
Murid Herpesvirus-4 Exploits Dendritic Cells to Infect B Cells
Dendritic cells (DCs) play a central role in initiating immune responses. Some persistent viruses infect DCs and can disrupt their functions in vitro. However, these viruses remain strongly immunogenic in vivo. Thus what role DC infection plays in the pathogenesis of persistent infections is unclear. Here we show that a persistent, B cell-tropic gamma-herpesvirus, Murid Herpesvirus-4 (MuHV-4), infects DCs early after host entry, before it establishes a substantial infection of B cells. DC-specific virus marking by cre-lox recombination revealed that a significant fraction of the virus latent in B cells had passed through a DC, and a virus attenuated for replication in DCs was impaired in B cell colonization. In vitro MuHV-4 dramatically altered the DC cytoskeleton, suggesting that it manipulates DC migration and shape in order to spread. MuHV-4 therefore uses DCs to colonize B cells.
Published in the journal:
. PLoS Pathog 7(11): e32767. doi:10.1371/journal.ppat.1002346
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002346
Summary
Dendritic cells (DCs) play a central role in initiating immune responses. Some persistent viruses infect DCs and can disrupt their functions in vitro. However, these viruses remain strongly immunogenic in vivo. Thus what role DC infection plays in the pathogenesis of persistent infections is unclear. Here we show that a persistent, B cell-tropic gamma-herpesvirus, Murid Herpesvirus-4 (MuHV-4), infects DCs early after host entry, before it establishes a substantial infection of B cells. DC-specific virus marking by cre-lox recombination revealed that a significant fraction of the virus latent in B cells had passed through a DC, and a virus attenuated for replication in DCs was impaired in B cell colonization. In vitro MuHV-4 dramatically altered the DC cytoskeleton, suggesting that it manipulates DC migration and shape in order to spread. MuHV-4 therefore uses DCs to colonize B cells.
Introduction
Dendritic cells (DCs) act as sentinels against infection: they encode pathogen-responsive receptors, abound at pathogen entry sites, and orchestrate both innate and adaptiveimmune responses [1], [2]. Virus-infected DCs are generally immunogenic [3]–[5], and DC infection may be important for optimal T cell priming [6]. However several persistent viruses, which might be expected to limit their exposure to host immunity, efficiently infect DCs [7]–[10]. The infected DCs may function abnormally in vitro [11], [12], but the corresponding in vivo infections remain potently immunogenic. Therefore how DC infection benefits these persistent viruses, or whether it instead benefits the host, is unclear.
Murid Herpesvirus-4 (MuHV-4) is a gamma-herpesvirus that readily allows in vivo analysis of host colonization [13], [14]. Like Epstein-Barr virus and the Kaposi's Sarcoma-associated Herpesvirus, MuHV-4 persists in B cells [15], [16]. It also acutely infects macrophages and dendritic cells [17]–[19]. Myeloid infection provides MuHV-4 with a site of persistence in B cell-deficient mice, but the antibody deficiency of these mice leads to a somewhat atypical chronic lytic infection [20]; in immunocompetent mice, infected macrophages and dendritic cells are hard to detect long-term [19]. Therefore myeloid infection seems more likely to be important for establishing MuHV-4 host colonization than for maintaining it. Epstein-Barr virus and Kaposi's Sarcoma-associated Herpesvirus can also infect myeloid cells in vitro [9], [21]. While they seem rarely to do so in vivo, their clinical presentations post-date infection by at least 1 month [22], so early host colonization is rarely studied. Thus acute myeloid cell infection may not be unique to MuHV-4.
The difficulty of curing established gamma-herpesvirus infections makes early events in host colonization important to understand. It has been suggested that incoming Epstein-Barr virus infects B cells and so establishes latency directly [23]. This would argue against an important role for myeloid infection. However, good supporting evidence for direct B cell infection is lacking. Indeed vaccination to block Epstein-Barr virions binding to B cells failed to reduce the incidence of infection [24]. And while DNA from lung-inoculated, replication-deficient MuHV-4 has been found associated with B cells [25], [26], viral genome-positive B cells did not reach the spleen, so whether the detected DNA was a viable primary infection or merely adsorbed debris was unclear. That fibroblast-propagated MuHV-4 infects mice well [27] and B cells poorly [28] would argue against B cells being a significant primary target. Natural host entry probably occurs via the upper respiratory tract rather than the lung [29], but MuHV-4 lacking thymidine kinase or ribonucleotide reductase fails to infect by this route and neither enzyme is required for replication in B cells [30], [31]. Therefore here too B cells would seem an unlikely primary target.
How then does MuHV-4 reach B cells? HIV can infect T cells via DCs [32], [33], and DCs also communicate with B cells [34], so exploiting DC/lymphocyte interactions could be a common theme among lymphotropic viruses. However, in vivo evidence is again sparse. HIV-infected DCs are hard to find in vivo, and most HIV taken up by DCs in vitro is degraded. Thus it has been possible only to hypothesize that DCs contribute to host entry [35]. MuHV-4 allows more comprehensive analysis, and this is what we undertook here. As removing DCs causes immunosuppression [36], we used DC-specific cre recombinase expression to inhibit MuHV-4 replication in DCs or to mark genetically viruses that had replicated in a DC, while leaving normal DC functions intact.
Results
Direct visualization of MuHV-4 spread
That MuHV-4 colonizes B cells is indisputable, but where it first infects them is unclear. To identify the likely anatomical site we used viral luciferase expression [29] to track host colonization after upper respiratory tract infection (Fig. 1). Live imaging signals were evident in noses 3 days after inoculation and in the neck after 10 days (Fig. 1a). Signals were present in both sites after 6 days, indicating that the virus spread from nose to neck at 3–6 days post-infection.

Ex vivo imaging of dissected tissues at the peak of neck infection - day 10 - (Fig. 1b) established that this signal came entirely from the superficial cervical lymph nodes (SCLN), which receive lymphatic drainage from the nose [37]. No other tissues were luciferase+ at this time. Imaging dissected mice at day 6 (Fig. 1c) revealed luciferase expression in the nasal turbinates and in the SCLN, but not in intervening sites. Nor was luciferase expression evident in other lymph nodes, or in the nasal-associated lymphoid tissue that lies on either side of the palate. Therefore MuHV-4 appeared to move directly from the nose to the SCLN.
