
IL-1β Processing in Host Defense: Beyond the Inflammasomes
Stimulation and release of proinflammatory cytokines is an essential step for the activation of an effective innate host defense, and subsequently for the modulation of adaptive immune responses. Interleukin-1β (IL-1β) and IL-18 are important proinflammatory cytokines that on the one hand activate monocytes, macropages, and neutrophils, and on the other hand induce Th1 and Th17 adaptive cellular responses. They are secreted as inactive precursors, and the processing of pro-IL-1β and pro-IL-18 depends on cleavage by proteases. One of the most important of these enzymes is caspase-1, which in turn is activated by several protein platforms called the inflammasomes. Inflammasome activation differs in various cell types, and knock-out mice defective in either caspase-1 or inflammasome components have an increased susceptibility to several types of infections. However, in other infections and in models of sterile inflammation, caspase-1 seems to be less important, and alternative mechanisms such as neutrophil-derived serine proteases or proteases released from microbial pathogens can process and activate IL-1β. In conclusion, IL-1β/IL-18 processing during infection is a complex process in which the inflammasomes are only one of several activation mechanisms.
Published in the journal:
. PLoS Pathog 6(2): e32767. doi:10.1371/journal.ppat.1000661
Category:
Review
doi:
https://doi.org/10.1371/journal.ppat.1000661
Summary
Stimulation and release of proinflammatory cytokines is an essential step for the activation of an effective innate host defense, and subsequently for the modulation of adaptive immune responses. Interleukin-1β (IL-1β) and IL-18 are important proinflammatory cytokines that on the one hand activate monocytes, macropages, and neutrophils, and on the other hand induce Th1 and Th17 adaptive cellular responses. They are secreted as inactive precursors, and the processing of pro-IL-1β and pro-IL-18 depends on cleavage by proteases. One of the most important of these enzymes is caspase-1, which in turn is activated by several protein platforms called the inflammasomes. Inflammasome activation differs in various cell types, and knock-out mice defective in either caspase-1 or inflammasome components have an increased susceptibility to several types of infections. However, in other infections and in models of sterile inflammation, caspase-1 seems to be less important, and alternative mechanisms such as neutrophil-derived serine proteases or proteases released from microbial pathogens can process and activate IL-1β. In conclusion, IL-1β/IL-18 processing during infection is a complex process in which the inflammasomes are only one of several activation mechanisms.
The Role of IL-1β and IL-18 in Host Defense
The main cellular innate host defense mechanisms are the phagocytosis and killing of bacteria and fungi by neutrophilic granulocytes, monocytes, and macrophages [1],[2], and the lysis of viral-infected cells by natural killer (NK) cells [3]. Upon recognition of a microorganism, proinflammatory cytokines such as tumor necrosis factor (TNF), interferon-γ (IFNγ), interleukin (IL)-18, and IL-1β are secreted. These cytokines activate neutrophils and macrophages to phagocytose the invading pathogen and to release toxic oxygen and nitrogen radicals. TNF is an essential component of the host defense, as demonstrated by the important infectious complications in patients treated with anti-TNF biological agents [4]. Similarly, IFNγ activates both neutrophils and macrophages for intracellular killing of bacteria or fungi. Patients with defects in the IL-12/IFNγ activation pathways are at increased risk of severe mycobacterial and Salmonella infections [5], and recombinant IFNγ is an established therapy in patients with chronic granulomatous disease [6]. However, in addition to TNF and IFNγ, the proinflammatory cytokines of the IL-1 family, most notably IL-1β and IL-18, also have very important roles for antimicrobial host defense. IL-1α and IL-1β, which bind and activate the same receptor [7], activate the release of other proinflammatory cytokines such as TNF and IL-6, and induce a Th17 bias in the cellular adaptive responses [8]. In vivo, IL-1 is largely responsible for the acute phase response, which includes fever, acute protein synthesis, anorexia, and somnolence [7], while IL-18 is essential for the induction of IFNγ and Th1 responses [9]. Through these mechanisms, cytokines of the IL-1 family are a crucial component of the host defense against infections.
IL-1β and IL-18 Processing and Release: The Inflammasomes
Much interest has been generated regarding the processing and release of bioactive IL-1β since the discovery of an entire group of disorders called autoinflammatory syndromes that specifically respond to the blockade of the IL-1 receptor with the IL-1 receptor antagonist (IL-1Ra), or with neutralization of IL-1β by the monoclonal anti-IL-1β antibodies. These syndromes are characterized by attacks of sterile inflammation of joints, serositis, fever, and skin lesions. Some of the best known diseases in this group include familial Mediterranean fever (FMF) [10], cryopyrin-associated periodic syndromes (also known as cryopyrinopathies, which include familial cold auto-inflammatory syndrome [FCAS] [11], Muckle-Wells syndrome [MWS] [12], and neonatal onset multisystem inflammatory disease [NOMID] [13]), hyperimmunoglobulin D syndrome (HIDS) [14], TNF receptor–associated periodic syndrome (TRAPS), and adult-onset Still's disease [15]. Blood monocytes from patients with some of these disorders, especially cryopyrinopathies, readily release more IL-1β than monocytes from unaffected controls, revealing a loss of the tight control that regulates the processing and release of active IL-1β. An abnormal production of IL-1β has been therefore proposed to be the underlying cause of these diseases.
Several mechanisms control the production and activity of IL-1β, including the processing of the 31-kDa inactive IL-1β precursor form into the bioactive 17-kDa IL-1β [16], and the release from secretory lysosomes through K+-dependent mechanisms [17],[18]. In addition, control over IL-1 activity is exerted by the IL-1 receptor antagonist (IL-1Ra) or the type II decoy receptors [19]. Processing of bioactive IL-1β (and that of IL-18) depends on activation of caspase-1 by protein complexes termed the inflammasomes [20]. Several protein platforms/inflammasomes have been described for the activation of caspase-1, and each of them include members of the NOD-like receptor (NLR) family of proteins [21]. Through CARD–CARD and pyrin domain–pyrin domain interactions, a large macromolecular complex is formed to represent a scaffold for the recruitment and activation of pro-caspase-1. It is believed, yet not proven, that caspase-1 activation in the inflammasome is induced by the formation of oligomers and proximity between caspase-1 molecules.
