
Global mRNA Degradation during Lytic Gammaherpesvirus Infection Contributes to Establishment of Viral Latency
During a lytic gammaherpesvirus infection, host gene expression is severely restricted by the global degradation and altered 3′ end processing of mRNA. This host shutoff phenotype is orchestrated by the viral SOX protein, yet its functional significance to the viral lifecycle has not been elucidated, in part due to the multifunctional nature of SOX. Using an unbiased mutagenesis screen of the murine gammaherpesvirus 68 (MHV68) SOX homolog, we isolated a single amino acid point mutant that is selectively defective in host shutoff activity. Incorporation of this mutation into MHV68 yielded a virus with significantly reduced capacity for mRNA turnover. Unexpectedly, the MHV68 mutant showed little defect during the acute replication phase in the mouse lung. Instead, the virus exhibited attenuation at later stages of in vivo infections suggestive of defects in both trafficking and latency establishment. Specifically, mice intranasally infected with the host shutoff mutant accumulated to lower levels at 10 days post infection in the lymph nodes, failed to develop splenomegaly, and exhibited reduced viral DNA levels and a lower frequency of latently infected splenocytes. Decreased latency establishment was also observed upon infection via the intraperitoneal route. These results highlight for the first time the importance of global mRNA degradation during a gammaherpesvirus infection and link an exclusively lytic phenomenon with downstream latency establishment.
Published in the journal:
. PLoS Pathog 7(7): e32767. doi:10.1371/journal.ppat.1002150
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002150
Summary
During a lytic gammaherpesvirus infection, host gene expression is severely restricted by the global degradation and altered 3′ end processing of mRNA. This host shutoff phenotype is orchestrated by the viral SOX protein, yet its functional significance to the viral lifecycle has not been elucidated, in part due to the multifunctional nature of SOX. Using an unbiased mutagenesis screen of the murine gammaherpesvirus 68 (MHV68) SOX homolog, we isolated a single amino acid point mutant that is selectively defective in host shutoff activity. Incorporation of this mutation into MHV68 yielded a virus with significantly reduced capacity for mRNA turnover. Unexpectedly, the MHV68 mutant showed little defect during the acute replication phase in the mouse lung. Instead, the virus exhibited attenuation at later stages of in vivo infections suggestive of defects in both trafficking and latency establishment. Specifically, mice intranasally infected with the host shutoff mutant accumulated to lower levels at 10 days post infection in the lymph nodes, failed to develop splenomegaly, and exhibited reduced viral DNA levels and a lower frequency of latently infected splenocytes. Decreased latency establishment was also observed upon infection via the intraperitoneal route. These results highlight for the first time the importance of global mRNA degradation during a gammaherpesvirus infection and link an exclusively lytic phenomenon with downstream latency establishment.
Introduction
A number of viruses from diverse evolutionary lineages down-regulate cellular gene expression through a variety of mechanisms [1], [2], [3], [4]. For certain viruses this host shutoff activity is a consequence of viral gene expression strategies, such as influenzavirus cap snatching [5] or picornavirus translation factor cleavage to facilitate alternate mechanisms of ribosome engagement [6]. For others, the contribution of host shutoff towards viral replication and gene expression is not as apparent, although proposed roles include resource reallocation and immune evasion [6], [7]. In many cases, the precise in vivo function has been difficult to delineate, in part due to the multifunctional nature of the viral proteins driving host shutoff and the lack of appropriate model systems. Within this latter category are the gammaherpesviruses, which direct host shutoff primarily through the induction of host mRNA degradation.
Gammaherpesviruses include the human pathogens Epstein-Barr virus (EBV/HHV-4) and Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) [8], the etiologic agents of some of the most common AIDS-associated cancers, such as Burkitt's lymphoma, nasopharyngeal carcinoma, and primary effusion lymphoma. The related murine gammaherpesvirus 68 (MHV68) is often used as a model for human gammaherpesviruses because, unlike KSHV and EBV, it is genetically tractable, readily replicates in tissue culture without the use of chemicals or overexpression to overcome latency, and infects small rodents including laboratory mice [9], [10], [11]. MHV68 has therefore been instrumental in the identification of factors that contribute to in vivo replication and pathogenesis of the gammaherpesviruses.
All herpesviruses exhibit a biphasic lifecycle. In the latent state few viral genes are expressed, and the viral genome is maintained in the nucleus of the host cell until stimulating signals induce lytic reactivation [12], [13], [14]. Most gammaherpesvirus-induced diseases are associated with latency. During the lytic cycle the vast majority of viral genes are expressed, and the virus undergoes active replication to produce progeny virions. At this stage during productive replication, gammaherpesviruses induce global mRNA degradation via the product of the alkaline exonuclease gene, termed SOX in KSHV, muSOX (ORF37) in MHV68, and BGLF5 in EBV [1], [15], [16]. SOX and its homologs are conserved in all subfamilies of herpesviruses, where they were first identified as DNA exonucleases critical for the resolution of branched structures that arise during viral DNA replication [17], [18], [19], [20]. Only in members of the gammaherpesvirus subfamily does the SOX protein have an additional mRNA turnover function. Although the precise mechanism by which these proteins cause mRNA degradation remains unclear, biochemical evidence suggests that at least EBV BGLF5 and KSHV SOX possess some intrinsic ribonuclease (RNase) activity in vitro [21], [22]. In addition to mRNA turnover, SOX promotes the nuclear relocalization of cytoplasmic poly(A) binding protein (PABPC), which leads to aberrant polyadenylation and nuclear retention of mRNAs [23], [24]. The failure to repopulate the cytoplasm with newly transcribed mRNAs is thought to increase the overall magnitude of the host gene expression blockade [23].
The contribution of host shutoff towards the gammaherpesviral lifecycle is unknown, and efforts to delete muSOX from MHV68 yielded virus unable to replicate, presumably due to an essential role for the DNase activity [16]. Previous studies found the DNase and host shutoff functions of SOX and BGLF5 to be genetically separable [25], [26], making it possible to evaluate the role of each function of the protein in isolation. We report herein the identification of a functionally similar MHV68 muSOX point mutant that renders the virus selectively defective for host shutoff, enabling the analysis of the role of this phenotype during infection of cultured cells and in vivo. The mutant virus exhibited little to no defect during multi-step growth curves in tissue culture or during acute replication in the mouse lung following intranasal inoculation. Unexpectedly, the virus accumulated to significantly reduced levels in the lymph nodes at 10 days post infection and was highly attenuated at the downstream stage of latency establishment. Once trafficking was bypassed via an intraperitoneal infection, the host shutoff defective virus still was attenuated in establishing latency in the spleen. Our results identify for the first time a key role for the lytic mRNA turnover activity in establishing viral latency, emphasizing the important interplay between these seemingly disparate stages of the viral lifecycle.
Results
Identification of a Host Shutoff Defective muSOX Mutant
We sought to evaluate the contribution of muSOX-induced mRNA turnover towards the gammaherpesvirus lifecycle through the generation of a muSOX mutant lacking mRNA turnover activity but retaining the conserved DNase function. We engineered mutations in the sites necessary for host shutoff activity in muSOX homologs from other gammaherpesviruses (EBV BGLF5 [25] and KSHV SOX [26]), as well as in several sites selectively conserved amongst these homologs that lay outside the putative DNase domains [27]. However, none of the mutants generated had the desired single-function phenotype (Table S1).
