
A NIMA-Related Kinase Suppresses the Flagellar Instability Associated with the Loss of Multiple Axonemal Structures
Cilia are specialized projections found on the surface of eukaryotic cells. They play crucial sensory functions, as well as motile functions needed for clearing airways or propelling cells. Ciliary motility is perturbed in the inherited disease, Primary Ciliary Dyskinesia (PCD). Two coiled coil domain-containing (CCDC39 and CCDC40) proteins are needed for the assembly of multiple key structures/complexes that are required for generating ciliary motility. Using the unicellular green alga, Chlamydomonas, we have identified a kinase (CNK11) that when mutated is able to partially rescue the short flagella phenotype of the ccdc39 and ccdc40 mutants as well as mutants lacking axonemal dyneins or the N-DRC complex. In addition, CCDC40 is required for tubulin polyglutamylation at the proximal end of flagella. We suggest that substructures like dynein arms and the N-DRC, which are needed for motility, play a second role in stabilizing the axonemal microtubules and are needed for proper length control. The polyglutamylase, TTLL9, and the kinase, CNK11, play roles in stabilizing the axonemal microtubules based on their ability to partially rescue the short flagella phenotypes of multiple mutants.
Published in the journal:
. PLoS Genet 11(9): e32767. doi:10.1371/journal.pgen.1005508
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005508
Summary
Cilia are specialized projections found on the surface of eukaryotic cells. They play crucial sensory functions, as well as motile functions needed for clearing airways or propelling cells. Ciliary motility is perturbed in the inherited disease, Primary Ciliary Dyskinesia (PCD). Two coiled coil domain-containing (CCDC39 and CCDC40) proteins are needed for the assembly of multiple key structures/complexes that are required for generating ciliary motility. Using the unicellular green alga, Chlamydomonas, we have identified a kinase (CNK11) that when mutated is able to partially rescue the short flagella phenotype of the ccdc39 and ccdc40 mutants as well as mutants lacking axonemal dyneins or the N-DRC complex. In addition, CCDC40 is required for tubulin polyglutamylation at the proximal end of flagella. We suggest that substructures like dynein arms and the N-DRC, which are needed for motility, play a second role in stabilizing the axonemal microtubules and are needed for proper length control. The polyglutamylase, TTLL9, and the kinase, CNK11, play roles in stabilizing the axonemal microtubules based on their ability to partially rescue the short flagella phenotypes of multiple mutants.
Introduction
Defects in ciliary assembly and function cause a wide range of human diseases and syndromes called ciliopathies. Primary ciliary dyskinesia (PCD) is diagnosed by defects in ciliary motility, and is associated with a genetically heterogeneous group of recessive disorders [1]. Mutations causing PCD have been identified in genes encoding axonemal dynein subunits [2, 3], dynein assembly factors [4–6], and dynein docking/adaptor factors [7, 8]. The nexin-dynein regulatory complex (N-DRC) is an axonemal structure critical for the regulation of dynein motors and for connecting doublet microtubules to each other. Loss-of-function mutations in DRC1 (CCDC164/PF3) and DRC3 (CCDC65) cause severe defects in assembly of the N-DRC structure and result in defective ciliary movement in humans and Chlamydomonas reinhardtii [6, 9, 10]. PF2, which encodes DRC4, was used to identify 11 proteins in the N-DRC [10]. Mutations in CCDC39 and CCDC40 cause altered ciliary beating with the disorganization of the axoneme that includes the displacement of the peripheral outer doublets, as well as central pair microtubules, radial spokes and inner dynein arm defects [11–15]. Loss-of-function mutations in CCDC39 and CCDC40 in Chlamydomonas lead to short flagella, irregularly spaced radial spokes, absence or reduction of N-DRC components and inner dynein arm proteins [16, 17]. CCDC39 and CCDC40 mutations in children lead to earlier and more severe lung disease than in PCD patients with outer dynein arm mutations [18].
In Chlamydomonas, there are many mutations that can lead to short flagella. Partial reduction in IFT proteins (IFT144 (FLA15) and IFT139 (FLA17)) or motors such as the kinesin-2 motor FLA10 or cytoplasmic dynein result in short flagella [19–21]. Changes in the cytoplasmic pool of tubulins and flagellar precursor proteins also affect flagellar length [22, 23]. In addition, the simultaneous loss of multiple substructures, such as the dynein arms, radial spokes, and the N-DRC, result in short flagella [24–26]. LeDizet and Piperno isolated a suppressor (ssh1) that increased the flagellar length in double mutant strains that lacked outer and inner dynein arms without restoring the missing structures [26]. A recent study identified a deletion in the TPG2 gene as the causative mutation in the ssh1 strain [27]. TPG2 encodes FAP234, a flagellar protein that forms a complex with a tubulin polyglutamylase TTLL9/TPG1 [28, 29]. Tubulin polyglutamylation adds multiple glutamates to both α - and β-tubulin subunits along microtubules in cilia/flagella, basal bodies, and neuron axons [30–32]. Several tubulin tyrosine ligase-like (TTLL) proteins carry out the polyglutamylation process. Tubulin polyglutamylation can affect microtubule assembly, stability, and motility [32]. In Chlamydomonas, tpg1 affects polyglutamylation of α-tubulin specifically and shows a flagellar motility defect [29]. Both tpg1 and tpg2 mutations suppress the short flagella phenotype found in mutants that lack multiple axonemal dynein species [27].
NIMA-related protein kinases have been found in eukaryotes and their functions are related to regulation of cell cycle, cilia length, and microtubule stability [33–38]. Currently, there are 11 NIMA-related protein kinases identified in Chlamydomonas [33] and only two of them have been functionally characterized [35, 36]. A null mutant of the NIMA-like protein kinase CNK2 in Chlamydomonas has slightly longer flagella and defective flagellar disassembly. The cnk2-1 mutant has decreased tubulin turnover at the flagellar tip, which suggests that a reduced rate of flagellar disassembly is compensated by reduced rate of assembly [36]. The CNK2 protein, together with a MAP kinase (LF4), respond to flagellar length signals and block assembly and promote disassembly, respectively [36]. Thus, they provide input to the balance between assembly and disassembly of axonemal microtubules and flagellar length.
In this study, we identify a novel NIMA-related protein kinase CNK11 that rescues the short flagella phenotype found in several N-DRC mutants, as well as mutants lacking dynein arms. In addition, we discovered that the polyglutamylation defect caused by tpg1 could not rescue the CCDC40 mutant. Instead, the CCDC40 mutation in the tpg1 background has narrower distribution of polyglutamylated tubulin at the proximal end of flagella. The microtubule stabilizing drug paclitaxel is able to rescue the short flagella phenotype in CCDC39/CCDC40 mutants but this rescue fails in the presence of cnk11 or tpg1.
Results
Identification of the causative mutations in pf7, pf8 and fla12
The pf7 and pf8 mutants were first isolated as mutants with no flagella or short flagella with a motility defect [17, 39] (Fig 1), and mapped to chromosome 17 [40, 41]. Using whole genome sequencing in combination with our SNP and short insertion/deletion library, we identified the causative mutations in both pf7 and pf8 mutant strains [42] (Table 1). A nonsense mutation in Cre17.g698365 (CCDC40) is responsible for the pf7 mutant phenotype; a nonsense mutation in Cre17.g701250 (CCDC39) leads to the pf8 mutation (Table 2). We performed BAC rescue to confirm they are the causative mutations (Fig 1A). Forty-one independent transformants that contain BAC DNA 17F4, which carries the CCDC40 gene, showed restoration of both flagellar length and motility in pf7; 20 independent transformants that contain BAC DNA 31N18, which carries the CCDC39 gene, restored flagellar length and motility in pf8. For each rescue, we analyzed 16 independent transformants and the transformed BAC DNA cosegregates with rescue in all transformants. Independently, Oda et al. showed that pf7 and pf8 encode CCDC40 and CCDC39 [16].



The fla12 mutant was isolated as a temperature-sensitive flagellar assembly mutant [43] that was previously mapped to chromosome 17 [40]. The fla12 cells shorten their flagella gradually and become immotile after the temperature is raised from 21°C to 32°C (Fig 1B). We used the same whole genome sequencing approach to identify a L845P change in CCDC39 in fla12 (Tables 1 and 2). The transgene that rescued the pf8 mutant was introduced into fla12 by meiotic crosses. In 12 independent progeny, the transgene restores normal flagellar length and motility in all strains at 32°C.