Several major inflammasome complexes that activate caspase-1 have been described to date. The most intensely studied has been the inflammasome formed by the NLR family member NLRP3, which forms complexes that include the adapter protein ASC for the activation of caspase-1 (Figure 1A). Mutations in NLRP3 have been described in the cryopyrin-associated periodic syndromes (CAPS; cryopyrin is a name previously used for NLRP3), whereas specific NLRP-3 polymorphisms have been associated with Crohn's disease [22]. A large number of stimuli have been described to activate the NLRP3 inflammasome: some of them of bacterial origin (muramyl dipeptide [MDP], bacterial RNA, double-stranded RNA), some of them are danger-associated molecular patterns (uric acid crystals, amyloid-β), but also exogenous compounds such as asbestos, silica, or alum adjuvant [23]–[27]. The precise mechanism leading to the activation of the NLRP3 is still unclear. The diverse molecular structure of these compounds most likely precludes the direct stimulation of the NLRP3 inflammasome. A unifying hypothesis proposes that common intracellular activities such as induction of hypokalemia, reactive oxygen species, or calcium-dependent phospholipase 2 are indirectly activating the inflammasome [28]. However, stimulation of cells solely with ATP, a known inducer of potassium efflux through P2X7-mediated mechanisms, is unable to activate caspase-1, and cell priming with lipopolysaccharide (LPS) is necessary for ATP to induce inflammasome activation. In this context, induction of NF-kB-dependent transcription of NLRP3 by Toll-like receptor (TLR) ligands [29] or proinflammatory cytokines [30] seems to be the critical checkpoint needed for cell priming prior to inflammasome activation by ATP. In addition, formation of pores by pannexin-1 is one mechanism through which microbial products (e.g., MDP) can be delivered into the cytoplasm to activate the inflammasome [31].

The only inflammasome that has been reconstituted biochemically is the NLRP1 inflammasome. A study using purified NLRP1, ASC, and caspase-1 has shown that NLPR1 forms oligomers in the presence of MDP [32]. However, no evidence has been presented that MDP can actually bind NLRP1, although another study suggested the involvement of NOD2/NLRP1 complexes in this process [33]. NLRP-1 polymorphisms have been associated with vitiligo and autoimmune diseases [34].
In addition to the NLRPs, another NLR member, NLRC4/IPAF, forms an inflammasome that activates caspase-1 in response to intracellular flagellin in an ASC-independent manner [35],[36]. Caspase-1 activation by flagellin/NLRC4 is independent of TLR5, suggesting that flagellin recognition is mediated by two systems: extracellular sensing by TLR5, and intracytoplasmatic sensing by NLRC4 (Figure 1B). Finally, a newly described mechanism involving recognition of bacterial DNA by the intracellular sensor AIM2 suggests the existence of a specific inflammasome complex that induces caspase-1 activation upon sensing nucleic acids [37],[38]. This is a particularly important finding, as intracellular detection of DNA from invading pathogens is likely to be central for an effective host defense. AIM2-dependent activation of IL-1β has been suggested to be critical for the activation of host defense against vaccinia virus and Francisella tularensis [37], and it is to be expected that similar effects will be identified in the future for other pathogens.
Through these studies on the structure and function of the various inflammasomes, a dogma has emerged during the last few years in which production and release of IL-1β and IL-18 is the result of two independent signals: one signal is induced through pattern recognition receptors ( e.g., TLRs) to activate transcription of pro-IL-1β and pro-IL-18, and one signal is mediated by the NLR-containing inflammasomes (and independent of the TLRs) that induce cleavage of cytokine precursors into the active IL-1β and IL-18 forms through caspase-1 activation.
Differential Role of the Inflammasome in Monocytes and Macrophages
Despite the progress made in understanding the process of IL-1β synthesis, controversy surrounded the capacity of TLR ligands such as LPS to activate caspase-1 and cause the release of active IL-1β. By using transfected cell lines and/or NLRP3 knock-out mice, a broad panel of exogenous and endogenous stimuli have been proposed to activate the NLRP3 inflammasome (see above), but purified TLR ligands such as LPS were not among these inflammasome stimuli. Therefore, based on defective responses of the monocyte-like leukemia cell line THP-1 to LPS stimulation, a concept has arisen that IL-1β production induced by LPS is due to contamination with non-LPS ligands such as peptidoglycans [23], while LPS by itself is ineffective as a stimulator of IL-1β release. A second signal, such as MDP or ATP, is required, and this would induce activation of caspase-1 followed by IL-1β processing and release [39]. However, this model is derived from data in THP-1 cells [23] and in primary mouse macrophages [31], and it is inconsistent with many studies showing abundant production and release of IL-1β from blood monocytes by TLR ligands such as purified LPS, lipopeptides, and lipoteichoic acid, as well as cytokines such as TNFα and IL-1 itself [40],[41]. In addition, several studies reported that synthetic products, which exclude contamination with NLRP1 or NLRP3 ligands, stimulate IL-1β release [42],[43].
