinetic parameters and compared them to those of wild type Pol IV. As summarized in Table 3, the catalytic efficiency (kpol/Kd) of Pol IV-T120P was ~2.5-fold higher than wild type Pol IV for incorporation of dC opposite template dG, and ~0.5-fold lower than wild type Pol IV for incorporation of dT opposite template dA. Both Pol IV and Pol IV-T120P were able to incorporate the other three dNTPs opposite a template dG or dA. However, in all cases the efficiency of misincorporation was significantly less (<10%) than that measured for correct incorporation. The small differences in catalytic efficiency between wild type Pol IV and Pol IV-T120P were attributable to effects of the T120P substitution on both dNTP binding (Kd) and Pol turnover (kpol) (Table 3). Thus, despite the fact that residue T120 is well removed from the catalytic center of Pol IV (Fig 1), its substitution with a proline nevertheless exerts a modest effect on Pol IV catalysis. These findings, taken together with those discussed above, confirm that Pol IV-T120P retains essentially wild type Pol activity when replicating undamaged DNA, despite its inability to impede E. coli growth when expressed at elevated levels.

Mutant Pol IV proteins are impaired for TLS in vivo
Whereas Pol IV plays an important role in tolerating MMS-induced DNA damage by accurately bypassing lesions including N3-mdA, Pol V (umuDC) contributes to MMS-induced mutations by mediating error-prone bypass of apurinic/apyrimidinic (AP) sites generated by either DNA glycosylases involved in the repair of alkylated bases, or their spontaneous decay [41,58]. Consistent with one published result [41], loss of Pol IV function resulted in a ~5-fold increase in the frequency of Pol V-dependent MMS-induced mutagenesis (Fig 4A). In contrast, expression of Pol IV at ~4-fold higher than SOS-induced levels from a low copy number plasmid (pRM102) reduced the frequency of MMS-induced mutagenesis ~5-fold compared to the pWSK29 empty vector control (Fig 4B). Taken together, we interpret these results to mean that Pol IV is limiting for accurate bypass of MMS-induced DNA lesions in vivo, and that when Pol IV is present, it leads to a reduction in the number of AP sites encountered by the replisome, thereby minimizing the Pol V-dependent mutator phenotype. In light of these findings, we asked whether any of the Pol IV mutants were able to reduce MMS-induced mutagenesis when expressed at ~4-fold higher than SOS-induced levels. In addition to the Pol IV mutants identified in the screen, we also analyzed Pol IVR and Pol IVC. As summarized in Fig 4B, overexpression of Pol IVR or Pol IVC failed to reduce the frequency of MMS-induced mutagenesis (p<0.0001 based on Student’s t-test). Importantly, these results demonstrate a biologically important role for the β clamp rim and cleft in supporting Pol IV TLS function. Like Pol IVR and Pol IVC, each of the other Pol IV mutants were also unable to reduce the frequency of MMS-induced mutagenesis compared to the strain expressing wild type Pol IV from pRM102 (p<0.0001 based on Student’s t-test). Taken together, these findings suggest that these mutant Pol IV proteins are impaired for gaining access to MMS-induced DNA adducts in vivo and/or mediating their bypass following recruitment, effectively shifting the bypass burden to Pol V.
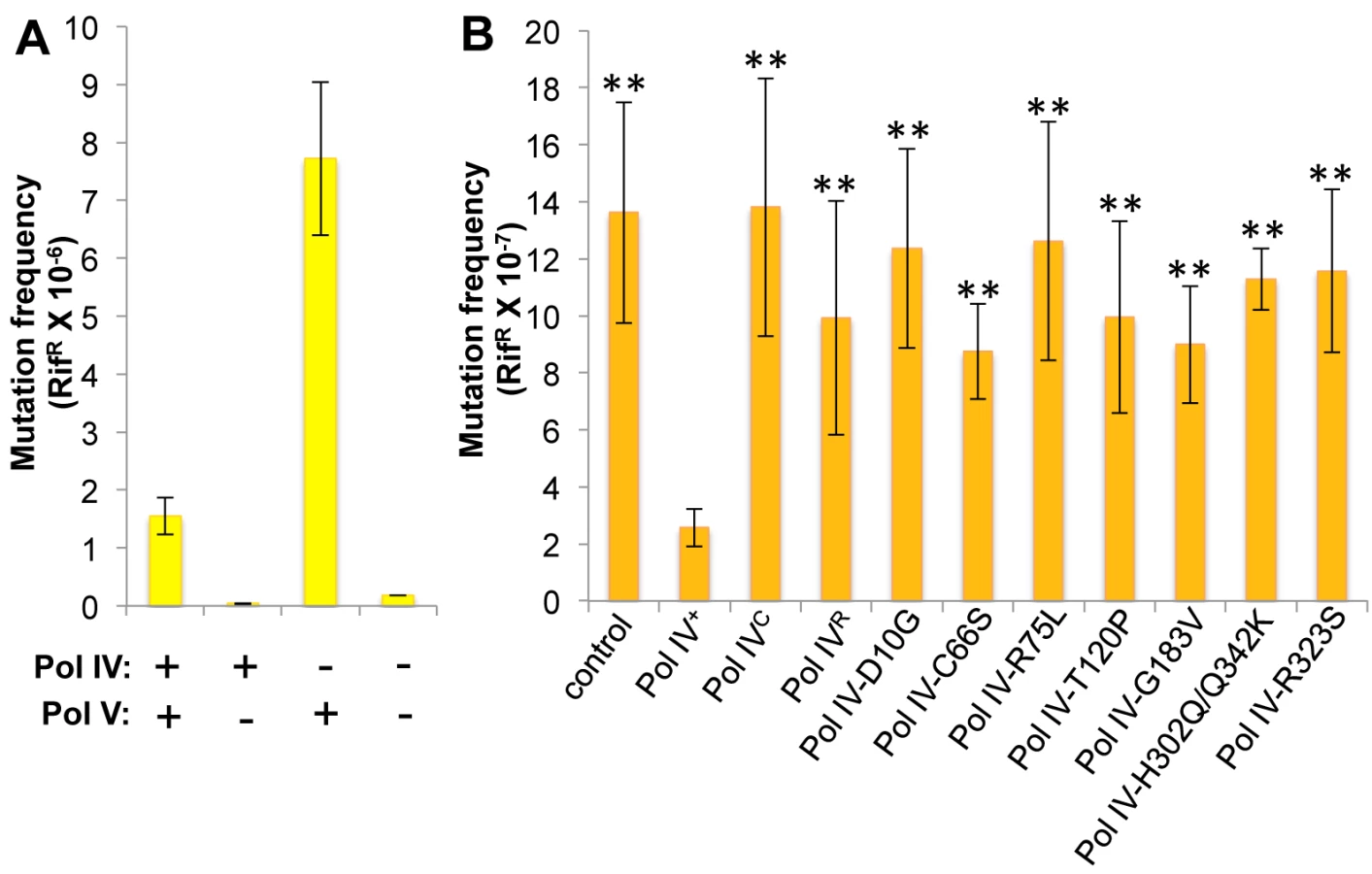
Pol IV-T120P is impaired for tolerating MMS-induced DNA damage in vivo
Since the Pol IV-T120P mutant retained complete catalytic activity while replicating undamaged DNA in vitro (Fig 3 and Table 3), but failed to accurately tolerate MMS-induced lesions in vivo when expressed at an elevated level (Fig 4), we more rigorously analyzed the TLS activity of Pol IV-T120P in vivo under physiologically relevant conditions. To this end, the dinB+ allele was replaced with dinB89, which encodes the Pol IV-T120P mutation (see Table 2), and the ability of the resulting strain to tolerate MMS-induced DNA damage was measured. As controls, strains lacking dinB (Pol IV) and/or umuDC (Pol V), as well as Pol IV-D103N (dinB80), which lacks detectable Pol activity were used. In addition, we also constructed and analyzed Pol IVR (dinB82) and Pol IVC (dinB81) strains to gain insight into the biological importance of these contacts to Pol IV TLS. We first examined MMS sensitivity by spotting serial dilutions of respective cultures onto plates supplemented with 0, 3 or 4.5 mM MMS. Each of the Pol IV mutants displayed modest sensitivity to 3 mM MMS, with the ΔdinB, Pol IV-D103N, and Pol IVC strains being slightly more sensitive than the Pol IVR and Pol IV-T120P strains (Fig 5). At 4.5 mM MMS, the ΔdinB, Pol IV-D103N, and Pol IVC strains were between ~100 - to 1,000-fold more sensitive than the wild type Pol IV strain. The Pol IVR and Pol IV-T120P strains were similar to each other, and were only slightly less sensitive than the ΔdinB strain (Fig 5). Taken together, these results demonstrate a biologically important role in Pol IV TLS for the β clamp rim as well as residue T120 of Pol IV. In contrast to ΔdinB, the ΔumuDC strain failed to display MMS sensitivity, or to exacerbate sensitivity of the ΔdinB strain, consistent with published reports [41]. Finally, MMS sensitivity of each of the Pol IV mutant strains was fully complemented by pRM102, which expresses wild type Pol IV (S5A Fig).

