
A Role for the Chemokine RANTES in Regulating CD8 T Cell Responses during Chronic Viral Infection
RANTES (CCL5) is a chemokine expressed by many hematopoietic and non-hematopoietic cell types that plays an important role in homing and migration of effector and memory T cells during acute infections. The RANTES receptor, CCR5, is a major target of anti-HIV drugs based on blocking viral entry. However, defects in RANTES or RANTES receptors including CCR5 can compromise immunity to acute infections in animal models and lead to more severe disease in humans infected with west Nile virus (WNV). In contrast, the role of the RANTES pathway in regulating T cell responses and immunity during chronic infection remains unclear. In this study, we demonstrate a crucial role for RANTES in the control of systemic chronic LCMV infection. In RANTES−/− mice, virus-specific CD8 T cells had poor cytokine production. These RANTES−/− CD8 T cells also expressed higher amounts of inhibitory receptors consistent with more severe exhaustion. Moreover, the cytotoxic ability of CD8 T cells from RANTES−/− mice was reduced. Consequently, viral load was higher in the absence of RANTES. The dysfunction of T cells in the absence of RANTES was as severe as CD8 T cell responses generated in the absence of CD4 T cell help. Our results demonstrate an important role for RANTES in sustaining CD8 T cell responses during a systemic chronic viral infection.
Published in the journal:
. PLoS Pathog 7(7): e32767. doi:10.1371/journal.ppat.1002098
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002098
Summary
RANTES (CCL5) is a chemokine expressed by many hematopoietic and non-hematopoietic cell types that plays an important role in homing and migration of effector and memory T cells during acute infections. The RANTES receptor, CCR5, is a major target of anti-HIV drugs based on blocking viral entry. However, defects in RANTES or RANTES receptors including CCR5 can compromise immunity to acute infections in animal models and lead to more severe disease in humans infected with west Nile virus (WNV). In contrast, the role of the RANTES pathway in regulating T cell responses and immunity during chronic infection remains unclear. In this study, we demonstrate a crucial role for RANTES in the control of systemic chronic LCMV infection. In RANTES−/− mice, virus-specific CD8 T cells had poor cytokine production. These RANTES−/− CD8 T cells also expressed higher amounts of inhibitory receptors consistent with more severe exhaustion. Moreover, the cytotoxic ability of CD8 T cells from RANTES−/− mice was reduced. Consequently, viral load was higher in the absence of RANTES. The dysfunction of T cells in the absence of RANTES was as severe as CD8 T cell responses generated in the absence of CD4 T cell help. Our results demonstrate an important role for RANTES in sustaining CD8 T cell responses during a systemic chronic viral infection.
Introduction
During many chronic infections, virus spreads rapidly from the site of initial infection to distal tissues. T cells, on the other hand, must first become activated in the LNs and spleen and then gain the ability to migrate to infected organs. Chemokines play a key role in orchestrating all stages of this T cell response from recruitment of naïve T cells to inflamed lymphoid tissue, migration of T cells within lymphoid organs, movement of activated T cells from lymphoid tissues to effector sites, and the movement of effector T cells within non-lymphoid tissues [1]. While chemokine receptor-ligand pairs such as CCR7-CCL19/21 and CXCR5-CXCL13 are important for migration of T cells into and within lymphoid tissues, others such as CCR4-CCL17/22 and CCR10-CCL27/28 are important for T cell migration into peripheral tissues [2].
One chemokine that has been shown to play a role in immune responses to viral infections is the beta chemokine RANTES (regulated on activation normal T cell expressed and secreted). While RANTES was originally considered a T cell-specific chemokine, it is now known to be expressed by a number of other cell types including epithelial cells and platelets and acts as a potent chemoattractant for many cell types such as monocytes, NK cells [3], memory T cells [4], eosinophils [5] and DCs [6]. A receptor for RANTES, CCR5, is a G protein coupled receptor that, in addition to being the major receptor for RANTES, can also bind MIP1α (CCL3) and MIP1β (CCL4). While the importance of these and many other chemokine∶chemokine receptor pathways has been examined following acute infection or immunization, the role of specific chemokines in regulating T cell responses to chronic viral infections is less clearly defined.
One role for chemokines in regulating T cell responses is the regulation of spatial organization and cellular interactions within lymphoid tissues. For the initiation of an immune response, rare antigen-specific lymphocytes must come into contact with peptide-presenting APCs. Castellino et al showed that antigen-specific interactions of CD4 T cells with antigen-bearing DCs leads to the local production of MIP1α and MIP1β that then recruits naïve CD8 T cells to the same peptide-presenting DC activated by the CD4 T cell [7]. Thus, these chemokines can contribute to the provision of CD4 T cell help for optimal CD8 T cell priming. While Castellino et al found only a modest effect of RANTES neutralization in their protein immunization system, the relative importance of MIP-1α, MIP-1β and RANTES during infection is unknown. Given the overlap in the function of MIP-1α, MIP-1β and RANTES, these studies suggest a potential role for RANTES early in T cell responses to infection possibly via CD4 help. The importance of CD4 T cell help has long been appreciated for a number of chronic viral infections including LCMV, HCV and HIV [8], [9], [10]. When CD4 T cells are transiently depleted at the time of infection with LCMV clone 13, the mice become viremic for life in contrast to untreated mice that control viremia in 2–3 months [8]. Moreover, the CD8 T cells in the CD4 depleted mice are more severely exhausted [11]. Thus, chemokines play important roles during immune responses including aiding in the organization of tissues and in regulating cell-cell interactions.
RANTES regulates protective immunity to viral infections. For example, lymphocytes and epithelial cells produce RANTES in response to infection with respiratory syncytial virus [12] or influenza virus [13], [14], [15], [16], [17]. During respiratory infections, the RANTES∶CCR5 pathway has been shown to be important for DC migration to the dLN [18], survival of alveolar macrophages [19] and the accelerated recruitment of effector and memory T cells to the lung after challenge [20]. Evidence that chemokines can also regulate acute systemic infections arose from the infection of mice lacking CCR5 with west nile virus (WNV), which resulted in markedly higher viral titers in the central nervous system [21]. Humans with the CCR5-Δ32 genotype (a 32-base pair deletion in the CCR5 open reading frame of the CCR5 gene) also have a risk for more aggressive disease following WNV infection [22]. Thus, the RANTES∶CCR5 pathway can influence immune responses in multiple ways during acute viral infections.