These apparent discrepancies have been resolved by a study from our group showing that synthesis and release of IL-1β differ between human monocytes and macrophages. Monocytes have constitutively activated caspase-1, leading to release of active IL-1β after a single stimulation event with bacterial ligands such as LPS, whereas macrophages (and THP-1 cells) need two distinct stimuli: one stimulus induces transcription and translation, and a second stimulus is needed for caspase-1 activation with subsequent IL-1β processing and secretion [44] (Figure 2). Although caspase-1 is constitutively activated in human monocytes, that is still dependent on inflammasome components, as the inhibition of ASC by siRNA results in a significant reduction of both caspase-1 activation and processing of IL-1β [44]. A crucial functional aspect in relation to the constitutive inflammasome activation in monocytes relates to the release of endogenous ATP by monocytes. Endogenous ATP from monocytes can in turn activate the NLRP3 inflammasome and induce IL-1β secretion through P2X7. In contrast, macrophages completely lack the capacity to release ATP [45].

Consistent with the failure of in vitro–differentiated macrophages to release IL-1β is the long known defect in IL-1β synthesis of the alveolar macrophages. Wewers and colleagues proposed a post-transcriptional defect in freshly obtained alveolar macrophages [46]. Recently, they reported differences in pyrin expression between monocytes and macrophages, and suggested that pyrin induces IL-1β release [47]. Monocytes from patients with FMF who have mutations in pyrin release more IL-1β upon stimulation than cells from control subjects, suggesting a failure to suppress the activation of caspase-1 [10].
These data imply a paradigm shift in our understanding of the inflammasome. The demonstration of a role for ASC and NLRP3 in the constitutive activation of caspase-1, independent of stimulation by TLRs or inflammasome ligands, uncouples caspase-1 activation from pathogen-associated molecular pattern (PAMP) recognition in human primary monocytes. This new model, in which the inflammasome components ASC and NLRP3 form a protein platform responsible for the constitutive activation of caspase-1, explains why IL-1β induction in monocytes by a very diverse panel of stimuli (including TLR ligands) is caspase-1 dependent, although these stimuli need not themselves be involved in inflammasome activation. In addition, a role of ASC and NLRP3 in caspase-1 activation in the monocyte, independently of “classical” inflammasome stimuli, explains the resistance to experimental endotoxemia in ASC−/ − and NLRP-3−/ − mice [48],[49]. In contrast, macrophages need two signals in order to produce IL-1β, in a model close to the current concept in the literature: one signal is mediated by TLRs to induce gene transcription, and a second signal to induce inflammasome activation for the processing of IL-1β.
The single (TLR ligand only) stimulation in monocytes compared with the double (TLR ligand/ATP) stimulation in macrophages (Figure 2) likely represents an adaptation of each cell type to their respective environments. Circulating monocytes function in the surveillance of an essentially pathogen-free environment, so they must respond promptly to any danger signal (especially of microbial origin). On the other hand, macrophages are confined to an environment (e.g., alveolar space, mucosal surfaces) in which they are constantly exposed to (colonizing) microbial stimuli. An easily inducible response of macrophages to release IL-1β for each encounter with such exogenous stimuli would result in chronic deleterious inflammatory reactions. Thus, repeated bouts of inflammation are likely reduced by the requirement of a second stimulus for the activation of the inflammasome and release of active IL-1β. Such second stimuli would be available at sites of infection, trauma, or necrosis where ATP levels are elevated and can trigger the P2X7 receptor [50]. In addition, second signals can come from the cathelicidin-derived peptide LL37 from infiltrating neutrophils [51], or the release of bacterial toxins [25]. One situation in which caspase-1 activation seems to be constitutively activated also in macrophages is represented by the absence of the autophagy gene ATG16L1, although the precise mechanism responsible for this effect is not yet elucidated [52]. However, the association of ATG16L1 polymorphisms with Crohn's disease makes this aspect potentially important for the pathophysiology of this important disease [53],[54].
The Role of Caspase-1 and the Inflammasome in Antimicrobial Host Defense
Due to their role in the processing of IL-1β and IL-18, caspase-1 and the inflammasome components are bound to have important roles in the host defense against pathogenic microorganisms. In vitro studies have shown that Bacillus anthracis toxin activates IL-1β through the Nalp1b inflammasome in the mouse [55]. Similarly, NLRP3 activation of caspase-1 has been linked to a variety of microorganisms ranging from the bacteria Listeria monocytogenes [56],[57] and Staphylococcus aureus [58] to viruses such as influenza, adenoviruses, and Sendai virus [59]. The interaction of NLRC4 with microorganisms was probably one of the best characterized, demonstrating the involvement of NLRC4 (independently of ASC) in the activation of caspase-1 by Salmonella typhimurium [35],[36],[48], Legionella pneumophila [60], Pseudomonas aeruginosa [61],[62], and Shigella flexneri [63].