We next measured the frequency of MMS-induced mutagenesis. The strain lacking Pol IV displayed a ~7-fold increase in Pol V-dependent MMS-induced mutagenesis (Fig 6A), as expected [41]. Frequencies for the Pol IV-D103N (dinB80), Pol IV-T120P (dinB89), Pol IVC (dinB81) and Pol IVR (dinB82) strains were ~4-, ~5-, ~6 - and ~3-fold elevated, respectively, relative to the wild type Pol IV control (Fig 6B; p≤0.0001 based on Student’s t-test). These results demonstrate the importance of position T120 in Pol IV, as well as the ability of Pol IV to interact with the β clamp rim and cleft to carry out TLS in vivo. Importantly, the ability of Pol IV to inhibit mutagenesis by Pol V was not affected by the presence of the zaf-3633::cat cassette (p = 0.7 based on Student’s t-test comparing the Pol IV+ strains MG1655 and MKS103). As with MMS sensitivity, wild type Pol IV expressed from plasmid pRM102 restored the frequency of MMS-induced mutagenesis for these dinB mutants to the wild type Pol IV level (S5B Fig).

To determine if the MMS phenotypes of the Pol IV-T120P strain were due to a catalytic TLS defect which rendered Pol IV-T120P incapable of bypassing MMS-induced lesions, we used a primer extension assay to measure the ability of purified Pol IV-T120P to catalyze in vitro bypass of the model MMS-induced lesions O6-methylguanine (O6-mdG), 3-deaza-3-methyladenine (3d-medA), which is a stable mimic of N3-mdA [59], as well as an AP site. As summarized in Table 3, both wild type Pol IV and Pol IV-T120P were able to bypass template O6-mdG and 3d-medA. While there were some differences in substrate binding (Kd) and/or turnover (kpol), Pol IV-T120P was as efficient or more so than wild type Pol IV. Pol IV and Pol IV-T120P each incorporated either dC or dT opposite template O6-mdG with roughly equivalent efficiencies. In both cases, bypass was considerably less efficient than that observed for template dG, due to a reduction in both Kd and kpol, with Pol IV-T120P slightly outperforming wild type Pol IV. Both Pols were capable of incorporating low levels of dA or dG opposite template O6-mdG. However, the amount of incorporation was less than 10% compared to that for the insertion of dC or dT opposite the alkylated lesion. 3d-medA was easier for both Pol IV and Pol IV-T120P to bypass, again with Pol IV-T120 outperforming wild type Pol IV by a factor of ~2-fold, attributable in large part to stronger substrate binding (Kd). Both Pols incorporated dA, dC or dG opposite template 3d-medA. The level of incorporation was significantly less than that measured for incorporation of dT. Finally, even though Pol IV-T120P was marginal in regard to its ability to bypass the AP site, inserting dA, it was nevertheless more efficient than wild type Pol IV (Table 3). Taken together, these results demonstrate that Pol IV-T120P is proficient in vitro for TLS past a variety of DNA adducts commonly induced by MMS, suggesting that the inability of Pol IV-T120P to cope with MMS-induced DNA damage in vivo was the result of its inability to gain access to the lesions.
Pol IVCD-T120P fails to displace an actively replicating Pol III from β clamp in vitro
We previously utilized a single molecule primer extension assay to demonstrate exchange of Pol III and Pol IV on β clamp at the 3’ primer terminus in vitro [12]. The distinct polymerization rates of Pol III and Pol IV allowed us to unambiguously assign individual DNA synthesis events to each respective Pol and to measure their respective processivities when incubated alone or together (Fig 7A). At 300 nM Pol IV, a 60-fold molar excess over Pol III that simulates levels found in SOS-induced cells [3], Pol IV actively displaced Pol III from the DNA template as inferred from Pol III processivity measurements ([12,16,17,38,45]; see Fig 7B). This ability of Pol IV to reduce Pol III processivity was dependent on the Pol IV CBM, arguing that Pol III displacement involves at a minimum a conformational exchange of the two Pols on the β clamp. Using this approach, we asked whether Pol IV-T120P was likewise able to displace Pol III from the β clamp, as inferred by a reduction in its processivity. As summarized in Fig 7B, a 60-fold molar excess of Pol IV-T120P over Pol III was as efficient as wild type Pol IV at inhibiting Pol III processivity. Together, these results suggest one of two possibilities: either (i) the T120P mutation does not impact the ability of Pol IV to displace Pol III from β clamp, or (ii) efficient recruitment of Pol IV to the Pol III-β clamp complex through its interactions with β clamp masks the Pol IV-T120P-dependent defect in Pol IV displacement of Pol III from β clamp. To distinguish between these two models, we analyzed the ability of Pol IVCD and Pol IVCD-T120P, both of which lack the Pol IVLF β clamp-binding domain, to impede Pol III processivity using the same single molecule assay. Furukohri and colleagues previously demonstrated that a ~900 - to 1,800-fold molar excess of Pol IVCD over Pol III (890 nM Pol IV compared to 0.5–1.0 nM Pol III) was able to disrupt the Pol III-β clamp complex assembled in vitro on a primed DNA substrate [16]. Similarly, we found that 900 nM Pol IVCD (a 180-fold molar excess over Pol III) disrupted Pol III synthesis, reducing its processivity to one-half of that observed in the absence of Pol IVCD (Fig 7C, p <0.01, determined using the Wilcoxon rank-sum test). This reduction in Pol III processivity most likely results from the ability of Pol IVCD to displace Pol III from the β clamp assembled on DNA [12,16,45]. Importantly, an equivalent concentration of Pol IVCD-T120P failed to reduce processivity of Pol III. Taken together, these findings support the view that residue T120 of Pol IV plays an important role in displacing Pol III from the β clamp, and demonstrate that the Pol IVLF domain contributes to this ability. Finally, these biochemical results are remarkably similar to the growth phenotypes observed for strains overproducing Pol IVCD-T120P (pDB23) or full length Pol IV-T120P (pDB33) from the arabinose promoter (Fig 2), which demonstrate the ability of the Pol IVLF domain to mask the phenotype of the T120P mutation in vivo.