Host colonization measured by viral DNA loads and recoverable infectivity
We next tracked infection by quantitative PCR (Q-PCR) of viral DNA (Fig. 2a). In agreement with the luciferase imaging, viral genomes were detected in the nose at day 3 post-inoculation and not consistently in the SCLN until day 5. Recovering replication-competent virus by infectious centre assay (Fig. 2b) was less sensitive but showed similar trends, arguing that the DNA signals in the SCLN were due to infection rather than just accumulated viral debris. Both upper and lower respiratory tract infections showed viral genomes being most abundant in the peripheral epithelial site at day 5 post-inoculation, and most abundant in lymphoid tissue at day 14 (Fig. 2c). At day 5 viral genomes were more abundant in draining lymph nodes than in spleens, whereas by day 14 these sites were equivalent. These results were consistent with epithelial infection seeding first to its draining lymph nodes, and then disseminating to the spleen, presumably via B cells [38].

Kinetic comparison of DC and B cell infections
Previous analyses of MuHV-4 genome loads in different cell types have focussed on peak splenic titers, when most infected cells are B cells [18], [19]. We reasoned that if B cells only became infected once virus reached lymph nodes, then early on viral genome loads might be lower in lymph node B cells than in the cells first responsible for taking the virus there. Antigen transport of lymph nodes is a major function of DCs. We therefore tested whether DC infection might precede B cell infection, by separating these sub-populations from acutely infected lymph nodes on affinity columns and determining their viral genome loads by Q-PCR (Fig. 2d). At days 11 and 14 after virus inoculation into either noses or lungs, viral genome loads were higher in lymph node B220+ cells (B cells) than in CD11c+ cells (DCs), but at days 5 and 8 they were higher in DCs.
Further analysis (Fig. 2e) identified significantly higher viral genome loads also in CD11c-CD11b+ lymph node cells (macrophages) than in B cells early after virus inoculation into the lungs, but not after virus inoculation into the nose. At 8 days after virus inoculation into the upper respiratory tract, 105 cells purified from the SCLN of pooled pairs of mice yielded 20.0±7.6 infectious centres for macrophages, 53.3±26.2 for DCs and 14.7±4.3 for B cells (mean ± SEM, n = 3). Each population yielded <1 p.f.u. per 105 cells by plaque assay. Therefore after upper respiratory tract infection, a predominantly latent infection of DCs appeared to precede that of B cells. SCLN suspensions are typically 50% B cells and <5% DCs, so at all time points B cells accounted for most of the recoverable viral DNA. However virus seeds to the SCLN at a low level, is rapidly passed to B cells, which proliferate, and seeds asynchronously between individual mice. Thus by the time DC infection is readily detected, some B cell infection and amplification has inevitably occurred. The kinetic changes in relative genome load argued strongly for DC infection preceding B cell infection: the genome+ DCs at day 14 may have acquired infection from B cells, but this was unlikely at day 5 because B cell viral loads then were low.
Identification of MuHV-4-infected DCs
We sought next to visualize infected cells directly, using MuHV-4 that expresses eGFP from an intergenic EF1α promoter [39] (Fig. 3). At day 11 post-infection - that is after the onset of virus-driven lymphoproliferation - flow cytometry identified viral eGFP expression mainly in B cells (Fig. 3a): approximately 1% of CD19+ lymph nodes cells were eGFP+. The eGFP+ cells were also positive for surface immunoglobulin and MHC class II. Most were CD69+, consistent with MuHV-4 up-regulating this acute activation marker on B cells [40]. B cells are normally syndecan-4+ [41], so a surprising finding was that most eGFP+ cells were syndecan-4-. Syndecan-4 is a carrier of heparan sulfate, on which MuHV-4 infection strongly depends [42], so MuHV-4 may down-regulate syndecan-4 on infected B cells to prevent super-infection.

No clear CD19-eGFP+ population was identifiable by flow cytometry. However, immunohistochemistry at day 11 post-infection identified both eGFP+B220+ and eGFP+CD11c+ cells in lymph nodes (Fig. 3b). At day 7 (Fig. 3c) eGFP+ cells in the SCLN were 64.0±13.0% CD11c+, 14.1±5.1% B220+ and 21.9±12.3% neither (mean ± SEM, n = 6 mice, counting >100 eGFP+ cells per mouse). At day 11, SCLN eGFP+ cells were 12.0±2.8% CD11c+, 78.0±2.4% B220+, and 10.0±3.1% neither. Therefore both PCR of viral DNA and immunostaining of virus-expressed eGFP showed DC infection early in lymph node colonization, before B cell infection was well established. Our failure to detect a clear population of infected DCs by flow cytometry possibly reflected that these cells are difficult to isolate intact.
A marker virus for assaying exposure to cre recombinase
Cre-lox recombination allows transient infection events to be recorded by a permanent genetic mark [43]. We used this technology to identify MuHV-4 that had replicated in cre recombinase+ cells, by inserting a loxP-flanked eCFP expression cassette between the 3′ ends of ORFs 57 and 58 (Fig. 4a). The MuHV-4 BAC/eGFP cassette is also flanked by loxP sites [44], so BAC-derived viruses retain a single loxP site at the genome left end. To avoid recombination between this site and those flanking eCFP, we incorporated a point mutation into the spacer region of the latter. We then transfected BAC DNA into BHK-21 cells, passed the recovered eGFP+eCFP+ virus once through cre+ NIH-3T3 cells (2 p.f.u./cell) and selected eGFP-eCFP+ virus clones from the mixed progeny (Fig. 4b).

LoxP-eCFP MuHV-4 (eGFP-eCFP+) showed no in vivo growth deficit (Fig. S1). In vitro it rapidly lost eCFP expression when passed through cre+ NIH-3T3 cells (Fig. 4c). We quantitated this loss by counting plaques under phase contrast and typing each plaque as eCFP+ or eCFP- under ultra-violet illumination (Fig. 4d). LoxP-eCFP MuHV-4 also lost eCFP expression in cre+DCs - derived from CD11c-cre transgenic mouse bone marrow by growth in GM-CSF - albeit less dramatically than in cre+ fibroblasts (Fig. 4c). Thus eCFP loss provided a minimum estimate of the proportion of virions passing through a DC.
Functional evidence for MuHV-4 reaching B cells via DCs
We looked for a possible selective effect of eCFP excision by infecting non-transgenic (cre-) mice with different mixtures of eCFP+ and eCFP- derivatives of the loxP-eCFP virus, and 20 days later typing splenic infectious centres for eCFP expression (Fig. 4e). No marked difference in eCFP+ frequency was observed between the input and recovered viruses. ECFP loss therefore provided an unbiased marker of in vivo virus exposure to cre. A limitation of eCFP-based analysis was that even non-transgenic mice yielded 6.1±2.8% eCFP- plaques (mean±SD, n = 12). PCR across the ORF57/58 junction identified loxP recombination in 6/6 eCFP- plaques recovered from CD11c-cre mice infected with loxP-eCFP MuHV-4. 3/3 eCFP- plaques recovered from non-transgenic mice (which were rare) did not show loxP recombination, and were presumably due to cassette silencing: that viral expression cassettes are not always active even in lytically infected cells is well-established [45]. Therefore we took 10% eCFP- plaques (>1 SD above the background) as indicating significant virus exposure to cre recombinase.