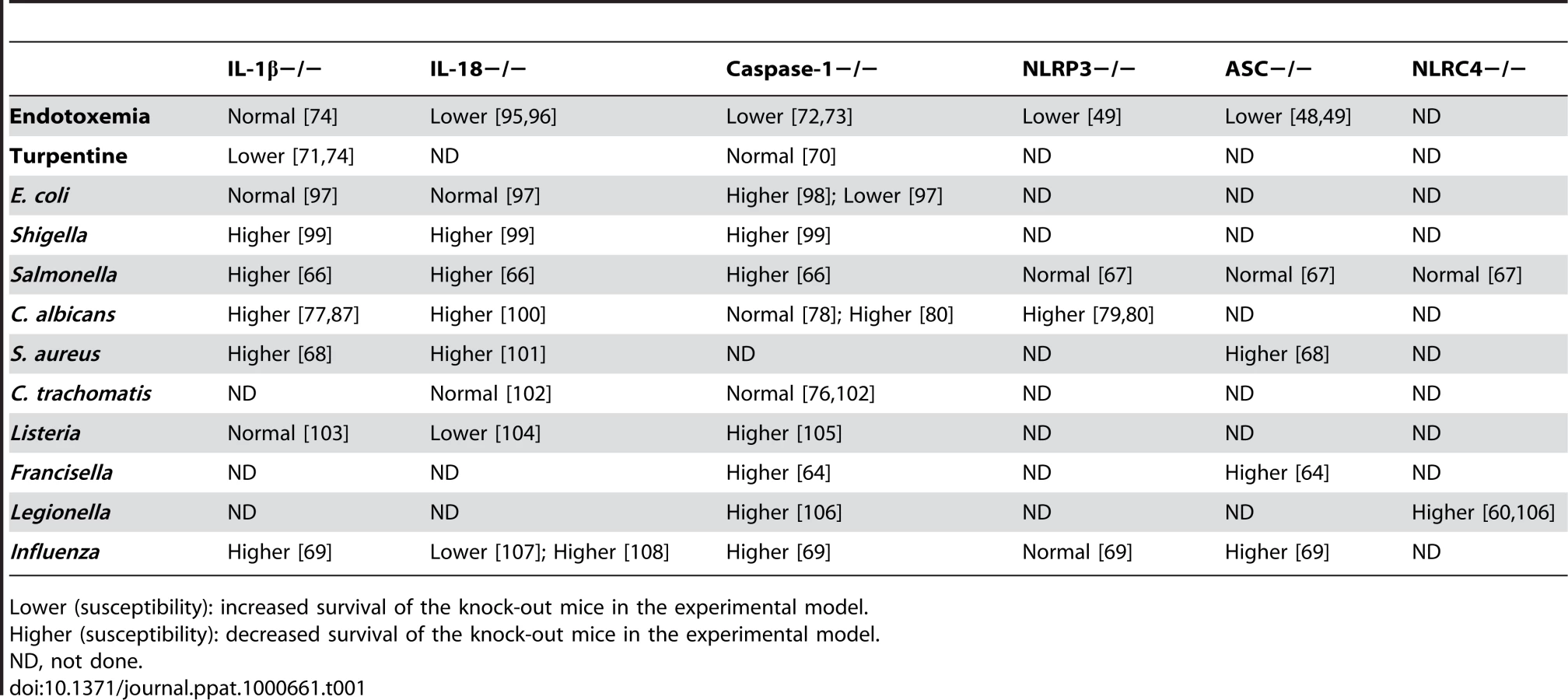
In vivo experimental models of infection have also demonstrated that the lack of caspase-1 in knock-out mice leads to an increased susceptibility to a variety of infections, including those with F. tularensis [64], L. pneumophila [65], Shigella [63], Salmonella [66],[67], and P. aeruginosa [62] (Table 1). What all of these infections have in common is that caspase-1 activity, and thus IL-1β production, is dependent on the assembly of an inflammasome complex [20], although they may differ in their specific inflammasome components. Experimental infections with some of these pathogens have been also investigated in knock-out mice lacking components of the inflammasome. In this respect, ASC-deficient mice have been shown to be more susceptible to infections with some bacteria (Francisella and Staphylococcus) [64],[68], as well as to influenza viruses [69], demonstrating its importance in host defense mechanisms. As in some models ASC−/ − mice are clearly more susceptible to infections than NLRP3−/ − mice (e.g., influenza [69]), one may suggest a more important role for ASC in antimicrobial defense. Alternatively, the partial redundancy between different NLRs may explain the more pronounced phenotype of ASC knock-out mice compared to single NLR-deficient mice.
Inflammasome-Independent IL-1β Activation
Despite the importance of inflammasome activation in certain experimental models of inflammation, certain in vivo infection models in mice deficient in inflammasome components show intriguing results that question the importance of the inflammasome (Table 1). One category of results shows that although IL-1β is definitely important for inflammatory reactions in antimicrobial defense, components of the inflammasome or even caspase-1 can be dispensable. In models of sterile inflammation induced by turpentine, IL-1β−/ − mice are protected against inflammation, while caspase-1−/ − mice are not [70],[71]. This is in clear contrast with LPS models in which caspase-1−/ − mice are protected (Table 1) [72],[73]. It appears therefore that caspase-1 and inflammasome activation is important in some, but not all, types of IL-1β-driven inflammation [74]. Furthermore, caspase-1 seems not to be involved in the host defense against certain types of microorganisms such as Chlamydia trachomatis [75],[76], although IL-1β is involved in the inflammatory responses induced by these microorganisms [77]. These data argue for inflammasome-independent activation of IL-1β in certain infectious processes.
An interesting case regarding the involvement of the inflammasome in host defense is represented by the fungal pathogen Candida albicans. IL-1 plays an important role for the host defense and survival of mice during disseminated candidiasis [75],[85]. Surprisingly, caspase-1-deficient mice have been reported to have a normal resistance to disseminated candidiasis [78], suggesting activation of IL-1β by alternative mechanisms (see below). However, NLRP3−/ − and ASC−/ − mice have been reported to be more susceptible to both systemic [79] and mucosal [80] Candida infections, opening the intriguing possibility of biological functions of inflammasome components that are not related to caspase-1 activation. Indeed, an earlier study on the function of ASC has reported its interaction with NF-κB and an influence on gene transcription [81]. Whether ASC or NLRP3 have underestimated roles that are independent of inflammasome activation remains to be studied. These studies have shown a role of inflammasome components in experimental models of Candida infection in mice, and in line with this the activation of IL-1β in human monocytes is dependent on caspase-1. However, in contrast to mouse macrophages, caspase-1 is constitutively activated in human monocytes and thus does not need fungal recognition by the NLRs in the inflammasome [82].
Alternative Mechanisms of IL-1β Processing
Shortly after the discovery of IL-1β, when it was apparent that cleavage of the pro-cytokine is needed for activation, a question arose as to whether other enzymes apart from caspase-1 would also be capable of processing pro-IL-1β. Indeed, subsequent studies have identified neutrophil - and macrophage-derived serine proteases such as proteinase-3 (PR3), elastase, and cathepsin-G as enzymes that can process pro-IL-1β into 21-kDa active fragments [7],[83]. Similarly, processing of pro-IL-18 by PR3 can also lead to active fragments [84]. The crucial role played by neutrophil-dependent, inflammasome-independent activation of pro-IL-1β has been elegantly confirmed recently by the group of Greten and colleagues [85] (Figure 3).