Discussion
With the goal of gaining new insights into the relationship between the physiological function of Pol IV in TLS and its ability when overexpressed to impede E. coli growth, we exploited the hypersensitivity of the dnaN159 strain to elevated levels of Pol IV to identify 13 novel Pol IV mutants that were unable to impede growth (Table 2). These Pol IV mutants were deficient in stimulating reversion of the CC108 lacZ –1 frameshift reporter when expressed at SOS-induced levels (S2 Fig), and for conferring UV sensitivity upon the dnaN159 strain (S3 Fig), indicating that they were unable to effectively compete with Pol III for access to the replication fork. Likewise, these mutations failed to impede growth of the dnaN+ strain when introduced into Pol IVCD and overproduced from the arabinose promoter (Fig 2). Finally, despite the fact that all but one of the mutant Pol IV proteins (Pol IV-D10G) retained appreciable catalytic activity in vitro (Fig 3), they were nevertheless impaired for tolerating MMS-induced lesions in vivo (Fig 4). These results, taken together with those discussed below, support the view that overexpressed levels of Pol IV impede E. coli growth by actively replacing Pol III at the replication fork via a mechanism that is similar to that used under physiological conditions to coordinate high fidelity processive Pol III replication with potentially mutagenic Pol IV TLS. In contrast to an earlier study [47], we failed to observe an ability of overproduced levels of Pol IV to mediate cell death in either the dnaN159 or dnaN+ strains via excessive incorporation of oxidized precursors (S2 Table and S4 Fig). Thus, Pol IV appears to be able to impede E. coli growth by either incorporating lethal levels of 8-oxo-dG or by displacing Pol III. This view is consistent with the finding that under the conditions used in this study lethality was independent of Pol IV catalytic activity [38,45]. However, our finding that several Pol IV mutants identified in this work were impaired for catalytic activity in vitro (Fig 3) suggests that the ability of Pol IV to replace Pol III at the replication fork is dependent at least in part on residues in Pol IV that contribute to catalytic activity. Alternatively, the ability of Pol IV to persist at the replication fork after replacing Pol III likely contributes to the killing, and would rely on Pol IV processivity, which, with the exception of Pol IV-T120P, was impaired in the Pol IV mutants analyzed here.
The Pol IV-T120P mutant was remarkable in that it was comparable to wild type Pol IV for replication of undamaged DNA in vitro, as well as for catalyzing bypass of 3d-medA, O6-mdG and an AP site (Fig 3 and Table 3), yet it was nevertheless unable to tolerate MMS-induced DNA lesions in vivo (Figs 4–6), presumably due to its inability to access these lesions. Consistent with this conclusion, the T120P mutation abrogated the ability of Pol IVCD to inhibit Pol III processivity in vitro (Fig 7). Based on previously published results [12,16,45], inhibition of Pol III processivity is the result of Pol IV displacing Pol III from the face of the β clamp. Our finding that the Pol IV-T120P strain was impaired for tolerating MMS-induced DNA damage indicates that the ability of Pol IV to inhibit Pol III processivity is required for the TLS function of Pol IV in vivo. Furthermore, our finding that the strain expressing Pol IV-T120P (dinB89) was almost as deficient as the isogenic ΔdinB, Pol IV-D103N (dinB80) and Pol IVC (dinB81) strains for tolerating MMS-induced lesions indicates that the ability of Pol IV to displace Pol III from the face of the β clamp is critical to Pol IV TLS function in vivo (Figs 5 and 6). TLS has been suggested to take place at either the replication fork via a Pol switch [3,12,15,17,18,20], or in ssDNA gaps generated in part by Pol III skipping over DNA lesions to continue replication downstream of the blockage [18,39]. However, to date, the extent to which these two mechanisms are used in vivo was unknown. Our finding that the T120P mutation specifically interferes with the ability of Pol IV to switch with Pol III, taken together with its inability to cope with MMS-induced DNA damage in vivo (Figs 4–6), suggests that a significant fraction of Pol IV-mediated TLS in vivo relies on a Pol III-Pol IV switch. Consistent with this conclusion, SOS-induced levels of Pol IV slowed the rate of DNA replication in vivo by ~12%, arguing that Pol IV frequently replaces Pol III at the replication fork following SOS induction [14], possibly via a Pol III-Pol IV switch. While it remains to be determined whether the reduced rate of replication in response to SOS induction represents a biologically important checkpoint effector function of Pol IV, as previously suggested [13,45], the fact that strains lacking Pol IV function fail to exhibit enhanced sensitivity to agents that generate classes of DNA lesions other than those that Pol IV is capable of bypassing, such as UV photoproducts [60], argues against such a model. Finally, Benson and colleagues [61] identified two Pol IV mutants (V7G and F292Y) based on their inability to impede growth of an E. coli strain expressing a mutant Pol IIIα allele (dnaE915). Both of these mutants retained the ability to bypass 3d-medA in vitro, but their abilities to cope with MMS-induced DNA damage in vivo was not examined. While V7 is in close proximity to T120 (S6A Fig), neither it nor F292 is surface exposed (S6B Fig), suggesting that these mutations may affect the structure of Pol IV. Regardless, it is possible that the V7G and/or F292Y mutations impair the Pol III-Pol IV switch.
We previously described results supporting an important role for the Pol IV-β clamp rim interaction in mediating the Pol III-Pol IV switch in vitro [17,38]. In contrast to our findings, Gabbai and colleagues, utilizing a different assay that may more accurately represent the structure and composition of the replisome, concluded that the Pol IV-β clamp rim contact stimulated, but was not required for a Pol III-Pol IV switch in vitro [18]. In light of this finding, they suggested that direct competition between Pol III and Pol IV for the β clamp cleft represented an alternative mechanism for their switching. Previous efforts to define the role of the β clamp rim in Pol IV function in vivo utilized multi-copy plasmids expressing higher than physiological levels of Pol IVR and Pol IVC to complement the NFZ-sensitive phenotype of a ΔdinB strain [17,62]. Under these conditions, the Pol IVR strain was indistinguishable from the wild type Pol IV strain, suggesting the Pol IV-β clamp rim interaction was dispensable for Pol IV function in vivo. However, using strains expressing the Pol IVC or Pol IVR mutants from the chromosomal dinB locus, we confirmed an essential role for the β clamp cleft in Pol IV TLS, and provide compelling evidence for a biologically important role for the β clamp rim in contributing to Pol IV TLS (Figs 4–6). These findings, taken together with those discussed above regarding the T120P mutation, support the model that Pol IV TLS function in vivo relies on its ability to bind to both the rim and cleft of the β clamp, as well as its ability to inhibit Pol III processivity, possibly via a direct interaction of Pol IV with one or more subunits of Pol III. Since both the Pol IV-β clamp and the postulated Pol IV-Pol III interactions are required for Pol IV TLS function in vivo, it is conceivable that the postulated Pol III-Pol IV interaction could act to relax the requirement for the Pol IV-β clamp rim interaction in vitro, potentially explaining the apparent discrepancy between our published results and those of Gabbai and colleagues regarding the importance of the β clamp rim to the Pol III-Pol IV switch in vitro.