In addition to the role of the RANTES∶CCR5 pathway in coordinating spatial interactions during immune responses, CCR5 is a co-receptor for HIV [23], [24]. Humans with the CCR5-Δ32 genotype have slower progression with HIV infection [25] and therapeutic strategies targeting RANTES and CCR5 are being used for treatment against HIV infection [26]. For example, the CCR5 inhibitor maraviroc, in combination with other antiretroviral agents, is indicated for patients with CCR5-tropic strains of HIV. While the benefit of maraviroc in patients with CCR5-tropic strains of HIV is clear (maraviroc can reduce viral loads), how the therapeutic targeting of the CCR5 pathway affects immune responses to other pathogens is unclear.
The role of the RANTES∶CCR5 pathway in respiratory infections, WNV infection and HIV infection suggests that the function of this pathway could be important during other viral infections and that the effect of RANTES during HIV infection might be complex. For example, the CCR5-Δ32 is beneficial during HIV infection because of a direct impediment to viral entry, however, this same mutation is detrimental during WNV infection [27] as well as tick-borne encephalitis [28]. Subjects with the CCR5-Δ32 mutation also have reduced DTH responses [25]. In contrast to acute infections with WNV, influenza virus and Sendai virus, little information exists on how RANTES impacts the T cell function or control of chronic viral infection where viral entry is not affected by CCR5 or RANTES. Thus, we used the mouse model of acute or chronic LCMV infection to investigate the role of RANTES in sustaining CD8 T cell responses during chronic infection. RANTES expression is upregulated during acute LCMV infection [29], [30] but very little is known about the expression or role of RANTES during chronic LCMV infection. Here we demonstrate that RANTES is upregulated to a much higher degree during chronic LCMV infection compared to acute LCMV infection. Unlike acute LCMV infection where RANTES deficiency had little impact on T cell responses or viral control, the absence of RANTES during chronic LCMV infection led to more severe CD8 T cell exhaustion including compromised cytokine production, higher inhibitory receptor expression and reduced cytotoxicity. The loss of IFNγ production coincided with a decrease in Tbet expression similar to levels seen in CD4-depleted mice during chronic LCMV infection. This increase in T cell dysfunction in the absence of RANTES corresponded to a substantially reduced ability to control chronic infection compared to WT mice but was not due to an intrinsic requirement of CD8 T cells to produce or respond to RANTES directly. These results suggest that manipulation of the RANTES pathway may hinder immune responses to, and thus control of, chronic infection with some pathogens.
Materials and Methods
Mice
C57BL/6 and Ly5.1 mice were purchased from the National Cancer Institute (NCI). CCR5−/− mice were purchased from Jackson laboratories (Bar Harbor, Maine). RANTES−/− mice were a gift from Michael Holtzman, Washington University St Louis and bred in-house at AALAC-approved animal care facility at the Wistar Institute, Philadelphia, PA. P14 mice were maintained at the Wistar Institute and crossed to the RANTES−/− mice.
Viruses
For primary infections, mice were infected with either LCMV Armstrong (2×105 pfu) i.p. or LCMV clone 13 (2×106 pfu) i.v. For re-infections, mice were infected intranasally (i.n.) with recombinant influenza virus expressing the LCMV GP33 epitope (x31-GP33, 1.6×105 TCID50). Prior to i.n. infection, mice were anaesthetized by intraperitoneal injection of ketamine hydrochloride and xylazine (Phoenix Scientific) in 0.2 ml of PBS. Recombinant influenza strains were obtained from Dr. Richard J. Webby and were propagated in specific-pathogen-free eggs and stored at −80°C before use.
Adoptive transfer
For adoptive transfer experiments, single-cell suspensions of CD8 T cells were equalized for the number of antigen-specific CD8 T cells and adoptively transferred by i.v. injection into the tail vein. CD8 T cells were purified (>90% purity) from whole lymphocytes using magnetic beads (CD8+ T cell isolation kit, MACS beads; Miltenyi Biotec) and the CD8 T cells stained with tetramer and the numbers of LCMV-specific CD8 T cells normalized before being transferred i.v. For the P14 experiments, LNs were isolated from P14 WT or P14 RANTES−/− mice. The number of P14 cells was equalized and a total of 1,000 P14 cells were transferred into C57BL/6 mice at a 50∶50 ratio. Mice were infected the following day with LCMV clone 13.
Bone-marrow chimeras
Ly5.1 mice from NCI were irradiated with 950 RADS. The following day, bone-marrow cells from Ly5.1 WT mice and Ly5.2 RANTES−/− mice or Ly.2 CCR5−/− mice were depleted of T, B and NK cells with MACs magnetic beads and adoptively transferred i.v. at a 1∶1 ratio. A total of 1–5×106 BM cells were transferred per mouse. Mice were fed antibiotics for 2 weeks following irradiation and allowed to reconstitute for eight weeks before use.
Isolation of lymphocytes from tissues
Mice were euthanized and the hepatic vein cut. The liver was perfused by injecting PBS into the left heart ventricle. Livers were incubated in 0.25 mg/ml collagenase D (Roche Diagnostics) and 1 U/ml DNase I (Roche Diagnostics) at 37°C for 30 min. Digested livers were homogenized using a cell strainer, applied to a 44/56% Percoll gradient, centrifuged at 850 g for 20 mins at 4°C and the lymphocyte population was harvested from the interface. Red blood cells were lysed using ACK lysing buffer (Quality Biological) before cells were washed and counted. Spleens were homogenized using a cell strainer. Red blood cells were lysed using ACK lysing buffer and the cells washed and counted.
Flow cytometry and intracellular cytokine staining
Lymphocytes isolated from different tissues were stained using standard techniques and analyzed by flow cytometry. Virus-specific CD4 and CD8 T cells were analyzed at the peak of the response (LCMV day 8) and in the memory/chronic phase (>day 30). Virus-specific T cells were quantified in tissues using MHC-I and MHC-II tetramer staining. MHC class I peptide tetramers were made and used as described [31]. MHC-II tetramer was obtained from the NIH Tetramer Core Facility (Emory University, Atlanta, GA). For examination of cytokine production, 1×106 splenocytes were cultured in the absence or presence of the indicated peptide (0.2 µg/ml for CD8 peptides and 2 µg/ml for GP66-77) and brefeldin A for 5 h at 37°C. Intracellular cytokine staining was carried out using the BD cytofix/cytoperm kit followed by antibodies for IFNγ, TNFα, IL-2 and MIP-1α. Samples were collected using the LSR II flow cytometer (Becton Dickinson). For CD107a staining, the antibody was added during the stimulation as described [32].