The inflammasome-independent activation of pro-IL-1β in situations when neutrophils are the major cell population in the inflammatory infiltrate can explain many of the apparently puzzling observations reviewed above. It is the neutrophils that form the backbone of the inflammatory infiltrates during disseminated candidiasis [86], and this explains the dependency of host defense against Candida on IL-1β [87], most likely activated by neutrophil-derived PR3, rather than caspase-1 [78]. Similarly, inflammatory infiltrates during arthritis consist of both macrophages and neutrophils. Indeed, we have observed a minimal role of caspase-1 during the acute inflammation of arthritis that is characterized by an overwhelming neutrophil infiltrate. In this phase of the inflammation, serine proteases such as PR3 played a more important role [88]. In contrast, during the chronic phase of inflammation when macrophages are the main component of the infiltrate, caspase-1 seems to have a more significant effect [88].
Neutrophil-derived serine proteases are not the only alternative mechanism of activation of pro-IL-1β. One very interesting phenomenon has recently been reported during infection with C. albicans, in which a fungus-derived protease can lead to processing and activation of host-derived pro-IL-1β and thus activation of the immune system [89]. This may represent an indirect pathway of Candida recognition by the innate immune system (Figure 3), reminiscent of the detection of the fungus Beauveria bassiana in Drosophila through maturation of the protease Persephone by a fungal-derived enzyme, leading to Toll activation [90]. Considering the production of a vast array of proteases by practically all pathogenic microorganisms, it is to be expected that similar IL-1β activation pathways are active during other infections.
Conclusions and Perspectives: IL-1β Processing beyond the Inflammasomes
A wealth of information regarding the mechanisms of pathogen recognition and activation of innate immunity has been accumulated during the last few years, and has contributed greatly to a better understanding of the host defense against pathogenic microorganisms. One of the most exciting areas of advancement was represented by the description of the inflammasomes and the mechanisms that lead to the processing and activation of cytokines of the IL-1 family.
There is no doubt that these discoveries have contributed to a better understanding of inflammatory processes. Beautifully designed studies have also shown that caspase-1-dependent mechanisms of IL-1β and IL-18 activation have important consequences during inflammation. However, as this review has tried to bring to light, the role played by the inflammasomes should not deter the acknowledgement of other mechanisms leading to IL-1β processing that may be just as important.
As shown above, inflammasome activation is not the same in all cell types, and caspase-1 activation is not the only mechanism leading to the processing of pro-IL-1β into active fragments. Neutrophil-derived serine proteases and pathogen-released enzymes can also process and activate IL-1β, and these processes have important effects during inflammation and infection. Further characterization of these alternative mechanisms can lead to the design of improved therapeutic strategies. In this respect, any inflammatory condition in which neutrophils are involved (e.g., rheumatoid arthritis, Crohn's disease, or gout) is unlikely to respond to caspase-1 inhibition alone, and a combination of caspase-1 and PR3 inactivation may be necessary. Beneficial therapeutic effects of the IL-1 receptor antagonist (IL-1Ra, anakinra) have often been presented as proof-of-principle for a role of the inflammasome in certain clinical conditions such as gout [91],[92], in which neutrophils play a crucial role [93]. This assumption is clearly too preliminary: IL-1Ra will block the effects of IL-1β irrespective of the mechanisms that led to its activation, apart from the fact that IL-1α effects are also blocked.
A similar generalization based on IL-1Ra effects is currently the dogma regarding the pathophysiology of autoinflammatory syndromes: many of the autoinflammatory syndromes are considered defects of inflammasome activation. While this is likely the case for some diseases, for example CAPS, in which NLRP3 mutations are the cause of the disease [94], this relation is much less clear in other autoinflammatory disorders. It is better to consider the disorders in which anakinra is active as “IL-1Ra responsive diseases” or perhaps “IL-1 related diseases” instead of immediately considering them “inflammasome-related diseases”. While caspase-1 is an obvious candidate to be investigated, neglecting to investigate other IL-1β-activating mechanisms (or IL-1α secretion) would be an oversight.
To conclude, the description of the inflammasomes has taught us a lot, but we should not fall in the trap of our own success. Caspase-1 activation is just one of the mechanisms of activation of one of the two active IL-1 molecules, and it is unlikely that an entire class of PAMP recognition receptors (the NLR receptors) have evolved only to be devoted to a single immunological function. While acknowledging the importance of the inflammasomes for our understanding of inflammatory reactions, we should consider in our endeavors the alternative pathways of IL-1β/IL-18 activation, and also actively examine alternative roles of the NLR class of receptors.