We therefore designed an unbiased screen to investigate a large number of mutants following the previous methodology used to identify the single-function SOX variants (Figure 1A) [26]. Briefly, the muSOX gene was amplified by error-prone PCR under conditions designed to introduce 1–4 mutations per gene. The PCR product was then cloned into a mammalian expression vector, and clones were screened for the ability to repress expression of a GFP reporter in a fluorescence-based assay in 293T cells. Mutants that did not deplete GFP fluorescence were categorized as host shutoff defective and further analyzed for wild-type protein expression levels by Western blotting, as even minor changes to the muSOX primary amino acid sequence often significantly reduced its protein expression. Finally, candidate mutants were tested for retention of DNase activity using an in vitro assay established previously for SOX and BGLF5 [25], [26]. After screening approximately 150 candidates, we identified a clone that lacked mRNA degradation activity of the GFP reporter, but retained wild-type protein expression levels and DNase activity. Sequencing revealed this clone to have only a single non-silent mutation, the amino acid substitution R443M, which we modified to R443I for purposes of screening by restriction digest. MuSOX R443I failed to suppress GFP protein expression in the fluorescent-based assay for host shutoff (Figure 1B), which was a consequence of its inability to degrade GFP mRNA (Figure 1C). R443I is expressed at slightly higher levels than wild-type (WT) muSOX (Figure 1D), as expected of a mutant that cannot promote turnover of its own mRNA. Importantly, WT and R443I muSOX both degrade linear DNA with very similar kinetics (Figure 1E). We conclude that the amino acid substitution R443I produces a single-function muSOX mutant selectively defective for host shutoff activity.

Generation and in vitro Characterization of MHV68.ΔHS
To investigate the role of muSOX-induced host shutoff during viral infection, we engineered a host shutoff defective MHV68 virus by replacing WT muSOX with muSOX R443I using bacterial artificial chromosome (BAC)-based homologous recombination [28]. The R443I mutation introduced an additional PsiI restriction site within the muSOX gene, allowing us to screen for successful recombination via restriction digest (Figure 2A). We also generated a mutant rescue (MR) virus by replacing the R443I mutant with WT muSOX to ensure that any observed phenotypes could be attributed to the R443I mutation rather than to unexpected secondary mutations elsewhere in the viral genome. Restriction digests with PsiI and EcoRI confirmed that both recombinant viruses were successfully isolated and did not have any unexpected recombination events (Figures 2B & 2C). These viruses shall henceforth be referred to as MHV68.ΔHS (host shutoff defective virus) and MHV68.MR (mutant rescue virus). Sequencing of muSOX and the surrounding genomic regions in both recombinants confirmed that only the desired changes were introduced.

We next verified that the R443I mutation in MHV68.ΔHS conferred a host shutoff defect during viral infection. We assayed levels of several endogenous mRNAs by real-time quantitative PCR (RT-qPCR) following infection of NIH 3T3 fibroblasts with wild-type MHV68 (MHV68.WT), MHV68.ΔHS, or MHV68.MR. WT and MR infections reduced endogenous β-actin (actB), tubulin-β (tubb5), rplp2, and gapdh mRNA levels to 20–40% of the mock infected sample. In contrast, mRNA levels remained significantly higher in MHV68.ΔHS infected cells (Figure 3A–3D). Thus, the single amino acid change R443I in muSOX is sufficient to suppress virus-induced endogenous mRNA turnover. The somewhat lower endogenous mRNA levels in MHV68.ΔHS compared to mock infected cells could be due either to incomplete inactivation of the muSOX host shutoff function, or to the contribution of one or more additional viral genes towards host shutoff.

An additional phenotype linked to host shutoff caused by muSOX and its homologs is the relocalization of cytoplasmic poly(A) binding protein (PABPC) into the nuclei of infected cells [24]. We observed clear nuclear relocalization of PABPC in cells infected with MHV68.WT, whereas in mock infected or cells infected with MHV68.ΔHS, PABPC remained cytoplasmic (Figure 3E). Thus, MHV68.ΔHS is defective for two hallmarks of host shutoff, mRNA depletion and PABPC relocalization. These results also demonstrate for the first time that muSOX-induced host shutoff is necessary to drive PABPC relocalization during a viral infection.
Previous studies with MHV68 mutants have shown that muSOX is critical for replication in cultured cells [16], [19]. To determine the contribution of host shutoff activity towards this defect, we performed a multi-step growth curve in murine fibroblasts. 3T3 cells were infected at a multiplicity of infection (MOI) of 0.1 plaque forming units (pfu) per cell and MHV68.WT, MHV68.ΔHS, and MHV68.MR all replicated to equivalent titers and with similar kinetics (Figure 4). We sequenced the muSOX gene from MHV68.ΔHS samples at 5 days post-infection (dpi) and found no evidence for reversion or secondary mutations (data not shown). Therefore, muSOX-dependent mRNA degradation had little effect on viral titers in 3T3 cells through multiple replication cycles.

MHV68.ΔHS Causes Larger Plaques to Develop and Increased Expression of Lytic Cycle Proteins
Interestingly, we noticed that plaques derived from MHV68.ΔHS infections differed morphologically from those obtained upon MHV68.WT or MHV68.MR infections (Figure 5A). To quantify these differences, we measured 75 plaques from 5 independent MHV68.ΔHS and MHV68.MR infections and found that, indeed, MHV68.ΔHS plaques were generally larger (p-value <0.01) and had a broader frequency distribution (Figure 5B). This seemed unlikely to be a consequence of more rapid lytic replication, given the above growth curve results. In addition, the altered plaque size does not appear to be caused by enhanced cell-to-cell spread of the mutant virus, as we observed no significant differences in the ratio of extracellular to intracellular virus produced in 3T3 cells infected with MHV68.WT, MHV68.ΔHS, and MHV68.MR (data not shown).

We and others have consistently observed that plaques formed upon infection with wild-type (or mutant rescue) MHV68 are often quite heterogeneous in size. One possible explanation for this is asynchronous entry into the lytic cycle, in which smaller plaques arise from later entry into the lytic cycle. In this regard, we sought to determine whether the large plaque phenotype of MHV68.ΔHS might be due to enhanced entry into the lytic cycle, signified by an increased percentage of infected cells expressing early lytic proteins relative to the wild-type virus. To test this hypothesis, we acquired a version of MHV68 (MHV68-YFP) which constitutively expresses YFP from a CMV promoter, regardless of whether infected cells are in the lytic or latent phase [29]. Thus, YFP expression serves as a general marker of infection, whereas lytically infected cells are identified by immunofluorescence-based detection of the early lytic protein muSOX. NIH 3T3 cells were infected at an MOI of 1 with either MHV68-YFP or a version of MHV68-YFP bearing the muSOX R443I mutation (MHV68-YFP.ΔHS), and the percentage of cells co-expressing muSOX and YFP was calculated at 18 hours post infection. A similar number of YFP-expressing cells were detected in cultures infected with MHV68-YFP and MHV68-YFP.ΔHS, indicating equivalent efficiency of initial infection (Figure 5C & data not shown). However, there was an approximately three-fold increase in the percent of cells co-expressing both YFP and muSOX following infection with MHV68-YFP.ΔHS compared to MHV68-YFP (30.1% versus 11.0%; Figure 5C). A similar trend was observed when the YFP - and muSOX-expressing cell populations were analyzed by flow cytometry following infection with these viruses (Figure S1). Also in agreement with these findings were increased levels of the viral ORF54 transcript following a MHV68.ΔHS infection relative to MHV68.WT or MHV68.MR (Figure S2). Thus, enhanced and/or more rapid expression of lytic genes occurs in cells infected with MHV68.ΔHS, perhaps escalating entry into the lytic cycle and leading to larger plaques.