RANTES ELISA
Activated CD8 and CD4 T cells were sorted using a FACSAria (BD Biosciences). Cells were stimulated with PMA/ionomycin for five hours and the supernatant used for ELISAs. The RANTES ELISA was purchased from Peprotech (Rocky Hill, NJ) and carried out according to the manufacturer's instructions.
RT-PCR
DbGP33-specific CD8 T cells and IAbGP66-specific CD4 T cells were sorted on a FACSAria (BD Biosciences). RNA extraction was performed with Trizol (Invitrogen). cDNA was generated using the High Capacity cDNA Archive Kit (Applied Biosystems). Relative quantification real-time PCR was performed on an ABI Prism 7000 with primers purchased from Applied Biosystems. HPRT was used as an endogenous control. Results are expressed relative to naïve cells.
Luminex assay
C57Bl/6 mice were infected with LCMV Armstrong or clone 13 and bled at day 8 and day 32 p.i. Serum samples were sent to Glaxo Smithkline for examination of RANTES protein by the luminex assay.
In vitro cytotoxicity assay
Protocol was similar to [33]. Ly5.1+ splenocytes were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE); half with 100 nM CFSE and half with 1.25 µM CFSE. The CFSE-labeled cells were then pulsed with 2 µg/ml of GP33-44 or OVA257-264 peptide, respectively, for 90 mins at 37°C and then rinsed three times in RPMI with 10% fetal calf serum. The peptide pulsed targets were incubated with magnetic bead purified Ly5.2+ CD8+ T cells from spleens of WT or RANTES−/− mice with a 2∶1 effector∶target ratio for 18 h. Cells were washed and stained with Ly5.1 and Live/Dead fixable red dead cell stain kit from Invitrogen (Carlsbad, CA). The killing efficiency was determined as previously described [33].
Statistical analysis
Data were analyzed using a two-tailed Student's t-test and a p value of ≤0.05 was considered significant.
Ethics statement
All animal experiments were performed in accordance to NIH guidelines, the Animal Welfare Act, and US federal law. The experiments were approved by the Wistar Institutes Institutional Animal Care and Use (IACUC) committee, animal welfare assurance number A3432-01. The Wistar Animal Care and Use Program is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC).
Results
Antiviral T cell responses are similar in RANTES−/− and WT mice during acute LCMV infection
Infection of mice with the Armstrong strain of LCMV results in an acute infection that is cleared within 8–10 days. CD8 T cells are important for the control of acute LCMV infection and competent CD4 T cell help is required for optimal memory CD8 T cells to develop [34], [35], [36]. We infected both WT and RANTES−/− mice with LCMV Armstrong to determine whether RANTES played a role in regulating T cell responses to this infection. WT and RANTES−/− mice were equally capable of clearing infection with LCMV Armstrong (data not shown). LCMV-specific CD8 T cells expanded similarly in the blood and resulted in comparable absolute numbers of antiviral memory CD4 and CD8 T cells (figure 1A and B). Moreover, the expression of CD62L and CD127 on virus-specific memory T cells on day 52 p.i. was similar in the presence or absence of RANTES (figure 1C) suggesting that the pattern of memory T cell differentiation was unchanged in the absence of this chemokine. Virus-specific memory CD4 and CD8 T cells from RANTES−/− mice were also able to co-produce multiple cytokines equally well (figure 1D, E and F) again showing that there was little, if any, influence of RANTES deficiency on the pattern of differentiation of anti-viral CD4 and CD8 T cell responses during acute LCMV infection.

Memory CD8 T cells from RANTES−/− mice generate efficient secondary responses
Given the role of the beta chemokines in regulating ‘helped’ CD8 T cell memory [7], we tested whether the memory CD8 T cells formed during acute LCMV infection could generate an anamnestic response, a key feature of optimal memory CD8 T cells. WT or RANTES−/− mice were infected with LCMV Armstrong to generate GP33-specific memory CD8 T cells. CD8 T cells were isolated from WT and RANTES−/− mice on day 52 p.i and equal numbers of DbGP33-specific CD8 T cells were adoptively transferred to congenically marked WT recipient mice. These recipient mice were then infected intranasally with influenza virus expressing the LCMV GP33 epitope (figure 2A). The ability of donor WT or RANTES−/− memory GP33-specific CD8 T cells to expand upon rechallenge was assessed on day 10 p.i. Both WT and RANTES−/− GP33-specific CD8 T cells expanded vigorously and to a similar degree (figure 2C). Moreover, the RANTES−/− memory cells formed secondary effector CD8 T cells that were phenotypically and functionally similar to WT secondary effectors (figure 2D–F). Thus, memory CD8 T cells generated in the absence of RANTES were fully functional, responded efficiently to local infection rechallenge and showed evidence of having received CD4 T cell help during priming.