We then infected CD11c-cre transgenic mice with loxP-eCFP MuHV-4 in the upper respiratory tract. The infectious virus recovered from noses showed little eCFP loss above the 10% cut-off (Fig. 4f). However eCFP loss from SCLN virus was significantly greater. Therefore DCs contributed relatively little to peripheral viral lytic replication, and much more to SCLN colonization. Splenic virus showed no further eCFP loss, consistent with the spread from the SCLN to here being accomplished by B cells. Substantial eCFP expression loss from the viruses of flow cytometrically sorted B cells (Fig. 4f) established that a significant proportion of the virus establishing long-term latency had passed through a DC.
The relative inefficiency of cre-mediated eCFP excision in CD11c-cre mice made it difficult to estimate exactly what proportion of the virus in B cells had passed through a DC. To answer this better we exposed DCs grown from CD11c-cre bone marrow in vitro to MuHV-4 (5 p.f.u./cell, 6 h, 37°C), then washed them in pH = 3 buffer to inactivate non-endocytosed virions and 24 h later - the earliest new infectivity can be recovered from MuHV-4-exposed DCs - titrated cell supernatants by plaque assay. ECFP typing under ultra-violet illumination identified 14.2±3.0% of plaques as eCFP- (mean ± SD of 10 cultures, counting at least 100 plaques for each culture). Thus assuming that an overnight infection of bone marrow-derived DCs equates roughly in cre exposure to DC-mediated in vivo virus transfer, essentially all the virus derived from B cells after upper respiratory tract infection had passed through a DC.
The virus recovered from lungs after lower respiratory tract infection (Fig. 4g) showed only modest eCFP loss. Unlike after upper respiratory tract infection, there was no increased eCFP loss upon reaching the draining lymph nodes (MLN). Therefore lung infection appeared to provide MuHV-4 with an alternative, DC-independent route to lymphoid tissue. Viruses recovered from spleens after lung infection showed more eCFP loss. However, nose infections inevitably accompany lung infections, so the splenic virus would also have come from the SCLN. Thus while DCs took virus to lymph nodes from the upper respiratory tract, they were less important when a more invasive entry point was used.
Cre-mediated virus inactivation
As a second approach to establishing the functional importance of DCs for host colonization, we generated a MuHV-4 mutant in which cre-mediated recombination would cause a lethal genomic deletion. Thus we inserted between ORFs 8 and 9 a loxP site compatible with that remaining at the genome left end after BAC cassette excision (Fig. 5a). Complete recombination between loxP sites would now excise both the BAC/eGFP cassette and the left end of the viral genome, including the essential ORFs 6, 7 and 8 [46]. It would also excise the M2 latency gene [47], the viral tRNA/miRNAs [48] and a likely promoter element for ORF73 [49], and so would probably also compromise the capacity of the viral genome to persist as a latent episome. Thus in contrast to eCFP excision from loxP-eCFP MuHV-4, which had no effect on viral fitness and so provided an index of virus exposure to cre recombinase, 8/9-loxP MuHV-4 would tell us the functional consequence of impaired DC infection.

To excise just the BAC cassette from 8/9-loxP MuHV-4 we passed it once through NIH-3T3-cre cells (2 p.f.u./cell) and selected viable eGFP- clones. These grew normally in cre- cells, but were severely attenuated for growth in cre+ fibroblasts (Fig. 5b). They were moderately attenuated for growth in cre+ DCs. To confirm the mechanism of attenuation we infected cre- and cre+ fibroblasts with wild-type or 8/9-loxP MuHV-4, then isolated infected cell DNA, digested it with HinDIII and probed it with a HinDIII-N viral genomic clone that spans the ORF8/ORF9 junction [50]. Cre-mediated deletion of M1-ORF8 was predicted to remove the left end of the HinDIII-N locus and join its remainder to the viral terminal repeats, thereby changing the probed fragment from 3632 bp to >20 kb (as MuHV-4 has up to 30 copies of its 1.2 kb terminal repeat unit), with a 1.2 kb submolar ladder due to variation in terminal repeat copy number. This is precisely what we observed (Fig. 5c).
In vivo consequences of virus inactivation in DCs
8/9-loxP MuHV-4 showed a significant defect in SCLN colonization at day 7 after inoculation into the noses of CD11c-cre mice relative to non-transgenic controls, whereas wild-type MuHV-4 showed no difference (Fig. 6a). An independently derived 8/9-loxP mutant showed the same phenotype, while a revertant virus did not (Fig. S2). Therefore DC-specific virus attenuation reduced virus spread to lymph nodes. A similar cre-dependent defect was observed in the colonization of B cells, purified from SCLN by flow cytometric sorting (Fig. 6b). 8/9-loxP virus lytic titers, by contrast, were not significantly different between the noses of cre+ and cre- mice (Fig. 6c), consistent with the limited loss of intergenic eCFP expression seen in noses (Fig. 4f).

Surprisingly, by day 15 post-infection (spleens and SCLN) the substantial early defect in SCLN colonization (Fig. 6d) of cre+ mice by 8/9-loxP MuHV-4 was no longer evident. MuHV-4 single gene knockouts [51], [52] have shown the same phenomenon of early replication defects not translating into differences in long-term latent loads. This possibly reflects that lower virus loads elicit less host response, such that mutant and wild-type viruses differ more in their speed of colonization than in their final set points. A different situation arises when mutant and wild-type viruses compete for a limited latency niche. To test this we co-infected cre+ and cre- mice with a 1∶100 mix of EF1a-eGFP and 8/9-loxP viruses (Fig. 6e), and determined their relative levels of host colonization 30 days later by typing spleen and lymph node infectious centres as eGFP- or eGFP+. The viruses recovered from non-transgenic mice showed the expected eGFP+:eGFP- ratio of 1∶100. In contrast, those recovered from CD11c-cre mice showed ratios of 1∶3–1∶4, a 25-30-fold bias against 8/9-loxP MuHV-4. Therefore in the face of virus competition, an attenuation of DC infection substantially reduced host colonization.