Suppressor/revertant screen of pf7, pf8, fla12 and pf7; pf8
Chlamydomonas offers the ability to use suppressor analysis to find genes that restore function to motility mutants [44, 45]. After UV mutagenesis of the pf7 mutant, we screened for swimming cells and recovered 31 independent strains. PCR/enzyme digestion and Sanger sequencing revealed that all 31 strains are intragenic revertants (Table 3). Using the same strategy, we isolated 34 revertants of pf8 and 4 revertants of fla12 (Table 3), all are intragenic events.

Subsequently, we performed two independent UV mutagenesis screens on pf7; pf8 double mutants to isolate extragenic suppressors. In contrast to nitrogen-starved, autolysin-treated cells that assemble ~ 2 μm flagella (Fig 1A), the pf7; pf8 mutant cells in nitrogen-containing medium are mostly aflagellate. The first UV mutagenesis screen led to isolation of three independent strains (pf7; pf8; cnk11-1, pf7; pf8; cnk11-2, and pf7; pf8; cnk11-3) and the second UV mutagenesis screen identified three additional strains (pf7; pf8; cnk11-4, pf7; pf8; cnk11-5, and pf7; pf8; sup2D). All six strains show partial suppression of the aflagellate phenotype of pf7; pf8, but do not suppress the motility defect (Fig 1A). None of them is linked to either pf7 or pf8. They each contain one suppressor mutation based on crosses to the pf7; pf8 parent; the aflagellate phenotype segregates 2 : 2. The suppressor mutations in five of the strains (cnk11-1 to cnk11-5) are tightly linked to one another (S1 Table). Whole genome sequencing (Table 1) revealed that the five strains each carry a mutation in Cre07.g339100. The causative mutation in the sixth suppressor, sup2D, is currently under analysis.
In the five strains carrying mutations in Cre07.g339100, two nonsense mutations (cnk11-1 and cnk11-2), a frame shift (cnk11-3 and cnk11-5), and a missense mutation (cnk11-4, Table 2) were identified (Fig 2). Using dCAPs markers designed to each mutation (S2 Table), we observed linkage between suppression and the mutant allele in each strain. Cre07.g339100 encodes a 2903 aa (amino acid) protein with a NIMA-like protein kinase (NEK) domain (aa 582–921). This protein is different from the 11 NEKs (CNK1—CNK10, and FA2) that have been previously annotated in Chlamydomonas [33]. Thus, we name it CNK11 (Chlamydomonas NIMA-like protein kinase 11). Using the conserved protein kinase domain, we constructed a phylogenetic tree with using the kinase domains found in 77 NEKs from Arabidopsis, Aspergillus, C. elegans, Chlamydomonas, Dictyostelium, Drosophila, human, mouse, Trypanosoma, rice, Xenopus and zebrafish (S1 Fig and S3 Table). The tree reveals that CNK11 is phylogenetically different from any of the known NEK classes.

The cnk11 suppressor fails to restore N-DRC proteins and other axonemal proteins in pf7 and pf8
In a previous report [16], Oda et al. showed that the pf7 and pf8 single mutants assemble reduced amounts of two N-DRC proteins; DRC2 and DRC4. In our analysis of isolated axonemes, we found that the single mutants assemble reduced amount of DRC1, DRC4, DRC5, DRC7, and DRC11 and completely lack DRC2, DRC3, and CCDC39 (Fig 3A). In addition, the amount of each N-DRC proteins in a pf7; pf8 double mutant and a pf2; pf7; pf8 triple mutant is comparable to that found in the single mutants, with the exception that no DRC4 protein is found in the triple mutant. This is expected given that the pf2 mutant lacks DRC4 (Fig 3A) [10]. The similarity of the single and double mutants is also expected given the co-assembly of CCDC39 and CCDC40 [16]. Transformation of wild-type PF7 or PF8 into the corresponding mutant restores the N-DRC proteins (Fig 3A).

In axonemes from the pf7; pf8; cnk11-1 mutant, the N-DRC proteins are not restored (Fig 3A). This suggests that cnk11 mutants do not suppress the flagellar length defect of pf7; pf8 via assembly of N-DRC proteins (Fig 3A). In addition to the N-DRC proteins, we asked if the pf7 and pf8 mutants affect other axonemal proteins (Fig 3A). Tektin, which is a microtubule binding protein, is diminished in the ida6 (DRC1) and pf3 (DRC2) mutants, which lack the inner dynein arms species e [46]. Tektin is missing in the single, double, and triple mutants (Fig 3A). Because loss of tektin is associated with the loss of dynein e, we suggest that these mutants are likely to lack dynein e as well. The proximally localized minor dynein heavy chain, DHC11 [47], is missing in the single, double, and triple mutants. DIC3/ IC140, which is the intermediate chain for the I1/f inner dynein arm [48, 49], is reduced in pf8, the double and triple mutants, but not in the pf7 mutant. This is one of the few difference found between pf7 and pf8, and was independently validated [16]. DLE2/centrin, which is part of the b, e, and g inner dynein arm complex [50, 51], is slightly reduced in the single mutants. There is no reduction of RSP16, which is one of the radial spoke proteins [52]; or DIC2/IC69, an intermediate chain in the outer dynein arm [53, 54]. DII1/p28 is only slightly reduced based on 2 independent preparations. Similar results were observed by Oda and colleagues [16]. The ribbon proteins, Rib72 and Rib43a, were first identified by their insolubility in various extraction protocols [55, 56]. There is no loss of these two proteins in the pf7 or pf8 mutant compared to wild-type or other N-DRC mutants. LF5, which is a CDKL5 homolog involved in length control that localizes to the proximal 1 μm of the flagella [57], is increased in the single, double, and triple mutants. Since we load equal amounts of protein in each sample, the increase is likely to be due to increased representation of proteins at the proximal end where LF5 localizes. The pf7 and pf8 rescued strains resemble wild-type axonemes and restore all proteins to wild-type levels. The triple mutant pf7; pf8; cnk11-1 shows similar losses to the pf7; pf8 preparations. It indicates that the cnk11 mutant suppresses the pf7; pf8 length phenotype by means other than restoration of axonemal proteins.
In N-DRC mutants such as pf2 and pf3, the presence of 0.1 mM ATP leads to splaying of individual outer doublet microtubules in the medial and distal regions of the isolated full-length axonemes [10]. The proximal end of these axonemes remained intact. In contrast, wild-type axonemes remain intact throughout the whole length. Bower and colleagues concluded that the N-DRC provides some but not all of the resistance to microtubule sliding between doublets. This helps to maintain optimal alignment of doublets for productive flagellar motility [10]. Given pf7 and pf8 axonemes either lack or have reduced amounts of most N-DRC proteins tested, we examined isolated axonemes exposed to 0.1 mM ATP (Fig 4). To verify the proximal end showed splaying, we used antibodies to LF5 that localizes to the proximal end [57]. We observed little or no splaying of the doublet microtubules in wild-type axonemes. In the mutants, we observed splaying in the medial and distal regions of the single, double, and triple mutants that was similar to the splaying observed in N-DRC mutants [10]. The doublets in the proximal 1 μm end remain intact, similar to the observation found in N-DRC mutants. Thus, we conclude that CCDC39 and CCDC40 are not required for holding the microtubules together in the proximal region.

The fla12 mutant has normal IFT movement and its temperature-sensitivity is partially suppressed by cnk11
Given the temperature-sensitive fla12 mutant carries a missense mutation in CCDC39, we asked whether the temperature shift affects axonemal proteins and flagellar length in this mutant. Four hours after temperature shift from 21°C to 32°C, fla12 cells contain less CCDC39, DRC1, DRC3, DRC4, DRC7, and tektin while maintaining normal levels of DIC2 and DIC3 (Fig 3B). This suggests that the missense mutation affects the thermal stability of CCDC39, which in turn leads to reduction of N-DRC and axonemal proteins as observed in the null CCDC39 mutant (Fig 3A).
At 21°C, the fla12 mutant assembles slightly shorter flagella (~6.4 μm) when compared to wild-type (~8.5 μm). Eight hours after the temperature shift from 21°C to 32°C, the average flagellar length of fla12 is ~1.6 μm. The flagellar length of the fla12; cnk11-3 double mutant (~6.5 μm) is similar to the single fla12 mutant at 21°C. However after temperature shift, the flagellar length is ~3.9 μm, which is significantly longer than fla12 cells at the same time point (Fig 1B). This indicates that similar to the partial suppression of the short flagella phenotype of pf7; pf8 double mutant, cnk11 can partially suppress the temperature-sensitive short flagella phenotype of fla12.
