
Deletion of AIF Ortholog Promotes Chromosome Aneuploidy and Fluconazole-Resistance in a Metacaspase-Independent Manner
Apoptosis is a form of programmed cell death critical for development and homeostasis in multicellular organisms. Apoptosis-like cell death (ALCD) has been described in several fungi, including the opportunistic human pathogen Cryptococcus neoformans. In addition, capsular polysaccharides of C. neoformans are known to induce apoptosis in host immune cells, thereby contributing to its virulence. Our goals were to characterize the apoptotic signaling cascade in C. neoformans as well as its unique features compared to the host machinery to exploit the endogenous fungal apoptotic pathways as a novel antifungal strategy in the future. The dissection of apoptotic pathways revealed that apoptosis-inducing factor (Aif1) and metacaspases (Mca1 and Mca2) are independently required for ALCD in C. neoformans. We show that the apoptotic pathways are required for cell fusion and sporulation during mating, indicating that apoptosis may occur during sexual development. Previous studies showed that antifungal drugs induce ALCD in fungi and that C. neoformans adapts to high concentrations of the antifungal fluconazole (FLC) by acquisition of aneuploidy, especially duplication of chromosome 1 (Chr1). Disruption of aif1, but not the metacaspases, stimulates the emergence of aneuploid subpopulations with Chr1 disomy that are resistant to fluconazole (FLCR) in vitro and in vivo. FLCR isolates in the aif1 background are stable in the absence of the drug, while those in the wild-type background readily revert to FLC sensitivity. We propose that apoptosis orchestrated by Aif1 might eliminate aneuploid cells from the population and defects in this pathway contribute to the selection of aneuploid FLCR subpopulations during treatment. Aneuploid clinical isolates with disomies for chromosomes other than Chr1 exhibit reduced AIF1 expression, suggesting that inactivation of Aif1 might be a novel aneuploidy-tolerating mechanism in fungi that facilitates the selection of antifungal drug resistance.
Published in the journal:
. PLoS Pathog 7(11): e32767. doi:10.1371/journal.ppat.1002364
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002364
Summary
Apoptosis is a form of programmed cell death critical for development and homeostasis in multicellular organisms. Apoptosis-like cell death (ALCD) has been described in several fungi, including the opportunistic human pathogen Cryptococcus neoformans. In addition, capsular polysaccharides of C. neoformans are known to induce apoptosis in host immune cells, thereby contributing to its virulence. Our goals were to characterize the apoptotic signaling cascade in C. neoformans as well as its unique features compared to the host machinery to exploit the endogenous fungal apoptotic pathways as a novel antifungal strategy in the future. The dissection of apoptotic pathways revealed that apoptosis-inducing factor (Aif1) and metacaspases (Mca1 and Mca2) are independently required for ALCD in C. neoformans. We show that the apoptotic pathways are required for cell fusion and sporulation during mating, indicating that apoptosis may occur during sexual development. Previous studies showed that antifungal drugs induce ALCD in fungi and that C. neoformans adapts to high concentrations of the antifungal fluconazole (FLC) by acquisition of aneuploidy, especially duplication of chromosome 1 (Chr1). Disruption of aif1, but not the metacaspases, stimulates the emergence of aneuploid subpopulations with Chr1 disomy that are resistant to fluconazole (FLCR) in vitro and in vivo. FLCR isolates in the aif1 background are stable in the absence of the drug, while those in the wild-type background readily revert to FLC sensitivity. We propose that apoptosis orchestrated by Aif1 might eliminate aneuploid cells from the population and defects in this pathway contribute to the selection of aneuploid FLCR subpopulations during treatment. Aneuploid clinical isolates with disomies for chromosomes other than Chr1 exhibit reduced AIF1 expression, suggesting that inactivation of Aif1 might be a novel aneuploidy-tolerating mechanism in fungi that facilitates the selection of antifungal drug resistance.
Introduction
Apoptosis is a form of programmed cell death critical for development and homeostasis in multicellular organisms [1]. The mechanisms of apoptosis are complex and energy-dependent. Numerous pro-and anti-apoptotic signals have to be integrated to activate apoptotic effectors only when necessary. Mitochondria play an important role in apoptosis by releasing several proteins, such as cytochrome c, which triggers the proteolytic maturation of caspases [2]. Caspases are a family of cysteine proteases that are recognized as key components of the apoptotic machinery. Caspases proteolytically cleave specific substrates, leading to the ordered dismantling of intracellular components during apoptotic death [3]. Caspase-independent apoptotic pathways involve the translocation of two nucleases, apoptosis-inducing factor (AIF) and endonuclease G (EndoG), from the mitochondria to the nucleus [4].
Features typical of apoptotic cell death, including chromatin fragmentation and condensation and translocation of phosphatidylserine to the outer layer of the cellular membrane, were described for the first time in fungi in a Saccharomyces cerevisiae cdc48 mutant [5]. Heterologous expression of mammalian pro - or anti-apoptotic Bax or Bcl2 in yeast causes or prevents apoptosis-like cell death (ALCD), respectively [6]–[8]. Subsequently, several fungal species have been reported to undergo ALCD as a consequence of interactions with other organisms and during development and aging (reviewed by [9]–[13]). The identification and functional analysis of core components of the apoptotic machinery has revealed partial conservation, along with substantial differences in function and mode of action between fungal and human proteins. Consequently, apoptotic pathways are considered suitable targets for novel antifungal therapies.
Cryptococcus neoformans var. neoformans undergoes ALCD when co-cultivated with the bacterium Staphylococcus aureus and also in response to oxidative stress induced by hydrogen peroxide [14]. Conversely, purified capsular polysaccharides from C. neoformans are known to induce apoptosis in host immune cells, including rat splenocytes [15], [16], activated human T cells [17]–[19], and murine macrophages [20]–[22]. Apoptosis induction is a strategy that C. neoformans may deploy to evade host innate immune responses and contributes to the powerful immunosuppression that accompanies cryptococcosis. Because C. neoformans both induces and undergoes apoptosis in response to different signals, its apoptotic machinery must have unique features compared to the apoptotic-signaling cascade of the host, which are potential new therapeutic targets.
C. neoformans has emerged as an important pathogen of immunocompromised and immunocompetent patients, estimated to cause 1 million new cryptococcal disease cases globally each year, with up to 600,000 fatalities yearly [23]. Cryptococcosis is a systemic mycosis that commonly involves the lungs and central nervous system. Untreated cryptococcal meningitis has a mortality rate of 100%. The recommended therapy for cryptococcal meningitis, which is intravenous amphotericin B (AMB) combined with flucytosine [24], is associated with high toxicity and serious adverse reactions in patients. Moreover, the high costs associated with AMB therapies limit its availability in resource-limited regions such as sub-Saharan Africa. Instead, a less effective agent, fluconazole (FLC), is widely used in these areas, and mortality remains high. FLC is a triazole antifungal drug that inhibits the fungal cytochrome P450 lanosterol 14α-demethylase (encoded by ERG11), thereby blocking the production of ergosterol, the main sterol of fungal membranes.
Here we studied the apoptotic pathways of C. neoformans aiming to identify unique features to specifically induce ALCD of this pathogen as a novel treatment. We found an unexpected connection between ALCD and antifungal drug resistance: the elimination of the apoptosis-inducing factor (Aif1) stabilizes aneuploidy and promotes the generation of FLC-resistance. Selection of FLCR populations with Aif1 defects occurs in vivo, and the inactivation of Aif1-mediated ALCD in C. neoformans may explain cases of FLC treatment failures in the clinical setting. In addition, apoptotic pathways are involved in sexual development and the formation of spores, which serve as infectious propagules of C. neoformans.
Results
Characterization of apoptotic pathways in Cryptococcus neoformans
ALCD has been described in several fungal species and is induced by different compounds and stress conditions (reviewed by [9], [11]–[13]). In particular, hydrogen peroxide has been reported to trigger ALCD in C. neoformans var. neoformans [14], and so we tested if similar conditions would induce cell death in C. neoformans var. grubii, which is responsible for more than 90% of cryptococcal infections worldwide [25]. Treatment of the H99 sequence reference strain with 2 mM hydrogen peroxide for 3 hours at 37oC induced characteristic apoptotic markers, such as DNA fragmentation indicated by TUNEL (TdT-mediated dUTP nick end labeling). TUNEL detects DNA fragmentation by using terminal deoxynucleotidyl transferase (TdT) to incorporate dUTP tagged with FITC into the blunt ends of double-stranded DNA breaks, and is the standard method for identification and quantification of apoptotic cells [26]. While TUNEL-FITC positive cells were only rarely observed in untreated controls (Figure 1A, upper panels), ∼45% of wild-type cells showed DNA degradation after treatment with hydrogen peroxide (Figure 1B).

We also used Annexin V-FITC to bind to phosphatidylserine (PS), a plasma membrane phospholipid that is externalized to the outer layer during apoptosis. Annexin V-FITC does not bind cells with an intact plasma membrane, but can falsely detect the PS present in the inner membrane of lysed (necrotic) cells. Therefore, simultaneous staining with the vital dye propidium iodide (PI) allows the discrimination of early apoptotic (FITC+, PI−) and late apoptotic or necrotic yeast cells (FITC+, PI+). The percentage of WT cells staining positive with Annexin V-FITC but negative with PI (apoptotic cells) rose to ∼42% after treatment with hydrogen peroxide (Figure S1A, green bars).
To find potential regulators of ALCD in C. neoformans, functional homologs of the major apoptotic machinery identified from S. cerevisiae were used to search the H99 genome database. The following homologs were identified as first reciprocal BLASTp hits: cytochrome c oxidase subunit 1 (COX1, CNAG_09009); inhibitor of apoptosis protein (IAP1, CNAG_04708), and the caspase-independent nucleases apoptosis-inducing factor (AIF1, CNAG_04521) and endonuclease G (ENDOG CNAG_02204). Two metacaspases were identified in the H99 genome using the S. cerevisiae Mca1p: MCA1 (CNAG_04636; 60% identity and 4e-103) and MCA2 (CNAG_06787, 44% identity and 1e-88). Interestingly, no apparent high-temperature resistance A homolog (HtrA2, also called Nma111p in yeast for nuclear mediator of apoptosis) was found. C. neoformans does not have apparent homologs of the apoptotic protease activating factor (APAF) and the poly(ADP-ribose) polymerase (PARP), which are present in filamentous fungi but absent in S. cerevisiae. Of note, multiple attempts to disrupt the COX1 gene were unsuccessful (Toffaletti and Perfect, unpublished observations) and it may therefore be an essential gene in C. neoformans.