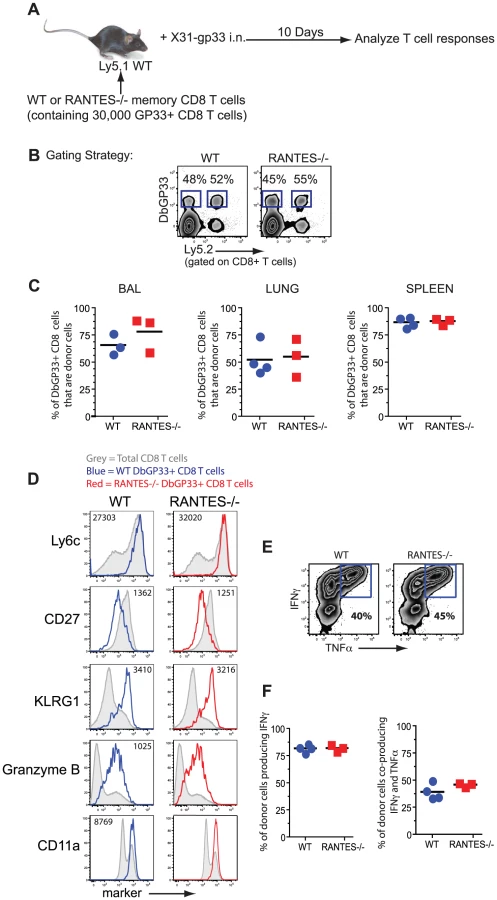
Memory CD8 T cells generated in the absence of RANTES can protect from LCMV clone 13 infection
Memory CD8 T cells generated in response to LCMV Armstrong are able to protect from infection with the more virulent strain LCMV clone 13. To determine whether memory CD8 T cells generated in the absence of RANTES were able to protect from LCMV clone 13 infection, we adoptively transferred equal numbers of either WT or RANTES−/− memory CD8 T cells into naïve WT or RANTES−/− mice and then challenged with LCMV clone 13. As a control, a cohort of WT mice did not receive any cells. After 9 days, the mice were sacrificed and the viral loads examined (figure 3a). The mice that did not receive any cells had high viral titers in the serum and kidneys (figure 3b). In contrast, both WT and RANTES−/− mice that received either WT or RANTES−/− memory CD8 T cells were protected against chronic infection. Thus, RANTES was not required for memory CD8 T cells to protect from LCMV clone 13 infection.

RANTES is highly expressed during chronic LCMV infection
Infection of naïve adult mice with LCMV clone 13 results in a chronic infection with viremia lasting 2–3 months. In contrast to LCMV Armstrong infection, during clone 13 infection the virus-specific CD8 T cells lose the ability to perform effector functions efficiently. This “exhaustion” is hierarchical and progressive with virus-specific CD8 T cells gradually losing the ability to produce IL-2, proliferate robustly, kill efficiently, make TNFα and, in severe exhaustion, produce IFNγ [31]. These exhausted CD8 T cells also express inhibitory receptors such as PD-1, LAG-3, 2B4 and CD160 [37], [38]. These receptors are actively involved in restraining CD8 T cell function during chronic infection and blockade of these pathways can reinvigorate antiviral T cell responses [32], [38].
To begin to address the role of RANTES during chronic infection we first measured RANTES protein in serum. During LCMV clone 13 infection, RANTES levels are increased in the serum at day 8 and day 32 p.i. compared to naïve mice and mice infected with LCMV Armstrong (figure 4A). RANTES expression was also examined at day 6 p.i., when virus was still present in both sets of mice. Both LCMV Armstrong and LCMV clone 13 induced RANTES expression early p.i. (figure 4B and [39] [30]) but high amounts of circulating RANTES were sustained only during LCMV clone 13 infection. Both LCMV-specific CD8 T cells and CD4 T cells upregulated RANTES mRNA expression, with a high amount of RANTES mRNA maintained in LCMV-specific CD8 T cells past day 30 following LCMV Armstrong or clone 13 infection (figure 4C). Given that RANTES transcription can continue in the absence of protein production [40] and that RANTES protein can be stored in granules in the absence of secretion [41], we also measured secreted RANTES protein. CD8+CD44hi T cells and CD4+CD44hi T cells were sorted from mice infected eight days previously with LCMV Armstrong or LCMV clone 13 and RANTES secretion measured after 5 hours of stimulation with PMA/ionomycin. CD8 T cells from LCMV Armstrong - or clone 13-infected mice secreted high levels of RANTES protein following stimulation with PMA/ionomycin (figure 4D). CD4 T cells also secreted RANTES, though the amounts were lower compared to CD8 T cells (figure 4D). Thus, while the high amounts of circulating RANTES found in mice with chronic LCMV infection could come from many cell types, T cells clearly have the potential to contribute to this circulating chemokine production particularly in the presence of persisting antigen. Expression of the main receptor for RANTES, CCR5, is also upregulated on LCMV-specific CD4 and CD8 T cells during both acute and chronic LCMV infection suggesting that not only do T cells produce RANTES upon infection but they also have an increased ability to bind RANTES (figure 4E).

CD8 T cell function is reduced during chronic LCMV infection in the absence of RANTES
Given the high circulating amounts of RANTES during LCMV clone 13 infection we next investigated whether RANTES had any role during chronic infection. WT and RANTES−/− mice were infected with LCMV clone 13 and T cell responses examined eight days later. While the total number of DbGP33 and DbGP276 tetramer positive CD8 T cells as well as IAbGP66 tetramer specific CD4 T cells were similar in WT and RANTES−/− mice, the total number of GP33 - and GP276-specific CD8 T cells producing IFNγ was significantly reduced in RANTES−/− mice (figure 5A and B). This difference in functionality was only observed in virus-specific CD8 T cells but not CD4 T cells as GP66-specific CD4 T cells from WT and RANTES−/− mice had similar cytokine co-production profiles (figure 5B, C and D). Thus, CD8 T cell responses (but not CD4 responses) are functionally compromised at day 8 p.i. in the absence of RANTES during LCMV clone 13 infection.

To determine whether the absence of RANTES led to a change in the development of T cell exhaustion, we examined later timepoints during clone 13 infection. In contrast to day 8 p.i., at day 30 p.i. the number of virus-specific CD8 T cells in the RANTES−/− mice determined by tetramer staining was significantly reduced compared to WT mice (figure 6A). The reduced LCMV-specific CD8 T cell responses in the spleen were unlikely to be due to enhanced migration to peripheral tissues since the LCMV-specific CD8 T cell response was not increased in the liver (figure 6E and F). Even though both WT and RANTES−/− CD8 T cells were highly dysfunctional at this time, exhaustion was substantially more severe in the absence of RANTES (figure 6B and 6C). Indeed, LCMV GP33 - and GP276-specific CD8 T cells were significantly less polyfunctional (i.e. more exhausted) in RANTES−/− compared to WT mice (figure 6B) suggesting that the absence of RANTES led to more severe exhaustion of virus-specific CD8 T cells. Similar to day 8 p.i., the LCMV-specific CD4 T cell response was unaffected by the absence of RANTES in terms of numbers of tetramer-specific CD4 T cells and production of IFNγ (figure 6G). A second hallmark of T cell exhaustion is elevated expression of inhibitory receptors. RANTES−/− virus-specific CD8 T cells had higher expression of PD1, LAG3 and 2B4 indicating that by multiple parameters virus-specific CD8 T cells are more exhausted in the absence of RANTES (figure 6D).