Intraflagellar transport (IFT) was monitored previously in numerous flagellar assembly mutants [58]. Analysis of the fla12; pf15 double mutant suggested that the velocity of anterograde and retrograde IFT was increased over control velocities, and the frequency of IFT particles was also higher. We reanalyzed IFT by TIRF (Total Internal Reflection Fluorescence) microscopy using GFP-tagged IFT20 [59] in the fla12 mutant. Data obtained from 4 FLA12 and 5 fla12 cells indicate that anterograde (Fig 5C) and retrograde (Fig 5D) IFT velocities in fla12; IFT20-GFP cells are identical to those in FLA12; IFT20-GFP cells. Thus, we conclude that there is no IFT velocity defect in the fla12 mutant. There are at least two possible explanations for the disagreement between these two studies. In the fla12; pf15 study, the pf15 mutation disrupts the p80 subunit of katanin [60]. It is possible that there is some synthetic interaction between katanin and CCDC39 that affects IFT velocity and number. Alternatively, the multiple backcrosses of fla12 before the TIRF study could have removed another mutation that affected IFT.

The cnk11 mutation suppresses the short flagella phenotype found in N-DRC and dynein arm deficient mutants
To ask about the specificity of the cnk11 suppressor, we introduced the cnk11-1 mutation into the pf2 (DRC4) [10] and pf3 (DRC1) [61] mutants through meiotic crosses. Mutants in DRC4 are missing N-DRC proteins as well as dyneins a and c [10]. The pf2 mutant has an average flagellar length of ~5.2 μm (Fig 6A). In comparison, the pf2; cnk11-1 double mutant has an average flagellar length of ~8.6 μm (Fig 6A), which is comparable to the average flagellar length found in wild-type CC-125 cells (~8.9 μm; Fig 1A) and significantly longer than pf2 flagella. The pf3 mutant obtained from the Chlamydomonas Resource Center (CC-1026) has an average flagellar length of 7.4 μm (Fig 6A), slightly shorter than in wild-type cells. Mutants in DRC1 are missing N-DRC proteins, tektin, and RSP13 [10]. PCR on progeny from a meiotic cross between CC-1026 and cnk11-1 revealed that over 8 kb of genomic DNA on chromosome 7 is deleted. The deleted region includes most of the CNK11 gene and at least half of the adjacent gene Cre07.g339104 (Fig 2). Therefore, the strain CC-1026 should be annotated as pf3; cnk11-6. A meiotic cross between pf3; cnk11-6 to wild-type CC-124 cells allowed the isolation of a pf3; CNK11 strain. The average flagellar length of pf3; CNK11 cells is ~4.8 μm (Fig 6A), which is comparable to the length observed in pf2 cells. In addition, the pf3; CNK11 cells have flagellar lengths that are more variable (ranging from <1 μm to >8 μm) than the pf2 (mostly 3~7 μm), pf7, and pf8 (both mostly 1~3 μm) cells (S2 Fig). In conclusion, the cnk11 mutant rescues the short flagella phenotype of CCDC39 and CCDC40 mutants as well as two N-DRC mutants.
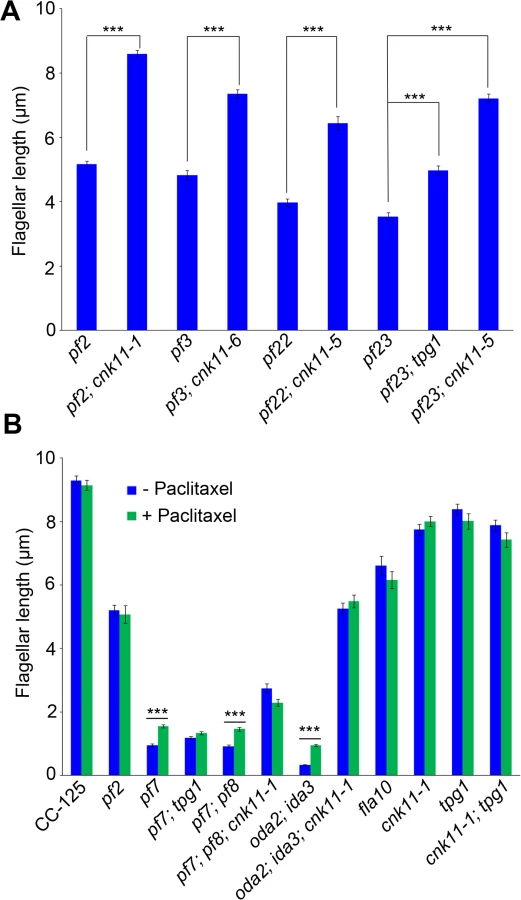
The pf22 and pf23 mutants were first isolated as paralyzed flagella mutants and both have short flagella [62]. The PF22 gene encodes a conserved cytoplasmic protein (DNAAF3) that is essential for the assembly of both outer and several inner dynein arms [4]. The pf23 mutant lacks inner dyneins a, c, d, and f [25, 63]. The outer dynein arm mutant, oda2, and inner dynein arm mutant, ida3, both display slow motility with normal flagellar lengths. The oda2; ida3 double mutant is paralyzed with very short flagella (Fig 6B) as has been observed for many oda; ida double mutants [26]. The N-DRC is not affected in any of these mutants. To ask whether the cnk11 suppressor can restore normal flagellar length in these mutants, we introduced cnk11 mutations into pf22, pf23, and oda2; ida3 mutants. The average flagellar length of pf22 is ~4.0 μm and ~6.4 μm for pf22; cnk11-5 (Fig 6A). The average flagellar length of pf23 is ~3.5 μm and ~7.2 μm for pf23; cnk11-5 (Fig 6A). The oda2; ida3 cells have an average flagellar length of ~0.5 μm; and, the oda2; ida3; cnk11-1 triple mutant has an average flagellar length of ~5.9 μm (Fig 6B). Thus, cnk11 mutations rescue the short flagella mutant phenotype of dynein arm deficient mutants, which lack multiple axonemal dynein species and presumably have unstable axonemal microtubules. Similar to the effect of cnk11 on pf7 and pf8, the cnk11 mutations do not rescue the motility defects found in the dynein arm deficient mutants. In addition, the cnk11 mutations do not rescue the temperature-sensitivity flagellar assembly of the kinesin-2 motor mutant, fla10, or the IFT mutants, fla15 and fla17, after 8 hrs at 32°C.
Chemical suppression of the short flagellar length phenotype
In human cell lines, knockdown of NEK4, a NIMA-like kinase, confers paclitaxel resistance and show defects in repolymerizing microtubules after nocadozole treatment [37]. In Arabidopsis thaliana, nek4, nek5, and nek6 all show hypersensitivity to paclitaxel [38]. Various NEK proteins play a role in microtubule stability. We tested the cnk11-1 allele on paclitaxel media with concentrations from 5 to 60 μM. The mutant strain behaved identically to the wild-type controls and we did not observe resistance or hypersensitivity as judged by cell division and cell size [64].
We then asked whether the addition of 10 μM paclitaxel for 30 minutes, a dosage that does not causes arrest of cell division in wild-type cells [64], would have any effect on flagellar length. In wild-type and cnk11-1 cells, which have normal flagellar lengths, there is no change (Fig 6B). We examined pf2, and the temperature-sensitive kinesin mutant fla10-1 which has about half-length flagella when grown at 28°C [19]. The addition of paclitaxel has no effect on either mutant (Fig 6B). In the short flagella mutants pf7, pf7; pf8, and oda2; ida3, paclitaxel conferred increased flagellar length. In contrast, paclitaxel does not lead to further elongation of flagella of these mutants when the cnk11 mutation is present (Fig 6B). This suggests that CNK11 and paclitaxel could act via the same mechanism to stabilize axonemal microtubules in these short flagella mutants.
The tpg1 mutant fails to suppress the short flagella phenotype in the CCDC40 mutant
Kubo et al. showed that both tpg1 and tpg2 can rescue the short flagella phenotype found in pf23 and pf28; pf30 [27]. PF28 is an allele of ODA2 (the gamma dynein heavy chain) and PF30 is an allele of IDA1 (1-alpha dynein heavy chain, I1/f complex). This result is similar to the effect of cnk11 on pf22, pf23, and oda2; ida3 (Fig 6). Therefore, we asked whether the tpg1 mutation can rescue the short flagella phenotype found in the pf7; pf8 double mutant. The TPG1 gene maps to chromosome 17 at 0.51 Mb, between the PF7 (chromosome 17 at 0.33 Mb) and PF8 (chromosome 17 at 0.74 Mb) genes. The short distance among these three genes makes it extremely hard to generate the pf7; pf8; tpg1 triple mutant by meiotic recombination since it would require two crossovers in an interval of only 4 map units. Given the pf7 mutant behaves similarly to the pf7; pf8 double mutant, we analyzed pf7 and pf7; tpg1 instead of pf7; pf8 and pf7; pf8; tpg1.