We generated two independent aif1, mca1 mca2, and aif1 mca1 mca2 null mutants in congenic α and a C. neoformans var. grubii backgrounds to address whether C. neoformans has parallel caspase-dependent and caspase-independent apoptotic pathways. After treatment with 2 mM hydrogen peroxide, the aif1 mutants showed a 41% reduction, and mca1 mca2 showed a 47% reduction in TUNEL-positive cells when compared to wild-type (Figure 1B), while the aif1 mca1 mca2 triple mutant showed a reduction of 79%. In addition, all mutant strains showed reduced externalization of PS revealed by Annexin V assay (Figure S1A) and did not show increased vacuolization observed during autophagic cell death (not shown). Finally, the aif1, mca1 mca2, and aif1 mca1 mca2 null mutants were more resistant to 2 mM hydrogen peroxide than the wild-type (Figure S2). Our results indicate that Aif1 and the two metacaspases independently promote ALCD in C. neoformans.
Aif1 inactivation potentiates fluconazole-resistance and aneuploidy
Amphotericin B (AMB) and fluconazole (FLC) are antifungal drugs widely used to treat major fungal diseases. Recent treatment guidelines recommend the more toxic AMB-based regimens for induction therapy instead of the better-tolerated FLC-based regimens to treat cryptococcal meningitis, because the latter is fungistatic and because Cryptococcus strains repeatedly exposed to FLC can acquire direct drug resistance [27]. AMB was shown to induce ALCD in Aspergillus fumigatus [28] and Candida albicans in vitro [29], and in biofilms of C. albicans, Candida krusei, and Candida parapsilosis [30]. To test if antifungal drugs would induce ALCD in C. neoformans, we first determined if the apoptotic mutants have increased resistance to AMB and FLC. Using Epsilometer test strips (Etest), the aif1 and mca1 mca2 mutants showed no significant difference in sensitivity to AMB (Table 1) and we could not detect ALCD induction by treating wild-type cells with the minimal inhibitory concentration of 0.125 µg/ml of AMB (Figure S1B). On the other hand, the aif1 and mca1 mca2 mutants showed increased resistance to FLC (Table 1). Even though FLC is a fungistatic agent, we tested if this azole was able to induce ALCD in C. neoformans. We could not detect ALCD induction by treating wild-type cells with up to 64 µg/ml of FLC (Figure S1B and data not shown) and therefore, the increased resistance of the aif1 and mca1 mca2 mutants is likely not attributable simply to a lack of cell death induction.

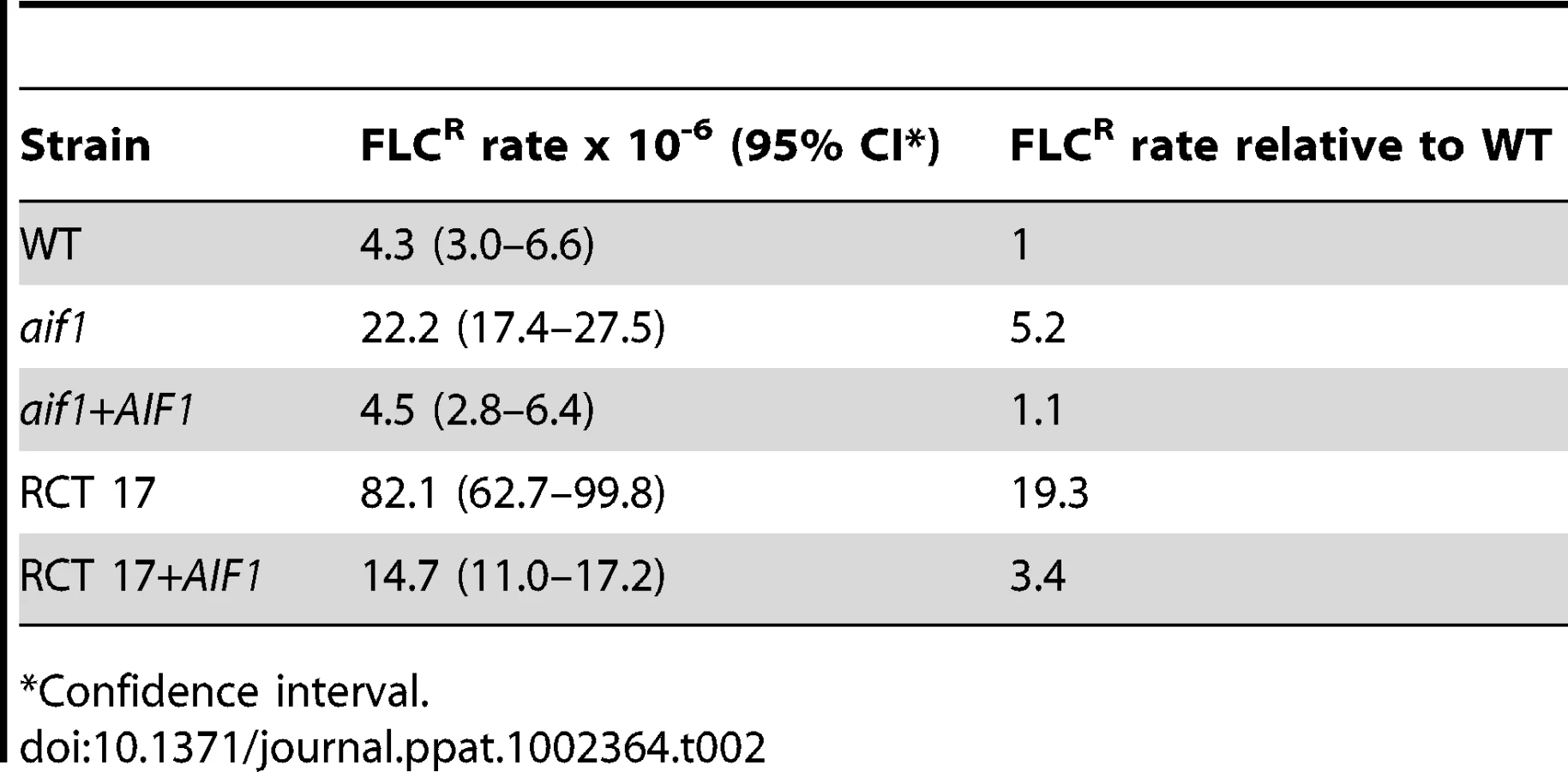
We also noticed that considerably more FLCR colonies grew within the zone of inhibition around the FLC strip in Etest assays of two independent aif1 deletion mutants, aif1::NAT and aif1::HYG (Figure 2A), a phenomenon called heteroresistance. Heteroresistance to azoles, or the presence of drug-resistant cells within a drug-sensitive population, was previously observed as an intrinsic resistance mechanism in all serotypes of C. neoformans [31]–[33] and C. gattii [34]. During these Etest experiments, resistant colonies within the zone of inhibition were observed up to 10 times more frequently in the aif1 deletion mutants (Figure 2A). Fluctuation analysis using 12 independent colonies estimated that the resistance rate to 32 µg/ml FLC is about 5 times higher in the aif1 mutant compared to H99 (Table 2). This is not due to a higher mutation rate in the aif1 strain because it showed similar 5-FOAR rates compared to the wild-type stain (5.1 and 4.5×10−8 respectively).

H99 was recently reported to be able to adapt to FLC concentrations higher than its minimal inhibitory concentration (MIC) by becoming aneuploid [35]. Chromosome 1 (Chr1) disomy was a common signature of FLCR strains, and two of the genes resident on this chromosome were shown to be important for FLC resistance: ERG11 (cytochrome P450 lanosterol 14α-demethylase), which is the target of FLC, and AFR1 (antifungal resistance 1, [36]), which is the major ATP binding cassette (ABC) transporter of azoles in C. neoformans [35]. Therefore, we determined the copy number of Chr1 in several FLCR colonies isolated from independent Etest assays (Table 3). We used a quantitative polymerase chain reaction (qPCR) assay and three probes distributed along Chr1: ERG11, ACT1, and AFR1 (Figure S3). A subset of the colonies was karyotyped by array comparative genomic hybridization (aCGH; Figure 2B-E and Figure S4). DNA content analyses by flow cytometry indicate that all strains described in Table 3 are haploid or near haploid (n+1) (data not shown).

In the wild-type H99 background, four out of eight colonies (50%) isolated from Etest halos had MICs greater than 48 µg/ml for FLC and also had Chr1 disomy (Table 3, Figure 2B). The four colonies that presented MICs less than 48 µg/ml were euploid (Table 3). Resistant colonies isolated from a second round of Etests presented higher MICs than the original four strains, and in all cases Chr1 was duplicated (Table 3, Figure 2C). Notably, all aif1 FLCR colonies (100%) isolated directly from Etest experiments became completely resistant to FLC and also had whole or partial Chr1 disomy (Table 3). From our aCGH analysis, it was clear that some FLCR colonies in the aif1 background had whole or partial chromosome duplications in addition to Chr1, which were not observed for the H99 wild-type background (Figure 2D-E, Table 3, Figure S4).
A complemented strain, in which the AIF1 gene with its native promoter and terminator was ectopically integrated into the genome, behaved like the wild-type strain in Etest assays (Figure 2A) and FLC fluctuation analysis (Table 2). Only two out of ten colonies (25%) isolated from Etest halos had MICs greater than 48 µg/ml for FLC and also had Chr1 disomy (Table 3), while the remaining colonies were euploid. In conclusion, aneuploidy and FLC resistance emerge at higher frequency in the absence of Aif1.