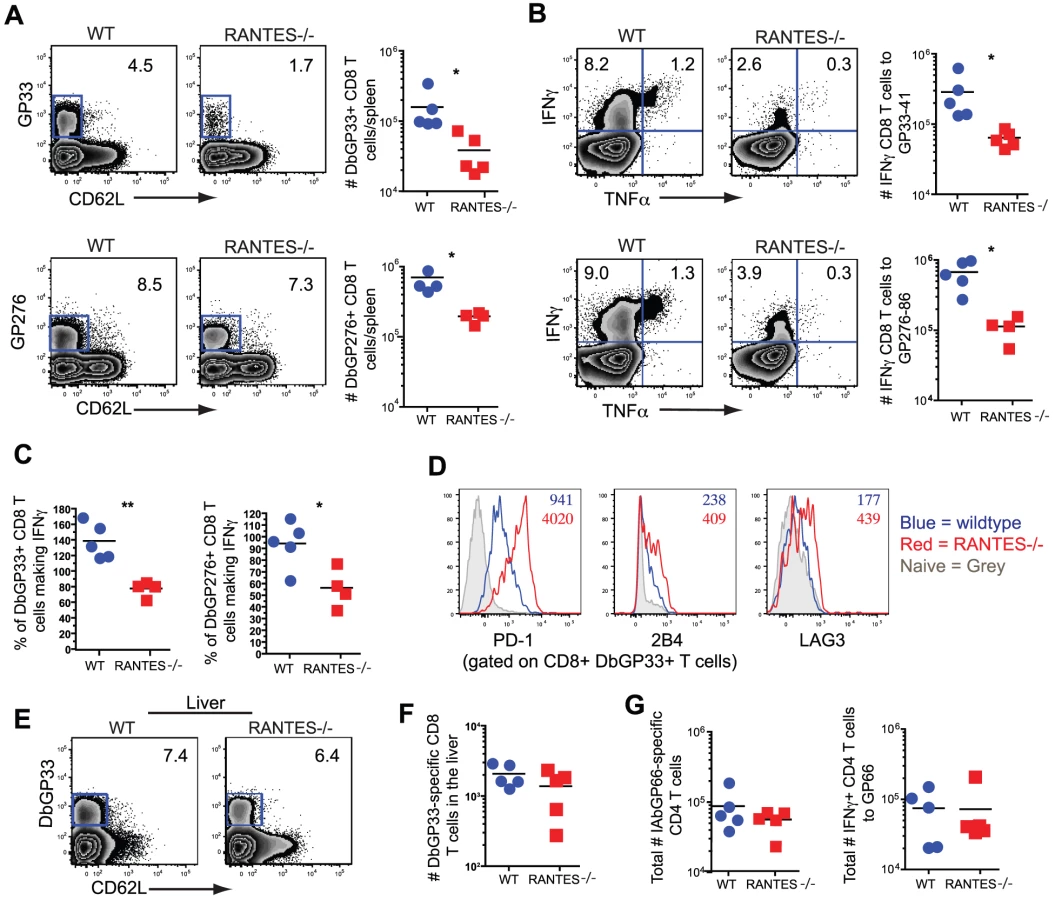
The cytotoxic ability of CD8 T cells is critical during chronic infections. While granzyme B levels were slightly higher in LCMV-specific CD8 T cells from RANTES−/− mice (figure 7A), the ability of these cells to kill was lower than WT T cells from chronically infected mice (figure 7B). Degranulation, as measured by surface CD107a staining, was also slightly lower in LCMV-specific CD8 T cells from RANTES−/− mice suggesting that granule contents might not be released as effectively by CD8 T cells from RANTES−/− mice leading to an accumulation of granzyme B intracellularly (figure 7C).

Given the reduced cytokine production and cytotoxicity in CD8 T cells from mice lacking RANTES, we examined whether these T cell defects had an impact on viral control. At day 8 p.i., viral titers in RANTES−/− mice were similar to WT mice in multiple tissues and sera (figure 8A). However, by day 30 p.i., RANTES−/− mice had higher viral load, consistent with a reduced and more dysfunctional CD8 T cell response (figure 8A). Moreover, when RANTES−/− mice were examined 3–4 months p.i., some of the RANTES−/− mice still had high levels of virus in the liver and were still viremic (figure 8A and B) while WT mice had controlled virus from the serum. These results demonstrate that the absence of RANTES compromises the ability to control viral replication, in some cases leading to a long-term failure to efficiently contain persisting infection.

Providing RANTES in trans is sufficient for normal CD8 T cell responses to chronic infection
Given the reduction in CD8 T cell responses and greater exhaustion in RANTES−/− mice we made mixed bone-marrow chimeras to determine whether the role of RANTES was T cell intrinsic or whether supplying RANTES in trans could prevent more severe CD8 T cell dysfunction. Congenically marked Ly5.1 mice were lethally irradiated and reconstituted with 50% Ly5.1 BM and 50% Ly5.2 RANTES−/− BM (figure 9A). These mice were infected with LCMV clone 13 and examined thirty days later. LCMV-specific CD8 T cell responses were similar for WT versus RANTES−/− CD8 T cells at this time point (figure 9B). Moreover, in a situation where ∼ half of the cells were able to produce RANTES, the RANTES−/− CD8 T cells were as functional as WT T cells in terms of the percentage of IFNγ-producers able to make TNFα and the MFI of IFNγ (right) (figure 9C). Finally, RANTES−/− T cells in the mixed chimeras had similar expression of the inhibitory receptors 2B4, PD-1 and LAG-3 as WT T cells (Figure 9D, E). We also used a non-bone marrow chimera TCR transgenic adoptive transfer system to determine whether the need for RANTES was T cell intrinsic. P14 mice bearing a T cell receptor specific for the DbGP33 epitope from LCMV were crossed to RANTES−/− mice. Equal numbers of WT and RANTES−/− P14 CD8 T cells were co-transferred into WT mice before infection with LCMV clone 13 and the CD8 T cell responses examined. Again, LCMV-specific CD8 T cells did not need to make RANTES themselves since the expression of PD-1 and the ability to make IFNγ and TNFα was similar between WT and RANTES−/− P14 cells in the same chronically infected mice (figure S1). Thus, the critical role of RANTES in sustaining T cell responses during chronic LCMV infection was not cell intrinsic. In other words, the defects in T cell responses to chronic viral infections observed in the complete absence of RANTES could be corrected by providing RANTES signals in trans.