We also evaluated whether increased cell death might contribute to the larger plaques observed following a MHV68.ΔHS infection. We first measured necrosis by quantifying levels of lactate dehydrogenase released into the media at 16 hpi, but detected no measurable release following infection with any of the viruses (Figure S3A). We next monitored apoptosis by several assays, including measuring caspase activity, and testing for loss of plasma membrane symmetry and increased membrane permeability. While there was a slight, but statistically significant, increase in caspase 3/7 activity following MHV68.ΔHS infection relative to MHV68.WT or MHV68.MR infection, in all three cases the level was below that observed in mock infected cells (Figure S3B), making it unlikely that this is a primary cause of the increased plaque size. Plasma membrane asymmetry was monitored using PE-conjugated Annexin V, which binds to a component of the plasma membrane normally found on the cytoplasmic surface, and membrane permeability was quantified by the nucleotide binding dye 7-Amino-Actinomycin (7-AAD). By flow cytometry we detected no significant increase in Annexin V binding or 7-AAD staining following infection with either MHV68.ΔHS or MHV8.MR relative to the uninfected control (Figure S3C). Collectively, these findings indicate that although a higher percentage of MHV68.ΔHS infected cells express viral lytic antigens, this does not appear to cause enhanced cell death, nor does it culminate in higher viral titers.
Host Shutoff is Dispensable for Acute Replication in vivo
The presumed natural route of MHV68 infection is through the upper respiratory tract [30], whereupon the virus undergoes lytic replication in the mucosal epithelial cells lining the mouse lung and nasal cavity. Following intranasal inoculation, viral loads in the lung peak 5–8 dpi before subsiding as the virus traffics to the draining lymph nodes and ultimately to the spleen [31]. A burst of lytic replication occurs in these sites of latency establishment [30], [32] which seeds subsequent latency, primarily in germinal center B cells, although also in macrophages and dendritic cells [33]. By 14–18 dpi splenomegaly develops, and as many as one out of 100 splenocytes harbor the latent viral genome [34].
Host shutoff manifests with delayed early kinetics during lytic replication, consistent with the onset of muSOX expression [16]. We therefore hypothesized that MHV68.ΔHS would be attenuated during lytic viral replication in the lung. We infected C57BL/6 mice intranasally with 5×104 pfu of MHV68.WT, MHV68.ΔHS, or MHV68.MR, and monitored viral titers in the lung at 3, 5, and 7 dpi by plaque assay. Viral titers were equivalent at 3 dpi for all three viruses, and at 5 and 7 dpi we observed only a very modest 2–3 fold defect in MHV68.ΔHS relative to MHV68.MR accumulation (Figure 6). Although MHV68.WT reached higher titers than MHV68.MR at 5 dpi, the two viruses accumulated to equivalent levels at all earlier and later timepoints (see subsequent sections). To ensure MHV68.ΔHS had not reverted back to WT or incorporated any compensatory mutations, we sequenced the muSOX gene from infected lung homogenate but found no changes. Thus, contrary to our hypothesis, host shutoff appears largely dispensable for acute replication of gammaherpesviruses in vivo.

MHV68.ΔHS is Attenuated in Latency Establishment
To evaluate the role of host shutoff during later stages of the viral lifecycle, mice were infected intranasally with 5×104 pfu of MHV68.WT, MHV68.ΔHS, or MHV68.MR, and the spleens were harvested at 17 dpi, during the peak of latency establishment. Interestingly, mice infected with MHV68.ΔHS did not display the characteristic splenomegaly and had spleens three to four times smaller than MHV68.WT and MHV68.MR infected mice (Figure 7A), indicating a role for host shutoff in viral pathogenesis.
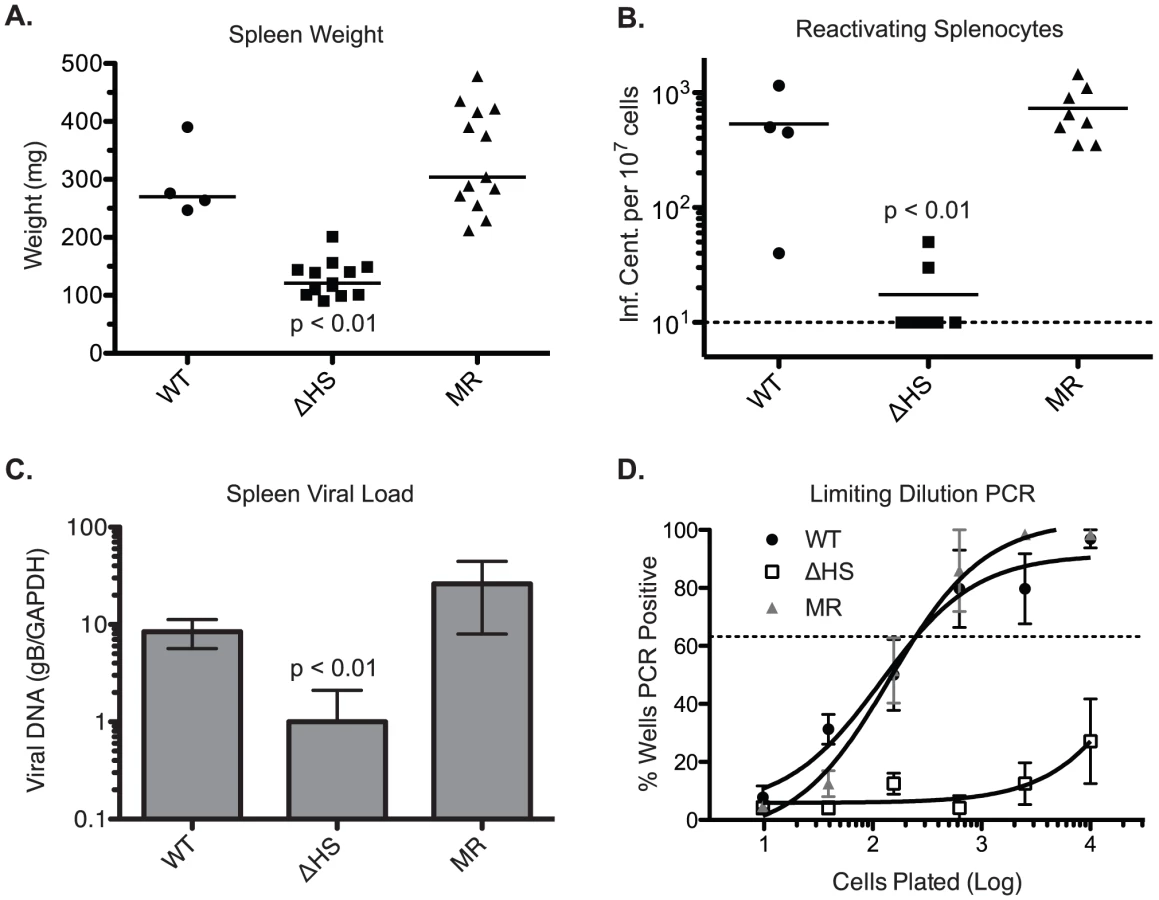
To evaluate the cause of MHV68.ΔHS attenuation, we first performed an infectious center assay to test whether the splenocytes from infected mice harbored latent virus capable of lytic reactivation. In this assay, equivalent numbers of splenocytes from infected mice are overlayed onto cultured mouse fibroblasts. Spontaneous lytic reactivation of the splenocytes leads to infection of the fibroblasts, which can be quantified by counting the resulting plaques [34]. Splenocytes from MHV68.ΔHS infected mice reactivated at a significantly lower frequency than those from MHV68.WT or MHV68.MR infections (Figure 7B). This reactivation frequency was independently assessed in a subset of the samples by a limiting dilution cytopathic effect assay, which yielded identical results (data not shown). Plaque assays on spleen homogenates confirmed that no preformed virus was present, indicating that the virus detected in these experiments originated from latently infected cells.