To our surprise, the tpg1 mutation does not rescue the short flagella phenotype found in pf7. Instead, the pf7; tpg1 mutant has a more severe flagella phenotype than the pf7 mutant. In nitrogen-free medium, ~85% of pf7 cells have flagella. In contrast, only ~48% of pf7; tpg1 cells have flagella. Measurement of flagellated cells in both strains showed no significant difference in the flagellar lengths between pf7 and pf7; tpg1 (Fig 6B).
We asked whether polyglutamylation of tubulin is altered in the pf7; tpg1 mutant by both immunoblots and immunofluorescence. In wild-type cells, tubulin in axonemal microtubules is polyglutamylated. A polyclonal antibody (Poly E) that specifically recognizes tubulin with three or more glutamates reveals much stronger signal intensity in α-tubulin than in β-tubulin in wild-type axonemes [29]. The signal intensity of polyglutamylated α-tubulin relative to polyglutamylated β-tubulin is significantly reduced in pf7 and pf8 (Fig 7A). Similar to the findings by Kubo et al. [29], we noticed significant reduction of α-tubulin polyglutamylation but not β-tubulin polyglutamylation in tpg1. A significant reduction of α-tubulin polyglutamylation was observed in both pf23; tpg1 and cnk11-1; tpg1 mutants, but not in the pf23 or cnk11-1 mutants. However in the pf7; tpg1 mutant, polyglutamylated α-tubulin remains (Fig 7A). The change of relative signal intensities between polyglutamylated α - and β-tubulins found in pf7, pf8, and pf7; tpg1 is not due to their short flagellar lengths, since the short flagellar length mutant oda2; ida3 has stronger signal intensity in α-tubulin than in β-tubulin, as found in wild-type axonemes (Fig 7A). Immunoblots with an anti-TPG2/FAP234 antibody [28] show that no TPG2 protein is detected in the axonemes of any strain carrying the tpg1 mutant (Fig 7A). It suggests that the presence of small amount of polyglutamylated α-tubulin in pf7; tpg1 is not due to the recruitment or recovery of the TPG1-TPG2 complex in the axoneme. The abundance of TPG2/FAP234 is not significantly affected by flagellar length or the pf7 and pf8 mutations.

By immunofluorescence, the polyglutamylated tubulin detected by the polyE antibody shows signal along the entire length of the axoneme in wild-type and cnk11-1 cells (Fig 7B). As observed previously, the signal in tpg1 is concentrated at the proximal end [29], and we observe that the polyglutamylated tubulin signal is only ~1.5 μm in length (Fig 7C). The cnk11-1; tpg1 and pf23; tpg1 double mutants have a similar stretch of polyglutamylated tubulin signal regardless of their flagellar lengths (Fig 7B). The pf7; tpg1 cells are strikingly different, the polyglutamylated tubulin signal is reduced to ~0.5 μm, which is significantly shorter than in the single or other double mutants (Fig 7B & 7C). This result is different from what we observed in the immunoblots, in which the polyE signal of α-tubulin is more abundant in pf7; tpg1 than in tpg1. It is likely that the difference is due to using isolated flagella that include both the microtubule axoneme and the flagellar membrane/matrix for the immunoblot and using axonemes that have the membrane/matrix fraction removed by detergent for immunofluorescence. Polyglutamylation of α-tubulin but not β-tubulin is associated with soluble tubulin heterodimers [65]. Thus the difference in polyE abundance between the two techniques is likely due to the removal of the soluble polyglutamylated α-tubulin in the immunofluorescence experiments. Combining the immunoblot and immunofluorescence results suggests that PF7/CCDC40 is needed for polyglutamylation at the proximal end of the microtubule axonemes.
Next we asked whether paclitaxel has any effect on tpg1. As might be expected for flagella with normal length, neither tpg1 nor cnk11-1; tpg1 is affected by treatment with paclitaxel for 30 minutes (Fig 6B). However, no increase in flagellar length is observed after paclitaxel treatment of the pf7; tpg1 mutant. We suggest that paclitaxel does not increase flagellar length in strains with the cnk11 or tpg1 mutations.
The cnk11-1 mutation increases tubulin turnover at the flagellar tip
Given that the NIMA-related kinase cnk2-1 mutant affects the disassembly rate of flagella, we asked whether cnk11-1 affects the rates of assembly and/or disassembly. We first compared the rates of flagellar assembly after flagella amputation by pH shock in wild-type (CC-125) and cnk11-1 cells (Fig 8A). Within 30 minutes following flagellar amputation, the assembly rate of cnk11-1 cells was ~0.23 μm/min, which is not significantly faster than the rate of wild-type cells (~0.20 μm/min). This is very similar to rates observed in the cnk2-1 cells by Hilton et al. [36]. However, the assembly rate in cnk11-1 cells reduced significantly within the next 90 minutes, and resulted in slightly shorter flagella than in wild-type cells (Fig 8A). We conclude that the overall assembly rate during flagellar regeneration is not affected in cnk11-1 cells.

Another way to measure the dynamic of flagellar assembly is to test the incorporation rate of new tubulin subunits at the flagellar tip. When a pair of Chlamydomonas cells mate, they form a quadriflagellate cell (QFC), which has two pairs of flagella. Tubulin subunits are added at the tip of the flagella, using subunits from the cytoplasm [19]. The two pairs of flagella can be distinguished by using one parent that carries an epitope-tagged HA-tubulin gene (Fig 8B insert, green), while the other parent lacks this gene. Both pairs of flagella are visualized with an antibody to acetylated α-tubulin (Fig 8B insert, magenta). The flagella from the parent with the tagged α-tubulin are stained with an antibody to the HA tag. Newly incorporated tubulin on the unlabeled flagella is visualized with the antibody to the HA tag. We mated two wild-type strains and tracked the incorporation of HA-tubulin subunits at 30, 60, 90 minutes after mating (Fig 8B, magenta). The length of incorporated HA-tubulin at the tip of flagella gradually increased along time and reached ~0.48 μm at 90 minutes. We mated two parents with the cnk11-1 mutation and observed more incorporation of HA-tubulin subunits at 60 and 90 minutes (Fig 8B, blue). The length of incorporated HA-tubulin at the tip was ~0.80 μm at 90 minutes, which suggests a rate that is nearly twice as fast as in wild-type QFCs. Since the length of the flagella did not increase, we suggest that the cnk11-1 mutation increases tubulin turnover at the flagellar tip.
Kubo et al. showed that the tpg2 mutant has slow tubulin turnover at the flagellar tip [27]. We performed the same assay on the tpg1 mutant. The incorporation length of HA-tubulin in tpg1 cells was ~0.16 μm at 90 minutes, significantly lower than that in wild-type or cnk11-1 cells (Fig 8B, purple). The incorporation length of HA-tubulin in cnk11-1; tpg1 cells was ~0.17 μm at 60 minutes but dropped to ~0.05 μm at 90 minutes (Fig 8B, black). Therefore, the faster turnover rate of HA-tubulin observed in cnk11-1 flagella is suppressed by the tpg1 mutation.
The addition of 1-isobutyl-3-methylxanthine (IBMX) causes gradual disassembly of flagella in wild-type cells. To ask whether flagellar disassembly is affected by cnk11, we compared the flagellar shortening rates in wild-type (CC-125) and cnk11-1 cells (Fig 8C). The disassembly rates of CC-125 and cnk11-1 cells within the first 30 minutes were both ~0.10 μm/min, similar to the rate Hilton et al. reported [36]. The disassembly rate of tpg1 (~0.11 μm/min) was similar to wild-type and cnk11-1. It was slightly reduced in cnk11-1; tpg1 (~0.09 μm/min, Fig 8C). Thus, neither cnk11-1 nor tpg1 mutation affects the flagellar disassembly rate.
Discussion
Regulation of ciliary and flagellar length is extremely important to the proper function of these organelles in different organisms. In humans, ciliary length defects are observed in multiple ciliopathies that include Bardet-Biedl syndrome, nephronophthisis, Joubert syndrome, polycystic kidney disease, and Meckel-Gruber syndrome [66]. In Chlamydomonas, flagellar length defects affect both motility and mating [67]. Flagellar length can be regulated by multiple factors, including the rates of flagellar assembly and disassembly [66, 68], availability of IFT proteins, motors, and structure proteins [20], as well as factors that affect the stability of axonemal microtubules [69].