CD8 T cells do not need intrinsic CCR5 signaling to respond to chronic LCMV infection
While intrinsic RANTES production was not required by the CD8 T cells, it remained possible that the CD8 T cells need to bind RANTES themselves. To test this idea we generated mixed bone marrow chimeras using Ly5.1 WT and Ly5.2 CCR5−/− BM (figure 10A). Upon reconstitution, mice were infected with LCMV clone 13 and CD8 T cell responses examined. This chimera system confirmed that WT LCMV-specific CD8 T cells expressed CCR5 during chronic LCMV infection (figure 10B). A similar response was observed for WT and CCR5−/− CD8 T cells in this setting as measured by the frequency of DbGP33 positive CD8 T cells (figure 10C). Expression of PD-1 and production of IFNγ was also similar for WT and CCR5−/− LCMV-specific CD8 T cells (figure 10D, E and F). Thus, it appears that the major impact of RANTES during chronic LCMV infection could be on a non-CD8 T cell and that more severe CD8 T cell exhaustion was a symptom rather than a cause of poor control of infection.

Absence of CD4 T cell help or RANTES results in reduced Tbet expression during chronic infection
The transient depletion of CD4 T cells at the time of infection with LCMV clone 13 results in life-long viremia and high viral titers throughout the mouse [8]. This deficiency coincides with more severe exhaustion of the CD8 T cell response demonstrated by further diminished cytokine production [31], [42]. Given the reduced cytokine potential of RANTES−/− mice, we examined how this dysfunction compared to CD4-depleted WT mice and whether CD4 T cell depletion of RANTES−/− mice could further increase the severity of exhaustion. When we compared CD8 T cell cytokine production, we found that the reduced IFNγ production in RANTES−/− mice was similar to that of WT mice depleted of CD4 T cells. Moreover, the depletion of CD4 T cells in RANTES−/− mice did not further decrease cytokine production (figure 11A). CD4-depletion of WT and RANTES−/− mice ablated any difference in cytokine potential of CD8 T cells (figure 11D) and resulted in similarly high viral titers in WT and RANTES−/− mice (figure 11E). This observation suggested that RANTES plays a role in mitigating the severity of exhaustion and that either RANTES−/− CD4 T cells provide little benefit to the CD8 T cell response in RANTES−/− mice or the higher viral load in RANTES−/− mice drives more severe CD8 T cell exhaustion despite the CD4 T cells.

Transcription factors have recently been demonstrated to play a key role in regulating CD8 T cell exhaustion during clone 13 infection. We have recently found that Tbet is downregulated in exhausted CD8+ T cells during chronic LCMV infection and this downregulation is accentuated in the absence of CD4 help (Kao et al. submitted) (figure 11F). This loss of Tbet results in more severe T cell exhaustion during chronic viral infection. We therefore next examined whether Tbet expression was impacted by the loss of RANTES. In chronically infected RANTES−/− mice Tbet expression was substantially lower than in WT mice (figure 11F). In fact, the loss of RANTES alone reduced Tbet expression in virus-specific CD8 T cells to levels seen in CD4 depleted WT mice. To determine whether this loss of Tbet was also seen earlier during clone 13 infection, we examined Tbet expression at day 8 p.i. Tbet expression was already slightly reduced by day 8 p.i. (this reduction reached significance with the DbGP276-specific CD8 T cells but only a trend in the DbGP33-specific CD8 T cells) (figure 11G). Reduced Tbet expression was consistent with the reduction in IFNγ production observed at this early time p.i.
Discussion
The role of chemokines in regulating immune responses during chronic viral infections is poorly understood. Here we investigated the importance of RANTES in response to a chronic infection where CCR5 is not a viral co-receptor. RANTES was more highly expressed during chronic LCMV infection compared to acute infection. While the absence of RANTES did not impact T cell responses following acute LCMV infection, a different scenario emerged during chronic LCMV infection. During chronic infection, CD8 T cells become exhausted and their dysfunction was characterized by a loss of cytokine production, reduced cytotoxicity and increased inhibitory receptor expression, all of which can hinder the ability to control the infection [31], [32], [43], [44]. In the absence of RANTES, CD8 T cell exhaustion was more severe with reduced virus-specific CD8 T cell numbers, cytokine production and higher expression of inhibitory receptors. The cytotoxic potential of virus-specific CD8 T cells responding to clone 13 infection in RANTES−/− mice was also reduced compared to WT controls. Consistent with the more severe exhaustion of the CD8 T cell response, mice lacking RANTES also had higher viral loads. Thus, the absence of RANTES resulted in the dysfunction of virus-specific CD8 T cells and poor viral control suggesting that RANTES has an important role in regulating and/or sustaining optimal immune responses during chronic viral infection.
There are a number of ways in which the absence of RANTES could result in the higher viral titers and reduced CD8 T cell function during clone 13 infection. First, slightly higher viral loads at the beginning of the response could lead to more severe CD8 T cell exhaustion. One possible mechanism for RANTES affecting viral load is via one of the main cell types infected by LCMV, macrophages. Macrophages play a key role in the immune defense against LCMV. Marginal zone macrophages and metallophilic macrophages may act as filters, controlling the spread of LCMV [45]. The increased tropism of LCMV clone 13 for macrophages and DCs is thought to result in the ability of the virus to persist [46]. The absence of RANTES could impact macrophage function or survival. For example, RANTES is essential to prevent apoptosis of macrophages infected with Sendai virus [19]. Thus, it will be important to investigate the role of RANTES in regulating DC and macrophage differentiation during persisting infections. A second possibility is that RANTES regulates the homing dynamics of the T cells, preventing T cell migration to the peripheral tissues or microenvironments and therefore limiting the ability of these cells to control the infection. However, during chronic LCMV infection the LCMV-specific CD8 T cells were found in spleen, blood and liver showing that the virus-specific T cells could still migrate to peripheral tissues at least at the level of the whole tissue. This observation does not rule out potential differences in movement within tissue, however, and a more detailed analysis of the migration dynamics of exhausted CD8 T cells in the absence of RANTES could be important. Third, CD4 T cell help could be reduced/absent in mice lacking RANTES. At least with LCMV Armstrong infection, CD4 T cell help appears to be intact as CD8 T cell memory cells are fully functional upon secondary challenge. Moreover, LCMV-specific CD4 T cell expansion and cytokine production in RANTES−/− mice were similar to WT mice in response to both LCMV Armstrong and LCMV clone 13. While the phenotype of the CD8 T cells in RANTES−/− mice was similar to CD4-depleted mice, the viral titers in mice lacking CD4 T cells was much higher suggesting that the CD4-depleted phenotype is more severe. Given that CD4 T cells also produce RANTES, it is possible that CD4 T cells are an important source of RANTES during LCMV clone 13 infection but that remains to be determined. A fourth possibility is that RANTES directly affects T cell activation/differentiation leading to reduced effector functions and that loss of RANTES directly results in functional defects in T cells leading to higher viral loads. While RANTES has been shown to act as a costimulator of T cells [47], [48], CCR5−/− T cells responded similarly to WT CD8 T cells in a competitive environment suggesting that the importance of RANTES during LCMV clone 13 infection was not due to direct costimulation of CD8 T cells, though other receptors capable of binding RANTES could have a role.