The reduction in lytic reactivation frequency could either be a consequence of a decreased number of latently infected splenocytes or, alternatively, a failure of latently infected cells to undergo lytic reactivation. To distinguish these possibilities, we first monitored viral DNA load in the spleens of infected mice using a previously described qPCR assay for the glycoprotein B (gB) gene [35] and found that MHV68.ΔHS infected splenocytes harbor fewer viral genomes than splenocytes infected with MHV68.WT or MHV68.MR (Figure 7C). We also analyzed the frequency of splenocytes harboring the viral genome by limiting dilution PCR. Briefly, serial dilutions of splenocytes were plated in a 96 well plate and subjected to nested PCR for the viral ORF50 gene [36]. Spiked template controls confirmed that in this assay we were achieving near single copy sensitivity for the viral genome, with a very low rate of false positives (data not shown). While approximately one out of 200 splenocytes latently harbor the MHV68.WT or MHV68.MR genome, the frequency of MHV68.ΔHS genome harboring splenocytes was less than one out of 10,000 splenocytes (Figure 7D). Thus, while host shutoff appears dispensable for acute replication in the lung, it plays an important role in the downstream events leading to efficient latency establishment in the spleen.
MHV68.ΔHS Exhibits Defects in Both Trafficking and Latency Establishment
During the normal course of an infection, the virus traffics from the site of lytic replication in the lung to the draining lymph nodes and spleen where latency is established. We reasoned that the decreased frequency of latently infected splenocytes during MHV68.ΔHS infection could be a consequence of a defect in trafficking, persistence at these sites, or a latency establishment defect. We quantified levels of virus in the cervical lymph node at 10 dpi, an intermediate time point after peak virus replication in the lung but before peak latency establishment in the spleen. Following an intranasal inoculation, high levels of MHV68 have been found in the cervical lymph node at this time point [30], [32]. Interestingly, we detected significantly lower levels of MHV68.ΔHS at this site relative to MHV68.MR (Figure 8A), suggesting that muSOX-induced mRNA degradation contributes to the ability of the virus to traffic to or persist at the sites of latency establishment.

To determine whether this was the root cause of the defect in latency establishment upon MHV68.ΔHS infection, we next infected mice via the intraperitoneal route, which bypasses the requirement for the virus to undergo acute replication and traffic through the lymph before reaching the spleen [37]. At 19 dpi, we found significantly less MHV68.ΔHS virus in the spleen relative to MHV68.MR by qPCR (Figure 8B), indicating that a defect in viral trafficking cannot fully account for the latency establishment defect observed during an intranasal infection. Collectively, these data indicate that muSOX-induced host shutoff plays important roles both in the ability of MHV68 to traffic to the lymph nodes, as well as establish latency in the spleen.
Discussion
All gammaherpesviruses studied to date block host gene expression through widespread mRNA degradation, yet the contribution of this function towards the viral lifecycle remained unknown. Here we analyzed the impact of host shutoff both in tissue culture and in vivo infections with the murine gammaherpesvirus MHV68, and we found this activity to be largely dispensable for acute lytic replication yet critical for the downstream viral accumulation in the lymph nodes and subsequent establishment of latency in the spleen. These results were unexpected given that MHV68-induced host shutoff is executed by muSOX, a viral protein whose expression has only been detected during the lytic cycle [38], [39], [40]. In addition, the role for muSOX-induced mRNA turnover clearly diverges from that of the alphaherpesvirus host shutoff factor vhs, which similarly induces global mRNA degradation but plays a critical role during acute replication in vivo [41], [42], [43]. The severe attenuation of HSV-1 and HSV-2 vhs mutants is hypothesized to be a consequence of ineffective immune evasion, as vhs blocks dendritic cell activation and down-regulates the type I interferon response [44], [45], [46]. In contrast, we find little difference between MHV68.MR and MHV68.ΔHS during acute replication in mice, suggesting that while alpha - and gammaherpesviruses block host gene expression via analogous mechanisms, the functional ramifications of this activity are distinct, and may relate to the unique in vivo biology of each class of virus.
Modeling the muSOX three-dimensional structure from the KSHV SOX or EBV BGLF5 crystal structure [21], [47] indicates that amino acid R443 is positioned on the outer surface, away from the catalytic core where nucleic acids are presumably cleaved (data not shown). Although SOX, BGLF5, and muSOX are functionally homologous and likely induce mRNA degradation through a similar mechanism, the identified amino acid mutations that selectively remove host shutoff activity are not conserved, nor are they positioned in the same three-dimensional region. Likely these different mutations alter the local structure of each protein thereby inhibiting or weakening the mRNA or co-factor interactions which contribute to overall mRNA degradation. In this regard, we anticipate that the residual host shutoff activity observed upon infection with the MHV68.ΔHS virus is at least partially due to incomplete inactivation of muSOX mRNA turnover activity by the R443I mutation.
Host shutoff has been hypothesized to play a role in the diversion of gene expression resources and machinery towards the virus during lytic replication [16], [48], as well as general immune evasion through the down-regulation of immune stimulatory factors [49]. Our failure to detect a significant replication defect for MHV68.ΔHS during a multi-step growth curve in cultured murine fibroblasts or in the mouse lung implies that resource reallocation is unlikely to be the primary role for muSOX-induced mRNA depletion. Likewise, the similar viral titers in the lungs of mice infected with MHV68.MR or MHV68.ΔHS argues against a role for muSOX in the general suppression of host innate immune responses Because MHV68.ΔHS still retains residual mRNA degradation activity, it is possible that some degree of host shutoff could be important for viral replication. However, the appearance of significant defects in downstream viral events even with a partial host shutoff defect further strengthens our conclusion that host shutoff plays a vital role in the lifecycle and pathogenesis of MHV68 in vivo.