MuHV-4 alters the DC cytoskeleton
DCs transport peripherally acquired antigens to lymph nodes even without viral infection [1]. Therefore MuHV-4 could reach lymph nodes simply by infecting DCs peripherally and remaining latent. However, viruses often alter cell behaviour to make host colonization more efficient. We looked for such effects using DCs grown from bone marrow stem cells with GM-CSF. After overnight MuHV-4 infection (3 p.f.u./cell), most bone marrow-derived DCs are latently infected - typically 20–30% express late lytic genes [45]. However almost all virus-exposed DCs showed cytoskeletal changes (Fig. 7a): actin was rapidly relocated to peripheral cytoplasmic projections, and after overnight infection the DCs had become flattened with many displaying long cytoplasmic processes. Phosphotyrosine and more specifically Y925-phosphorylated (activated) focal adhesion kinase (Fig. 7b) adopted peripheral, punctate distributions consistent with focal adhesion formation. MuHV-4-exposed DCs do not become non-specifically activated [44], [53], [54], and equivalent cytoskeletal changes were not induced by DC activation with lipopolysaccharide. Therefore these cytoskeletal changes were induced specifically by MuHV-4.
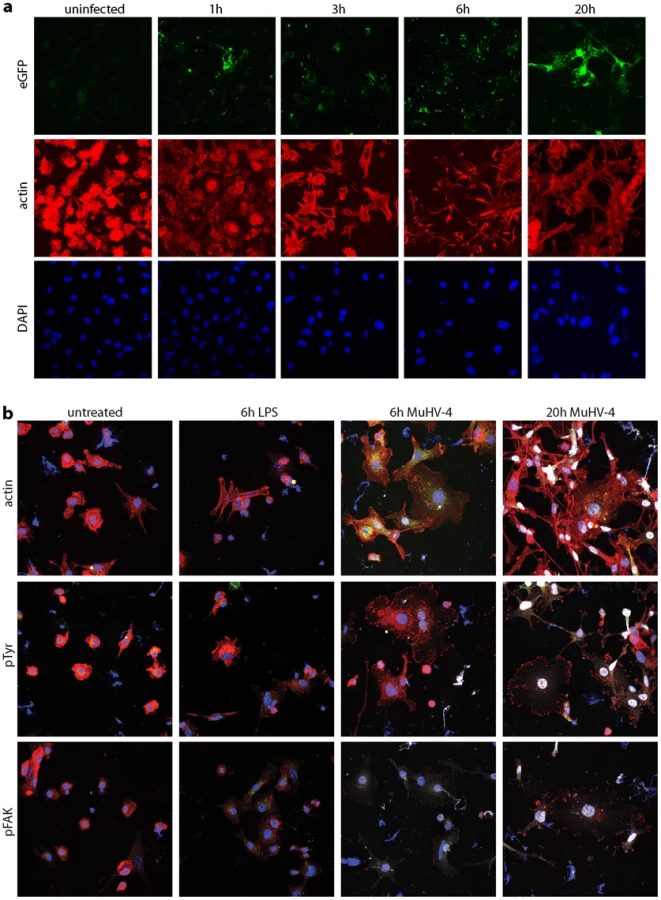
Real-time imaging (Video S1) showed that DCs became flattened and adherent within 6 h of exposure to MuHV-4, yet continued to show rapid changes in shape. This phenotype persisted at 20 h post-infection (Video S2), by which time the combination of adherence and dynamic remodelling and had led to the long cytoplasmic extensions seen in Fig. 7. Thus it appeared that infected DCs were motile but unable to detach from plastic. How these in vitro changes relate to in vivo DC migration is unclear, as tissue culture plastic provides a rather artificial surface, but it was clear that MuHV-4 actively manipulates the DC cytoskeleton.
Discussion
Lymphotropic viruses arrive at mucosal epithelia, whereas naive lymphocytes circulate through organized lymphoid tissue. Therefore lymphotropic viruses face a problem in reaching their target cells. Lymphatic channels normally provide a route for DCs and cell-free antigens to travel from epithelia to lymph node subcapsular sinuses. Small, soluble antigens can pass directly into B cell follicles via specialized conduits [55]–[57], but larger antigens - immune complexes [58], virus-sized particles [59], and cell-free virions [60], [61] - are first captured by subcapsular sinus macrophages. Thus viruses can enter lymph nodes via migratory DCs or subcapsular sinus macrophages.
Most analysis of DC migration has depended on indirect measures such as T cell priming [62]. In contrast, virion capture by subcapsular sinus macrophages has been observed directly [60], [61]. However, direct imaging has depended on injecting large virion numbers. Such high doses may reveal mainly high-capacity rather than high-efficiency capture pathways, and the tissue pressures created by injection tend to force material into and along lymphatics. Therefore the relevance to non-invasive infections of antigen injections must still be established. Here we analyzed lymph node colonization after a non-invasive infection. MuHV-4 does not establish a detectable cell-free viraemia [63], and depends for its in vivo propagation on cell/cell spread [64] more than on cell-free virion binding [65] or release [66]. Thus it might be expected to follow a cell-associated route to lymph nodes. Consistent with this idea, we identified a major role for DCs in passing infection to B cells. Thus MuHV-4 sets a new precedent for host exploitation by a lymphotropic virus.
We could not determine whether MuHV-4 reaches lymph nodes only via DCs, but the similar efficiencies of eCFP excision and 8/9-loxP virus attenuation between in vitro DC infection and in vivo host colonization argued that DC infection plays a predominant role. This would also be consistent with MuHV-4 still infecting the lymph nodes of B cell-deficient mice [67]. CD11c is a well-established DC marker, but is not exclusive to DCs [68]. Expression on activated T cells can be discounted here as MuHV-4 does not infect T cells. Lung macrophages express CD11c [68], as do subcapsular sinus macrophages at a low level [60]. However lysM-cre mice, which express cre in macrophages [69], showed substantial eCFP loss from loxP-eCFP MuHV-4 only after lower respiratory tract infection (unpublished data). Therefore while macrophages may feature prominently in MuHV-4 lung infection, DCs provided the major route of its transfer from nose to lymph nodes. Thus MuHV-4 exploits olfaction to enter the upper respiratory tract [29]; lymphocytes to persist [70]; and DCs to link them by virus transport.