Zdroje
1. MarodiL
KorchakHM
JohnstonRBJr
1991 Mechanisms of host defense against Candida species. 1. Phagocytosis by monocytes and monocyte-derived macrophages. J Immunol 146 2783 2789
2. TaylorPR
Martinez-PomaresL
StaceyM
LinHH
BrownGD
2005 Macrophage receptors and immune recognition. Annu Rev Immunol 23 901 944
3. OrangeJS
BallasZK
2006 Natural killer cells in human health and disease. Clin Immunol 118 1 10
4. LinJ
ZiringD
DesaiS
KimS
WongM
2008 TNFalpha blockade in human diseases: an overview of efficacy and safety. Clin Immunol 126 13 30
5. OttenhoffTH
VerreckFA
Lichtenauer-KaligisEG
HoeveMA
SanalO
2002 Genetics, cytokines and human infectious disease: lessons from weakly pathogenic mycobacteria and salmonellae. Nat Genet 32 97 105
6. GallinJI
FarberJM
HollandSM
NutmanTB
1995 Interferon-gamma in the management of infectious diseases. Ann Int Med 123 216 222
7. DinarelloCA
1996 Biologic basis for interleukin-1 in disease. Blood 87 2095 2147
8. ChungY
ChangSH
MartinezGJ
YangXO
NurievaR
2009 Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30 576 587
9. DinarelloCA
1999 IL-18: A Th1-inducing, proinflammatory cytokine and a new member of the IL-1 family. J Allergy Clin Immunol 103 11 24
10. ChaeJJ
WoodG
MastersSL
RichardK
ParkG
2006 The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci U S A 103 9982 9987
11. HoffmanHM
RosengrenS
BoyleDL
ChoJY
NayarJ
2004 Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet 364 1779 1785
12. HawkinsPN
LachmannHJ
AgannaE
McDermottMF
2004 Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum 50 607 612
13. AksentijevichI
NowakM
MallahM
ChaeJJ
WatfordWT
2002 De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 46 3340 3348
14. van der MeerJW
VossenJM
RadlJ
van NieuwkoopJA
MeyerCJ
1984 Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet 1 1087 1090
15. FitzgeraldAA
LeclercqSA
YanA
HomikJE
DinarelloCA
2005 Rapid responses to anakinra in patients with refractory adult-onset Still's disease. Arthritis Rheum 52 1794 1803
16. WilsonKP
BlackJA
ThomsonJA
KimEE
GriffithJP
1994 Structure and mechanism of interleukin-1 beta converting enzyme. Nature 370 270 275
17. AndreiC
MargioccoP
PoggiA
LottiLV
TorrisiMR
2004 Phospholipases C and A2 control lysosome-mediated IL-1 beta secretion: Implications for inflammatory processes. Proc Natl Acad Sci U S A 101 9745 9750
18. PerregauxD
GabelCA
1994 Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem 269 15195 15203
19. ColottaF
ReF
MuzioM
BertiniR
PolentaruttiN
1993 Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science 261 472 475
20. MartinonF
BurnsK
TschoppJ
2002 The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10 417 426
21. MartinonF
TschoppJ
2004 Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 117 561 574
22. VillaniAC
LemireM
FortinG
LouisE
SilverbergMS
2009 Common variants in the NLRP3 region contribute to Crohn's disease susceptibility. Nat Genet 41 71 76
23. MartinonF
AgostiniL
MeylanE
TschoppJ
2004 Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol 14 1929 1934
24. KannegantiTD
OzorenN
Body-MalapelM
AmerA
ParkJH
2006 Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440 233 236
25. MariathasanS
WeissDS
NewtonK
McBrideJ
O'RourkeK
2006 Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440 228 232
26. EisenbarthSC
ColegioOR
O'ConnorW
SutterwalaFS
FlavellRA
2008 Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453 1122 1126
27. DostertC
PetrilliV
Van BruggenR
SteeleC
MossmanBT
2008 Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320 674 677
28. PetrilliV
PapinS
DostertC
MayorA
MartinonF
2007 Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14 1583 1589
29. BauernfeindFG
HorvathG
StutzA
AlnemriES
MacDonaldK
2009 Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183 787 791
30. FranchiL
EigenbrodT
NunezG
2009 Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol 183 792 796
31. KannegantiTD
LamkanfiM
KimYG
ChenG
ParkJH
2007 Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 26 433 443
32. FaustinB
LartigueL
BrueyJM
LucianoF
SergienkoE
2007 Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell 25 713 724
33. HsuLC
AliSR
McGillivrayS
TsengPH
MariathasanS
2008 A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci U S A 105 7803 7808
34. JinY
MaillouxCM
GowanK
RiccardiSL
LaBergeG
2007 NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med 356 1216 1225
35. FranchiL
AmerA
Body-MalapelM
KannegantiTD
OzorenN
2006 Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol 7 576 582
36. MiaoEA
Alpuche-ArandaCM
DorsM
ClarkAE
BaderMW
2006 Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol 7 569 575
37. HornungV
AblasserA
Charrel-DennisM
BauernfeindF
HorvathG
2009 AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458 514 518
38. Fernandes-AlnemriT
YuJW
DattaP
WuJ
AlnemriES
2009 AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458 509 513
39. MartinonF
TschoppJ
2005 NLRs join TLRs as innate sensors of pathogens. Trends Immunol 26 447 454
40. DinarelloCA
CannonJG
WolffSM
1986 Tumor necrosis factor (cachectin) is an endogenous pyrogen and induces production of interleukin-1. J Exp Med 163 1433 1450
41. DinarelloCA
IkejimaT
WarnerSJC
OrencoleSF
LonnemannG
1987 Interleukin 1 induces interleukin 1. I. Induction of interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol 139 1902 1910
42. HurmeM
SeppalaIJ
1988 Differential induction of membrane-associated interleukin 1 (IL-1) expression and IL-1 alpha and IL-1 beta secretion by lipopolysaccharide and silica in human monocytes. Scand J Immunol 27 725 730
43. MillerKM