The first major defect observed in an in vivo MHV68.ΔHS infection is significantly lower levels of the virus in the lymph nodes at 10 dpi. Following an intranasal infection, wild-type MHV68 undergoes lytic replication in the lung and upper respiratory track, after which the virus drains to the lymph nodes and spleen where a variety of cell types are infected, including macrophages, dendritic cells, and B-cells [37], [50], [51], [52]. A burst of lytic replication occurs at these sites [30], [32], after which replicating virus is cleared and long-term latency is established. Lower levels of MHV68.ΔHS in the lymph node could be caused by a cell-type specific role for muSOX-dependent mRNA degradation in viral replication or immune evasion during viral transport to, and maintenance in, the lymphatic tissue. The observation that the EBV muSOX homolog (BGLF5) down-regulates HLA class I molecules and CD8+ T cell recognition in cultured cells may support this model [25]. The means by which the virus traffics to the sites of latency establishment remain unclear, yet it is likely that latently infected B cells carry the virus given that viremia is undetectable during an infection, and latently infected B cells are present in the lung very early after infection [37], [53], [54]. A failure of MHV68.ΔHS to establish latency in these cells might also cause their selective immune-based eradication and decreased accumulation in the lymph nodes. Alternatively, muSOX could influence the ability of the virus to reactivate from latency, as this process has been linked to efficient latency establishment. Mutations in a number of genes alter the frequency of lytic reactivation and lead to lower levels of latency establishment following an intranasal infection, such as v-cyclin (ORF72) [55], M1 [56], M2 [57], [58], and LANA (ORF73) [59], [60], but unlike muSOX these genes are all expressed during latency as well as during lytic replication [61]. Mutations in the lytic ORF36 gene encoding a protein kinase also lead to defects in latency establishment and reactivation [62], [63], which have been tracked to the requirement for ORF36 to inhibit the IRF-3 mediated type I interferon response [62] and its modification of the DNA-damage response protein H2AX [63]. Experiments are currently underway to examine MHV68.ΔHS replication, reactivation, and latency establishment in specific cell types relevant to in vivo infection.
The second major defect observed upon MHV68.ΔHS infection is a marked reduction in the number of infected splenocytes present during peak latency establishment relative to mice infected with wild-type or mutant rescue viruses. This defect could arise simply as a downstream consequence of the aforementioned impairment in trafficking. However, significantly reduced levels of viral DNA are detected in the spleen even after an intraperitoneal infection, which bypasses the need for acute replication in the lung and subsequent trafficking through the lymph nodes [37]. Thus, MHV68.ΔHS appears defective in both trafficking and latency establishment in the spleen. Such a phenotype might occur if MHV68.ΔHS-infected cells preferentially entered the lytic cycle, perhaps enabling more efficient clearance by the immune system. This model is supported by our observation that an increased percentage of cultured 3T3 cells infected with MHV68.ΔHS express lytic markers relative to those infected with MHV68.WT. However, if this represents enhanced entry into the lytic cycle, there must be a downstream defect that tempers subsequent viral output, as the host shutoff mutant virus does not replicate to higher titers in 3T3 cells than the wild-type virus. In this regard, host shutoff could be important for optimizing the balance of host or viral proteins in an infected cell, as has been reported in an EBV mutant lacking BGLF5, where altered levels of proteins inhibited viral maturation and egress [64]. One mechanism that could shift the balance of infection away from latency is if muSOX down-regulates factors that inhibit latency establishment; such negative regulators would then accumulate in the presence of the R443I mutant, driving cells into lytic replication. MuSOX activity may also be required for the virus to achieve the appropriate level of host and or viral factors packaged into the viral particle in order to efficiently establish latency in newly infected cells rather than enter the lytic cycle.
The large plaque phenotype observed with MHV68.ΔHS is similar to what is observed following infection with MHV68 constitutively expressing (or over-expressing) the lytic transcription factor RTA [65]. Also similar to MHV68.ΔHS, these viruses replicate to near wild-type levels in the lung but establish latency at lower levels in the spleen. The constitutive RTA-expressing viruses have been found to efficiently traffic to the spleen but fail to be maintained at this site [66]. This attenuation has been ascribed to the viruses producing excessive amounts of the RTA-inducible lytic genes, creating an intracellular environment favoring immediate entry into the lytic cycle rather than entering latency. Thus, during an infection, an important role for muSOX may be to ensure that factors driving lytic replication are not overrepresented. While it is tempting to speculate a connection between the increased percentage of lytically infected 3T3 cells and the latency establishment defect in vivo with MHV68.ΔHS, at present the precise relationship between these observations remains unclear.
Given the broad effects of host shutoff, the mechanism of MHV68.ΔHS attenuation is anticipated to deviate from the gene-for-gene interactions associated with many virulence factors. Further research in this area will uncover the likely means by which host shutoff influences latency establishment, as well as reveal novel connections between the lytic and latent stages of the gammaherpesviruses lifecycle.
Materials and Methods
PCR Mutagenesis
A hemagglutinin (HA) tag was introduced at the 5′ end of muSOX by PCR using the primers 5′-CGGAATTCATGGCTTACCCATACGATGTACCTGACTATGCGATGGAAG GGTCGATTATTC-3′ and 5′-ATAGTTTAGCGGCCGCTTAGGGGGTTATGGGTTTTCT-3′. HA-muSOX was then cloned into the EcoRI/NotI sites of pCDEF3 containing a T7 promoter to generate pCDEF3-T7-HA-muSOX. MuSOX was randomly mutagenized by PCR with the Genemorph kit (Strategene) according to the manufacturer's protocol using 35 ng of pCDEF3-T7-HA-muSOX as template, the above primers, and 30 PCR cycles to generate a pool of random mutants. The mutants were then cloned into the EcoRI/NotI sites of pCDEF3-T7. R443I was generated by QuickChange (Stratagene) using the primers 5′-GCTCATCATCACTCCTGT TATAATTCCATCTACTGTGCTGC-3′ and 5′-GCAGCACAGTAGATGGAATTATAACAG GAGTGATGATGAGC-3′.
Cells, Transfections, and Viruses
HEK293T, COS7, NIH 3T3, NIH 3T12, and Vero cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen). 293T cells were transfected with Effectene (Qiagen) following the manufacturer's protocol.