The structure defects in CCDC39 and CCDC40 mutants
In mutant screens performed by McVittie and others, three pf7 and five pf8 alleles were identified [17, 70]. The mutants show abnormalities in the organization of the axoneme and radial spokes [17]. Oda and colleagues localized CCDC39 and CCDC40 using tagged genes together with cryo-EM tomography to show that these proteins serve as docking sites along the doublet microtubules for axonemal structures, which include the radial spokes, the N-DRC and all of the inner dynein arms [16]. Our immunoblots with DLE2/centrin suggest that not all of the inner arms are missing in pf7 and pf8 since centrin associates with three inner dynein heavy chains (b, e, and g) and is only slightly reduced. Although the pf7 and pf8 mutants have paralyzed flagella, their dyneins are functional based on our sliding/splaying assays. Both single mutants and the double mutant show splaying of the microtubules (Fig 4) that is similar to the splaying observed in the N-DRC mutants [10]. Thus, the paralysis is likely to be due to the microtubule and radial spoke disorganization that regulate the coordinated behavior of the dynein arms, as hypothesized by both McVittie and Oda et al. [16, 17]. The splaying experiments also suggest that the link in the proximal 2 μm does not rely on CCDC39/40, DRC1, DRC4, or the inner dynein arm I1/f (Fig 4 and [10]). Bui and colleagues [71] identified rod-like circumferential interdoublet linkers in the proximal axoneme that are clearly structural different from the N-DRC structure. We assume that these structures are retained in the pf7 and pf8 mutants, but they have not been examined.
In all patients diagnosed with PCD with CCDC39 or CCDC40 mutations, the changes result in premature truncation of the protein, which suggests that null alleles are associated with the phenotype [11]. Unexpectedly, the long-term prognosis of children with CCDC39 or CCDC40 mutations is worse than for other PCD patients, and similar to patients with cystic fibrosis [18]. These alleles would be similar to the mutations in pf7 and pf8 that have premature termination alleles. We also identified a conditional allele, fla12, in the PF8/CCDC39 locus (Table 1). The leucine to proline change occurs in an unstructured region of the C-terminus of the protein and the leucine is not conserved in other organisms. At the permissive temperature (21°C) the flagella are slightly shorter. This missense mutation leads to reduced CCDC39 and other DRC proteins at the restrictive temperature (Fig 3B). After 8 hours at the restrictive temperature, the phenotype of fla12 cells resembles the phenotype of pf8 cells. The flagella are immotile and short. It is possible that missense alleles in CCDC39 in humans may have a less severe phenotype that only slightly alters the motility and would not have been grouped together with the more severe null alleles associated with PCD [11, 12, 14]. Our fla12 revertants (Table 3) indicate that the leucine can be replaced by a variety of amino acids. This suggests that the change to a proline undermines the protein function and leads to the short and paralyzed flagella at 32°C. Given that the speeds of anterograde and retrograde IFT in fla12 shows no difference compared to those found in wild-type cells, it suggests that IFT is unlikely to play a role in flagellar length control in this CCDC39 mutant.
The polyglutamylation defect in CCDC39 and CCDC40 mutants
Post-translational modification of tubulin, which includes polyglutamylation and polyglycination, affects axonemal microtubule stability. Suryavanshi et al. and Kubo et al. showed in Tetrahymena and Chlamydomonas that loss of polyglutamylation on the B-tubule is likely to affect the activity of inner arm dyneins [72, 73]. A decrease in tubulin polyglutamylation in mouse airway cilia changes the curvature of the cilia as well as the asymmetry of the beating [74]. Overexpression of the polyglutamylation enzyme (TTLL6) in Tetrahymena destabilizes axonemal microtubules [75]. Knockdown of the glycination enzymes (TTLL3 and TTLL8) causes instability and results in short or absent mouse ependymal cilia. Polyglutamylation changes the binding affinities of a number of microtubule associated proteins and motors [76], and promotes microtubule severing [77]. Thus, the presence of polyglutamylation may affect microtubule stability in a variety of ways. Polyglutamylation like acetylation of tubulin is associated with long-lived microtubules [76, 78].
Because the loss of CCDC39 and CCDC40 affects the level of polyglutamylation, we examined the tpg1 mutation in TTLL9. Loss of α-tubulin polyglutamylation in tpg1 causes a motility defect due to the loss of tektin but no change in flagellar length ([29] and Fig 6B). Thus, reduction in tubulin polyglutamylation in the pf7 and pf8 mutants cannot be solely responsible for the short flagella in the CCDC39/40 mutants. The tpg1 mutation in combination with either pf7 or inner dynein arm deficient mutant pf23 has very different consequences. The tpg1 mutant partially rescues the flagellar length defect in pf23 (Fig 6A) but leads to more aflagellate cells with pf7 and no change in the length of the remaining flagella. By immunoblots, the level of polyglutamylated α-tubulin in the flagella of pf23; tpg1 is significantly less than in the flagella of pf7; tpg1 (Fig 7A). By immunofluorescence, we show that localization of polyglutamylated tubulin at the proximal end of axoneme is reduced in pf7; tpg1, but not in pf23; tpg1, when compared to tpg1 (Fig 7B & 7C). One possibility is that CCDC39 and CCDC40 are required for the activity of one or more tubulin glutamylases at the proximal end of the flagella while the TPG1 is responsible to polyglutamylation of tubulin along the rest of the flagella. There are 10 TTLL proteins found in Chlamydomonas [29] and the flagellar proteome includes only the TTLL9/TPG1 protein [79]. However, a proteomic analysis of flagellar phosphoproteins indicates that at least 3 additional TTLL proteins are found in the flagella [80]. They include TTLL13, a homolog of human tubulin polyglutamylase TTLL6; Cre09.g403108, an ortholog of human tubulin polyglutamylases TTLL4 and TTLL5; and Cre03.g145447, a homolog of human monoglycylase TTLL3. The former two are good candidates to be involved in tubulin glutamylation in the flagella.
The involvement of protein kinases in flagellar length control
Multiple protein kinases affect flagellar length. In Chlamydomonas, three CDK-like kinases (LF2, LF5, and FLS1), one MAP kinase (LF4), and one NIMA-related kinase (CNK2), have been characterized [36, 57, 81–83]. Loss of LF2, LF4, LF5, and CNK2 increase flagellar length. Loss of FLS1, CNK2, and LF4 block flagellar disassembly and loss of CNK2 decreases incorporation of new tubulin at the flagellar tip. The direct targets of these kinases remain to be identified. A recent global phosphoproteomic study revealed that over 180 Chlamydomonas flagellar proteins are phosphorylated [80]. This set includes N-DRC proteins, IFT proteins, outer and inner dynein arm proteins, central pair proteins, radial spoke proteins, and CCDC39.
Our screen for suppressors of the pf7; pf8 double mutant was designed to find swimming cells. However, no restoration of motility was found. The cnk11 mutant alleles led to short stumpy flagella, which still have a motility defect. In addition, we found that the cnk11 mutant alleles rescue the flagellar length defect but not the motility defect of N-DRC mutants as well as the dynein arm deficient mutants (Fig 6A). These results, along with the fact that multiple DRC proteins and axonemal proteins are not restored in pf7; pf8; cnk11-1 and pf3; cnk11-6 mutants (Fig 3A), suggest that the cnk11 mutations partially increase flagellar length via a N-DRC - and dynein protein-independent mechanism. Thus, even though CCDC39 is found to be phosphorylated in the flagella [80], it is unlikely that it is the direct target of CNK11.
During flagellar assembly, a cytoplasmic pool of tubulin subunits are constantly transported to the tip of flagella via IFT [59]. Different from flagellar assembly, flagellar disassembly is not dependent on flagellar length [19]. It is affected by the rates of IFT, disassembly of axonemal microtubules, and disassembly of axoneme-associated protein [81]. Unlike the lf2, lf4, lf5, and cnk2 mutants, both cnk11-1 and tpg1 mutants have normal flagellar length (Figs 1A and 6B and [29]), flagellar assembly (Fig 8A and [29]), and flagellar disassembly (Fig 8C). One difference between cnk11-1 and tpg1 mutants is that cnk11-1 shows a faster than normal tubulin turnover rate at the flagellar tip and tpg1 has a slower than normal rate (Fig 8B). The double mutant shows a turnover rate similar to tpg1 (Fig 8B) and proximal end localized polyglutamylated tubulin similar to tpg1 (Fig 7B & 7C). It is unlikely that TPG1 and TPG2 are the direct targets of CNK11, given that they are not found in the flagellar phosphoproteome [80].
In conclusion, we found that CCDC39 and CCDC40 mutants that have short flagella and fail to assemble the N-DRC and several inner dynein arms. Post-translational modification such as polyglutamylation and phosphorylation can affect flagellar length via IFT-independent and structural protein-independent pathways. These modification, may function similarly to the microtubule stabilizing drug paclitaxel and stabilize the unstable axonemal microtubules found in short flagella mutants. Further analysis of other short flagella mutants, such as shf1, shf2, and shf3 [84], or pf21 [85], are likely to identify more genes involved in flagellar assembly and length control.