Aneuploid isolates are stable and do not show decreased stress resistance
FLC resistance acquired through Chr1 disomy was reported to be lost during passage in drug-free media, and the clones returned to their original euploid state [33], [35]. To test if FLCR aneuploid isolates would lose their adaptive phenotype upon removal of the selective pressure imposed by the drug, we chose to test two isolates with Chr1 disomy [n+(1)] and FLC resistance in wild-type H99, the aif1 mutant, and the aif1+AIF1 complemented strain. Upon growth in FLC-free liquid medium for about 20 generations, the isolates in the wild-type (CPS106 and CPS149) and complemented (CPS178 and CPS179) backgrounds returned to the native heteroresistant phenotype (Table 4). However, the isolates in the aif1 background (CPS18 and CPS24) did not lose the FLC resistance phenotype during nonselective growth (Table 4). These results indicate that the aneuploid FLCR cells are stabilized and tolerated in the absence of Aif1.

Aneuploid S. cerevisiae strains bearing an extra copy of various chromosomes displayed decreased resistance to stress, such as increased sensitivity to high temperatures and to protein synthesis inhibitors [37]. We tested whether the FLCR aneuploid isolates that we isolated had a similar phenotype. As shown in Figure 3A, none of the tested aneuploid strains (including isolates of different strain backgrounds and containing various chromosomal amplifications) showed increased sensitivity to high temperatures or to the protein synthesis inhibitor rapamycin. The only difference that we noticed in the aneuploid strains compared with euploid strains was the presence of elongated and larger cells (data not shown), which was also observed by Sionov et al. [35]. Because there was no difference between aneuploid strains in the AIF1 and aif1 backgrounds, we did not further investigate this phenotypic characteristic.

Increased aneuploidy in aif1 mutant is not due to a defect in mitotic checkpoint
We considered that the increased frequency of FLCR subpopulations in the aif1 mutant could be explained by two possible mechanisms. First, it is possible that Aif1-mediated ALCD would eliminate aneuploid cells from the population, and in the absence of Aif1, aneuploid cells persist within the population and emerge as FLCR isolates. Another hypothesis is that Aif1 has non-apoptotic functions related to proper chromosomal segregation.
To test if aif1 has mitotic spindle checkpoint defects, we examined its sensitivity to benomyl. In S. cerevisiae, hypersensitivity to the microtubule-destabilizing drug benomyl of “budding uninhibited by benzimidazole” (bub) mutants correlates with their mitotic checkpoint defect [38], [39]. As shown in Figure 3B, the aif1 mutant did not exhibit sensitivity to benomyl. To eliminate the possibility that benomyl is simply not taken up by C. neoformans, we deleted the gene CNAG_03184, which was the first reciprocal BLASTp hit with S. cerevisiae Bub1, in the H99 background. As expected, the bub1 mutant showed an increased sensitivity to benomyl, indicating that this drug is active in C. neoformans. In conclusion, because aif1 has an intact mitotic spindle checkpoint, we propose that Aif1-mediated ALCD eliminates aneuploid cells from the population.
Aneuploid isolates are resistant to fluconazole treatment in vivo
C. neoformans strains with FLC resistance levels greater than 32 µg/ml were previously reported to have higher virulence in mice [33]. To test if that would also be the case for isolates in the aif1 mutant background, we tested the virulence of two strains completely resistant to FLC using an inhalation murine model of cryptococcosis. Mice were intranasally infected with the indicated strains, and their survival was monitored and plotted against time. The aif1 mutant has a virulence level similar to wild-type (see below). As shown in Figure 3C, FLCR strains CPS18 [FLCR256 n+(1)] and CPS24 [FLCR256 n+dup(1)(3)] showed similar virulence to the H99 strain (untreated cohort of animals; p-values of 0.5739 and 0.5252, respectively).
Next, we tested the efficacy of antifungal therapy when infection is caused by FLCR strains. Treatment with 20 mg/kg/day of FLC was initiated 24 hours after infection and was continued for 14 days, as described by [33]. As shown in Figure S5, treatment with 20 mg/kg/day of FLC for 14 days modestly prolonged the survival of mice inoculated with wild-type H99 (p-value of 0.0043) and aif1 mutant (p-value of 0.0023) strains, but it did not prolong the survival of cohorts inoculated with the CPS18 (p-value of 0.1443) and CPS24 (p-value of 0.2237) resistant isolates. In a second set of experiments, we evaluated the efficacy of a higher FLC dosage, and animals received 100 mg/kg/day of FLC, without treatment interruption. All mice inoculated with wild-type H99 and aif1 mutant strains and treated with 100 mg/kg/day of FLC survived for 60 days (p-values <0.0001). On the other hand, while treatment with 100 mg/kg/day of FLC prolonged the survival of mice inoculated with the CPS18 and CPS24 resistant isolates (p-values <0.0001), 100% mortality was observed in these cohorts (Figure 3C).
Additionally, we examined if resistant strains would appear during FLC treatment and if aneuploidy would be stable with in vivo passage. We randomly selected three mice treated with 100 mg/kg/day of FLC for each strain. For CPS18 and CPS24 cohorts, the mice were sampled post-mortem between 30 and 35 days post-infection. For H99 and aif1 cohorts, we used asymptomatic mice sacrificed on day 60, when the experiment was terminated. Colonies recovered on YPD medium from lungs and brains were tested for resistance to FLC. About 8% of wild-type and 29% of aif1 colonies developed FLCR in vivo during treatment, while more than 96% of CPS18 and CPS24 colonies retained their initial FLC resistance acquired in vitro (Figure 4A). The aCGH analysis of a wild-type H99 FLCR colony (CPS129) selected randomly showed that this isolate acquired Chr1 disomy (Figure 4B). Ploidy analysis by FACS indicated that CPS129 was diploid [2n+(1), data not shown]. Increased ploidy has recently been described in C. neoformans to be associated with the formation of giant yeast cells in infected animals [40], [41]. The aCGH analysis of an aif1 FLCR colony (CPS134) showed duplication of chromosomes 1, 4, and 6 (Figure 4C), while FLCR colonies in the CPS18 (CPS145) and CPS24 (CPS146) backgrounds maintained their initial state of aneuploidy (Figure 4D and 4E). These in vivo results can be correlated to our in vitro experiments (Table 2 and Table 4), but it is clear that the interaction of C. neoformans with the murine host environment promotes genomic plasticity, which serves to generate new phenotypic adaptations in this pathogen with disease management consequences.

Clinical isolate RCT 17 shows increased fluconazole resistance associated with aneuploidy and reduced AIF1 expression
To evaluate and validate if defects in the apoptotic pathway of C. neoformans have any impact on azole resistance in the human clinical setting, we tested the susceptibility to FLC of seven clinical isolates taken directly from frozen cerebrospinal fluid (CSF) of individual patients with cryptococcal meningitis. Microevolution and phenotypic variability of C. neoformans isolates occurs in the laboratory through multiple in vitro passages (reviewed by [42]–[44]). Therefore, an important factor considered in our experimental design when testing clinical isolates was the use of colonies isolated directly from CSF. We also targeted initial clinical isolates, collected before the commencement of antifungal therapy. It is important to note that the RCT isolates originated from South Africa, an HIV endemic area, and FLC is sometimes used as a prophylactic antifungal treatment in HIV/AIDS patients, and thus we cannot completely rule out prior exposure to azoles in these isolates.
As shown in Table 1, the susceptibility to FLC of the seven direct clinical isolates varied, with MICs ranging from 4 to >256 µg/ml. Specifically, isolate RCT 17 showed FLCR colonies within the zone of inhibition around the FLC strip in Etest assays, similar to the aif1 mutants (Figure 5A). Moreover, such colonies were completely resistant to FLC and exhibit Chr1 disomy (Figure 5B, and Table 3). RCT 17 showed a FLC resistance rate that was about 20 times higher than H99 and 4 times higher than the aif1 strain (Table 2). The FLCR isolates CPS104 and CPS157 in the RCT 17 background did not revert to FLC sensitivity during growth on FLC-free medium for 20 generations (Table 4), showing that aneuploid FLCR cells are stable in the RCT17 background, as was observed for the aif1 mutant.

Given the similarities between the clinical isolate RCT 17 and the aif1 mutant, we hypothesized that the AIF1 gene might be defective in this clinical isolate. Quantitative RT-PCR (qRT-PCR) revealed that AIF1 expression is approximately 80% reduced in the RCT 17 isolate compared to the H99 strain (Figure 5C). DNA sequence analysis determined the presence of five nucleotide polymorphisms in the AIF1 promoter and coding sequence from RCT 17 compared to the H99 (data not shown). Of note, the primer pair used for the qRT-PCR was designed to encompass the boundaries of exons 1 and 2, a region that has no polymorphisms in the RCT 17 isolate. Complementation of the RCT 17 strain with the AIF1 gene and its native promoter and terminator restored normal AIF1 expression levels (Figure 5C), reduced the FLC heteroresistance rate (Figure S6A and Table 2), and reversed the stability of the aneuploid Chr1 in the absence of FLC (Table 4), indicating that low AIF1 expression levels are responsible for the RCT 17 aif1-like phenotypes.
Additionally, we evaluated AIF1 expression in two other clinical isolates from the USA that were reported to be disomic for other chromosomes besides Chr1. Isolates HC-4 and HC-6 show partial duplication for chromosomes 9 and 6, respectively (Figure 5D and 5E and Hu and Kronstad et al., manuscript in preparation). AIF1 expression is also reduced in isolates HC-4 and HC-6 (Figure 5C), indicating that downregulation of AIF1 is not restricted to cryptococcal cells under strong selective drug pressure and might be a novel aneuploidy-tolerating mechanism.