Pol IV interacts physically with both UmuD and RecA [63–65]. These interactions are proposed to improve the fidelity of Pol IV by enclosing its open active site [64]. Interestingly, Pol IV-C66A was previously reported to bind more tightly to both RecA and UmuD [65]. It is conceivable that we identified the Pol IV-C66S with our genetic assay because it was affected for interactions with UmuD and/or RecA. Alternatively, we may have identified Pol IV-C66S due to its reduced stability ([65]; see S1 Fig). In addition to C66, residues P166, F172 and L176 of Pol IV have also been demonstrated to interact with UmuD [64]. In contrast to C66, these residues are in close proximity to T120 (S6 Fig, panels C and D). Thus, it is possible that the T120P mutation also affects an interaction of Pol IV with UmuD. Finally, UmuD may contribute to the ability of Pol IV to switch with Pol III. Consistent with this possibility, UmuD interacts physically with the Pol IIIα and Pol IIIε subunits, as well as the β clamp [21,22,66]. However, the failure of Pol IVCD-T120P to impede Pol III processivity in vitro was independent of UmuD (Fig 7C).
Results discussed in this report provide several new insights into the mechanism by which the actions of Pol III are coordinately regulated with those of Pol IV, and when taken together with previously published findings [17,33,38,67], support a new model for the role of Pol III-Pol IV switching in Pol IV-mediated TLS in vivo. In addition to its interactions with the β clamp, an interaction of Pol IV with Pol III also appears to play a biologically important role in recruiting Pol IV to lesions (Figs 5 and 6). Biochemical interaction of Pol IVCD with Pol III holoenzyme is sufficient to mediate displacement of Pol III from the face of the β clamp in vitro [12,13,16,45]. Our single molecule assay reproduced this finding, and further demonstrated that position T120 of Pol IV is important for this function (Fig 7). Residue T120 is one helical turn from the start of α-helix 5 (see Fig 8) and its substitution with proline likely truncates the start of this helix. Thus, residue T120, and/or residues in its vicinity, presumably mediates a physical interaction with one or more subunits of the Pol III holoenzyme. Both the α catalytic and the ε proofreading subunits of Pol III contain a CBM that interacts with the β clamp cleft [68,69]. Although the Pol III holoenzyme binds both β clamp clefts, a single β clamp cleft is sufficient to support processive Pol III replication, as well as Pol III-Pol IV switching [17,36,67]. Since the Pol IIIα-β clamp cleft interaction is required for Pol III function both in vitro and in vivo, while the Pol IIIε-β clamp cleft appears to be dispensable [17,29,36,67,69], we suggest that a mechanism by which Pol IV initiates a switch with a stalled Pol III involves Pol IV first binding to the β clamp rim adjacent to the β clamp cleft that is bound by Pol IIIα. Based on an in silico model of the Pol III-β clamp-Pol IV complex, residue T120 of Pol IV is well positioned to contact Pol IIIα (Fig 8A), but not Pol IIIε (Fig 8B). Thus, Pol IV may be recruited to the replication fork through a combination of the Pol IVLF-β clamp rim and Pol IVCD-Pol III interactions. The Pol IVLF-β clamp rim interaction is likely too weak (~1.3 μM) on its own to recruit Pol IV to the replisome in the absence of SOS-induction when Pol IV levels are ~330 nM [3,70]. However, if a Pol IV-Pol III interaction contributes to Pol IV recruitment, the affinity of Pol IV for the replisome may be sufficiently high to enable a Pol III-Pol IV switch irrespective of SOS-induction, which increases Pol IV levels from ~330 nM to ~3.3 μM [3,70].

Pol IV can only switch with a stalled Pol III [15,38], suggesting that a conformational change in Pol IIIα contributes to the Pol III-Pol IV switch, possibly by unmasking a surface of Pol III that interacts with Pol IV leading to displacement of Pol III. It is currently unclear whether Pol IV is recruited to the replication fork in response to a specific conformation of the stalled Pol III replisome, or whether it is recruited through one or more Pol III-Pol IV interactions that are independent of a stalled Pol III. If a stalled Pol III replisome acts to recruit Pol IV, then a single Pol III-Pol IV interaction may be sufficient for both recruitment and Pol III displacement. However, if an actively replicating Pol III replisome recruits Pol IV irrespective of Pol III stalling, then distinct Pol III-Pol IV contacts would seemingly be required for Pol IV recruitment and Pol III-Pol IV switching. Finally, irrespective of the fact that the Pol IV-β clamp rim interaction is required for Pol IV-mediated TLS in vivo (Figs 5 and 6), the finding that Pol IVCD displaced Pol III from the face of the β clamp ([16]; see Fig 7) suggests that Pol IV may not have to simultaneously bind the β clamp rim as well as one or more subunits of Pol III in order to displace Pol III from the face of the β clamp. Irrespective of the mechanism, once Pol III is displaced, the C-terminal 6 residues of Pol IV are able to bind the cleft of the β clamp that was previously bound by Pol III, ultimately granting control of both the β clamp and the replication fork to Pol IV for TLS (Fig 8C). Although Pol IV displaces Pol III from the β clamp [12,16,45], we suggest that Pol IIIα-DnaXτ-DnaB interactions act to retain Pol III within the replisome complex until such time as Pol IV relinquishes its control of the DNA template [71], allowing Pol III to regain control of the replication fork after Pol IV leaves. However, when Pol IV is overexpressed, or when the replisome contains the mutant dnaN159-encoded β clamp protein, Pol IV repeatedly replaces Pol III on the clamp face. This repeated replacement could act to displace Pol III from the replisome, explaining the lethal phenotype observed for strains overexpressing Pol IV. While the Pol IV-β clamp rim interaction is presumably dispensable once Pol IV gains access to the β clamp cleft [17], processive Pol IV replication requires contact with residues 148HQDVR152 of β clamp [33]. Inasmuch as residues H148, Q149 and R152 of β clamp interact with the DNA template that it encircles [33,72], Pol IV may have to compete with DNA to gain access to 148HQDVR152 of β clamp, which, in turn, may act to reposition the β clamp on the DNA, possibly exposing additional surfaces on the β clamp that stabilize the Pol IV-β clamp complex, and/or enhance its catalytic activity. Finally, our finding that the Pol IVR, Pol IVC and Pol IV-T120P mutant strains were severely impaired for tolerating MMS-induced DNA damage is consistent with the view that Pol III-Pol IV switching plays a pivotal role in regulating access of Pol IV to the DNA in vivo. Further studies are required to determine how long Pol IV maintains control of the replication fork after switching with Pol III, as well as whether Pol III and/or other factors play a role in displacing Pol IV from the replication fork.
Materials and Methods
E. coli strains, plasmid DNAs and bacteriological techniques
Bacteria were cultured in either Luria Bertani (LB; 10 g/l tryptone, 5 g/l yeast extract, 10 g/l NaCl), or M9 minimal medium (12.9 g/L Na2HPO4•7H2O, 3 g/L KH2PO4, 0.5 g/L NaCl, 1 g/L NH4Cl) supplemented with 0.1 mM CaCl2, 2 mM MgCl2, 5 μg/ml thiamine, 0.5% casamino acids and 0.5% glucose or 0.2% arabinose, as indicated. For anaerobic growth, 100 mM KNO3 was added to the growth to act as the terminal electron acceptor. When required, the following antibiotics were used at the indicated concentrations: ampicillin (Amp), 150 μg/ml; tetracycline (Tet), 10 μg/ml; kanamycin (Kan), 40 μg/ml; chloramphenicol (Cam), 20 μg/ml; rifampicin (Rif), 50 μg/ml. E. coli strains were constructed using P1vir-mediated generalized transduction [73], or λRed-mediated recombineering [74], and are described in Table 1. Strain genotypes were verified using either diagnostic PCR or nucleotide sequence analysis (Roswell Park Biopolymer Facility, Buffalo, NY) of respective PCR-amplified alleles. Strains were made competent for transformation using CaCl2 as described [48]. Bacterial plasmid transformation frequency [38], UV sensitivity [38,48] and lacZ→Lac+ reversion [51,52,56] was measured as described in the indicated references. Plasmid DNAs are described in Table 1. Standard techniques were used for cloning. Site-directed mutagenesis was performed using the Quickchange kit (Stratagene). Synthetic oligonucleotide primers used for mutagenesis were purchased from either IDT or Operon, and their sequences are presented in S3 Table. All plasmid sequences were confirmed by nucleotide sequence (Roswell Park Biopolymer Facility, Buffalo, NY). Mutant dinB alleles were subcloned from pWSK29 into pET11a (Novagen) by NdeI and BamHI (Fermentas) restriction, followed by ligation to the similarly prepared pET11a backbone using T4 DNA ligase (Fermentas). Sensitivity to MMS (Sigma) was measured as described [41].