The green fluorescent protein (GFP)-expressing MHV68 bacterial artificial chromosome (BAC) infectious clone has been described elsewhere (RγHV68A98.01 [28]), and mutants were generated by allelic exchange as previously described [67]. To generate the MHV68.ΔHS BAC, a targeting region consisting of 566 nt upstream and 568 nt downstream of the mutation site was ligated into pGS284 between BglII and NotI restriction sites and electroporated into the S17λpir strain of E. coli. The MHV68-YFP BAC infectious clone was a generous gift from Dr. Samuel Speck (Emory University) [29], and MHV68-YFP.ΔHS was generated using the same methodology as MHV68.ΔHS. The targeting vector for the mutant rescue BAC (MHV68.MR) was generated by ligating the region around wild-type muSOX into pGS284. Targeting vector-containing cells were cross-streaked with BAC-containing GS500 cells and successful recombinants were identified by colony PCR and subsequent digest with PsiI. BAC DNA was isolated from positive clones using the Qiagen Large-Construct kit (Qiagen). BAC variants were verified by restriction digests with EcoRI and PsiI and sequencing of the region surrounding the recombination site. BAC-derived MHV68 virus was produced by transfecting 2 µg of BAC DNA into NIH 3T3 cells using SuperFect (Qiagen). Virus was then amplified in NIH 3T12 cells and titered by plaque assays on NIH 3T3 cells. Before infecting mice, the loxP-flanked BAC vector sequence was removed from the recombinant viruses by passaging the virus over Vero cells expressing Cre recombinase (kindly provided by Dr. Samuel Speck, Emory University) [59], and BAC removal was confirmed by PCR analysis.
Immunofluorescence Assays
MuSOX expression and PABPC or HA-muSOX localization were analyzed as described previously [16], [23]. Briefly, NIH 3T3, HEK293T, or COS7 cells were grown on coverslips, and infected with MHV68 variants or transfected with HA-muSOX variants. Cells were then stained with rabbit polyclonal anti-muSOX (1 : 25 dilution), mouse monoclonal anti-PABPC 10e10 (1∶25 dilution) (Santa Cruz Biotechnology), or mouse monoclonal anti-HA (1 : 500 dilution) (Abcam) and AlexaFluor 546 - or 488-conjugated goat anti-mouse or goat anti-rabbit secondary antibody (1∶1500). Coverslips were mounted in DAPI-containing Vectashield mounting medium (Vector Labs) to stain cell nuclei.
For flow cytometry analysis, NIH 3T3 cells were infected with the indicated virus and, 18 hpi, cells were washed with PBS and harvested via trypsin digestion. Cells were fixed in a 4% formaldehyde solution, permeabilized in 1% Triton X-100 and 0.1% sodium citrate in PBS, and incubated with anti-muSOX antibodies at a 1 : 12.5 dilution in 1% Triton X-100, 0.5% Tween, and 3% BSA in PBS. Stained cells were then washed in PBS and incubated with goat anti-rabbit antibodies conjugated to PE-Cy5.5 (Invitrogen) at a dilution of 1 : 150. Data were collected on an EPICS XL cytometer (Beckman-Coulter) and analyzed using FlowJo software (Tree Star).
Western Blots and Northern Blots
For Western blotting, cell lysates were prepared in RIPA buffer [50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% Nonidet P-40 (vol/vol), 0.1% SDS (w/vol)] containing Protease Inhibitor Cocktail (Roche) and quantified by Bradford assay (Bio-Rad). Equivalent amounts of each sample were resolved by SDS-PAGE, transferred to a PVDF membrane, and Western blotted with anti-HA 12CA5 monoclonal antibodies (Invitrogen; 1∶5,000) and HRP-conjugated goat anti-mouse secondary antibodies (Southern Biotechnology; 1∶5,000).
For Northern blotting, total RNA was harvested using RNA-BEE (Tel-Test) and resolved by agarose-formaldehyde gel electrophoresis. RNAs were transferred to a 0.45 µm nylon membrane and probed with 32P-labeled GFP DNA probes generated using the Rediprime II random prime labeling system (GE Healthcare).
DNase Assays
DNase activity of the muSOX variants was performed as described previously [16]. Briefly, muSOX variants were in vitro transcribed with the mMessage mMachine T7 Kit (Ambion) and translated using rabbit reticulocyte lysates (Promega). Translational product was incubated with linear DNA at 37°C for the indicated time period and then the DNA was phenol/chloroform extracted, and resolved by agarose gel electrophoresis. One-sixth each IVT reaction was also separated by SDS-PAGE. The gels were then fixed, dried, and visualized by autoradiography to verify equivalent protein expression.
Quantitative PCR
To quantify RNA, we isolated RNA from transfected cells using RNA-Bee (Tel-Test) or the Zymo Mini RNA II Isolation Kit (Zymo Research). Samples were treated with Turbo DNase (Ambion) according to the manufacturer's protocol to remove genomic DNA contamination. To generate cDNA, RNA samples were reverse transcribed using AMV RT (Promega) and an oligo dT or 18S-specific primer. 18S rRNA or GAPDH mRNA levels were quantified using TaqMan ribosomal RNA control reagents or TaqMan rodent GAPDH control reagents (Applied Biosystems). Other endogenous mRNA levels were quantified using Applied Biosystems Taqman Gene Expression Assays (actB-Mm01205647_g1, rplp2-Mm03059047_gH, tubb5-Mm00495806_g1). To quantify viral genomes, DNA was first isolated from spleen cells or cervical lymph node cells by the Qiagen QIAamp DNA Mini Kit following the manufacturer's protocol. Viral DNA levels were then measured using a previously described assay targeting the MHV68 ORF8 gene [35]. All qPCR reactions were performed with TaqMan Universal PCR Master Mix.
Cell Death Assays
To measure levels of lactate dehydrogenase (LDH) released into the media during an infection, NIH 3T3 cells were infected at an MOI of 5 for 16.5 h. Infection supernatant (60 µl) was mixed with 60 µl LDH detection reagent in triplicate in a 96-well plate as described previously [68]. Absorbance was read on an Elisa-reader at 490 nm wavelength. To detect caspase activity, NIH 3T3 cells were infected at an MOI of 5 for 24 h, whereupon caspase activity was measured using the Caspase-Glo 3/7 Assay System (Promega) following the manufacturer's protocol. To monitor Annexin V-PE and 7-AAD staining by flow cytometry, NIH 3T3 cells were infected at an MOI of 10 for 18 h. Cells were then washed with PBS and harvested via trypsin digestion. Infection supernatant, PBS wash, and cells were combined and pelleted by centrifugation. Cells were washed in PBS and then stained for Annexin V-PE (BD Biosciences, Material # 556422) and 7-AAD (BD Biosciences, Material # 559925) via the manufacturer's protocol using 3 µl of each dye. Data were collected on an EPICS XL cytometer (Beckman-Coulter) and analyzed using FlowJo software (Tree Star).
In vivo Infections and Experiments
Female C57BL/6J mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and infected when 4–6 weeks old. Mice were anesthetized with isoflourane and inoculated intranasally with 5×104 plaque forming units (pfu) in 20 µl DMEM (Invitrogen). For intraperitoneal infections, 1×103 pfu in 0.2 ml PBS were injected into the peritoneal cavity of mice. Lungs were harvested at 3, 5, or 7 dpi, homogenized with a tissue homogenizer for 1 minute at 24,000 rpm in 10 ml DMEM with 10% FBS, 100 U of penicillin per ml, and 100 mg of streptomycin per ml (Pen/Strep, Invitrogen). Spleens were harvested at 17 or 19 dpi. To identify any preformed infectious particles, half of each spleen was homogenized as above in 5 ml DMEM with 10% FBS, and Pen/Strep. The tissue homogenate was then assayed for viral particles by plaque assay on monolayers of NIH 3T3 cells overlaid with 1% agarose for 4 days. The cells were then fixed and stained with 0.04% methylene blue. Splenocytes were isolated from the other half of the spleen by dissociating the spleen and passing through a 40 µm cell strainer (BD-Falcon). Cells were then pelleted, resuspended in red blood cell lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA), and incubated at room temperature for 5 minutes. Cells were again pelleted and resuspended in RPMI medium with 10%FBS and Pen/Strep before counting. Cervical lymph nodes were harvested at 10 dpi and isolated in the same manner as splenocytes, but without lysing the red blood cells.