AndersonJM
1988 Human monocyte/macrophage activation and interleukin 1 generation by biomedical polymers. J Biomed Mater Res 22 713 731
44. NeteaMG
Nold-PetryCA
NoldMF
JoostenLA
OpitzB
2009 Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113 2324 2335
45. FerrariD
ChiozziP
FalzoniS
HanauS
Di VirgilioF
1997 Purinergic modulation of interleukin-1 beta release from microglial cells stimulated with bacterial endotoxin. J Exp Med 185 579 582
46. WewersMD
DareHA
WinnardAV
ParkerJM
MillerDK
1997 IL-1 beta-converting enzyme (ICE) is present and functional in human alveolar macrophages: macrophage IL-1 beta release limitation is ICE independent. J Immunol 159 5964 5972
47. SeshadriS
DuncanMD
HartJM
GavrilinMA
WewersMD
2007 Pyrin levels in human monocytes and monocyte-derived macrophages regulate IL-1beta processing and release. J Immunol 179 1274 1281
48. MariathasanS
NewtonK
MonackDM
VucicD
FrenchDM
2004 Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430 213 218
49. SutterwalaFS
OguraY
SzczepanikM
Lara-TejeroM
LichtenbergerGS
2006 Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24 317 327
50. FerrariD
PizziraniC
AdinolfiE
LemoliRM
CurtiA
2006 The P2X7 receptor: a key player in IL-1 processing and release. J Immunol 176 3877 3883
51. ElssnerA
DuncanM
GavrilinM
WewersMD
2004 A novel P2X7 receptor activator, the human cathelicidin-derived peptide LL37, induces IL-1 beta processing and release. J Immunol 172 4987 4994
52. SaitohT
FujitaN
JangMH
UematsuS
YangBG
2008 Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456 264 268
53. HampeJ
FrankeA
RosenstielP
TillA
TeuberM
2007 A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 39 207 211
54. RiouxJD
XavierRJ
TaylorKD
SilverbergMS
GoyetteP
2007 Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 39 596 604
55. BoydenED
DietrichWF
2006 Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 38 240 244
56. OzorenN
MasumotoJ
FranchiL
KannegantiTD
Body-MalapelM
2006 Distinct roles of TLR2 and the adaptor ASC in IL-1beta/IL-18 secretion in response to Listeria monocytogenes. J Immunol 176 4337 4342
57. WarrenSE
MaoDP
RodriguezAE
MiaoEA
AderemA
2008 Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J Immunol 180 7558 7564
58. FranchiL
KannegantiTD
DubyakGR
NunezG
2007 Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem 282 18810 18818
59. KannegantiTD
Body-MalapelM
AmerA
ParkJH
WhitfieldJ
2006 Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem 281 36560 36568
60. AmerA
FranchiL
KannegantiTD
Body-MalapelM
OzorenN
2006 Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem 281 35217 35223
61. FranchiL
StoolmanJ
KannegantiTD
VermaA
RamphalR
2007 Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur J Immunol 37 3030 3039
62. SutterwalaFS
MijaresLA
LiL
OguraY
KazmierczakBI
2007 Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med 204 3235 3245
63. SuzukiT
FranchiL
TomaC
AshidaH
OgawaM
2007 Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog 3 e111 doi:10.1371/journal.ppat.0030111
64. MariathasanS
WeissDS
DixitVM
MonackDM
2005 Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med 202 1043 1049
65. RenT
ZamboniDS
RoyCR
DietrichWF
VanceRE
2006 Flagellin-deficient Legionella mutants evade caspase-1 - and Naip5-mediated macrophage immunity. PLoS Pathog 2 e18 doi:10.1371/journal.ppat.0020018
66. RaupachB
PeuschelSK
MonackDM
ZychlinskyA
2006 Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect Immun 74 4922 4926
67. Lara-TejeroM
SutterwalaFS
OguraY
GrantEP
BertinJ
2006 Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J Exp Med 203 1407 1412
68. MillerLS
PietrasEM
UricchioLH
HiranoK
RaoS
2007 Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J Immunol 179 6933 6942
69. IchinoheT
LeeHK
OguraY
FlavellR
IwasakiA
2009 Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med 206 79 87
70. FantuzziG
KuG
HardingMW
LivingstonDL
SipeJD
1997 Response to local inflammation of IL-1beta converting enzyme-deficient mice. J Immunol 158 1818 1824
71. LabowM
ShusterD
ZetterstromM
NunesP
TerryR
1997 Absence of IL-1 signaling and reduced inflammatory response in IL-1 type I receptor-deficient mice. J Immunol 159 2452 2461
72. KuidaK
LippkeJA
KuG
HardingMW
LivingstonDJ
1995 Altered cytokine export and apoptosis in mice deficient in interleukin-β converting enzyme. Science 267 2000 2003
73. LiP
AllenH
BanerjeeS
FranklinS
HerzogL
1995 Mice deficient in IL-1β-converting enzyme are defective in production of nature IL-1-beta and resistant to endotoxic shock. Cell 80 401 411
74. FantuzziG
DinarelloCA
1996 The inflammatory response in interleukin-1beta-deficient mice: comparison with other cytokine-related knock-out mice. J Leuk Biol 59 489 493
75. LuH
ShenC
BrunhamRC
2000 Chlamydia trachomatis infection of epithelial cells induces the activation of caspase-1 and release of mature IL-18. J Immunol 165 1463 1469
76. ChengW
ShivshankarP
LiZ
ChenL
YehIT
2008 Caspase-1 contributes to Chlamydia trachomatis-induced upper urogenital tract inflammatory pathologies without affecting the course of infection. Infect Immun 76 515 522
77. BellocchioS
MontagnoliC
BozzaS
GazianoR
RossiG
2004 The contribution of Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol 172 3059 3069
78. MencacciA
BacciA
CenciE
MontagnoliC
FiorucciS
2000 Interleukin 18 restores defective Th1 immunity to Candida albicans in Caspase 1-deficient mice. Infect Immun 68 5126 5131
79. GrossO
PoeckH
BscheiderM
DostertC
HannesschlagerN
2009 Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature
80. HiseAG
TomalkaJ
GanesanS
PatelK
HallBA