Materials and Methods
Strains and culture conditions
The pf7 and pf8 mutant strains were obtained from the Chlamydomonas Resource Center as CC - 568 and CC-560. The strains from the stock center were aflagellate. Different media conditions were tried, but less than 20% of the cells assembled flagella. Both mutants were backcrossed to CC-124 three successive times to determine if reassorting the genome would increase the fraction of flagellated cells. After three backcrosses, strain pf8 2–4 was chosen. After 4 hours in nitrogen-free HSM medium, greater than 10% of cells had ~7 μm flagella. These were used for additional matings and for flagella preparations. After two backcrosses, strain pf7 2–2 was chosen. After 4 hours nitrogen-free medium, greater than 80% of cells had short (<4μm) flagella. Other strains and culture conditions are as reported previously [86]. Treatment of cells with paclitaxel was performed in yellow Lucite boxes to prevent breakdown of the paclitaxel by white light [64, 87].
Whole genome sequencing
Chlamydomonas genomic DNA preparation for whole genome sequencing was prepared as described previously [86]. Three micrograms of DNA was submitted to Genome Technology Access Core (Washington University School of Medicine) for library construction, Illumina sequencing, and initial data analysis. For multiplex Illumina sequencing, 7-nucleotide indexes were added to individual DNAs during the library construction before the samples were subjected to sequencing.
Illumina whole genome sequencing reads were aligned onto the Chlamydomonas version 5.3.1 genome assembly, and then aligned to JGI predicted exomes ([42] for details). SAMTools [88] were used for calling of SNPs/short indels from each strain. The SNPs/short indels from individual strains were compared to a SNP/short indel library (http://stormo.wustl.edu/dgranas/form.php) generated from 16 previously sequenced strains (15 in the cases of pf7 and pf8 because they were included as two of 16 original strains used to construct the library) [20, 42]. The unique SNPs/short indels in each strain were analyzed and filtered by SnpEff [89]. Only changes that have Phred quality scores of over 100 and rest within the coding region and splicing sites were retained. Whole genome sequencing reads of pf7 and pf8 can be found under NCBI BioProject Accession Number PRJNA245202. Whole genome sequencing reads of fla12, pf7; pf8; cnk11-1, pf7; pf8; cnk11-2, pf7; pf8; cnk11-3, pf7; pf8; cnk11-4, and pf7; pf8; cnk11-5 can be found under NCBI BioProject Accession Number PRJNA293107.
Revertant analysis
Revertant analysis was performed as previously reported [86]. Most of the mutant cells fail to oppose gravity and fall to the bottom of the tube. Swimming cells rise to the top of the tube and the upper 10 mL was transferred to fresh medium five times over the course of 13 days. Cultures were plated for individual colonies and one colony with swimming cells was kept from each tube. For the isolation of suppressors of the pf7; pf8 strains, we failed to recover any swimming cells. However, the nature of the pellet changed following the rounds of enrichment. Instead of large clumps of cells, the pellets were smooth and there were single cells. This was used to identify the suppressors.
Flagellar length measurement
Two day-old cells were resuspended in nitrogen-free medium for 4 hours before treated with freshly made autolysin and fixed in cold methanol. Cells were stained with anti-acetylated α-tubulin antibody followed by Alexa 594-conjugated goat anti-mouse secondary antibody. ImageJ was used to measure the flagellar length.
Axonemal isolation and immunoblots
Protocols are as described previously [20]. Antibodies used in this study are listed in S4 Table.
Axoneme reactivation
Cells were deflagellated by pH shock and the isolated flagella were resuspended in demembranating buffer as described [10]. Half of the resultant axonemes were treated with 0.1 mM ATP at room temperature for 4 minutes. Both ATP-treated and non-treated axonemes were fixed with 2% paraformaldehyde at room temperature for 10 minutes on poly-lysine-coated multi-well slides (Thermo Scientific). The slide was then immersed in cold methanol for 10 minutes at -20°C. The samples were allowed to air dry on the slide before the addition of blocking buffer (5% BSA, 1% fish gelatin). The primary antibodies used were LF5 (1 : 200 dilution) and acetylated α-tubulin (1 : 250 dilution) diluted in 20% blocking buffer. The secondary antibodies were Alexa 488-conjugated goat-anti-rabbit IgG (1 : 500 dilution) and Alexa 594-conjugated goat-anti-mouse IgG (1 : 500 dilution) diluted in 20% blocking buffer.
TIRF microscopy for IFT velocity measurements
Cells were imaged on manufacturer pre-cleaned fused silica chips (6W675-575 20C, Hoya Corporation USA, San Jose, CA), and sandwiched between the fused silica surface and a coverslip (1.8 x 1.8 cm2), resulting in a 25 μm thick water layer that allowed the 10 μm diameter Chlamydomonas cell body to move freely in solution. We used total internal reflection fluorescence (TIRF) microscopy to image the cells. The details of the imaging methods were reported previously [90]. Videos of individual surface-attached flagella were processed into kymographs. For visible IFT tracks in a kymograph, a minimum of 3 consecutive and clearly distinguishable IFT20::GFP intensity profiles were required for a track to be used. For each selected IFT track, the slope of the line through the centroid of the first and last IFT20::GFP intensity profiles in the track was used to determine the IFT velocity.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. Zariwala MA, Omran H, Ferkol TW: The emerging genetics of primary ciliary dyskinesia. Proceedings of the American Thoracic Society 2011, 8 : 430–433.
2. Olbrich H, Haffner K, Kispert A, Volkel A, Volz A, Sasmaz G, Reinhardt R, Hennig S, Lehrach H, Konietzko N, et al: Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nature genetics 2002, 30 : 143–144.
3. Zariwala MA, Leigh MW, Ceppa F, Kennedy MP, Noone PG, Carson JL, Hazucha MJ, Lori A, Horvath J, Olbrich H, et al: Mutations of DNAI1 in primary ciliary dyskinesia: evidence of founder effect in a common mutation. American journal of respiratory and critical care medicine 2006, 174 : 858–866.
4. Mitchison HM, Schmidts M, Loges NT, Freshour J, Dritsoula A, Hirst RA, O'Callaghan C, Blau H, Al Dabbagh M, Olbrich H, et al: Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nature genetics 2012, 44 : 381–389, S381–382.
5. Diggle CP, Moore DJ, Mali G, Zur Lage P, Ait-Lounis A, Schmidts M, Shoemark A, Garcia Munoz A, Halachev MR, Gautier P, et al: HEATR2 Plays a Conserved Role in Assembly of the Ciliary Motile Apparatus. PLoS genetics 2014, 10:e1004577.
6. Horani A, Druley TE, Zariwala MA, Patel AC, Levinson BT, Van Arendonk LG, Thornton KC, Giacalone JC, Albee AJ, Wilson KS, et al: Whole-Exome Capture and Sequencing Identifies HEATR2 Mutation as a Cause of Primary Ciliary Dyskinesia. American journal of human genetics 2012, 91 : 685–693.
7. Gao C, Wang G, Amack JD, Mitchell DR: Oda16/Wdr69 is essential for axonemal dynein assembly and ciliary motility during zebrafish embryogenesis. Developmental dynamics: an official publication of the American Association of Anatomists 2010, 239 : 2190–2197.
8. Ahmed NT, Gao C, Lucker BF, Cole DG, Mitchell DR: ODA16 aids axonemal outer row dynein assembly through an interaction with the intraflagellar transport machinery. J Cell Biol 2008, 183 : 313–322.
9. Wirschell M, Olbrich H, Werner C, Tritschler D, Bower R, Sale WS, Loges NT, Pennekamp P, Lindberg S, Stenram U, et al: The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nature genetics 2013, 45 : 262–268.
10. Bower R, Tritschler D, Vanderwaal K, Perrone CA, Mueller J, Fox L, Sale WS, Porter ME: The N-DRC forms a conserved biochemical complex that maintains outer doublet alignment and limits microtubule sliding in motile axonemes. Molecular biology of the cell 2013, 24 : 1134–1152.
11. Antony D, Becker-Heck A, Zariwala MA, Schmidts M, Onoufriadis A, Forouhan M, Wilson R, Taylor-Cox T, Dewar A, Jackson C, et al: Mutations in CCDC39 and CCDC40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms. Human mutation 2013, 34 : 462–472.
12. Blanchon S, Legendre M, Copin B, Duquesnoy P, Montantin G, Kott E, Dastot F, Jeanson L, Cachanado M, Rousseau A, et al: Delineation of CCDC39/CCDC40 mutation spectrum and associated phenotypes in primary ciliary dyskinesia. Journal of medical genetics 2012, 49 : 410–416.