Infectious Center Assay
The number of reactivating splenocytes was determined as described previously [34]. Briefly, 21,000 NIH 3T3 cells were plated per well in a 24-well plate the day before infection. 2.5×106, 5×105, or 1×105 splenocytes were overlayed onto the NIH 3T3 cells and incubated for 4 days. Cells were then fixed with 10% formaldehyde and stained with 0.04% methylene blue to visualize plaques.
Limiting Dilution PCR
The frequency with which cells latently harbor the viral genome was determined as described previously [36]. Briefly, 4-fold serial dilutions of splenocytes were plated in 96-well plates with 16 wells per dilution. Nested PCR for the ORF50 gene was performed and the resulting PCR product was run on an agarose gel. Wells positive for a PCR product by ethidium bromide staining contain at least one copy of the viral genome. Single-copy sensitivity was confirmed using serial dilutions of MHV68 BAC DNA.
Graph Design and Statistical Analysis
All graphs were designed and statistical analysis performed using the GraphPad Prism Software version 4 or 5. Data were analyzed for statistical significance using the Student's t-test or, for in vivo data, the Mann-Whitney nonparametric test. For limiting dilution PCR analysis, data were subject to nonlinear regression using the log(agonist) vs. response curve with a nonvariable slope.
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the University of California Berkeley (Permit Number: R292-0507). All animals were anesthetized prior to infection with isoflurane, and all efforts were made to minimize suffering.
Supporting Information
Zdroje
1. GlaunsingerBGanemD 2004 Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol Cell 13 713 723
2. KamitaniWNarayananKHuangCLokugamageKIkegamiT 2006 Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc Natl Acad Sci U S A 103 12885 12890
3. DarnellJEJrLevintowL 1960 Poliovirus protein: source of amino acids and time course of synthesis. J Biol Chem 235 74 77
4. KatzeMGKrugRM 1984 Metabolism and expression of RNA polymerase II transcripts in influenza virus-infected cells. Mol Cell Biol 4 2198 2206
5. PlotchSJBouloyMUlmanenIKrugRM 1981 A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23 847 858
6. ThompsonSRSarnowP 2000 Regulation of host cell translation by viruses and effects on cell function. Curr Opin Microbiol 3 366 370
7. WatheletMGOrrMFriemanMBBaricRS 2007 Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J Virol 81 11620 11633
8. McGeochDJDolanARalphAC 2000 Toward a comprehensive phylogeny for mammalian and avian herpesviruses. J Virol 74 10401 10406
9. BlaskovicDStancekovaMSvobodovaJMistrikovaJ 1980 Isolation of five strains of herpesviruses from two species of free living small rodents. Acta Virol 24 468
10. VirginHWLatreillePWamsleyPHallsworthKWeckKE 1997 Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol 71 5894 5904
11. WuTTBlackmanMASunR 2010 Prospects of a novel vaccination strategy for human gamma-herpesviruses. Immunol Res 48 122 146
12. MoserJMUptonJWGrayKSSpeckSH 2005 Ex vivo stimulation of B cells latently infected with gammaherpesvirus 68 triggers reactivation from latency. J Virol 79 5227 5231
13. IskraSKallaMDelecluseHJHammerschmidtWMoosmannA 2010 Toll-like receptor agonists synergistically increase proliferation and activation of B cells by epstein-barr virus. J Virol 84 3612 3623
14. GregorySMWestJADillonPJHilscherCDittmerDP 2009 Toll-like receptor signaling controls reactivation of KSHV from latency. Proc Natl Acad Sci U S A 106 11725 11730
15. RoweMGlaunsingerBvan LeeuwenDZuoJSweetmanD 2007 Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc Natl Acad Sci U S A 104 3366 3371
16. CovarrubiasSRichnerJMClydeKLeeYJGlaunsingerBA 2009 Host shutoff is a conserved phenotype of gammaherpesvirus infection and is orchestrated exclusively from the cytoplasm. J Virol 83 9554 9566
17. MartinezRSariskyRTWeberPCWellerSK 1996 Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates. J Virol 70 2075 2085
18. SheafferAKWeinheimerSPTenneyDJ 1997 The human cytomegalovirus UL98 gene encodes the conserved herpesvirus alkaline nuclease. J Gen Virol 78 Pt 11 2953 2961
19. MoormanNJLinCYSpeckSH 2004 Identification of candidate gammaherpesvirus 68 genes required for virus replication by signature-tagged transposon mutagenesis. J Virol 78 10282 10290
20. MorrisonJMKeirHM 1968 A new DNA-exonuclease in cells infected with herpes virus: partial purification and properties of the enzyme. J Gen Virol 3 337 347
21. BuissonMGeouiTFlotDTarbouriechNRessingME 2009 A bridge crosses the active-site canyon of the Epstein-Barr virus nuclease with DNase and RNase activities. J Mol Biol 391 717 728
22. BagnerisCBriggsLCSavvaREbrahimiBBarrettTE 2011 Crystal structure of a KSHV-SOX-DNA complex: insights into the molecular mechanisms underlying DNase activity and host shutoff. Nucleic Acids Res. E-pub ahead of print doi:10.1093/nar/gkr1111
23. KumarGRGlaunsingerBA 2010 Nuclear import of cytoplasmic poly(A) binding protein restricts gene expression via hyperadenylation and nuclear retention of mRNA. Mol Cell Biol 30 4996 5008
24. LeeYJGlaunsingerBA 2009 Aberrant herpesvirus-induced polyadenylation correlates with cellular messenger RNA destruction. PLoS Biol 7 e1000107
25. ZuoJThomasWvan LeeuwenDMiddeldorpJMWiertzEJ 2008 The DNase of gammaherpesviruses impairs recognition by virus-specific CD8+ T cells through an additional host shutoff function. J Virol 82 2385 2393
26. GlaunsingerBChavezLGanemD 2005 The exonuclease and host shutoff functions of the SOX protein of Kaposi's sarcoma-associated herpesvirus are genetically separable. J Virol 79 7396 7401
27. GoldsteinJNWellerSK 1998 The exonuclease activity of HSV-1 UL12 is required for in vivo function. Virology 244 442 457
28. AdlerHMesserleMWagnerMKoszinowskiUH 2000 Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J Virol 74 6964 6974
29. CollinsCMBossJMSpeckSH 2009 Identification of infected B-cell populations by using a recombinant murine gammaherpesvirus 68 expressing a fluorescent protein. J Virol 83 6484 6493
30. MilhoRSmithCMMarquesSAlenquerMMayJS 2009 In vivo imaging of murid herpesvirus-4 infection. J Gen Virol 90 21 32