2009 An Essential Role for the NLRP3 Inflammasome in Host Defense against the Human Fungal Pathogen Candida albicans. Cell Host Microbe 5 487 497
81. HasegawaM
ImamuraR
KinoshitaT
MatsumotoN
MasumotoJ
2005 ASC-mediated NF-kappaB activation leading to interleukin-8 production requires caspase-8 and is inhibited by CLARP. J Biol Chem 280 15122 15130
82. van de VeerdonkFL
JoostenLA
DevesaI
Mora-MontesHM
KannegantiTD
2009 Bypassing pathogen-induced inflammasome activation for the regulation of interleukin-1beta production by the fungal pathogen Candida albicans. J Infect Dis 199 1087 1096
83. CoeshottC
OhnemusC
PilyavskayaA
RossS
WieczorekM
1999 Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci U S A 96 6261 6266
84. SugawaraS
UeharaA
NochiT
YamaguchiT
UedaH
2001 Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J Immunol 167 6568 6575
85. GretenFR
ArkanMC
BollrathJ
HsuLC
GoodeJ
2007 NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 130 918 931
86. KullbergBJ
Van 't WoutJW
Van FurthR
1990 Role of granulocytes in enhanced host resistance to Candida albicans induced by recombinant interleukin-1. Infect Immun 58 3319 3324
87. VonkAG
NeteaMG
van KriekenJH
IwakuraY
van der MeerJW
2006 Endogenous interleukin (IL)-1 alpha and IL-1 beta are crucial for host defense against disseminated candidiasis. J Infect Dis 193 1419 1426
88. JoostenL
NeteaM
FantuzziG
KoendersM
HelsenM
2009 Inflammatory arthritis in caspase-1 gene deficient mice: crucial role of proteinase 3 for caspase-1-independent production of bioactive IL-1beta. Arthritis Rheum in press
89. BeausejourA
GrenierD
GouletJP
DeslauriersN
1998 Proteolytic activation of the interleukin-1beta precursor by Candida albicans. Infect Immun 66 676 681
90. GottarM
GobertV
MatskevichAA
ReichhartJM
WangC
2006 Dual detection of fungal infections in Drosophila via recognition of glucans and sensing of virulence factors. Cell 127 1425 1437
91. SoA
De SmedtT
RevazS
TschoppJ
2007 A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther 9 R28
92. PopeRM
TschoppJ
2007 The role of interleukin-1 and the inflammasome in gout: implications for therapy. Arthritis Rheum 56 3183 3188
93. Popa-NitaO
Rollet-LabelleE
ThibaultN
GilbertC
BourgoinSG
2007 Crystal-induced neutrophil activation. IX. Syk-dependent activation of class Ia phosphatidylinositol 3-kinase. J Leukoc Biol 82 763 773
94. HoffmanHM
MuellerJL
BroideDH
WandererAA
KolodnerRD
2001 Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 29 301 305
95. NeteaMG
FantuzziG
KullbergBJ
StuytR
PulidoEJ
2000 Neutralization of interleukin-18 reduces neutrophil tissue accumulation and protects mice against lethal Escherichia coli and Salmonella typhimurium endotoxemia. J Immunol 164 2644 2649
96. HochholzerP
LipfordGB
WagnerH
PfefferK
HeegK
2000 Role of interleukin-18 (IL-18) during lethal shock: decreased lipopolysaccharide sensitivity but normal superantigen reaction in IL-18-deficient mice. Infect Immun 68 3502 3508
97. SarkarA
HallMW
ExlineM
HartJ
KnatzN
2006 Caspase-1 regulates Escherichia coli sepsis and splenic B cell apoptosis independently of interleukin-1beta and interleukin-18. Am J Respir Crit Care Med 174 1003 1010
98. JoshiVD
KalvakolanuDV
HebelJR
HasdayJD
CrossAS
2002 Role of caspase 1 in murine antibacterial host defenses and lethal endotoxemia. Infect Immun 70 6896 6903
99. SansonettiPJ
PhaliponA
ArondelJ
ThirumalaiK
BanerjeeS
2000 Caspase-1 activation of IL-1beta and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity 12 581 590
100. StuytRJ
NeteaMG
VerschuerenI
FantuzziG
DinarelloCA
2002 Role of interleukin-18 in host defense against disseminated Candida albicans infection. Infect Immun 70 3284 3286
101. WeiX-q
LeungBP
NiedbalaW
PiedrafitaD
FengG-j
1999 Altered immune responses and susceptibility to Leishmania major and Staphylococcus aureus infection in IL-18-deficient mice. J Immunol 163 2821 2828
102. LuH
YangX
TakedaK
ZhangD
FanY
2000 Chlamydia trachomatis mouse pneumonitis lung infection in IL-18 and IL-12 knockout mice: IL-12 is dominant over IL-18 for protective immunity. Mol Med 6 604 612
103. ZhengH
FletcherD
KozakW
JiangM
HofmannK
1995 Resistance to fever induction and impaired acute-phase response in interleukin-1β deficient mice. Immunity 3 9 17
104. LochnerM
KastenmullerK
NeuenhahnM
WeighardtH
BuschDH
2008 Decreased susceptibility of mice to infection with Listeria monocytogenes in the absence of interleukin-18. Infect Immun 76 3881 3890
105. TsujiNM
TsutsuiH
SekiE
KuidaK
OkamuraH
2004 Roles of caspase-1 in Listeria infection in mice. Int Immunol 16 335 343
106. CaseCL
ShinS
RoyCR
2009 Asc and Ipaf Inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect Immun 77 1981 1991
107. Van Der SluijsKF
Van EldenLJ
ArensR
NijhuisM
SchuurmanR
2005 Enhanced viral clearance in interleukin-18 gene-deficient mice after pulmonary infection with influenza A virus. Immunology 114 112 120
108. LiuB
MoriI
HossainMJ
DongL
TakedaK
2004 Interleukin-18 improves the early defence system against influenza virus infection by augmenting natural killer cell-mediated cytotoxicity. J Gen Virol 85 423 428
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 2
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Caspase-1 Activation via Rho GTPases: A Common Theme in Mucosal Infections?
- Kaposi's Sarcoma Associated Herpes Virus (KSHV) Induced COX-2: A Key Factor in Latency, Inflammation, Angiogenesis, Cell Survival and Invasion
- IL-1β Processing in Host Defense: Beyond the Inflammasomes
- Reverse Genetics in Predicts ARF Cycling Is Essential for Drug Resistance and Virulence