Genetic assay to identify Pol IV mutants
Spontaneous dinB mutations unable to impede growth of the dnaN159 lexA51(Def) E. coli strain MS105 were identified by selecting AmpR transformants using 200 ng of plasmid pJH110. This strain displayed a transformation efficiency of ~106 colony forming units/μg of supercoiled pWSK29 plasmid DNA. In instances where multiple colonies were obtained from a single transformation reaction, a single CFU was selected from the plate for further analysis to avoid sibling mutations. Plasmids were isolated using the Qiagen mini-prep kit as per the manufacturer’s recommendation. Purified plasmids were analyzed by agarose gel electrophoresis. Those of the appropriate size were retransformed into MS105 to verify their inability to impede growth. Those that transformed MS105 with an efficiency similar to that of the pWSK29 control plasmid were then analyzed by Western blotting using polyclonal anti-Pol IV antibodies as described [4,17].
Construction of mutant dinB strains
Strains MKS103-MKS107 were constructed using λ recombineering as described [74]. Briefly, the 2,329 bp lafU’ zaf-3633::cat dinB80 yafN’ DNA cassette was PCR amplified from plasmid pMKS100, pMKS101, pMKS102, pMKS103 or pMKS104 using primers P1 and P4 (S3 Table), and electroporated into E. coli strain MG1655 containing pKD46 as described [75]. Chloramphenicol resistant colonies were selected on LB agar plates supplemented with chloramphenicol, and subsequently confirmed to contain the desired dinB allele by diagnostic PCR using primers MKS055 and MKS046, which anneal 589 bp upstream of primer P1 and 498 bp downstream of primer P4, respectively. The remaining primers listed in S3 Table were used for nucleotide sequence verification of the lafU’–zaf-3633::cat–dinB–yafN’ cassette from 91 bp upstream of the start of primer P1 to 163 bp downstream from the end of primer P4, except for a 153 bp internal segment of the cat gene corresponding to amino acid residues L45-D96, prior to using P1vir to transduce the linked zaf-3633::cat and dinB alleles into a fresh isolate of strain MG1655.
Measure of MMS-induced mutation frequency
Cultures of LB media (mock samples to measure spontaneous mutagenesis) and LB media containing the indicated concentration of freshly added MMS (1.5 mM or 2.0 mM) were inoculated with 200 μl of an exponential culture (OD600 ~0.5) of the indicated strain. Cultures were incubated overnight at 37°C with aeration before plating appropriate dilutions onto LB media with or without Rif. Plates were incubated overnight at 37°C before counting colonies. MMS-induced mutation frequency was calculated as described [33].
In vitro DNA primer extension assay
Wild type and Pol IV mutant proteins [34], SSB [76], the γ3δδ’ form of the DnaX clamp loader [36] and β clamp [77] were purified as described in the indicated references. Primer extension assays were performed as described previously [17,34,56] using the 32P-radiolabeled PAGE purified 30-mer/100-mer DNA template. Briefly, reactions (20 μl) contained replication buffer (20 mM Tris-HCl [pH 7.5], 8.0 mM MgCl2, 0.1 mM EDTA, 5 mM DTT, 1 mM ATP, 5% glycerol, and 0.8 μg/ml BSA), 1 nM 30-mer/100-mer template, 133 μM dNTPs (Fermentas), 90 nM SSB, 1 nM γ3δδ’ DnaX clamp loader complex, 10 nM β clamp and 1 nM Pol IV. The reactions were pre-incubated for 3 min at 37°C to permit loading of β clamp prior to initiating replication by addition of dNTPs. Reactions were next incubated at 37°C for 5 min, then quenched by the addition of 25 mM EDTA and incubation at 95°C for 3 minutes. Aliquots of each reaction were then electrophoresed through an 8% UREA-PAGE at 60 watts for 3,332 volt hours, as described [56]. Replication products were visualized using a Bio-Rad imaging screen K and a Bio-Rad Personal Molecular Imager FX.
Kinetic analyses of Pol IV and Pol IV-T120P in vitro primer extension activity
Kinetic studies using wild type Pol IV or Pol IV-T120P were performed at 25°C in assay buffer (25 mM TrisOAc [pH 7.5], 150 mM KOAc, 10 mM β-mercaptoethanol, 1 mg/ml bovine serum albumin, and 10 mM MgCl2). The kinetic parameters (kpol, Kd, and kpol/Kd) for nucleotides were measured as previously described [78]. Briefly, a typical assay was performed by pre-incubating DNA substrate (200 nM) with a 2-fold molar excess of DNA polymerase (400 nM) in assay buffer. Reactions were initiated by adding variable concentrations of nucleotide substrate (1–500 μM). At variable times, 5 μl aliquots of the reaction were removed and immediately quenched by adding an equal volume of 200 mM EDTA. Polymerization reactions were monitored by electrophoresis through 20% sequencing gels as described [79]. Gel images were obtained with a Packard PhosphorImager by using the OptiQuant software supplied by the manufacturer. Product formation was quantified by measuring the ratio of 32P-labeled extended and un-extended primer. This ratio was corrected for substrate in the absence of polymerase (zero point). Corrected ratios were multiplied by the concentration of primer/template used in the assay to yield total product. Observed rate constants were obtained using the following equation: y = A*(1-ekobs*t)+C, where A is the burst amplitude in product formation, kobs is the observed rate constant of the reaction, t is the time, and C is the end-point in product formation. Data for the dependency of rate constant as a function of nucleotide concentration were fit to the Michaelis-Menten equation: kobs = kpol*(dNTP)/(Kd+[dNTP]), where kobs is the rate constant of the reaction (s−1), kpol is the maximal rate constant of polymerization, Kd is the apparent dissociation constant for dNTP, and [dNTP] is the concentration of nucleotide substrate.