Apoptotic pathways are required for sporulation during mating in C. neoformans
During the genetic manipulations of our apoptotic mutants, we noticed that they displayed defective mating phenotypes. Mating in C. neoformans is initiated by fusion between α and a cells to produce dikaryotic filaments, and the process culminates in the formation of basidia decorated with four spore chains. Spores formed by mating represent infectious propagules for C. neoformans. Mating filament production was reduced but not abolished when aif1, mca1 mca2, and aif1 mca1 mca2 independent mutants of opposite mating types were subjected to bilateral mutant crosses (Figure S7A). In unilateral crosses (mutant × wild-type), the aif1, mca1 mca2, and aif1 mca1 mca2 mutants had delayed formation of filaments compared with a wild-type cross, but were still fertile (data not shown). Bilateral crosses of the aif1 mca1 mca2 triple mutant showed more severe defects in filamentation, indicating that both Aif1 and the metacaspases have a role in C. neoformans mating. In confrontation assays between the aif1, mca1 mca2, and aif1 mca1 mca2 mutants and a crg1 mutant strain, which is hypersensitive to mating pheromone and produces abundant conjugation tubes when confronted with wild-type cells of the opposite mating type [45], no conjugation tubes were produced, indicating a decrease in pheromone secretion by the aif1, mca1 mca2 and aif1 mca1 mca2 mutants (Figure S7B).
Decreased hyphal growth during mating can be a result of defective fusion between α and a cells. To determine if this was the case in the aif1 and metacaspase mutants, cell fusion assays using opposite mating type strains with dominant genetic markers in bilateral crosses were conducted. The formation of double-resistant diploids by cell fusion was quantified after 48 h of incubation on mating medium. Many diploid isolates were produced from the wild-type control cross compared with the bilateral mutant crosses (Figure S8). We also examined fusion in unilateral crosses between the mca1 mca2 mutant and wild-type strains. The number of diploid isolates produced from these crosses was considerably lower than those with wild-type strains, demonstrating that one copy of each metacaspase gene is not sufficient for fusion (Figure S8). Hence, Aif1 and the metacaspases are required for the cell fusion step of mating in C. neoformans.
Additionally, a significant reduction in basidiospore production was observed in bilateral mutant crosses (Figure 6A). The aif1 and mca1 mca2 mutants produce defective basidia with aberrant spore chains (Figure 6A-D), although these strains were not sterile, and we were able to isolate recombinant spores in which the mutations were recovered in the opposite mating type. Sporulation in the aif1 mca1 mca2 bilateral mutant cross was impaired and most basidia produced few spores, in contrast to the four long spore chains observed in a wild-type cross (Figure 6). Basidiospore production defects were also observed in unilateral crosses between aif1 mca1 mca2 mutant and wild-type strains (data not shown). We conclude that ALCD is required for the proper sporulation of C. neoformans.

The aif1 and metacaspase mutants show decreased competitive fitness but have no defects in virulence
Apoptosis plays an essential role in maintaining homeostasis in multicellular organisms by eliminating permanently damaged cells. In the unicellular yeast S. cerevisiae, ALCD has been proposed to eliminate the less fit individuals from a monoclonal population of cells, thereby improving the chance of survival of the fitter clones in the genetic pool [46], [47]. To determine whether the apoptotic pathways are relevant to the ability of C. neoformans populations to compete for resources in a common environment, the fitness of apoptotic null mutants was measured in pair-wise growth competition assays with wild-type. The same amount of cells from wild-type and apoptotic null mutant strains grown to exponential phase were mixed in liquid YPD medium and incubated at 30°C with agitation. Each strain was identified based on differing genetic markers, including a control sample using two wild-type strains. To reduce cell fusion and mating during the experiment, all strains used were of the α mating type. Aliquots of each culture were taken at the indicated times and plated on selective media to calculate the survival ratios. As shown in Figure 7A, the apoptotic defective mutants were outcompeted by the wild-type cells. Because the aif1, mca1 mca2, and aif1 mca1 mca2 mutants had similar division rates as wild-type in liquid YPD medium at 30°C during solo culture (Figure S9), we eliminated the possibility that the wild-type cells were simply more fit to begin with, and thus concluded that wild-type cells exhibit enhanced fitness during co-culture with apoptotic mutant cells.

To determine whether the apoptotic null mutants had any growth defect in the host environment, we performed virulence assays in a murine inhalation model of cryptococcosis. Mice were intranasally infected with wild-type and mutant strains, and their survival was monitored and plotted against time (Figure 7B). Compared to the wild-type strain, the apoptotic defective mutants showed no significant difference in virulence (Figure 7B), fungal burden, or organ dissemination pattern (data not shown). Therefore, inactivation of ALCD had no apparent impact on the virulence of C. neoformans in these tested conditions.
Discussion
Apoptosis was long thought to be restricted to metazoans, but a growing body of evidence has established that it appears to be a common feature of diverse organisms. During the past decade, ALCD has been described in the majority of phylogenetic lineages including eubacteria, protists, plants, and fungi [11], [48], [49]. While the body of evidence supporting apoptotic fungal cell death is growing, the molecular mechanisms underlying fungal apoptotic pathways and their physiological relevance are poorly understood. Here, we show that Aif1 and metacaspases are independently required for apoptosis in the human pathogen C. neoformans. While both Aif1 and the metacaspases are important for sexual development, only Aif1 is necessary for ploidy maintenance (Figure 8, further discussed below).

We found that the aif1, mca1 mca2, and aif1 mca1 mca2 of C. neoformans have impaired sexual reproduction with a consequent reduction in formation of spores as infectious propagules (Figure 6). Similarly, the formation of ascospores in Podospora anserina was severely impaired in mutants with defective metacaspases [50] and the apoptosis-inducing factor homolog Amid2 [51]. Apoptotic markers, such as positive TUNEL reactions, were observed during sporulation in the basidiomycete Coprinopsis cinerea [52]. Furthermore, analysis of sporulation-defective mutants revealed the existence of a state in which, although meiosis was successfully completed, basidiospore formation was impaired due to lack of apoptosis [52].
Wild-type C. neoformans cells were observed to outgrow the aif1, mca1 mca2, and aif1 mca1 mca2 mutants in competitive growth assays. Likewise, in S. cerevisiae, the metacaspase yca1 and aif1 deletion mutants also show a decreased competitive fitness phenotype [47], [53]. ALCD was proposed to contribute to the fitness and adaptability of yeast populations by removing damaged or less fit clones. The fact that the aif1, mca1 mca2, and aif1 mca1 mca2 mutants cannot outcompete wild-type cells for resources in a common environment might explain, at least partially, why natural selection has preserved the apoptotic pathways in unicellular yeasts. It is also reasonable to consider that yeast apoptotic proteins may also participate in vital non-death functions. In support of this hypothesis, Yca1 was shown to have non-death roles in the timing of the yeast cell cycle [54] and the removal of insoluble protein aggregates [55].
It is interesting to highlight that the evolutionary implications of removing ALCD depend on selective pressure. For example, under conditions to which wild-type cells are well adapted, the aif1 mutant is not able to outcompete for environmental resources. On the other hand, under a strong selective pressure such as drug treatment, AIF1 inactivation promotes the stabilization of aneuploidy and thus rapid adaptation to the stress of FLC. Such observations can be compared to the known roles of apoptosis in multicellular eukaryotes; while apoptosis effectively promotes tissue homeostasis, its inactivation promotes tumorigenesis and the emergence of drug-resistant cancer cells.
Links between Aif1, aneuploidy, and the emergence of drug resistance
Alteration in gene copy number is a major mechanism for environmental adaptation of asexual yeast populations. In S. cerevisiae, aneuploidy was shown to confer improved growth and fitness advantages under stressful conditions [37], [56], [57]. Aneuploidy was also associated with FLC resistance in Candida species [58]–[61]. In C. albicans, a specific segmental aneuploidy, isochromosome 5L [i(5L)], is commonly found in FLCR isolates, and the loss of i(5L) is correlated with reduced FLC resistance [58]. Gain of i(5L), which is comprised of two identical chromosome 5 left arms flanking a centromere, amplifies two genes that contribute additively and independently to FLC resistance: ERG11 (the target of FLC) and TAC1 (a transcription factor that activates expression of the drug efflux pumps CDR1 and CDR2 [59]).
The acquisition of aneuploidy was recently reported in strains of C. neoformans that display heteroresistance to FLC [35]. Heteroresistance in C. neoformans is defined as heterogeneous FLC susceptibility within a population with resistant subpopulations being able to adapt to higher concentrations of the drug in a stepwise and reversible manner [31]–[33]. Heteroresistance is considered different from trailing phenomena or incomplete growth inhibition by azoles observed in Candida species. Trailing growth can cause the MICs of azoles for some isolates to be low (susceptible) after 24 h of growth but much higher (resistant) after 48 h [62]. The relevance of such discordant interpretive categories at the two time points is as yet unclear, but current evidence suggests that the lower MIC correlates most closely with the outcome in vivo [62]. Bacterial populations were reported to produce persister cells that neither grow nor die in the presence of microbicidal antibiotics. Persisters are largely responsible for high levels of biofilm tolerance to antimicrobials, but do not exhibit an increased MIC. C. albicans persister cells that survived killing by AMB were detected only in biofilms and not in exponentially growing or stationary-phase planktonic populations [63].
Even though we used Etest assays to isolate FLCR colonies in this study, our results support previous findings in which the H99 strain exposed to FLC in liquid cultures became resistant in a step-wise pattern to increasing concentrations of the drug after acquisition of specific chromosomal duplications [33], [35]. In our experiments, Chr1 disomy was observed in H99 derivatives that became resistant to more than 48 µg/ml of FLC and was more common on second Etest passages, while disruption of aif1 stimulated the selection of completely FLCR (MIC >256 µg/ml) subpopulations. Amplification of chromosomes other than Chr1, particularly Chr4, was observed in aif1 derivatives, but not in the H99 or AIF1 complemented derivatives isolated in Etest experiments (Table 3). In the aif1 derivatives, segmental chromosomal duplications were also observed; isolate CPS24 had the left arm of Chr1 duplicated, which harbors ERG11, and the end of the right arm of chromosome 3 (Figure 2E and Figure S3A). C. gattii isolates resistant to 64 µg/ml of FLC showed different patterns of chromosomal amplifications, including disomy and segmental duplication of several chromosomes besides Chr1 [64]. Clinical isolate RCT 52 had a FLC MIC >256 µg/ml (Table 1) but did not show any chromosomal amplification (data not shown), indicating that not all FLCR isolates are aneuploid. These findings emphasize the existence of FLC resistance mechanisms that are independent of Chr1 disomy.