Cells migrate by forming cytoplasmic protrusions, adhering these to the extracellular matrix, then detaching and contracting their trailing edges [71]. Actin, focal adhesion kinase and tyrosine phosphorylation all play central roles, so the redistributions of these markers in infected DCs, independent of viral lytic gene expression, was consistent with MuHV-4 inducing latently infected DCs to migrate. ORF27/ORF58-dependent actin rearrangements [72] could then promote further virion spread upon lytic reactivation. However, in vitro DC migration was prevented by infected DCs not detaching from plastic, and we cannot exclude that the infected SCLN DCs were resident and infected through antigen capture, rather than being migratory. Therefore virus-induced DC migration needs further investigation; current evidence establishes only that DC infection is important for establishing B cell infection in lymph nodes.
MuHV-4-infected DCs may also promote the amplification of B cell infection by secreting the M3 chemokine binding protein to protect in trans against CD8+ T cell attack [73]. In both this setting and that of virus transfer, the B cell latency defects of MuHV-4 lacking its MHC class I down-regulation gene K3 [51] or its bcl-2 homolog M11 [52] could reflect that these genes function in DCs [74], [75]. Exploiting DCs presumably brings gamma-herpesviruses advantages of efficiency and stealth. Whether it also creates possibilities for therapeutic intervention remains to be seen.
Materials and Methods
Mice
All animal experiments were approved by the Cambridge University ethical review board and by the UK Home Office (PPL 80/1992), and carried out in accordance with the Animals (Scientific Procedures) Act 1986. C57BL/6 and BALB/c mice were obtained from Harlan U.K. C57BL/6 back-crossed CD11c-cre mice, which express cre recombinase in DCs [76], were obtained from Jackson Laboratories and maintained as heterozygote × C57BL/6 non-transgenic crosses. Mice were typed by PCR of tail DNA, using the primers 5′-ACTTGGCAGCTGTCTCCAAG, 5′-GCGAACATCTTCAGGTTCTG (transgene-specific) and 5′ CAAATGTTGCTTGTCTGGTG, 5′ - GTCAGTCGAGTGCACAGTTT (internal control). Mice were infected with MuHV-4 when 6–12 weeks old. Intranasal infections with anaesthesia were in 30 µl; those without were in 5 µl. For luciferase imaging, mice were injected intraperitoneally with luciferin (2 mg/mouse), anaesthetized with isoflurane, then scanned with an IVIS Lumina (Caliper Life Sciences). To image specific tissues mice were killed and dissected after luciferin injection. All experiments conformed to local animal ethics regulations and Home Office Project Licence 80/1992.
Cells
Baby hamster kidney (BHK-21) cells, NIH-3T3 cells and NIH-3T3-CRE cells [50] were propagated in Dulbecco's modified Eagle medium (Invitrogen Corporation) supplemented with 2 mM glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin and 10% fetal calf serum (complete medium). Dendritic cells were derived from bone marrow progenitors of CD11c-cre mice or non-transgenic litter-mates. Bone marrow cells were depleted of adherent cells (30 min, 37°C) and then cultured in RPMI with 10% fetal calf serum, 50 µM 2-mercaptoethanol, 100 U/ml penicillin, 100 mg/ml streptomycin and 7.5 ng/ml GM-CSF (PeproTech). The medium was changed every 2 d, and non-adherent cells harvested after 7 d. These were >90% GR-1-CD11c+ by flow cytometry.
Bead-based cell separation
Lymph nodes were removed post-mortem, finely minced, digested (20 min) with type II collagenase (1 mg/ml, Worthington Biochemicals) plus DNaseI (20 µg/ml, Boehringer Mannheim), then incubated (5 min) in 100 mM EDTA to disrupt cell/cell conjugates. Debris was removed by filtration (100 µm). B cells, DCs and macrophages were then separated with antibody-coated magnetic beads (Miltenyi Biotec). First B cells were selected with anti-CD45R, then DCs were selected with anti-CD11c, then macrophages were selected with anti-CD11b. Each population was >90% pure by flow cytometry.
Viruses
Luciferase+ [29], EF1a-eGFP+ (May and Stevenson, 2010), and gM-eGFP+ [77] MuHV-4 reporter viruses have been described. To insert a floxed eCFP expression cassette into the MuHV-4 genome, the eCFP coding sequence was amplified by PCR (Pfu ploymerase, Promega Corporation) from pMSCV-IRES-eCFP using EcoRI and KpnI-restricted primers which also incorporated modified loxP sites. Specifically the GCATACAT spacer region was changed to GTATACAT [78] = loxP*. The loxP*-flanked eCFP coding sequence was then cloned as an EcoRI/KpnI-restricted fragment into the corresponding sites of pSP73-M3-pA [79]. This placed it between a 500 bp MuHV-4 M3 promoter and a bovine growth hormone polyadenylation site. The M3-driven eCFP expression cassette was then excised from pSP73-M3-pA with BglII+XhoI, blunted with Klenow fragment DNA polymerase and cloned into the blunted MfeI site (genomic co-ordinate 77176) [80] of a BglII MuHV-4 genomic clone (co-ordinates 75338–78717), again in pSP73. This placed it between the 3′ ends of ORFs 57 and 58. The eCFP expression cassette plus genomic flanks was then subcloned as a BglII fragment into the BamHI site of the pST76K-SR shuttle vector and recombined into a MuHV-4 BAC [44].
To insert a loxP site into the MuHV-4 genome between ORF8 (genomic coordinates 16526–19054) and ORF9 (genomic coordinates 19217–22300), we amplified by PCR 2 genomic flanks around genomic coordinate 19055, generating the loxP site from overlapping 3′ extensions of the inner primers. The 2 PCR products were then mixed and re-amplified with the outer primers to generate a single product. A SmaI/BglII-restricted fragment, corresponding to genomic coordinates 18614–19424 with the new loxP site at 19055, was then excised from this product, cloned into the SmaI/BamHI sites of pST76K-SR, and recombined into the MuHV-4 BAC. Mutant viruses were identified by restriction enzyme mapping and by DNA sequencing across the insertion site. We also derived a revertant virus by shuttle vector-mediated reconstitution of the original genome region without a loxP site. BAC DNA was reconstituted into infectious virus by transfection into BHK-21 cells (Fugene-6, Roche Diagnostics). The floxed BAC/GFP cassette was removed by virus passage through NIH 3T3-CRE cells, followed by plaque purification. Virus stocks were prepared in BHK-21 cells. Infected cell debris was removed by centrifugation (400×g, 3 min), and virions then recovered from supernatants by ultracentrifugation (38000×g, 90 min).