Single-molecule primer extension assay
Primer extension by Pol III and Pol IV was observed on single DNA molecules within custom microfluidic flow cells, as previously described [12]. Briefly, primed, single-stranded DNA substrates were derived from 7.2 kb phage M13mp18 DNA (New England Biolabs) end-labeled with digoxigenin and biotin. DNAs were bound to the streptavidin-coated flow cell surface on one end, and to anti-digoxigenin-coupled 2.8 μm-diameter beads on the other. Laminar flow through the flow cell exerted a constant ~3 pN force on the bead, and, by extension, uniformly throughout the tether. Conversion of entropically coiled ssDNA to extended dsDNA by a Pol at this constant force was observed as motion of the bead using dark-field microscopy. Bead positions were tracked by fitting beads to 2D Gaussians, and their motions were converted into DNA synthesis as a function of time. Resolution is determined by thermal fluctuations of the tethered bead (σ ~70 bp) and the choice of exposure time (0.5 s). All experiments were performed in replication buffer (50 mM Hepes-KOH [pH 7.9], 12 mM Mg[OAc]2, 80 mM KCl, 0.1 mg/ml BSA, 5 mM DTT) supplemented with 5 nM Pol IIIαεθ, 30 nM β, 15 nM of the τ3δδ’ψχ form of the DnaX clamp loader complex, 760 μM dNTPs and 1 mM ATP. Pol IV, Pol IV-T120P, Pol IVCD and Pol IVCD-T120P were additionally included at the indicated concentrations. A cutoff of 45 bp/s was used to distinguish Pol III (faster) from Pol IV (slower) events. This cutoff captured ~95% of events in experiments with each polymerase alone. The Pol III replisome components used in the single molecule experiments were purified as previously described: β [80]; α, δ and δ’ [81]; ε and θ [82]; and τ and χψ [83]. The Pol III core αεθ and the clamp loader assembly with stoichiometry τ3δδ’χψ were then assembled and purified [83].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, et al. (2006) DNA repair and mutagenesis. Washington, D. C.: ASM Press.
2. Sale JE, Lehmann AR, Woodgate R (2012) Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol 13 : 141–152. doi: 10.1038/nrm3289 22358330
3. Sutton MD (2010) Coordinating DNA polymerase traffic during high and low fidelity synthesis. Biochim Biophys Acta 1804 : 1167–1179. doi: 10.1016/j.bbapap.2009.06.010 19540941
4. Sanders LH, Devadoss B, Raja GV, O'Connor J, Su S, et al. (2011) Epistatic roles for Pseudomonas aeruginosa MutS and DinB (DNA Pol IV) in coping with reactive oxygen species-induced DNA damage. PLoS One 6: e18824. doi: 10.1371/journal.pone.0018824 21533111
5. Petrosino JF, Galhardo RS, Morales LD, Rosenberg SM (2009) Stress-induced beta-lactam antibiotic resistance mutation and sequences of stationary-phase mutations in the Escherichia coli chromosome. J Bacteriol 191 : 5881–5889. doi: 10.1128/JB.00732-09 19648247
6. Norton MD, Spilkia AJ, Godoy VG (2013) Antibiotic resistance acquired through a DNA damage-inducible response in Acinetobacter baumannii. J Bacteriol 195 : 1335–1345. doi: 10.1128/JB.02176-12 23316046
7. Moyano AJ, Lujan AM, Argarana CE, Smania AM (2007) MutS deficiency and activity of the error-prone DNA polymerase IV are crucial for determining mucA as the main target for mucoid conversion in Pseudomonas aeruginosa. Mol Microbiol 64 : 547–559. 17493134
8. Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, et al. (2009) Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev 73 : 134–154. doi: 10.1128/MMBR.00034-08 19258535
9. Stallons LJ, McGregor WG (2010) Translesion synthesis polymerases in the prevention and promotion of carcinogenesis. J Nucleic Acids 2010.
10. Pillaire MJ, Selves J, Gordien K, Gourraud PA, Gentil C, et al. (2010) A 'DNA replication' signature of progression and negative outcome in colorectal cancer. Oncogene 29 : 876–887. doi: 10.1038/onc.2009.378 19901968
11. Betous R, Rey L, Wang G, Pillaire MJ, Puget N, et al. (2009) Role of TLS DNA polymerases eta and kappa in processing naturally occurring structured DNA in human cells. Mol Carcinog 48 : 369–378. doi: 10.1002/mc.20509 19117014
12. Kath JE, Jergic S, Heltzel JM, Jacob DT, Dixon NE, et al. (2014) Polymerase exchange on single DNA molecules reveals processivity clamp control of translesion synthesis. Proc Natl Acad Sci U S A 111 : 7647–7652. doi: 10.1073/pnas.1321076111 24825884
13. Indiani C, Langston LD, Yurieva O, Goodman MF, O'Donnell M (2009) Translesion DNA polymerases remodel the replisome and alter the speed of the replicative helicase. Proc Natl Acad Sci U S A 106 : 6031–6038. doi: 10.1073/pnas.0901403106 19279203
14. Tan KW, Pham TM, Furukohri A, Maki H, Akiyama MT (2015) Recombinase and translesion DNA polymerase decrease the speed of replication fork progression during the DNA damage response in Escherichia coli cells. Nucleic Acids Res 43 : 1714–1725. doi: 10.1093/nar/gkv044 25628359
15. Indiani C, McInerney P, Georgescu R, Goodman MF, O'Donnell M (2005) A sliding-clamp toolbelt binds high - and low-fidelity DNA polymerases simultaneously. Mol Cell 19 : 805–815. 16168375
16. Furukohri A, Goodman MF, Maki H (2008) A dynamic polymerase exchange with Escherichia coli DNA polymerase IV replacing DNA polymerase III on the sliding clamp. J Biol Chem 283 : 11260–11269. doi: 10.1074/jbc.M709689200 18308729
17. Heltzel JM, Maul RW, Scouten Ponticelli SK, Sutton MD (2009) A model for DNA polymerase switching involving a single cleft and the rim of the sliding clamp. Proc Natl Acad Sci U S A 106 : 12664–12669. doi: 10.1073/pnas.0903460106 19617571
18. Gabbai CB, Yeeles JTP, Marians KJ (2014) Replisome-mediated translesion synthesis and leading strand template lesion skipping are competing bypass mechanisms. J Biol Chem 289 : 32811–32823. doi: 10.1074/jbc.M114.613257 25301949
19. Kannouche PL, Wing J, Lehmann AR (2004) Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 14 : 491–500. 15149598
20. Ikeda M, Furukohri A, Philippin G, Loechler E, Akiyama MT, et al. (2014) DNA polymerase IV mediates efficient and quick recovery of replication forks stalled at N2-dG adducts. Nucleic Acids Res 42 : 8461–8472. doi: 10.1093/nar/gku547 24957605
21. Sutton MD, Opperman T, Walker GC (1999) The Escherichia coli SOS mutagenesis proteins UmuD and UmuD' interact physically with the replicative DNA polymerase. Proc Natl Acad Sci U S A 96 : 12373–12378. 10535929
22. Sutton MD, Narumi I, Walker GC (2002) Posttranslational modification of the umuD-encoded subunit of Escherichia coli DNA polymerase V regulates its interactions with the beta processivity clamp. Proc Natl Acad Sci U S A 99 : 5307–5312. 11959982
23. Kannouche P, Fernandez de Henestrosa AR, Coull B, Vidal AE, Gray C, et al. (2002) Localization of DNA polymerases eta and iota to the replication machinery is tightly co-ordinated in human cells. EMBO J 21 : 6246–6256. 12426396
24. Okazaki R, Arisawa M, Sugino A (1971) Slow joining of newly replicated DNA chains in DNA polymerase I-deficient Escherichia coli mutants. Proc Natl Acad Sci U S A 68 : 2954–2957. 4943548
25. McHenry CS (2011) Bacterial replicases and related polymerases. Curr Opin Chem Biol 15 : 587–594. doi: 10.1016/j.cbpa.2011.07.018 21855395
26. O'Donnell M, Langston L, Stillman B (2013) Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb Perspect Biol 5.
27. Dalrymple BP, Kongsuwan K, Wijffels G, Dixon NE, Jennings PA (2001) A universal protein–protein interaction motif in the eubacterial DNA replication and repair systems. Proc Natl Acad Sci U S A 98 : 11627–11632. 11573000
28. Becherel OJ, Fuchs RP, Wagner J (2002) Pivotal role of the beta-clamp in translesion DNA synthesis and mutagenesis in E. coli cells. DNA Repair (Amst) 1 : 703–708.