13. Nakhleh N, Francis R, Giese RA, Tian X, Li Y, Zariwala MA, Yagi H, Khalifa O, Kureshi S, Chatterjee B, et al: High prevalence of respiratory ciliary dysfunction in congenital heart disease patients with heterotaxy. Circulation 2012, 125 : 2232–2242.
14. Merveille AC, Davis EE, Becker-Heck A, Legendre M, Amirav I, Bataille G, Belmont J, Beydon N, Billen F, Clement A, et al: CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nature genetics 2011, 43 : 72–78.
15. Becker-Heck A, Zohn IE, Okabe N, Pollock A, Lenhart KB, Sullivan-Brown J, McSheene J, Loges NT, Olbrich H, Haeffner K, et al: The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nature genetics 2011, 43 : 79–84.
16. Oda T, Yanagisawa H, Kamiya R, Kikkawa M: Cilia and flagella. A molecular ruler determines the repeat length in eukaryotic cilia and flagella. Science 2014, 346 : 857–860.
17. McVittie A: Flagellum mutants of Chlamydomonas reinhardii. J Gen Microbiol 1972, 71 : 525–540.
18. Davis SD, Ferkol TW, Rosenfeld M, Lee H-S, Dell SD, Sagel SD, Milla C, Zariwala MA, Pittman JE, Shapiro AJ, et al: Clinical Features of Childhood Primary Ciliary Dyskinesia by Genotype and Ultrastructural Phenotype. American journal of respiratory and critical care medicine 2015, 191 : 316–324.
19. Marshall WF, Qin H, Rodrigo Brenni M, Rosenbaum JL: Flagellar length control system: testing a simple model based on intraflagellar transport and turnover. Molecular biology of the cell 2005, 16 : 270–278.
20. Lin H, Nauman NP, Albee AJ, Hsu S, Dutcher SK: New mutations in flagellar motors identified from whole genome sequencing in Chlamydomonas reinhardtii. Cilia 2013, 2 : 14.
21. Iomini C, Li L, Esparza JM, Dutcher SK: Retrograde intraflagellar transport mutants identify complex A proteins with multiple genetic interactions in Chlamydomonas reinhardtii. Genetics 2009, 183 : 885–896.
22. Wang L, Piao T, Cao M, Qin T, Huang L, Deng H, Mao T, Pan J: Flagellar regeneration requires cytoplasmic microtubule depolymerization and kinesin-13. Journal of cell science 2013, 126 : 1531–1540.
23. Kannegaard E, Rego EH, Schuck S, Feldman JL, Marshall WF: Quantitative analysis and modeling of katanin function in flagellar length control. Molecular biology of the cell 2014, 25 : 3686–3698.
24. King SJ, Dutcher SK: Phosphoregulation of an inner dynein arm complex in Chlamydomonas reinhardtii is altered in phototactic mutant strains. J Cell Biol 1997, 136 : 177–191.
25. Piperno G, Ramanis Z, Smith EF, Sale WS: Three distinct inner dynein arms in Chlamydomonas flagella: molecular composition and location in the axoneme. J Cell Biol 1990, 110 : 379–389.
26. LeDizet M, Piperno G: The light chain p28 associates with a subset of inner dynein arm heavy chains in Chlamydomonas axonemes. Molecular biology of the cell 1995, 6 : 697–711.
27. Kubo T, Hirono M, Aikawa T, Kamiya R, Witman GB: Reduced tubulin polyglutamylation suppresses flagellar shortness in Chlamydomonas. Molecular biology of the cell 2015, 26 : 2810–2822.
28. Kubo T, Yanagisawa HA, Liu Z, Shibuya R, Hirono M, Kamiya R: A conserved flagella-associated protein in Chlamydomonas, FAP234, is essential for axonemal localization of tubulin polyglutamylase TTLL9. Molecular biology of the cell 2014, 25 : 107–117.
29. Kubo T, Yanagisawa H-a, Yagi T, Hirono M, Kamiya R: Tubulin Polyglutamylation Regulates Axonemal Motility by Modulating Activities of Inner-Arm Dyneins. Current Biology 2010, 20 : 441–445.
30. Song Y, Brady ST: Post-translational modifications of tubulin: pathways to functional diversity of microtubules. Trends in Cell Biology 2015, 25 : 125–136.
31. Janke C, Chloë Bulinski J: Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol 2011, 12 : 773–786.
32. Wloga D, Gaertig J: Post-translational modifications of microtubules. Journal of cell science 2010, 123 : 3447–3455.
33. Parker JD, Bradley BA, Mooers AO, Quarmby LM: Phylogenetic analysis of the Neks reveals early diversification of ciliary-cell cycle kinases. PLoS One 2007, 2:e1076.
34. O'Regan L, Blot J, Fry A: Mitotic regulation by NIMA-related kinases. Cell Division 2007, 2 : 25.
35. Mahjoub MR, Qasim Rasi M, Quarmby LM: A NIMA-related kinase, Fa2p, localizes to a novel site in the proximal cilia of Chlamydomonas and mouse kidney cells. Molecular biology of the cell 2004, 15 : 5172–5186.
36. Hilton LK, Gunawardane K, Kim JW, Schwarz MC, Quarmby LM: The kinases LF4 and CNK2 control ciliary length by feedback regulation of assembly and disassembly rates. Curr Biol 2013, 23 : 1–7.
37. Doles J, Hemann MT: Nek4 status differentially alters sensitivity to distinct microtubule poisons. Cancer research 2010, 70 : 1033–1041.
38. Motose H, Takatani S, Ikeda T, Takahashi T: NIMA-related kinases regulate directional cell growth and organ development through microtubule function in Arabidopsis thaliana. Plant Signaling & Behavior 2012, 7 : 1552–1555.
39. McVittie A: Studies on flagella-less, stumpy, and short flagellum mutants of Chlamydomonas reinhardii. Proceedings of the Royal Society of London Series B, Containing papers of a Biological character Royal Society 1969, 173 : 59–60.
40. Ramanis Z, Luck DJ: Loci affecting flagellar assembly and function map to an unusual linkage group in Chlamydomonas reinhardtii. Proc Natl Acad Sci U S A 1986, 83 : 423–426.
41. Dutcher SK: Genetic properties of linkage group XIX in Chlamydomonas reinhardtii. Basic Life Sci 1986, 40 : 303–325.
42. Lin H, Miller ML, Granas DM, Dutcher SK: Whole Genome Sequencing Identifies a Deletion in Protein Phosphatase 2A That Affects Its Stability and Localization in Chlamydomonas reinhardtii. PLoS genetics 2013, 9:e1003841.
43. Adams GM, Huang B, Luck DJ: Temperature-sensitive, assembly-defective flagella mutants of Chlamydomonas reinhardtii. Genetics 1982, 100 : 579–586.
44. Huang B, Ramanis Z, Luck DJ: Suppressor mutations in Chlamydomonas reveal a regulatory mechanism for Flagellar function. Cell 1982, 28 : 115–124.
45. Porter ME, Power J, Dutcher SK: Extragenic suppressors of paralyzed flagellar mutations in Chlamydomonas reinhardtii identify loci that alter the inner dynein arms. J Cell Biol 1992, 118 : 1163–1176.
46. Yanagisawa HA, Kamiya R: A tektin homologue is decreased in chlamydomonas mutants lacking an axonemal inner-arm dynein. Molecular biology of the cell 2004, 15 : 2105–2115.
47. Yagi T, Uematsu K, Liu Z, Kamiya R: Identification of dyneins that localize exclusively to the proximal portion of Chlamydomonas flagella. Journal of cell science 2009, 122 : 1306–1314.
48. Yang P, Sale WS: The Mr 140,000 intermediate chain of Chlamydomonas flagellar inner arm dynein is a WD-repeat protein implicated in dynein arm anchoring. Molecular biology of the cell 1998, 9 : 3335–3349.
49. Perrone CA, Yang P, O'Toole E, Sale WS, Porter ME: The Chlamydomonas IDA7 locus encodes a 140-kDa dynein intermediate chain required to assemble the I1 inner arm complex. Molecular biology of the cell 1998, 9 : 3351–3365.
50. King SM, Kamiya R: Axonemal Dyneins: Assembly, strucure and force generation. In The Chlamydomonas Sourcebook, Second Edition Volume 3: Cell Motility and Behavior. Edited by Witman GB. Amsterdam: Elsevier; 2009 : 131–208
51. Salisbury JL, Baron AT, Sanders MA: The centrin-based cytoskeleton of Chlamydomonas reinhardtii: distribution in interphase and mitotic cells. J Cell Biol 1988, 107 : 635–641.