CD8 T cell activation/differentiation is clearly negatively impacted by the absence of RANTES since IFNγ production by CD8 T cells was reduced even at day 8 p.i. in RANTES−/− mice and this reduced IFNγ production was even more dramatic at the chronic stage of disease. Given that IFNγ has been shown to regulate the ability to clear LCMV infection [44], [49], [50], this initial decrease in IFNγ at the early stage of infection could result in a reduced ability to control viral replication, leading to further CD8 T cell exhaustion. Our data supports a role for RANTES in allowing the efficient activation and differentiation of CD8 T cells that are required to help control clone 13 infection. Interestingly, RANTES was not required for memory CD8 T cells to clear clone 13 infection. This observation, along with the similar T cell response to acute LCMV infection supports a role for RANTES during a sustained infection and further supports the model that minor defects early in the response to a rapidly disseminating infection are magnified as the infection persists leading to more severe T cell dysfunction and pathogen persistence.
Transcription factors that regulate effector functions of CD8 T cells during LCMV infection include Tbet and eomesodermin [51], [52]. Tbet expression was reduced in the absence of RANTES during LCMV clone 13 infection. How the absence of RANTES regulates the expression of Tbet, however, is currently unclear. These findings do suggest that the CD8 T cells responding to clone 13 in RANTES−/− mice have differential expression of transcription factors compared to those from WT mice and perhaps these differences in transcription factor regulation impact their effector functions. Determining whether this effect can be directly attributed to RANTES or is a byproduct of higher viral load requires further investigation.
Interestingly, while CD8 T cell numbers and function were clearly reduced in the absence of RANTES, the CD4 T cells were not as sensitive to the loss of RANTES. CD4 T cells were unaffected in terms of numbers and the ability to produce IFNγ. Thus, the absence of RANTES had differential effects on CD4 versus CD8 T cells. These observations are somewhat surprising given that both CD4 and CD8 T cells produce RANTES and express the main receptor, CCR5. Further, this observation suggests that at least some aspects of CD4 and CD8 T cell exhaustion are regulated differently during chronic LCMV infection. Perhaps the differential effects of RANTES on CD4 versus CD8 T cells could be due to differences in expression of the other receptors for RANTES.
The CCR5-Δ32 mutation is found at a high frequency in European populations and is thought to have arisen through selective pressure during Yersinia pestis or variola major infection [53]. While absence of CCR5 can clearly be protective against HIV, CCR5 plays a role in protecting against WNV and tick-borne encephalitis. CCR5 may also play a protective role in the response against yellow fever virus; viscerotropic disease following yellow fever virus (YFV) vaccination in one subject was associated with the CCR5-Δ32 polymorphism as well as an additional mutation in the RANTES promoter [54]. The dichotomy of protection versus susceptibility of various infections and the use of CCR5 inhibitors suggests the need for more research on subjects with the CCR5-Δ32 mutation in terms of susceptibility to infection with different pathogens.
Understanding the role of RANTES during chronic infection is highly relevant due to the interest in CCR5 inhibitors for the treatment of HIV. CCR5 inhibitors prevent the entry of the R5-tropic stains of HIV virus into the cell [26]. While CCR5 inhibitors can be of tremendous benefit to those infected with the CCR5-tropic stain of HIV, our data suggests that blocking the RANTES pathway could negatively influence ongoing immune responses to other persisting infections. Many patients infected with HIV are also co-infected with other pathogens and the effect of the RANTES∶CCR5 pathway on these co-infections is not well understood. As many as 30% of HIV-infected patients in western Europe and the USA are coinfected with hepatitis C virus (HCV) and complications from HCV coinfection have emerged as a significant cause of morbidity and mortality [55], [56], [57]. Given the role of RANTES in regulating responses to the flaviviruses WNV and YFV, and that serum levels of CC-chemokines are increased in patients infected with chronic hepatitis [58], it will be interesting to determine whether RANTES also plays a role in regulating T cell responses to another member of the flavivirus family HCV.
Our data suggest that therapeutic interventions targeting the RANTES pathway could have negative effects on the ability to control some chronic infections and indicates the need for further research into any link between the CCR5-Δ32 mutation and persistent infections. These observations also suggest that blocking the RANTES∶CCR5 receptor pathway could alter the development and or quality of antiviral immune responses to chronic viral infection and, therefore, CCR5 inhibitors that block only HIV binding but not the RANTES∶CCR5 pathway may be more ideal.