29. Dohrmann PR, McHenry CS (2005) A bipartite polymerase-processivity factor interaction: only the internal beta binding site of the alpha subunit is required for processive replication by the DNA polymerase III holoenzyme. J Mol Biol 350 : 228–239. 15923012
30. Maul RW, Sanders LH, Lim JB, Benitez R, Sutton MD (2007) Role of Escherichia coli DNA polymerase I in conferring viability upon the dnaN159 mutant strain. J Bacteriol 189 : 4688–4695. 17449610
31. Lopez de Saro FJ, Georgescu RE, Goodman MF, O'Donnell M (2003) Competitive processivity-clamp usage by DNA polymerases during DNA replication and repair. EMBO J 22 : 6408–6418. 14633999
32. Bunting KA, Roe SM, Pearl LH (2003) Structural basis for recruitment of translesion DNA polymerase Pol IV/DinB to the beta-clamp. EMBO J 22 : 5883–5892. 14592985
33. Heltzel JM, Scouten Ponticelli SK, Sanders LH, Duzen JM, Cody V, et al. (2009) Sliding clamp-DNA interactions are required for viability and contribute to DNA polymerase management in Escherichia coli. J Mol Biol 381 : 74–91.
34. Maul RW, Ponticelli SK, Duzen JM, Sutton MD (2007) Differential binding of Escherichia coli DNA polymerases to the beta-sliding clamp. Mol Microbiol 65 : 811–827. 17635192
35. Sutton MD, Duzen JM (2006) Specific amino acid residues in the beta sliding clamp establish a DNA polymerase usage hierarchy in Escherichia coli. DNA Repair (Amst) 5 : 312–323.
36. Scouten Ponticelli SK, Duzen JM, Sutton MD (2009) Contributions of the individual hydrophobic clefts of the Escherichia coli beta sliding clamp to clamp loading, DNA replication and clamp recycling. Nucleic Acids Res 37 : 2796–2809. doi: 10.1093/nar/gkp128 19279187
37. Beuning PJ, Sawicka D, Barsky D, Walker GC (2006) Two processivity clamp interactions differentially alter the dual activities of UmuC. Mol Microbiol 59 : 460–474. 16390442
38. Heltzel JM, Maul RW, Wolff DW, Sutton MD (2012) Escherichia coli DNA polymerase IV (Pol IV), but not Pol II, dynamically switches with a stalled Pol III* replicase. J Bacteriol 194 : 3589–3600. doi: 10.1128/JB.00520-12 22544274
39. Yeeles JT, Marians KJ (2011) The Escherichia coli replisome is inherently DNA damage tolerant. Science 334 : 235–238. doi: 10.1126/science.1209111 21998391
40. Jarosz DF, Godoy VG, Delaney JC, Essigmann JM, Walker GC (2006) A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature 439 : 225–228. 16407906
41. Bjedov I, Dasgupta CN, Slade D, Le Blastier S, Selva M, et al. (2007) Involvement of Escherichia coli DNA polymerase IV in tolerance of cytotoxic alkylating DNA lesions in vivo. Genetics 176 : 1431–1440. 17483416
42. Baxter JC, Sutton MD (2012) Evidence for roles of the Escherichia coli Hda protein beyond regulatory inactivation of DnaA. Mol Microbiol 85 : 648–668. doi: 10.1111/j.1365-2958.2012.08129.x 22716942
43. Ona KR, Courcelle CT, Courcelle J (2009) Nucleotide Excision Repair Is a Predominant Mechanism for Processing Nitrofurazone-Induced DNA Damage in Escherichia coli. J Bacteriol 191 : 4959–4965. doi: 10.1128/JB.00495-09 19465649
44. Maul RW, Sutton MD (2005) Roles of the Escherichia coli RecA protein and the global SOS response in effecting DNA polymerase selection in vivo. J Bacteriol 187 : 7607–7618. 16267285
45. Uchida K, Furukohri A, Shinozaki Y, Mori T, Ogawara D, et al. (2008) Overproduction of Escherichia coli DNA polymerase DinB (Pol IV) inhibits replication fork progression and is lethal. Mol Microbiol 70.
46. Pillaire M-J, Bétous R, Hoffmann J-S (2014) Role of DNA polymerase κ in the maintenance of genomic stability. Molecular & Cellular Oncology 1: e29902.
47. Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC (2012) Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336 : 315–319. doi: 10.1126/science.1219192 22517853
48. Sutton MD (2004) The Escherichia coli dnaN159 mutant displays altered DNA polymerase usage and chronic SOS induction. J Bacteriol 186 : 6738–6748. 15466025
49. Sutton MD, Duzen JM, Maul RW (2005) Mutant forms of the Escherichia coli beta sliding clamp that distinguish between its roles in replication and DNA polymerase V-dependent translesion DNA synthesis. Mol Microbiol 55 : 1751–1766. 15752198
50. Kim SR, Maenhaut-Michel G, Yamada M, Yamamoto Y, Matsui K, et al. (1997) Multiple pathways for SOS-induced mutagenesis in Escherichia coli: an overexpression of dinB/dinP results in strongly enhancing mutagenesis in the absence of any exogenous treatment to damage DNA. Proc Natl Acad Sci U S A 94 : 13792–13797. 9391106
51. Cupples CG, Cabrera M, Cruz C, Miller JH (1990) A set of lacZ mutations in Escherichia coli that allow rapid detection of specific frameshift mutations. Genetics 125 : 275–280. 2199309
52. Cupples CG, Miller JH (1989) A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc Natl Acad Sci U S A 86 : 5345–5349. 2501784
53. Sharma A, Kottur J, Narayanan N, Nair DT (2013) A strategically located serine residue is critical for the mutator activity of DNA polymerase IV from Escherichia coli. Nucleic Acids Res 41 : 5104–5114. doi: 10.1093/nar/gkt146 23525461
54. Boudsocq F, Kokoska RJ, Plosky BS, Vaisman A, Ling H, et al. (2004) Investigating the role of the little finger domain of Y-family DNA polymerases in low fidelity synthesis and translesion replication. J Biol Chem 279 : 32932–32940. 15155753
55. Ling H, Boudsocq F, Woodgate R, Yang W (2001) Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell 107 : 91–102. 11595188
56. Sanders LH, Rockel A, Lu H, Wozniak DJ, Sutton MD (2006) Role of Pseudomonas aeruginosa dinB-encoded DNA polymerase IV in mutagenesis. J Bacteriol 188 : 8573–8585. 17041045
57. Wagner J, Gruz P, Kim SR, Yamada M, Matsui K, et al. (1999) The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol Cell 4 : 281–286. 10488344
58. Nieminuszczy J, Sikora A, Wrzesinski M, Janion C, Grzesiuk E (2006) AlkB dioxygenase in preventing MMS-induced mutagenesis in Escherichia coli: effect of Pol V and AlkA proteins. DNA Repair (Amst) 5 : 181–188.