After about 20 generations of non-selective growth, more than 90% of FLCR colonies in the H99 background and 75% in the complemented strain returned to being FLC sensitive, while less than 10% in the aif1 background lost resistance to FLC upon removal of the drug pressure (Table 4). The lack of benomyl sensitivity in the aif1 mutant excludes the possibility that the observed increased aneuploidy in this strain is a result of a defective mitotic spindle checkpoint. Our results show that the lack of a functional Aif1 was able to stabilize the aneuploid state selected by FLC treatment, and we propose that aif1 is an aneuploidy-tolerating mutation in C. neoformans.
Torres et al. [56] described the existence of aneuploidy-tolerating mutations in yeast, such as the deubiquitinating enzyme Ubp6, which improves growth rates in aneuploid yeast strains by attenuating the protein stoichiometry imbalances caused by chromosomal amplifications. We did not observe that FLCR aneuploid strains display an increased sensitivity to stresses, such as reported in S. cerevisiae by Torres et al. [37]. The results shown in Figure 3A suggest that aneuploidy does not inevitably result in decreased resistance to stress, and are in agreement with a recent study by Pavelka et al. [57], which showed that aneuploidy directly confers phenotypic variation that can result in a growth advantage under stress conditions that are suboptimal for euploid cells. The conflicting results in S. cerevisiae were explained by the different methods used to generate the aneuploid strains; while Torres et al. [37] constructed their aneuploid strains through selection with a combination of drug and nutrient markers, Pavelka et al. [57] generated aneuploid strains through random meiotic segregation, suggesting an impact of both karyotype and continuous selection with drugs and nutrient markers in the phenotypic variation conferred by aneuploidy [57]. Interestingly, as in Pavelka et al. [57], we observed that certain aneuploid strains were able to grow better in the presence of drugs such FLC and rapamycin (strain WT FLCR256, Figure 3A).
Rancati et al. [65] reported that S. cerevisiae cells lacking MYO1, which encodes the only myosin-II normally required for cytokinesis, rapidly acquired aneuploidy that led to specific changes in the transcriptome that restored growth and cytokinesis. Therefore, one possibility is that aif1 mutants have more stable Chr1 aneuploidy because genes present on Chr1L might complement Aif1 functions. It should be noted that no growth defect (besides the decreased competitiveness with wild-type) was observed for aif1 mutant strains, and that Chr1 aneuploidy did not appear to improve aif1 mutant strains growth. We speculate that apoptosis orchestrated by Aif1 eliminates aneuploid cells from the population.
Why didn't the metacaspase mutants have an increased rate of aneuploidy and consequent resistance to FLC? Based on the fact that Aif1 and the metacaspases are independently required for apoptosis in C. neoformans, it is possible to speculate that the elimination of aneuploid cells from the population is regulated by a caspase-independent apoptotic pathway. In yeast, the Yca1 caspase-like protease participates in approximately 40% of the investigated cell death scenarios, while Aif1 and endonuclase G execute caspase-independent cell death [66]. It is also possible that ploidy maintenance is a non-death function of Aif1.
FLCR colonies isolated from the H99 and aif1 strains during our in vivo experiments showed more complex patterns of ploidy increase and amplifications (Figure 4B and 4C). The interaction of C. neoformans with mammalian hosts has been shown to promote genomic plasticity in this pathogen (reviewed by [67]). Comparative hybridization studies characterized disomy for chromosome 13 in two independent clinical isolates [68]. Additionally, the formation of giant cells with diameters up to 100 µm was observed during murine cryptococcal infection [40], [41]. These giant yeast cells were found mainly in lung tissue and were polyploid and uninucleate, suggesting that they arise from DNA endoreplication without concomitant cell division. It will be interesting to investigate if ALCD is also important in ploidy increase and the formation of giant yeast cells. In preliminary analysis of FLC Etests using a diploid wild-type strain, we did not observe an increase in heteroresistance while a diploid aif1 strain still showed increased heteroresistance (Figure S6B).
In our in vivo virulence experiments, we evaluated the efficacy of FLC therapy for wild-type and FLCR strains using a murine model of cryptococcal meningitis. Treatment with 100 mg/kg/day of FLC had antifungal activity against H99 and aif1 strains (but did not clear infection in the brains of the animals) and doubled the survival time of mice infected with FLCR strains. Pharmacodynamic studies, using the area under the concentration-time curve (AUC), showed that FLC therapy in doses of 100 mg/kg/day given to mice mimics doses of 400 mg/day given to humans [69]. Therefore, the use of higher dosages of FLC might be beneficial in patients, and may help to reduce morbidity and mortality due to cryptococcal meningitis in Africa and other resource-limited regions.
We found that RCT 17, one of seven direct clinical isolates, had increased heteroresistance to FLC, similar to the heteroresistance observed in the aif1 mutants (Table 1, Figure 5A). Furthermore, FLCR colonies isolated from the RCT 17 background also became completely resistant to FLC through the acquisition of aneuploidy (Figure 5B, and Table 3) and did not revert to being FLC-sensitive during growth on FLC-free medium for 20 generations (Table 4). AIF1 expression was shown to be downregulated in RCT 17 by qRT-PCR (Figure 5C), and complementation with AIF1 with its native promoter and terminator recovered normal expression levels and reverted the increased heteroresistance to FLC (Table 2 and Table 3). AIF1 expression was also reduced in the HC-4 and HC-6 clinical isolates (Figure 5C), which show disomies for chromosomes other than Chr1 and are not resistant to FLC (Table 1, Figure 5D and 5E). Therefore, we propose that the inactivation of AIF1 is a novel aneuploidy-tolerating mechanism in fungi that might not be restricted to cells under strong selective FLC drug pressure. Future investigations will test the mechanisms that result in AIF1 downregulation.
Our results suggest that triggering fungal ALCD should be further investigated as a new antifungal approach. Potential antifungal targets include those that can help another antifungal agent by blocking resistance mechanism(s) in vivo used by the microorganisms. This principle has previously been demonstrated by beta-lactamase inhibitors in antibacterial combination drugs [70] but has yet to be applied in therapies including new antifungal agents. An AIF1-inducing agent could have a profound effect on the ability of C. neoformans to become resistant in vivo to FLC, and the addition of such inducer to the antifungal regimen could make FLC fungicidal in the host.
In conclusion, our results bring to light the importance of the Aif1 in maintaining the euploidy of yeast populations, possibly through the elimination of aneuploid cells via apoptosis. Inactivation of AIF1 might increase genomic plasticity of C. neoformans during infection and promote the generation of phenotypic adaptations such as new virulence traits and antifungal drug resistance.
Materials and Methods
Ethics statement
This study was carried out in strict accordance with The National Research Council's Guide for the Care and Use of Laboratory Animals, Public Health Service Policy on Humane Care and Use of Laboratory Animals, and AAALAC accreditation guidelines. The protocol was approved by the Duke University and Duke University Medical Center Institutional Animal Care and Use Committee (protocol number: A266–08–10). All efforts were made to minimize suffering. The clinical isolates were collected and stored in the Duke Infectious Disease Repository under an IRB-approved protocol entitled “Database and specimen repository for infectious diseases-related studies” (#CR3_Pro00005314). The specimens were de-identified discarded samples collected as part of routine clinical practice and were exempt from patient written informed consent.
Strains and growth conditions
C. neoformans strains used in this study are listed in Table S1. YL99a strain was obtained by an extra round of H99 and KN99a backcrossing. The tested clinical isolates were taken directly from the CSF of patients with cryptococcal meningitis from USA (HC isolates, Duke patients) and from South Africa (RCT isolates, obtained from Tihana Bicanic and Tom Harrison). The indicated mating types were determined by crosses and confirmed with PCR, and the serotyping of clinical isolates was determined by PCR using STE20 mating-type - and serotype-specific primer pairs [71]. Strains were maintained in −80°C glycerol stocks and grown on YPD medium (1% yeast extract, 2% Bacto Peptone, and 2% dextrose). To perform mating assays, cells of opposite mating type were mixed in water, spotted on 5% V8 juice agar medium (pH 5.0) or Murashige and Skoog (MS) medium minus sucrose and incubated at room temperature in the dark [72]–[75]. For spot assays, cultures grown in liquid YPD were diluted to 2×106 cells/ml and serially diluted tenfold. Then, 5 µl of each culture was spotted onto YPD or YPD plus 2 mM H2O2 (Sigma) or YPD plus 20 ng/ml rapamycin (LC Laboratories) plates and incubated at the indicated temperatures. For benomyl sensitivity assays, 5 µl of fivefold serial dilutions were spotted onto YPD plates with DMSO (control) or with 10 or 20 µg/ml benomyl (Sigma). Growth differences were imaged following incubation of the plates for 72 h.
Generation of gene disruption and complemented strains
Strains with gene disruptions were generated using an overlap PCR approach and biolistic transformation in serotype A congenic strains H99 and KN99a as previously described [76]. For gene deletions, the 5' and 3' flanking regions of apoptotic genes and BUB1 were amplified from H99 genomic DNA and the dominant selectable markers NAT, NEO, or HYG were amplified with the universal M13F and M13R primers from plasmids pJAF13, pJAF12 and pJAF15 respectively. A second overlap PCR amplified a full-length deletion cassette containing the three previous fragments. The products of overlap PCR were purified, precipitated onto gold microcarrier beads (0.6 mm, Bio-Rad), and the H99 strain was biolistically transformed [5], [26]. Stable transformants were selected for resistance to nourseothricin (100 µg/ml), G418 (100 µg/ml), or hygromycin B (300 µg/ml), screened by PCR, and confirmed by Southern blot. The α mca1 mutant background was used for disruption of MCA2 by biolistic transformation. The resulting α mca1 mca2 mutant was crossed to strain KN99a to generate single and double mutants with opposite mating types (see Table S1). The aif1 mca1 mca2 triple mutants are progeny obtained from the cross of α aif1with a mca1 mca2 (see Table S1). For complementation, an overlap PCR product with the NEO marker and the wild-type AIF1 gene containing its native promoter and terminator from strain H99 was generated. The PCR product was biolistically transformed into aif1 mutant and RCT17 backgrounds. Transformants were selected for resistance to G418 (100 µg/ml) and confirmed by PCR. The sequences of primers used are listed in Table S2.