52. Yang C, Compton MM, Yang P: Dimeric novel HSP40 is incorporated into the radial spoke complex during the assembly process in flagella. Molecular biology of the cell 2005, 16 : 637–648.
53. Mitchell DR, Kang Y: Identification of oda6 as a Chlamydomonas dynein mutant by rescue with the wild-type gene. J Cell Biol 1991, 113 : 835–842.
54. King SM, Otter T, Witman GB: Characterization of monoclonal antibodies against Chlamydomonas flagellar dyneins by high-resolution protein blotting. Proceedings of the National Academy of Sciences 1985, 82 : 4717–4721.
55. Linck RW, Norrander JM: Protofilament ribbon compartments of ciliary and flagellar microtubules. Protist 2003, 154 : 299–311.
56. Norrander JM, deCathelineau AM, Brown JA, Porter ME, Linck RW: The Rib43a protein is associated with forming the specialized protofilament ribbons of flagellar microtubules in Chlamydomonas. Molecular biology of the cell 2000, 11 : 201–215.
57. Tam LW, Ranum PT, Lefebvre PA: CDKL5 regulates flagellar length and localizes to the base of the flagella in Chlamydomonas. Molecular biology of the cell 2013, 24 : 588–600.
58. Iomini C, Babaev-Khaimov V, Sassaroli M, Piperno G: Protein particles in Chlamydomonas flagella undergo a transport cycle consisting of four phases. J Cell Biol 2001, 153 : 13–24.
59. Wren KN, Craft JM, Tritschler D, Schauer A, Patel DK, Smith EF, Porter ME, Kner P, Lechtreck KF: A differential cargo-loading model of ciliary length regulation by IFT. Curr Biol 2013, 23 : 2463–2471.
60. Dymek EE, Lefebvre PA, Smith EF: PF15p is the chlamydomonas homologue of the Katanin p80 subunit and is required for assembly of flagellar central microtubules. Eukaryot Cell 2004, 3 : 870–879.
61. Wirschell M, Olbrich H, Werner C, Tritschler D, Bower R, Sale WS, Loges NT, Pennekamp P, Lindberg S, Stenram U, et al: The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nature genetics 2013, 45 : 262–268.
62. Huang B, Piperno G, Luck DJ: Paralyzed flagella mutants of Chlamydomonas reinhardtii. Defective for axonemal doublet microtubule arms. Journal of Biological Chemistry 1979, 254 : 3091–3099.
63. Kamiya R: Exploring the function of inner and outer dynein arms with Chlamydomonas mutants. Cell Motility and the Cytoskeleton 1995, 32 : 98–102.
64. Esparza JM, O'Toole E, Li L, Giddings TH Jr., Kozak B, Albee AJ, Dutcher SK: Katanin localization requires triplet microtubules in Chlamydomonas reinhardtii. PLoS One 2013, 8:e53940.
65. Audebert S, Desbruyères E, Gruszczynski C, Koulakoff A, Gros F, Denoulet P, Eddé B: Reversible polyglutamylation of alpha - and beta-tubulin and microtubule dynamics in mouse brain neurons. Molecular biology of the cell 1993, 4 : 615–626.
66. Avasthi P, Marshall WF: Stages of ciliogenesis and regulation of ciliary length. Differentiation 2012, 83:S30–S42.
67. Silflow CD, Lefebvre PA: Assembly and Motility of Eukaryotic Cilia and Flagella. Lessons from Chlamydomonas reinhardtii. Plant Physiology 2001, 127 : 1500–1507.
68. Wilson NF, Iyer JK, Buchheim JA, Meek W: Regulation of flagellar length in Chlamydomonas. Seminars in Cell & Developmental Biology 2008, 19 : 494–501.
69. Cao M, Li G, Pan J: Chapter 17—Regulation of Cilia assembly, Disassembly, and Length by Protein Phosphorylation. In Methods in Cell Biology. Volume Volume 94. Edited by Roger DS: Academic Press; 2009 : 333–346
70. Randall J, STARLING D: Chapter 3—Genetic determinants of flagellum phenotype in Chlamydomonas reinhardii. In The Genetics of Algae. Volume 12. Edited by Lewin RA: University of California Press; 1976 : 49–62
71. Bui KH, Yagi T, Yamamoto R, Kamiya R, Ishikawa T: Polarity and asymmetry in the arrangement of dynein and related structures in the Chlamydomonas axoneme. The Journal of Cell Biology 2012, 198 : 913–925.
72. Kubo T, Yagi T, Kamiya R: Tubulin polyglutamylation regulates flagellar motility by controlling a specific inner-arm dynein that interacts with the dynein regulatory complex. Cytoskeleton 2012, 69 : 1059–1068.
73. Suryavanshi S, Eddé B, Fox LA, Guerrero S, Hard R, Hennessey T, Kabi A, Malison D, Pennock D, Sale WS, et al: Tubulin Glutamylation Regulates Ciliary Motility by Altering Inner Dynein Arm Activity. Current Biology 2010, 20 : 435–440.
74. Ikegami K, Sato S, Nakamura K, Ostrowski LE, Setou M: Tubulin polyglutamylation is essential for airway ciliary function through the regulation of beating asymmetry. Proceedings of the National Academy of Sciences 2010, 107 : 10490–10495.
75. Wloga D, Dave D, Meagley J, Rogowski K, Jerka-Dziadosz M, Gaertig J: Hyperglutamylation of tubulin can either stabilize or destabilize microtubules in the same cell. Eukaryot Cell 2010, 9 : 184–193.
76. Yu I, Garnham CP, Roll-Mecak A: Writing and Reading the Tubulin Code. Journal of Biological Chemistry 2015, 290 : 17163–17172.
77. Lacroix B, van Dijk J, Gold ND, Guizetti J, Aldrian-Herrada G, Rogowski K, Gerlich DW, Janke C: Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. The Journal of Cell Biology 2010, 189 : 945–954.
78. LeDizet M, Piperno G: Cytoplasmic microtubules containing acetylated alpha-tubulin in Chlamydomonas reinhardtii: spatial arrangement and properties. The Journal of Cell Biology 1986, 103 : 13–22.
79. Pazour GJ, Agrin N, Leszyk J, Witman GB: Proteomic analysis of a eukaryotic cilium. The Journal of Cell Biology 2005, 170 : 103–113.
80. Wang H, Gau B, Slade WO, Juergens M, Li P, Hicks LM: The Global Phosphoproteome of Chlamydomonas reinhardtii Reveals Complex Organellar Phosphorylation in the Flagella and Thylakoid Membrane. Molecular & Cellular Proteomics 2014, 13 : 2337–2353.
81. Hu Z, Liang Y, He W, Pan J: Cilia Disassembly with Two Distinct Phases of Regulation. Cell Reports 2015, 10 : 1803–1810.
82. Tam L - W, Wilson NF, Lefebvre PA: A CDK-related kinase regulates the length and assembly of flagella in Chlamydomonas. The Journal of Cell Biology 2007, 176 : 819–829.
83. Berman SA, Wilson NF, Haas NA, Lefebvre PA: A Novel MAP Kinase Regulates Flagellar Length in Chlamydomonas. Current Biology 2003, 13 : 1145–1149.
84. Kuchka MR, Jarvik JW: Short-Flagella Mutants of Chlamydomonas reinhardtii. Genetics 1987, 115 : 685–691.
85. McVittie A: Genetic studies on flagellum mutants of Chlamydomonas reinhardii. Genetics Research 1972, 19 : 157–164.
86. Dutcher SK, Li L, Lin H, Meyer L, Giddings TH Jr., Kwan AL, Lewis BL: Whole-Genome Sequencing to Identify Mutants and Polymorphisms in Chlamydomonas reinhardtii. G3 2012, 2 : 15–22.
87. Palombella AL, Dutcher SK: Identification of the gene encoding the tryptophan synthase beta-subunit from Chlamydomonas reinhardtii. Plant Physiol 1998, 117 : 455–464.
88. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S: The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25 : 2078–2079.
89. Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM: A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6 : 80–92.
90. DeSantis MC, DeCenzo SH, Li JL, Wang YM: Precision analysis for standard deviation measurements of immobile single fluorescent molecule images. Optics express 2010, 18 : 6563–6576.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 9
Nejčtenější v tomto čísle
- Arabidopsis AtPLC2 Is a Primary Phosphoinositide-Specific Phospholipase C in Phosphoinositide Metabolism and the Endoplasmic Reticulum Stress Response
- Bridges Meristem and Organ Primordia Boundaries through , , and during Flower Development in
- KLK5 Inactivation Reverses Cutaneous Hallmarks of Netherton Syndrome
- XBP1-Independent UPR Pathways Suppress C/EBP-β Mediated Chondrocyte Differentiation in ER-Stress Related Skeletal Disease