59. Plosky BS, Frank EG, Berry DA, Vennall GP, McDonald JP, et al. (2008) Eukaryotic Y-family polymerases bypass a 3-methyl-2'-deoxyadenosine analog in vitro and methyl methanesulfonate-induced DNA damage in vivo. Nucleic Acids Res 36 : 2152–2162. doi: 10.1093/nar/gkn058 18281311
60. Lee MC, Franco M, Vargas DM, Hudman DA, White SJ, et al. (2014) A ΔdinB mutation that sensitizes Escherichia coli to the lethal effects of UV - and X-radiation. Mutat Res 763–764 : 19–27. doi: 10.1016/j.mrfmmm.2014.03.003 24657250
61. Benson RW, Cafarelli TM, Rands TJ, Lin I, Godoy VG (2014) Selection of dinB alleles suppressing survival loss upon dinB overexpression in Escherichia coli. J Bacteriol 196 : 3023–3035. doi: 10.1128/JB.01782-14 24914188
62. Wagner J, Etienne H, Fuchs RP, Cordonnier A, Burnouf D (2009) Distinct beta-clamp interactions govern the activities of the Y family PolIV DNA polymerase. Mol Microbiol 74 : 1143–1151. doi: 10.1111/j.1365-2958.2009.06920.x 19843218
63. Cafarelli TM, Rands TJ, Godoy VG (2014) The DinB*RecA complex of Escherichia coli mediates an efficient and high-fidelity response to ubiquitous alkylation lesions. Environ Mol Mutagen 55 : 92–102. doi: 10.1002/em.21826 24243543
64. Godoy VG, Jarosz DF, Simon SM, Abyzov A, Ilyin V, et al. (2007) UmuD and RecA directly modulate the mutagenic potential of the Y family DNA polymerase DinB. Mol Cell 28 : 1058–1070. 18158902
65. Cafarelli TM, Rands TJ, Benson RW, Rudnicki PA, Lin I, et al. (2013) A single residue unique to DinB-like proteins limits formation of the polymerase IV multiprotein complex in Escherichia coli. J Bacteriol 195 : 1179–1193. doi: 10.1128/JB.01349-12 23292773
66. Chaurasiya KR, Ruslie C, Silva MC, Voortman L, Nevin P, et al. (2013) Polymerase manager protein UmuD directly regulates Escherichia coli DNA polymerase III alpha binding to ssDNA. Nucleic Acids Res 41 : 8959–8968. doi: 10.1093/nar/gkt648 23901012
67. Sutton MD, Duzen JM, Scouten Ponticelli SK (2010) A single hydrophobic cleft in the Escherichia coli processivity clamp is sufficient to support cell viability and DNA damage-induced mutagenesis in vivo. BMC Mol Biol 11 : 102. doi: 10.1186/1471-2199-11-102 21190558
68. Toste Rego A, Holding AN, Kent H, Lamers MH (2013) Architecture of the Pol III-clamp-exonuclease complex reveals key roles of the exonuclease subunit in processive DNA synthesis and repair. EMBO J 32 : 1334–1343. doi: 10.1038/emboj.2013.68 23549287
69. Jergic S, Horan NP, Elshenawy MM, Mason CE, Urathamakul T, et al. (2013) A direct proofreader-clamp interaction stabilizes the Pol III replicase in the polymerization mode. EMBO J 32 : 1322–1333. doi: 10.1038/emboj.2012.347 23435564
70. Kim SR, Matsui K, Yamada M, Gruz P, Nohmi T (2001) Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol Genet Genomics 266 : 207–215. 11683261
71. Kim S, Dallmann HG, McHenry CS, Marians KJ (1996) Coupling of a replicative polymerase and helicase: a tau-DnaB interaction mediates rapid replication fork movement. Cell 84 : 643–650. 8598050
72. Georgescu RE, Kim SS, Yurieva O, Kuriyan J, Kong XP, et al. (2008) Structure of a sliding clamp on DNA. Cell 132 : 43–54. doi: 10.1016/j.cell.2007.11.045 18191219
73. Miller JH (1999) A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria: Cold Spring Harbor Press.
74. Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97 : 6640–6645. 10829079
75. Benson RW, Cafarelli TM, Godoy VG (2011) SOE-LRed: A simple and time-efficient method to localize genes with point mutations onto the Escherichia coli chromosome. J Microbiol Methods 84 : 479–481. doi: 10.1016/j.mimet.2010.12.020 21185880
76. Lusetti SL, Hobbs MD, Stohl EA, Chitteni-Pattu S, Inman RB, et al. (2006) The RecF protein antagonizes RecX function via direct interaction. Mol Cell 21 : 41–50. 16387652
77. Duzen JM, Walker GC, Sutton MD (2004) Identification of specific amino acid residues in the E. coli beta processivity clamp involved in interactions with DNA polymerase III, UmuD and UmuD'. DNA Repair (Amst) 3 : 301–312.
78. Frey MW, Nossal NG, Capson TL, Benkovic SJ (1993) Construction and characterization of a bacteriophage T4 DNA polymerase deficient in 3'—>5' exonuclease activity. Proc Natl Acad Sci U S A 90 : 2579–2583. 8464864
79. Capson TL, Peliska JA, Kaboord BF, Frey MW, Lively C, et al. (1992) Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry 31 : 10984–10994. 1332748
80. Oakley AJ, Prosselkov P, Wijffels G, Beck JL, Wilce MC, et al. (2003) Flexibility revealed by the 1.85 A crystal structure of the beta sliding-clamp subunit of Escherichia coli DNA polymerase III. Acta Crystallogr D Biol Crystallogr 59 : 1192–1199. 12832762
81. Wijffels G, Dalrymple BP, Prosselkov P, Kongsuwan K, Epa VC, et al. (2004) Inhibition of protein interactions with the beta 2 sliding clamp of Escherichia coli DNA polymerase III by peptides from beta 2-binding proteins. Biochemistry 43 : 5661–5671. 15134440
82. Hamdan S, Bulloch EM, Thompson PR, Beck JL, Yang JY, et al. (2002) Hydrolysis of the 5'-p-nitrophenyl ester of TMP by the proofreading exonuclease (epsilon) subunit of Escherichia coli DNA polymerase III. Biochemistry 41 : 5266–5275. 11955076
83. Tanner NA, Hamdan SM, Jergic S, Loscha KV, Schaeffer PM, et al. (2008) Single-molecule studies of fork dynamics in Escherichia coli DNA replication. Nat Struct Mol Biol 15 : 170–176. doi: 10.1038/nsmb.1381 18223657
84. Ozawa K, Horan NP, Robinson A, Yagi H, Hill FR, et al. (2013) Proofreading exonuclease on a tether: the complex between the E. coli DNA polymerase III subunits alpha, epsilon, theta and beta reveals a highly flexible arrangement of the proofreading domain. Nucleic Acids Res 41 : 5354–5367. doi: 10.1093/nar/gkt162 23580545
85. Ho C, Kulaeva OI, Levine AS, Woodgate R (1993) A rapid method for cloning mutagenic DNA repair genes: isolation of umu-complementing genes from multidrug resistance plasmids R391, R446b, and R471a. J Bacteriol 175 : 5411–5419. 8366028
86. Wang RF, Kushner SR (1991) Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100 : 195–199. 2055470
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 9
Nejčtenější v tomto čísle
- Arabidopsis AtPLC2 Is a Primary Phosphoinositide-Specific Phospholipase C in Phosphoinositide Metabolism and the Endoplasmic Reticulum Stress Response
- Bridges Meristem and Organ Primordia Boundaries through , , and during Flower Development in
- KLK5 Inactivation Reverses Cutaneous Hallmarks of Netherton Syndrome
- XBP1-Independent UPR Pathways Suppress C/EBP-β Mediated Chondrocyte Differentiation in ER-Stress Related Skeletal Disease