Infectivity assays
Virus stocks were titered by plaque assay on BHK-21 cells [66]. Cell monolayers were incubated with virus dilutions (2 h, 37°C), overlaid with 0.3% carboxymethylcellulose, and 4 days later fixed in 4% formaldehyde and stained with 0.1% toluidine blue for plaque counting. Infectious virus in lungs and noses was measured by freeze-thawing the tissues, then homogenizing them in 1 ml complete medium prior to plaque assay. Latent virus was measured by infectious centre assay [66]: spleen cells were co-cultured with BHK-21 cells, then fixed and stained for plaque counting after 4 days. Plaque assay titers of freeze-thawed lymphoid homogenates were always <1% of infectious center assay titers, so the latter essentially measured reactivable latent virus. To distinguish eCFP+ and eCFP- (or eGFP+ and eGFP-) viruses, plaque or infectious centre assays were performed in limiting dilution format. Each well was then scanned under normal illumination, and for each positive well a chosen plaque scored as fluorescent or not under ultra-violet illumination. Thus each positive well was counted just once.
Viral genome quantitation
MuHV-4 genomic co-ordinates 4166–4252, corresponding to the M2 locus, was amplified from tissue DNA (50–80 ng) by quantitative PCR (Rotor Gene 3000, Corbett Research). The PCR products were quantitated by hybridization with a Taqman probe (genomic coordinates 4218–4189) and converted to genome copies by comparison with a standard curve of cloned plasmid template amplified in parallel [81]. Cellular DNA was quantitated in parallel by amplifying part of the adenosine phosphoribosyl transferase gene.
Southern blotting
Viral DNA was extracted by alkaline lysis [66], digested with HinDIII, electrophoresed and transferred to nylon membranes (Roche Diagnostics). A 32P-dCTP labelled probe (APBiotech) was generated by random primer extension (DECA prime II kit, Ambion). Membranes were hybridised with probe (65°C, 18 h), washed in 30 mM sodium chloride/3 mM sodium citrate/0.1% sodium dodecyl sulfate at 65°C and exposed to X-ray film.
Flow cytometry
Cells infected with eCFP+ or GFP+ viruses were trypsinized, washed in PBS and analysed directly for eGFP and eCFP fluorescence on a Fortessa flow cytometer (BD Biosciences). For specific staining of lymph node cells, IgG Fc receptors were blocked by pre-incubation (30 min, 4°C) with unlabelled rat anti-CD16/32 mAb, then stained for CD19, CD69, MHC class II or syndecan-4 with phycoerythrin-conjugated rat mAbs (all from BD Biosciences) and for surface immunoglobulin with Alexafluor 633-conjugated goat anti-mouse Ig(H+L) pAb (Invitrogen). After washing, cells were analysed on a FACS Calibur using Cellquest (BD Biosciences) and Weasel (Walter and Eliza Hall Institute of Medical Research). For flow cytometric sorting, spleen cells were pooled from pairs of mice and stained with phycoerythrin-conjugated rat anti-mouse CD19 mAb and Alexafluor 633-conjugated goat anti-mouse Ig(H+L) pAb. CD19+Ig+ cells were selected with a FACS Vantage (BD Biosciences). The recovered cells were >98% pure.
Immunofluorescence
DCs were plated overnight onto poly-D-lysine-coated coverslips after 7 days of culture, then infected or not with MuHV-4. After further culture, the cells were washed with PBS, fixed in 2% formaldehyde, permeabilized with 0.1% Triton-X-100, blocked with 3% BSA and stained for actin with Alexafluor568-conjugated phalloidin (Invitrogen), for phospho-tyrosine with mAb PY99 (Santa Cruz Biotechnology) plus Alexafluor568-conjugated goat anti-mouse pAb (Invitrogen), and for Y925-phosphorylated focal adhesion kinase with ab38512 (AbCam) plus Alexafluor568-conjugated goat anti-rabbit pAb (Invitrogen). EGFP fluorescence was visualized directly. After washing in PBS, the cells were mounted in Prolong Gold with DAPI (Invitrogen) and imaged with a Leica Confocal microscope.
Lymph nodes were removed post-mortem, fixed (24 h, 4°C) in 1% formaldehyde/10 mM sodium periodate/75 mM L-lysine, then equilibrated in 30% sucrose, and frozen in OCT mounting medium. 5 µm sections were blocked with 2% goat serum then stained for CD45R (mAb RA3-6B2, B cells) or for CD11c (mAb HL3, DCs) (BD Biosciences) plus Alexafluor568 or 488-conjugated goat anti-rat or anti-hamster IgG pAb (Invitrogen). Infected cells were detected with rabbit anti-eGFP pAb (Abcam) plus Alexafluor488 or 568-conjugated goat anti-rabbit IgG pAb (Invitrogen). The sections were mounted in Prolong Gold (Invitrogen) and imaged with a Leica Confocal microscope.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. SteinmanRM 1991 The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9 271 296
2. JoffreONolteMASpörriRReiseSousaC 2009 Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev 227 234 247
3. MacatoniaSETaylorPMKnightSCAskonasBA 1989 Primary stimulation by dendritic cells induces antiviral proliferative and cytotoxic T cell responses in vitro. J Exp Med 169 1255 1264
4. BhardwajNBenderAGonzalezNBuiLKGarrettMC 1994 Influenza virus-infected dendritic cells stimulate strong proliferative and cytolytic responses from human CD8+ T cells. J Clin Invest 94 797 807
5. ZinkernagelRM 2002 On cross-priming of MHC class I-specific CTL: rule or exception? Eur J Immunol 32 2385 2392
6. FreigangSProbstHCvan den BroekM 2005 DC infection promotes antiviral CTL priming: the ‘Winkelried’ strategy. Trends Immunol 26 13 18
7. KnightSCMacatoniaSECruickshankKRudgePPattersonS 1993 Dendritic cells in HIV-1 and HTLV-1 infection. Adv Exp Med Biol 329 545 549
8. SalioMCellaMSuterMLanzavecchiaA 1999 Inhibition of dendritic cell maturation by herpes simplex virus. Eur J Immunol 29 3245 3253
9. RappoccioloGJenkinsFJHenslerHRPiazzaPJaisM 2006 DC-SIGN is a receptor for human herpesvirus 8 on dendritic cells and macrophages. J Immunol 176 1741 1749
10. SinclairJ 2008 Manipulation of dendritic cell functions by human cytomegalovirus. Expert Rev Mol Med 10 e35