TUNEL assay
TdT-mediated dUTP nick end labeling (TUNEL) assays were performed as described previously [5], [26] with the following modifications. Log-phase cultures were diluted to an OD600 of 0.1 and treated with 2 mM H2O2 (Sigma) for 3 h at 37°C. Cells were fixed for 1 h at room temperature with 3.7% formaldehyde (Sigma) and washed with PBS. After treatment with 5% β-mercaptoethanol in SPM (1.2 M sorbitol; 50 mM K-phosphate, pH 7.3; and 1 mM MgCl2) for 1 h at 37°C with gentle agitation, cells were washed with SPM and then digested with 10 µl of zymolyaseTM (Zymo Research, 4 U/µL), 100 mg of lysing enzymes from Trichoderma harzianum (Sigma), and 0.1% bovine serum albumin (Sigma) in 1 ml of spheroplasting buffer (1 M sorbitol; 10 mM EDTA; and 100 mM sodium citrate, pH 5.8) for 40 min at 37°C with gentle agitation. Spheroplasts were gently harvested at 1600 rpm for 5 min at 4°C and washed three times with SPM to remove the enzymes. Samples were permeabilized with 0.5 ml of fresh prepared 0.1% Triton X-100, 0.1% sodium citrate solution in cold SPM for 2 min in ice, washed twice with SPM, and incubated with 50 µl TUNEL reaction mixture (In Situ Cell Death Detection Kit, Roche) for 60 min at 37°C in dark and humid conditions. Cells were washed with PBS and immediately analyzed by epifluorescence microscopy (see next section) or flow cytometry. TUNEL positive cells were quantified by collecting 10,000 events on the FL1 (FITC) channel of a FACSCalibur flow cytometer (Becton Dickinson) using CellQuest software (Becton Dickinson). Cell debris were excluded using the side and forward scatter dot-plot and a negative control (fixed and permeabilized wild-type cells incubated in 50 µl of TUNEL solution without terminal transferase) and a positive control [fixed and permeabilized wild-type cells treated with 0.1 µg/µl DNase I (Sigma) for 10 min at room temperature to induce DNA strand breaks prior to TUNEL labeling] were used to determine the percentage of TUNEL positive cells.
Annexin V assay
Exposed phosphatidylserine was detected by reaction with FITC-coupled Annexin V (Apoptosis Detection kit, Oncogene Research Products). Log-phase cultures diluted to an OD600 of 0.1 were treated with 2 mM H2O2 (Sigma) for 3 h at 37°C. Yeast cells were washed with PBS, treated with 5% β-mercaptoethanol, and spheroplasted as described above for the TUNEL assay. Spheroplasts were washed two times with SPM and once with Binding Buffer diluted to 1x with 2 M sorbitol, and double stained with FITC-Annexin V and propidium iodine (PI) for 20 min at room temperature in the dark dark as described by [77]. Annexin V-positive, PI-negative cells were quantified by collecting 10,000 events on the FL1 (FITC) and FL2 (PE) channels of a FACSCalibur flow cytometer (Becton Dickinson) using CellQuest software (Becton Dickinson).
Microscopy
To evaluate TUNEL assays, cells were mounted in ProLong Gold antifade reagent with DAPI (Molecular Probes). Brightfield, differential interference microscopy (DIC), and fluorescence images were captured with a Zeiss Axio Imager widefield fluorescence microscope (Carl Zeiss) equipped with an Orca cooled-CCD camera (Hamamatsu) using DAPI and FITC channels. Images were interfaced with MetaMorph software (Universal Imaging) and processed with PhotoShop (Adobe).
Mating structures were captured with an Eclipse E400 microscope (Nikon) equipped with a DXM1200F digital camera (Nikon). Images were interfaced with ACT-1 software (Nikon) and processed with PhotoShop (Adobe). For scanning electron microscopic analysis, mating plates were processed at the North Carolina State University Center for Electron Microscopy, Raleigh, NC, USA. Areas of interest were excised intact from the agar and fixed with 0.1 M sodium cacodylate (pH 6.8) buffer containing 3% glutaraldehyde for several days at 4°C. Samples were then washed for 1 h in cold sodium cacodylate buffer three times, dehydrated through a graded series of cold 30% and 50% ethanol for 1 h each, and held overnight in 70% ethanol. Dehydration was completed with 1-h incubations with cold 95% and 100% ethanol at 4°C, warming to room temperature in the 100% ethanol, followed by two additional 1-h washes with 100% ethanol at room temperature. The dehydrated samples were critical-point dried with liquid CO2 in a SAMDRI-795 (Tousimis) for 15 min and mounted on stubs with silver paint to ensure good adhesion and conductivity. Finally, the samples were coated with 50 Å of gold/palladium using a Hummer 6.2 sputter coater (Anatech) and held in a vacuum desiccator until viewed at 15 kV on a JSM 5900LV scanning electron microscope (JEOL), photographed with a Digital Scan Generator (JEOL) image acquisition system, and processed with PhotoShop (Adobe).
Etest
In vitro antifungal susceptibility to FLC and AMB was determined by Etest (AB Biodisk) according to the manufacturer's instructions. Cells grown to mid-log phase were washed in 0.85% NaCl and diluted to an OD600 of 0.5 (∼1 McFarland turbidity). Inocula were applied with cotton swabs in RPMI-1640 agar (Sigma) supplemented with 2% glucose and buffered to pH 7.0 with MOPS and allowed to dry completely before applying the Etest strip. Plates were incubated at 35°C for 72 h and the MIC was determined as the first growth inhibition ellipse.
Isolation of fluconazole resistant strains
Distinct colonies within the inhibition halo around the FLC strip during Etest assays were replica plated onto YPD agar and YPD agar plus 32 µg/ml FLC and incubated at 30°C. Isolates able to grow on the FLC plate after 2 days were inoculated into 5 ml YPD liquid and grown overnight at 30°C with agitation. Liquid cultures were tested by Etest assays and used to make gDNA (later used for ploidy analysis) and −80°C glycerol stocks. For virulence experiments and FLC resistance stability assays, −80°C glycerol stocks were inoculated into 5 ml YPD liquid and grown overnight at 30°C with agitation.
Array comparative genomic hybridization
Genomic DNA was extracted using alkyltrimethyl ammonium bromide buffer (CTAB) as described by [78]. The quality of the purified DNA was examined on an agarose gel and quantified in a Qubit fluorometer (Invitrogen) using the Quant-iT dsDNA BR kit according to Invitrogen's instructions. Then 2.5 µg of DNA was sonicated to generate ∼500-bp fragments, which were labeled with Perkin Elmer Cy3-dUTP (reference DNA from H99 wild-type strain) or Cy5-dUTP (strain of interest) using the Random Primer Reaction of BioPrime Array CGH Genomic Labeling System (Invitrogen). Labeled reference and sample DNA were combined in a 1∶1 ratio, purified using Microcon 30K Centrifugal Filter Units (Millipore), and concentrated in a Speed Vac Plus SC110 (Savant Instruments). Samples were resuspended in 50 µl of Long Oligo Hybridization Solution (Corning), denatured at 95°C for 5 min, and applied to the C. neoformans whole genome 70-mer oligonucleotide spotted array version II (Washington University Genome Sequencing Center) covered with a LifterSlip Microarray Coverslip (Erie Scientific Company). Arrays were hybridized at 42°C for 16 h and subsequently washed twice with 1X SSC, 0.3% SDS heated to 42°C, followed by two washes in 0.2X SSC, and two final washes with 0.05X SSC at room temperature for two minutes each. Slides were dried by centrifugation, scanned with a GenePix 4000B scanner (Axon Instruments), and analyzed using GenePix Pro 6.0 software (Molecular Devices). GeneSpring GX 7.3 (Agilent Technologies) was used to perform Lowess normalization of the data, and an S-plus script was used to remove exon-junction probes and to log2 transform the red/green ratios. The transformed data from experimental arrays and from four control hybridizations (Cy3-reference strain DNA x Cy5-reference strain DNA) were analyzed with CGH-Miner software [79], which uses the Clusters Along Chromosomes method to identify DNA copy number alterations. Raw data for the regions of gain/loss identified by the algorithm were then manually inspected to confirm the calls or eliminate occasional errors induced by noise. The plots of log2 data as a function of chromosomal nucleotide position were created using a sliding-window of 10 consecutive probes and a targeted false discovery rate (FDR, i.e., the expected proportion of false positive results) of 0.01. Vertical bars were plotted in red for amplifications and green for deletions that are statistically significant. Vertical bars in gray indicate no DNA copy number alteration or amplifications or deletions that did not meet the statistical settings.
Fluconazole resistance and reversion rates
FLC resistance rates were calculated by fluctuation analysis using the method of the median [80]. To measure FLC resistance rates, individual colonies of each strain were grown non-selectively in YPD agar from −80°C glycerol stocks. Twelve independent cultures from isolated colonies were grown in YPD liquid medium overnight at 30°C. Yeast cells were collected by centrifugation, washed with water, and serial dilutions were plated on YPD agar and YPD agar with 32 µg/ml FLC to determine the number of viable cells and the number of FLCR cells, respectively. The median number of FLCR cells in the different cultures was corrected for dilution factors, fractions plated, and number of viable cells. The numbers of FLCR cells were ranked, and the 3rd and 10th ranks were used to calculate the lower and upper limits of the 95% confidence interval [81]. Strains resistant to FLC were used to calculate the reversion rates after approximately 20 generations. Non-selectively grown cultures from -80°C glycerol stocks were incubated at 30°C for 24 h in YPD liquid. Culture dilutions were plated on YPD agar, and 100 individual colonies were replica plated onto YPD agar and YPD agar plus 32 µg/ml FLC, followed by incubation at 30°C. Colonies unable to grow on the FLC plate after 2 days were considered revertants.