31. Sunil-ChandraNPEfstathiouSArnoJNashAA 1992 Virological and pathological features of mice infected with murine gamma-herpesvirus 68. J Gen Virol 73 Pt 9 2347 2356
32. HwangSWuTTTongLMKimKSMartinez-GuzmanD 2008 Persistent gammaherpesvirus replication and dynamic interaction with the host in vivo. J Virol 82 12498 12509
33. SpeckSHGanemD 2010 Viral latency and its regulation: lessons from the gamma-herpesviruses. Cell Host Microbe 8 100 115
34. Sunil-ChandraNPEfstathiouSNashAA 1992 Murine gammaherpesvirus 68 establishes a latent infection in mouse B lymphocytes in vivo. J Gen Virol 73 Pt 12 3275 3279
35. WeinbergJBLutzkeMLAlfinitoRRochfordR 2004 Mouse strain differences in the chemokine response to acute lung infection with a murine gammaherpesvirus. Viral Immunol 17 69 77
36. WeckKEKimSSVirginHISpeckSH 1999 Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J Virol 73 3273 3283
37. FlanoEJiaQMooreJWoodlandDLSunR 2005 Early establishment of gamma-herpesvirus latency: implications for immune control. J Immunol 174 4972 4978
38. EbrahimiBDutiaBMRobertsKLGarcia-RamirezJJDickinsonP 2003 Transcriptome profile of murine gammaherpesvirus-68 lytic infection. J Gen Virol 84 99 109
39. JohnsonLSWillertEKVirginHW 2010 Redefining the genetics of murine gammaherpesvirus 68 via transcriptome-based annotation. Cell Host Microbe 7 516 526
40. Martinez-GuzmanDRickabaughTWuTTBrownHColeS 2003 Transcription program of murine gammaherpesvirus 68. J Virol 77 10488 10503
41. KwongADFrenkelN 1989 The herpes simplex virus virion host shutoff function. J Virol 63 4834 4839
42. StrelowLILeibDA 1995 Role of the virion host shutoff (vhs) of herpes simplex virus type 1 in latency and pathogenesis. J Virol 69 6779 6786
43. KoromMWylieKMMorrisonLA 2008 Selective ablation of virion host shutoff protein RNase activity attenuates herpes simplex virus 2 in mice. J Virol 82 3642 3653
44. SamadyLCostigliolaEMacCormacLMcGrathYCleverleyS 2003 Deletion of the virion host shutoff protein (vhs) from herpes simplex virus (HSV) relieves the viral block to dendritic cell activation: potential of vhs - HSV vectors for dendritic cell-mediated immunotherapy. J Virol 77 3768 3776
45. MurphyJADuerstRJSmithTJMorrisonLA 2003 Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J Virol 77 9337 9345
46. PasiekaTJLuBCrosbySDWylieKMMorrisonLA 2008 Herpes simplex virus virion host shutoff attenuates establishment of the antiviral state. J Virol 82 5527 5535
47. DahlrothSLGurmuDHaasJErlandsenHNordlundP 2009 Crystal structure of the shutoff and exonuclease protein from the oncogenic Kaposi's sarcoma-associated herpesvirus. FEBS J 276 6636 6645
48. SokoloskiKJChaskeyELWiluszJ 2009 Virus-mediated mRNA decay by hyperadenylation. Genome Biol 10 234
49. van GentMGriffinBDBerkhoffEGvan LeeuwenDBoerIG 2011 EBV lytic-phase protein BGLF5 contributes to TLR9 downregulation during productive infection. J Immunol 186 1694 1702
50. StevensonPGEfstathiouS 2005 Immune mechanisms in murine gammaherpesvirus-68 infection. Viral Immunol 18 445 456
51. FlanoEHusainSMSampleJTWoodlandDLBlackmanMA 2000 Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J Immunol 165 1074 1081
52. RosaGTGilletLSmithCMde LimaBDStevensonPG 2007 IgG fc receptors provide an alternative infection route for murine gamma-herpesvirus-68. PLoS ONE 2 e560
53. BartonEMandalPSpeckSH 2011 Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu Rev Immunol 29 351 397
54. RajcaniJBlaskovicDSvobodovaJCiamporFHuckovaD 1985 Pathogenesis of acute and persistent murine herpesvirus infection in mice. Acta Virol 29 51 60
55. van DykLFVirginHWtSpeckSH 2000 The murine gammaherpesvirus 68 v-cyclin is a critical regulator of reactivation from latency. J Virol 74 7451 7461
56. EvansAGMoserJMKrugLTPozharskayaVMoraAL 2008 A gammaherpesvirus-secreted activator of Vbeta4+ CD8+ T cells regulates chronic infection and immunopathology. J Exp Med 205 669 684
57. MacraeAIUsherwoodEJHusainSMFlanoEKimIJ 2003 Murid herpesvirus 4 strain 68 M2 protein is a B-cell-associated antigen important for latency but not lymphocytosis. J Virol 77 9700 9709
58. HerskowitzJJacobyMASpeckSH 2005 The murine gammaherpesvirus 68 M2 gene is required for efficient reactivation from latently infected B cells. J Virol 79 2261 2273
59. MoormanNJWillerDOSpeckSH 2003 The gammaherpesvirus 68 latency-associated nuclear antigen homolog is critical for the establishment of splenic latency. J Virol 77 10295 10303
60. PadenCRForrestJCMoormanNJSpeckSH 2010 Murine gammaherpesvirus 68 LANA is essential for virus reactivation from splenocytes but not long-term carriage of viral genome. J Virol 84 7214 7224
61. MarquesSEfstathiouSSmithKGHauryMSimasJP 2003 Selective gene expression of latent murine gammaherpesvirus 68 in B lymphocytes. J Virol 77 7308 7318
62. HwangSKimKSFlanoEWuTTTongLM 2009 Conserved herpesviral kinase promotes viral persistence by inhibiting the IRF-3-mediated type I interferon response. Cell Host Microbe 5 166 178
63. TarakanovaVLStanitsaELeonardoSMBigleyTMGauldSB 2010 Conserved gammaherpesvirus kinase and histone variant H2AX facilitate gammaherpesvirus latency in vivo. Virology 405 50 61
64. FeederleRBannertHLipsHMuller-LantzschNDelecluseHJ 2009 The Epstein-Barr virus alkaline exonuclease BGLF5 serves pleiotropic functions in virus replication. J Virol 83 4952 4962
65. RickabaughTMBrownHJMartinez-GuzmanDWuTTTongL 2004 Generation of a latency-deficient gammaherpesvirus that is protective against secondary infection. J Virol 78 9215 9223
66. MayJSColemanHMSmillieBEfstathiouSStevensonPG 2004 Forced lytic replication impairs host colonization by a latency-deficient mutant of murine gammaherpesvirus-68. J Gen Virol 85 137 146
67. SmithGAEnquistLW 1999 Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J Virol 73 6405 6414
68. DeckerTLohmann-MatthesML 1988 A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods 115 61 69
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 7
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Requires Glycerol for Maximum Fitness During The Tick Phase of the Enzootic Cycle
- Comparative Genomics Yields Insights into Niche Adaptation of Plant Vascular Wilt Pathogens
- The Role of IL-15 Deficiency in the Pathogenesis of Virus-Induced Asthma Exacerbations
- “Persisters”: Survival at the Cellular Level