11. AndrewsDMAndoniouCEGranucciFRicciardi-CastagnoliPDegli-EspostiMA 2001 Infection of dendritic cells by murine cytomegalovirus induces functional paralysis. Nat Immunol 2 1077 1084
12. RafteryMJSchwabMEibertSMSamstagYWalczakH 2001 Targeting the function of mature dendritic cells by human cytomegalovirus: a multilayered viral defense strategy. Immunity 15 997 1009
13. StevensonPGSimasJPEfstathiouS 2009 Immune control of mammalian gamma-herpesviruses: lessons from murid herpesvirus-4. J Gen Virol 90 2317 2330
14. SpeckSHGanemD 2010 Viral latency and its regulation: lessons from the gamma-herpesviruses. Cell Host Microbe 8 100 115
15. Sunil-ChandraNPEfstathiouSNashAA 1992 Murine gammaherpesvirus 68 establishes a latent infection in mouse B lymphocytes in vivo. J Gen Virol 73 3275 3279
16. FlañoEKimIJWoodlandDLBlackmanMA 2002 Gamma-herpesvirus latency is preferentially maintained in splenic germinal center and memory B cells. J Exp Med 196 1363 1372
17. WeckKEBarkonMLYooLISpeckSHVirginHW 1996 Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J Virol 70 6775 6780
18. FlañoEHusainSMSampleJTWoodlandDLBlackmanMA 2000 Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J Immunol 165 1074 1081
19. MarquesSEfstathiouSSmithKGHauryMSimasJP 2003 Selective Gene Expression of Latent Murine Gammaherpesvirus 68 in B Lymphocytes. J Virol 77 7308 7318
20. GangappaSKapadiaSBSpeckSHVirginHW 2002 Antibody to a lytic cycle viral protein decreases gammaherpesvirus latency in B-cell-deficient mice. J Virol 76 11460 11468
21. Guerreiro-CacaisAOLiLDonatiDBejaranoMTMorganA 2004 Capacity of Epstein-Barr virus to infect monocytes and inhibit their development into dendritic cells is affected by the cell type supporting virus replication. J Gen Virol 85 2767 2778
22. HoaglandRJ 1964 The incubation period of infectious mononucleosis. Am J Public Health Nations Health 54 1699 1705
23. FaulknerGCKrajewskiASCrawfordDH 2000 The ins and outs of EBV infection.e Trends Microbiol 8 185 189
24. SokalEMHoppenbrouwersKVandermeulenCMoutschenMLéonardP 2007 Recombinant gp350 vaccine for infectious mononucleosis: a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J Infect Dis 196 1749 1753
25. MoserJMFarrellMLKrugLTUptonJWSpeckSH 2006 A gammaherpesvirus 68 gene 50 null mutant establishes long-term latency in the lung but fails to vaccinate against a wild-type virus challenge. J Virol 80 1592 1598
26. KayhanBYagerEJLanzerKCookenhamTJiaQ 2007 A replication-deficient murine gamma-herpesvirus blocked in late viral gene expression can establish latency and elicit protective cellular immunity. J Immunol 179 8392 8402
27. TibbettsSALohJvan BerkelVMcClellanJSJacobyMA 2003 Establishment and maintenance of gammaherpesvirus latency are independent of infective dose and route of infection. J Virol 77 7696 7701
28. JarousseNChandranBCoscoyL 2008 Lack of heparan sulfate expression in B-cell lines: implications for Kaposi's sarcoma-associated herpesvirus and murine gammaherpesvirus 68 infections. J Virol 82 12591 12597
29. MilhoRSmithCMMarquesSAlenquerMMayJS 2009 In vivo imaging of murid herpesvirus-4 infection. J Gen Virol 90 21 32
30. GillMBWrightDESmithCMMayJSStevensonPG 2009 Murid herpesvirus-4 lacking thymidine kinase reveals route-dependent requirements for host colonization. J Gen Virol 90 1461 1470
31. GillMBMayJSColacoSStevensonPG 2010 Important role for the murid herpesvirus 4 ribonucleotide reductase large subunit in host colonization via the respiratory tract. J Virol 84 10937 10942
32. PopeMBetjesMGRomaniNHirmandHCameronPU 1994 Conjugates of dendritic cells and memory T lymphocytes from skin facilitate productive infection with HIV-1. Cell 78 389 398
33. LekkerkerkerANvan KooykYGeijtenbeekTB 2006 Viral piracy: HIV-1 targets dendritic cells for transmission. Curr HIV Res 4 169 176
34. MacPhersonGKushnirNWykesM 1999 Dendritic cells, B cells and the regulation of antibody synthesis. Immunol Rev 172 325 334
35. PiguetVSteinmanRM 2007 The interaction of HIV with dendritic cells: outcomes and pathways. Trends Immunol 28 503 510
36. JungSUnutmazDWongPSanoGde los SantosK 2002 In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 17 211 220
37. OghisoYMatsuokaO 1979 Distribution of colloidal carbon in lymph nodes of mice injected by different routes. Jpn J Exp Med 49 223 234
38. UsherwoodEJStewartJPRobertsonKAllenDJNashAA 1996 Absence of splenic latency in murine gammaherpesvirus 68-infected B cell-deficient mice. J Gen Virol 77 2819 2825
39. MayJSStevensonPG 2010 Vaccination with murid herpesvirus-4 glycoprotein B reduces viral lytic replication but does not induce detectable virion neutralization. J Gen Virol 91 2542 2552
40. StevensonPGDohertyPC 1999 Non-antigen-specific B-cell activation following murine gammaherpesvirus infection is CD4 independent in vitro but CD4 dependent in vivo. J Virol 73 1075 1079
41. YamashitaYOritaniKMiyoshiEKWallRBernfieldM 1999 Syndecan-4 is expressed by B lineage lymphocytes and can transmit a signal for formation of dendritic processes. J Immunol 162 5940 5948
42. GilletLMayJSStevensonPG 2009 In vivo importance of heparan sulfate-binding glycoproteins for murid herpesvirus-4 infection. J Gen Virol 90 602 613
43. SacherTPodlechJMohrCAJordanSRuzsicsZ 2008 The major virus-producing cell type during murine cytomegalovirus infection, the hepatocyte, is not the source of virus dissemination in the host. Cell Host Microbe 3 263 272
44. AdlerHMesserleMWagnerMKoszinowskiUH 2000 Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J Virol 74 6964 6974
45. SmithCMGillMBMayJSStevensonPG 2007 Murine gammaherpesvirus-68 inhibits antigen presentation by dendritic cells. PLoS One 2 e1048
46. SongMJHwangSWongWHWuTTLeeS 2005 Identification of viral genes essential for replication of murine gamma-herpesvirus 68 using signature-tagged mutagenesis. Proc Natl Acad Sci USA 102 3805 3810