Virulence studies
Virulence assays were conducted using a murine inhalation model of cryptococcosis. Cohorts of 4 - to 6-week-old female A/JCr mice were anesthetized by intraperitoneal injection of Nembutal (37.5 mg/kg) and infected intranasally with 5×104 cells in 50 µl of PBS pipetted slowly into the nares. Inoculum concentration was confirmed by plating serial dilutions in YPD agar and counting colony forming units (CFU) after 2 days. Mice were monitored twice daily, and those that showed signs of severe morbidity (weight loss, extension of the cerebral portion of the cranium, abnormal gait, paralysis, seizures, convulsions, or coma) were sacrificed by CO2 inhalation. The survival rates of animals were plotted against time, and p-values were calculated with the Mann-Whitney U test. In the first experiment using FLC treatment (Figure S5), 10 mice per strain were treated with 20 mg/kg/day FLC intraperitoneally, while control groups of 10 mice received only saline. Treatment started 24 h after inoculation of yeasts and was continued for 14 days. In the second experiment using FLC treatment (Figure 3C), 10 mice of about 20 g each were treated with 100 mg/kg/day FLC in the sole source of drinking water as described by [35]. Water intake was recorded daily for each cage (all 5 mice were assumed to drink the same volume) and averaged 0.23 ml/g. Treatment started 24 h after infection and was continued through day 60 with water being replaced every 2 to 3 days. FLC concentrations for each cage were recalculated based on weights of the animals and the water intake measured during the preceding days. Three euthanized mice in each treated group were dissected on the days indicated post-infection; their brains and lungs were removed, weighed, and homogenized in 1 ml sterile PBS. Serial dilutions of the organ samples were plated on Sabouraud-dextrose agar plus 100 µg/ml chloramphenicol and incubated at 30°C. Up to 50 individual colonies were replica plated onto YPD agar and YPD agar plus 32 µg/ml FLC and incubated at 30°C. Colonies unable to grow on the FLC plate after 2 days were considered revertants. Isolates able to grow on FLC were further tested by aCGH, qPCR, and Etest assays as described above.
RNA extraction and cDNA synthesis
Yeast cells for RNA extraction were grown in YPD liquid culture overnight and were harvested the following day and washed. RNA preparations were isolated with the RNAeasy Mini Kit (Qiagen), including the DNase treatment step, and reverse transcriptase reactions were performed using AffinityScript RT-RNase (Stratagene).
Quantitative PCR and RT-PCR analysis
Quantitative PCR reactions were performed in an Applied Biosystems 7500 Real-Time PCR System using Brilliant SYBR Green qRT-PCR master mix (Stratagene). PCR thermal cycling conditions were an initial step at 95°C for 5 min followed by 40 cycles at 95°C for 15 s, 60°C for 20 s and 72°C for 20 s. In each assay, "no-template" controls were included and melting curve analysis was performed to confirm a single PCR product. All experiments were done in triplicate for both the gene of interest and the control gene (GPB1 or ACT1, see figure legends). Data were normalized to the control genes and relative expression was determined by the 2−ΔΔCT method. The sequences of primers used are listed in Table S2. Detection of Chr1 disomy by qPCR was adapted from the method described by [81].
Mating cell fusion assays
To perform the mating cell fusion assays, wild-type and mutant strains containing HYG, NAT, or NEO resistance genes were grown in YPD liquid overnight. Yeast cells were washed and adjusted to 5×106 cells/ml, mixed 1∶1, and grown on V8 medium (pH 5.0) in the dark for 48 h. The mating colonies were collected by scraping, resuspended in sterile water, and serial dilutions were plated onto YPD medium containing hygromycin, nourseothricin, and/or G418 to select for cell-cell fusion products that have two drug-resistance markers (one from either parent). Plates were incubated at 30°C for 3 days until colonies formed and were then counted. The average number of colonies in triplicate assays was calculated and the fusion efficiency in apoptotic-mutants was determined as a function of the wild-type.
Competitive fitness and growth curve assays
For competition assays, overnight cultures in YPD were washed three times with sterile water and adjusted to a density of 1×107 cells/ml. The following strains were paired in a 1∶1 ratio: H99 and CPS76 (H99 HYG); H99 and CPS3; H99 and YPH104; and H99 and CPS89. Each mixture was inoculated onto fresh YPD medium and incubated at 30°C for the indicated times. Fifty isolated colonies from each time point were replica plated onto selective media and survival rates were calculated. Growth curves were determined by diluting the strains to OD600 of 0.01 in YPD and incubation at 30°C without agitation. The turbidity of quadruplicate assays was measured at OD600 every hour with a microdilution plate reader (Sunrise, Tecan). Readings were corrected for background (YPD media, no cells added), averaged, and plotted versus time in Excel (Microsoft Office).
Statistical analysis
All quantitative data are reported as means ± standard deviation and are derived from at least three independent experiments. The significance of the data was assessed using two-tailed t-tests to compare mutants with the wild-type strain. For comparisons of mice survival data, the logrank test was performed using Prism 4 (GraphPad Software). A p-value of less than 0.05 was considered significant.
Accession numbers
AIF1 (CNAG_04521); BUB1 (CNAG_03184); COX1 (CNAG_09009); ENDOG (CNAG_02204); IAP1 (CNAG_04708); MCA1 (CNAG_04636), MCA2 (CNAG_06787), and AIF1 from RCT 17 strain (JN606083).
Supporting Information
Zdroje
1. BaehreckeEH 2002 How death shapes life during development. Nat Rev Mol Cell Biol 3 779 787
2. JiangXWangX 2004 Cytochrome C-mediated apoptosis. Annu Rev Biochem 73 87 106
3. KurokawaMKornbluthS 2009 Caspases and kinases in a death grip. Cell 138 838 854
4. CandeCCecconiFDessenPKroemerG 2002 Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? J Cell Sci 115 4727 4734
5. MadeoFFrohlichEFrohlichKU 1997 A yeast mutant showing diagnostic markers of early and late apoptosis. J Cell Biol 139 729 734
6. LigrMMadeoFFrohlichEHiltWFrohlichKU 1998 Mammalian Bax triggers apoptotic changes in yeast. FEBS Lett 438 61 65
7. ManonSChaudhuriBGuerinM 1997 Release of cytochrome c and decrease of cytochrome c oxidase in Bax-expressing yeast cells, and prevention of these effects by coexpression of Bcl-xL. FEBS Lett 415 29 32
8. LongoVDEllerbyLMBredesenDEValentineJSGrallaEB 1997 Human Bcl-2 reverses survival defects in yeast lacking superoxide dismutase and delays death of wild-type yeast. J Cell Biol 137 1581 1588
9. ButtnerSEisenbergTHerkerECarmona-GutierrezDKroemerG 2006 Why yeast cells can undergo apoptosis: death in times of peace, love, and war. J Cell Biol 175 521 525
10. GlassNLDementhonK 2006 Non-self recognition and programmed cell death in filamentous fungi. Curr Opin Microbiol 9 553 558
11. RamsdaleM 2008 Programmed cell death in pathogenic fungi. Biochim Biophys Acta 1783 1369 1380
12. HamannABrustDOsiewaczHD 2008 Apoptosis pathways in fungal growth, development and ageing. Trends Microbiol 16 276 283
13. SharonAFinkelsteinAShlezingerNHatamI 2009 Fungal apoptosis: function, genes and gene function. FEMS Microbiol Rev 33 833 854
14. IkedaRSawamuraK 2008 Bacterial and H2O2 stress-induced apoptosis-like events in Cryptococcus neoformans. Res Microbiol 159 628 634
15. ChiapelloLSAokiMPRubinsteinHRMasihDT 2003 Apoptosis induction by glucuronoxylomannan of Cryptococcus neoformans. Med Mycol 41 347 353
16. ChiapelloLSBaronettiJLAokiMPGeaSRubinsteinH 2004 Immunosuppression, interleukin-10 synthesis and apoptosis are induced in rats inoculated with Cryptococcus neoformans glucuronoxylomannan. Immunology 113 392 400
17. PericoliniECenciEMonariCPeritoSMosciP 2006 Indinavir-treated Cryptococcus neoformans promotes an efficient antifungal immune response in immunosuppressed hosts. Med Mycol 44 119 126
18. PericoliniEGabrielliEBistoniGCenciEPeritoS 2010 Role of CD45 signaling pathway in galactoxylomannan-induced T cell damage. PLoS One 5 e12720
19. PericoliniEGabrielliECenciEDe JesusMBistoniF 2009 Involvement of glycoreceptors in galactoxylomannan-induced T cell death. J Immunol 182 6003 6010
20. MonariCPericoliniEBistoniGCenciEBistoniF 2005 Influence of indinavir on virulence and growth of Cryptococcus neoformans. J Infect Dis 191 307 311
21. ChiapelloLSBaronettiJLGarroAPSpessoMFMasihDT 2008 Cryptococcus neoformans glucuronoxylomannan induces macrophage apoptosis mediated by nitric oxide in a caspase-independent pathway. Int Immunol 20 1527 1541
22. VillenaSNPinheiroROPinheiroCSNunesMPTakiyaCM 2008 Capsular polysaccharides galactoxylomannan and glucuronoxylomannan from Cryptococcus neoformans induce macrophage apoptosis mediated by Fas ligand. Cell Microbiol 10 1274 1285
23. ParkBJWannemuehlerKAMarstonBJGovenderNPappasPG 2009 Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23 525 530