47. HusainSMUsherwoodEJDysonHColecloughCCoppolaMA 1999 Murine gammaherpesvirus M2 gene is latency-associated and its protein a target for CD8(+) T lymphocytes. Proc Natl Acad Sci USA 96 7508 7513
48. BowdenRJSimasJPDavisAJEfstathiouS 1997 Murine gammaherpesvirus 68 encodes tRNA-like sequences which are expressed during latency. J Gen Virol 78 1675 1687
49. ColemanHMEfstathiouSStevensonPG 2005 Transcription of the murine gammaherpesvirus 68 ORF73 from promoters in the viral terminal repeats. J Gen Virol 86 561 574
50. EfstathiouSHoYMMinsonAC 1990 Cloning and molecular characterization of the murine herpesvirus 68 genome. J Gen Virol 71 1355 1364
51. StevensonPGMayJSSmithXGMarquesSAdlerH 2002 K3-mediated evasion of CD8(+) T cells aids amplification of a latent gamma-herpesvirus. Nat Immunol 3 733 740
52. de LimaBDMayJSMarquesSSimasJPStevensonPG 2005 Murine gammaherpesvirus 68 bcl-2 homologue contributes to latency establishment in vivo. J Gen Virol 86 31 40
53. FlañoEKayhanBWoodlandDLBlackmanMA 2005 Infection of dendritic cells by a gamma2-herpesvirus induces functional modulation. J Immunol 175 3225 3234
54. HochreiterRPtaschinskiCKunkelSLRochfordR 2007 Murine gammaherpesvirus-68 productively infects immature dendritic cells and blocks maturation. J Gen Virol 88 1896 1905
55. GretzJENorburyCCAndersonAOProudfootAEShawS 2000 Lymph-borne chemokines and other low molecular weight molecules reach high endothelial venules via specialized conduits while a functional barrier limits access to the lymphocyte microenvironments in lymph node cortex. J Exp Med 192 1425 1440
56. PapeKACatronDMItanoAAJenkinsMK 2007 The humoral immune response is initiated in lymph nodes by B cells that acquire soluble antigen directly in the follicles. Immunity 26 491 502
57. RoozendaalRMempelTRPitcherLAGonzalezSFVerschoorA 2009 Conduits mediate transport of low-molecular-weight antigen to lymph node follicles. Immunity 30 264 276
58. PhanTGGrigorovaIOkadaTCysterJG 2007 Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat Immunol 8 992 1000
59. CarrascoYRBatistaFD 2007 B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity 27 160 171
60. JuntTMosemanEAIannaconeMMassbergSLangPA 2007 Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature 450 110 114
61. GonzalezSFLukacs-KornekVKuligowskiMPPitcherLADegnSE 2010 Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat Immunol 11 427 434
62. RandolphGROchandoJPartida-SánchezS 2008 Migration of Dendritic Cell Subsets and their Precursors. Ann. Rev. Immunol. 26 293 316
63. Sunil-ChandraNPEfstathiouSArnoJNashAA 1992 Virological and pathological features of mice infected with murine gamma-herpesvirus 68. J Gen Virol 73 2347 2356
64. MayJSWalkerJColacoSStevensonPG 2005 The murine gammaherpesvirus 68 ORF27 gene product contributes to intercellular viral spread. J Virol 79 5059 5068
65. GilletLMayJSColacoSStevensonPG 2007 Glycoprotein L disruption reveals two functional forms of the murine gammaherpesvirus 68 glycoprotein H. J Virol 81 280 291
66. de LimaBDMayJSStevensonPG 2004 Murine gammaherpesvirus 68 lacking gp150 shows defective virion release but establishes normal latency in vivo. J Virol 78 5103 5112
67. NashAADutiaBMStewartJPDavisonAJ 2001 Natural history of murine gamma-herpesvirus infection. Philos Trans R Soc Lond B Biol Sci 356 569 579
68. HumeDA 2011 Applications of myeloid-specific promoters in transgenic mice support in vivo imaging and functional genomics but do not support the concept of distinct macrophage and dendritic cell lineages or roles in immunity. J Leukoc Biol 89 525 538
69. ClausenBEBurkhardtCReithWRenkawitzRForsterI 1999 Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8 265 277
70. UsherwoodEJRossAJAllenDJNashAA 1996 Murine gammaherpesvirus-induced splenomegaly: a critical role for CD4 T cells. J Gen Virol 77 627 630
71. Le ClaincheCCarlierMF 2008 Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol Rev 88 489 513
72. GillMBEdgarRMayJSStevensonPG 2008 A gamma-herpesvirus glycoprotein complex manipulates actin to promote viral spread. PLoS One 3 e1808
73. RiceJde LimaBStevensonFKStevensonPG 2002 A gamma-herpesvirus immune evasion gene allows tumor cells in vivo to escape attack by cytotoxic T cells specific for a tumor epitope. Eur J Immunol 32 3481 3487
74. StevensonPG 2004 Immune evasion by gamma-herpesviruses. Curr Opin Immunol 16 456 462
75. MountAMMassonFKupresaninFSmithCMMayJS 2010 Interference with dendritic cell populations limits early antigen presentation in chronic γ-herpesvirus-68 infection. J Immunol 185 3669 3676
76. CatonMLSmith-RaskaMRReizisB 2007 Notch-RBP-J signaling controls the homeostasis of CD8 - dendritic cells in the spleen. J Exp Med 204 1653 1664
77. MayJSSmithCMGillMBStevensonPG 2008 An essential role for the proximal but not the distal cytoplasmic tail of glycoprotein M in murid herpesvirus 4 infection. PLoS One 3 e2131
78. HoessRHWierzbickiAAbremskiK 1986 The role of the loxP spacer region in P1 site-specific recombination. Nucleic Acids Res 14 2287 2300
79. MayJSColemanHMBonameJMStevensonPG 2005 Murine gammaherpesvirus-68 ORF28 encodes a non-essential virion glycoprotein. J Gen Virol 86 919 928
80. VirginHWLatreillePWamsleyPHallsworthKWeckKE 1997 Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol 71 5894 5904
81. GasparMGillMBLösingJBMayJSStevensonPG 2008 Multiple functions for ORF75c in murid herpesvirus-4 infection. PLoS One 3 e2781
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 11
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Multiple Candidate Effectors from the Oomycete Pathogen Suppress Host Plant Immunity
- The Splicing Factor Proline-Glutamine Rich (SFPQ/PSF) Is Involved in Influenza Virus Transcription
- A TNF-Regulated Recombinatorial Macrophage Immune Receptor Implicated in Granuloma Formation in Tuberculosis
- SH3 Domain-Mediated Recruitment of Host Cell Amphiphysins by Alphavirus nsP3 Promotes Viral RNA Replication