24. PerfectJRDismukesWEDromerFGoldmanDLGraybillJR 2010 Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis 50 291 322
25. ChayakulkeereeMPerfectJR 2006 Cryptococcosis. Infect Dis Clin North Am 20 507 544
26. SemighiniCPHarrisSD 2010 Methods to detect apoptotic-like cell death in filamentous fungi. Methods Mol Biol 638 269 279
27. PerfectJRCoxGM 1999 Drug resistance in Cryptococcus neoformans. Drug Resist Updat 2 259 269
28. MousaviSARobsonGD 2004 Oxidative and amphotericin B-mediated cell death in the opportunistic pathogen Aspergillus fumigatus is associated with an apoptotic-like phenotype. Microbiology 150 1937 1945
29. PhillipsAJSudberyIRamsdaleM 2003 Apoptosis induced by environmental stresses and amphotericin B in Candida albicans. Proc Natl Acad Sci U S A 100 14327 14332
30. Al-DhaheriRSDouglasLJ 2010 Apoptosis in Candida biofilms exposed to amphotericin B. J Med Microbiol 59 149 157
31. MondonPPetterRAmalfitanoGLuzzatiRConciaE 1999 Heteroresistance to fluconazole and voriconazole in Cryptococcus neoformans. Antimicrob Agents Chemother 43 1856 1861
32. YamazumiTPfallerMAMesserSAHoustonAKBoykenL 2003 Characterization of heteroresistance to fluconazole among clinical isolates of Cryptococcus neoformans. J Clin Microbiol 41 267 272
33. SionovEChangYCGarraffoHMKwon-ChungKJ 2009 Heteroresistance to fluconazole in Cryptococcus neoformans is intrinsic and associated with virulence. Antimicrob Agents Chemother 53 2804 2815
34. VarmaAKwon-ChungKJ 2010 Heteroresistance of Cryptococcus gattii to fluconazole. Antimicrob Agents Chemother 54 2303 2311
35. SionovELeeHChangYCKwon-ChungKJ 2010 Cryptococcus neoformans overcomes stress of azole drugs by formation of disomy in specific multiple chromosomes. PLoS Pathog 6 e1000848
36. SanguinettiMPosteraroBLa SordaMTorelliRFioriB 2006 Role of AFR1, an ABC transporter-encoding gene, in the in vivo response to fluconazole and virulence of Cryptococcus neoformans. Infect Immun 74 1352 1359
37. TorresEMSokolskyTTuckerCMChanLYBoselliM 2007 Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317 916 924
38. HoytMATotisLRobertsBT 1991 S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 66 507 517
39. LiRMurrayAW 1991 Feedback-control of mitosis in budding yeast. Cell 66 519 531
40. ZaragozaOGarcia-RodasRNosanchukJDCuenca-EstrellaMRodriguez-TudelaJL 2010 Fungal cell gigantism during mammalian infection. PLoS Pathog 6 e1000945
41. OkagakiLHStrainAKNielsenJNCharlierCBaltesNJ 2010 Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog 6 e1000953
42. FriesBCGoldmanDLCasadevallA 2002 Phenotypic switching in Cryptococcus neoformans. Microbes Infect 4 1345 1352
43. PerfectJR 2005 Cryptococcus neoformans: a sugar-coated killer with designer genes. FEMS Immunol Med Microbiol 45 395 404
44. GuptaGFriesBC 2010 Variability of phenotypic traits in Cryptococcus varieties and species and the resulting implications for pathogenesis. Future Microbiol 5 775 787
45. WangPCutlerJKingJPalmerD 2004 Mutation of the regulator of G protein signaling Crg1 increases virulence in Cryptococcus neoformans. Eukaryot Cell 3 1028 1035
46. FabrizioPPletcherSDMinoisNVaupelJWLongoVD 2004 Chronological aging-independent replicative life span regulation by Msn2/Msn4 and Sod2 in Saccharomyces cerevisiae. FEBS Lett 557 136 142
47. HerkerEJungwirthHLehmannKAMaldenerCFrohlichKU 2004 Chronological aging leads to apoptosis in yeast. J Cell Biol 164 501 507
48. LewisK 2000 Programmed death in bacteria. Microbiol Mol Biol Rev 64 503 514
49. LamE 2004 Controlled cell death, plant survival and development. Nat Rev Mol Cell Biol 5 305 315
50. HamannABrustDOsiewaczHD 2007 Deletion of putative apoptosis factors leads to lifespan extension in the fungal ageing model Podospora anserina. Mol Microbiol 65 948 958
51. BrustDHamannAOsiewaczHD 2010 Deletion of PaAif2 and PaAmid2, two genes encoding mitochondrial AIF-like oxidoreductases of Podospora anserina, leads to increased stress tolerance and lifespan extension. Curr Genet 56 225 235
52. LuBCGalloNKuesU 2003 White-cap mutants and meiotic apoptosis in the basidiomycete Coprinus cinereus. Fungal Genet Biol 39 82 93
53. BreslowDKCameronDMCollinsSRSchuldinerMStewart-OrnsteinJ 2008 A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods 5 711 718
54. LeeREPuenteLGKaernMMegeneyLA 2008 A non-death role of the yeast metacaspase: Yca1p alters cell cycle dynamics. PLoS One 3 e2956
55. LeeREBrunetteSPuenteLGMegeneyLA 2010 Metacaspase Yca1 is required for clearance of insoluble protein aggregates. Proc Natl Acad Sci U S A 107 13348 13353
56. TorresEMDephoureNPanneerselvamATuckerCMWhittakerCA 2010 Identification of aneuploidy-tolerating mutations. Cell 143 71 83
57. PavelkaNRancatiGZhuJBradfordWDSarafA 2010 Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468 321 325
58. SelmeckiAForcheABermanJ 2006 Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313 367 370
59. SelmeckiAGerami-NejadMPaulsonCForcheABermanJ 2008 An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol Microbiol 68 624 641
60. SelmeckiAMDulmageKCowenLEAndersonJBBermanJ 2009 Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet 5 e1000705
61. PolakovaSBlumeCZarateJAMentelMJorck-RambergD 2009 Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata. Proc Natl Acad Sci U S A 106 2688 2693
62. MarrKARustadTRRexJHWhiteTC 1999 The trailing end point phenotype in antifungal susceptibility testing is pH dependent. Antimicrob Agents Chemother 43 1383 1386
63. LaFleurMDKumamotoCALewisK 2006 Candida albicans biofilms produce antifungal-tolerant persister cells. Antimicrob Agents Chemother 50 3839 3846
64. D'SouzaCAKronstadJWTaylorGWarrenRYuenM 2011 Genome variation in Cryptococcus gattii, an emerging pathogen of immunocompetent hosts. mBio 2 e00342 00310
65. RancatiGPavelkaNFlehartyBNollATrimbleR 2008 Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 135 879 893
66. LengelerKBCoxGMHeitmanJ 2001 Serotype AD strains of Cryptococcus neoformans are diploid or aneuploid and are heterozygous at the mating-type locus. Infect Immun 69 115 122
67. KronstadJWAttarianRCadieuxBChoiJD'SouzaCA 2011 Expanding fungal pathogenesis: Cryptococcus breaks out of the opportunistic box. Nat Rev Microbiol 9 193 203
68. HuGLiuIShamAStajichJEDietrichFS 2008 Comparative hybridization reveals extensive genome variation in the AIDS-associated pathogen Cryptococcus neoformans. Genome Biol 9 R41
69. DrawzSMBonomoRA 2010 Three decades of beta-lactamase inhibitors. Clin Microbiol Rev 23 160 201
70. MadeoFCarmona-GutierrezDRingJButtnerSEisenbergT 2009 Caspase-dependent and caspase-independent cell death pathways in yeast. Biochem Biophys Res Commun 382 227 231
71. XueCTadaYDongXHeitmanJ 2007 The human fungal pathogen Cryptococcus can complete its sexual cycle during a pathogenic association with plants. Cell Host Microbe 1 263 273
72. KozubowskiLHeitmanJ 2010 Septins enforce morphogenetic events during sexual reproduction and contribute to virulence of Cryptococcus neoformans. Mol Microbiol 75 658 675
73. DavidsonRCBlankenshipJRKrausPRde Jesus BerriosMHullCM 2002 A PCR-based strategy to generate integrative targeting alleles with large regions of homology. Microbiology 148 2607 2615
74. FraserJAGilesSSWeninkECGeunes-BoyerSGWrightJR 2005 Same-sex mating and the origin of the Vancouver Island Cryptococcus gattii outbreak. Nature 437 1360 1364
75. FraserJASubaranRLNicholsCBHeitmanJ 2003 Recapitulation of the sexual cycle of the primary fungal pathogen Cryptococcus neoformans var. gattii: implications for an outbreak on Vancouver Island, Canada. Eukaryot Cell 2 1036 1045
76. DavidsonRCCruzMCSiaRAAllenBAlspaughJA 2000 Gene disruption by biolistic transformation in serotype D strains of Cryptococcus neoformans. Fungal Genet Biol 29 38 48
77. PitkinJWPanaccioneDGWaltonJD 1996 A putative cyclic peptide efflux pump encoded by the TOXA gene of the plant-pathogenic fungus Cochliobolus carbonum. Microbiology 142 (Pt 6) 1557 1565
78. LeaDECoulsonCA 1949 The distribution of the numbers of mutants in bacterial populations. J Genet 49 264 285
79. WangPKimYPollackJNarasimhanBTibshiraniR 2005 A method for calling gains and losses in array CGH data. Biostatistics 6 45 58
80. SpellRMJinks-RobertsonS 2004 Determination of mitotic recombination rates by fluctuation analysis in Saccharomyces cerevisiae. Methods Mol Biol 262 3 12
81. DingJCBauerMDiamondDMLealMAJohnsonD 1997 Effect of severity of meningitis on fungicidal activity of flucytosine combined with fluconazole in a murine model of cryptococcal meningitis. Antimicrob Agents Chemother 41 1589 1593
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 11
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Multiple Candidate Effectors from the Oomycete Pathogen Suppress Host Plant Immunity
- The Splicing Factor Proline-Glutamine Rich (SFPQ/PSF) Is Involved in Influenza Virus Transcription
- A TNF-Regulated Recombinatorial Macrophage Immune Receptor Implicated in Granuloma Formation in Tuberculosis
- SH3 Domain-Mediated Recruitment of Host Cell Amphiphysins by Alphavirus nsP3 Promotes Viral RNA Replication