Supporting Information
Zdroje
1. SallustoFMackayCRLanzavecchiaA 2000 The role of chemokine receptors in primary, effector, and memory immune responses. Annu Rev Immunol 18 593 620
2. EscheCStellatoCBeckLA 2005 Chemokines: key players in innate and adaptive immunity. J Invest Dermatol 125 615 628
3. LoetscherPSeitzMClark-LewisIBaggioliniMMoserB 1996 Activation of NK cells by CC chemokines. Chemotaxis, Ca2+ mobilization, and enzyme release. J Immunol 156 322 327
4. SchallTJBaconKToyKJGoeddelDV 1990 Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature 347 669 671
5. RotAKriegerMBrunnerTBischoffSCSchallTJ 1992 RANTES and macrophage inflammatory protein 1 alpha induce the migration and activation of normal human eosinophil granulocytes. J Exp Med 176 1489 1495
6. DieuMCVanbervlietBVicariABridonJMOldhamE 1998 Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med 188 373 386
7. CastellinoFHuangAYAltan-BonnetGStollSScheineckerC 2006 Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature 440 890 895
8. MatloubianMConcepcionRJAhmedR 1994 CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol 68 8056 8063
9. GrakouiAShoukryNHWoollardDJHanJHHansonHL 2003 HCV persistence and immune evasion in the absence of memory T cell help. Science 302 659 662
10. LechnerFWongDKDunbarPRChapmanRChungRT 2000 Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med 191 1499 1512
11. ZajacAJBlattmanJNMurali-KrishnaKSourdiveDJSureshM 1998 Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 188 2205 2213
12. SaitoTDeskinRWCasolaAHaeberleHOlszewskaB 1997 Respiratory syncytial virus induces selective production of the chemokine RANTES by upper airway epithelial cells. J Infect Dis 175 497 504
13. HaoXKimTSBracialeTJ 2008 Differential response of respiratory dendritic cell subsets to influenza virus infection. J Virol 82 4908 4919
14. CulleyFJPennycookAMTregoningJSHussellTOpenshawPJ 2006 Differential chemokine expression following respiratory virus infection reflects Th1 - or Th2-biased immunopathology. J Virol 80 4521 4527
15. MatsukuraSKokubuFKuboHTomitaTTokunagaH 1998 Expression of RANTES by normal airway epithelial cells after influenza virus A infection. Am J Respir Cell Mol Biol 18 255 264
16. ChanMCCheungCYChuiWHTsaoSWNichollsJM 2005 Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir Res 6 135
17. WareingMDLyonABLuBGerardCSarawarSR 2004 Chemokine expression during the development and resolution of a pulmonary leukocyte response to influenza A virus infection in mice. J Leukoc Biol 76 886 895
18. GraysonMHRamosMSRohlfingMMKitchensRWangHD 2007 Controls for lung dendritic cell maturation and migration during respiratory viral infection. J Immunol 179 1438 1448
19. TynerJWUchidaOKajiwaraNKimEYPatelAC 2005 CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat Med 11 1180 1187
20. KohlmeierJEMillerSCSmithJLuBGerardC 2008 The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity 29 101 113
21. GlassWGLimJKCholeraRPletnevAGGaoJL 2005 Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med 202 1087 1098
22. LimJKMcDermottDHLiscoAFosterGAKrysztofD 2010 CCR5 deficiency is a risk factor for early clinical manifestations of West Nile virus infection but not for viral transmission. J Infect Dis 201 178 185
23. DragicTLitwinVAllawayGPMartinSRHuangY 1996 HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381 667 673
24. DoranzBJRuckerJYiYSmythRJSamsonM 1996 A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 85 1149 1158
25. DolanMJKulkarniHCamargoJFHeWSmithA 2007 CCL3L1 and CCR5 influence cell-mediated immunity and affect HIV-AIDS pathogenesis via viral entry-independent mechanisms. Nat Immunol 8 1324 1336
26. FatkenheuerGPozniakALJohnsonMAPlettenbergAStaszewskiS 2005 Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med 11 1170 1172
27. GlassWGMcDermottDHLimJKLekhongSYuSF 2006 CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med 203 35 40
28. KindbergEMickieneAAxCAkerlindBVeneS 2008 A deletion in the chemokine receptor 5 (CCR5) gene is associated with tickborne encephalitis. J Infect Dis 197 266 269
29. AsensioVCCampbellIL 1997 Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J Virol 71 7832 7840
30. MuellerSNHosiawa-MeagherKAKoniecznyBTSullivanBMBachmannMF 2007 Regulation of homeostatic chemokine expression and cell trafficking during immune responses. Science 317 670 674
31. WherryEJBlattmanJNMurali-KrishnaKvan der MostRAhmedR 2003 Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 77 4911 4927
32. BarberDLWherryEJMasopustDZhuBAllisonJP 2006 Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439 682 687
33. HermansIFSilkJDYangJPalmowskiMJGileadiU 2004 The VITAL assay: a versatile fluorometric technique for assessing CTL - and NKT-mediated cytotoxicity against multiple targets in vitro and in vivo. J Immunol Methods 285 25 40
34. ShedlockDJShenH 2003 Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 300 337 339
35. SunJCBevanMJ 2003 Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 300 339 342
36. JanssenEMDroinNMLemmensEEPinkoskiMJBensingerSJ 2005 CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature 434 88 93
37. WherryEJHaSJKaechSMHainingWNSarkarS 2007 Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27 670 684
38. BlackburnSDShinHHainingWNZouTWorkmanCJ 2009 Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 10 29 37
39. ZhouSHalleAKurt-JonesEACernyAMPorpigliaE 2008 Lymphocytic choriomeningitis virus (LCMV) infection of CNS glial cells results in TLR2-MyD88/Mal-dependent inflammatory responses. J Neuroimmunol 194 70 82
40. EberleinJNguyenTTVictorinoFGolden-MasonLRosenHR 2010 Comprehensive assessment of chemokine expression profiles by flow cytometry. J Clin Invest 120 907 923
41. CatalfamoMKarpovaTMcNallyJCostesSVLockettSJ 2004 Human CD8+ T cells store RANTES in a unique secretory compartment and release it rapidly after TcR stimulation. Immunity 20 219 230
42. FullerMJKhanolkarATeboAEZajacAJ 2004 Maintenance, loss, and resurgence of T cell responses during acute, protracted, and chronic viral infections. J Immunol 172 4204 4214
43. WalshCMMatloubianMLiuCCUedaRKuraharaCG 1994 Immune function in mice lacking the perforin gene. Proc Natl Acad Sci U S A 91 10854 10858
44. LeistTPEpplerMZinkernagelRM 1989 Enhanced virus replication and inhibition of lymphocytic choriomeningitis virus disease in anti-gamma interferon-treated mice. J Virol 63 2813 2819
45. SeilerPAichelePOdermattBHengartnerHZinkernagelRM 1997 Crucial role of marginal zone macrophages and marginal zone metallophils in the clearance of lymphocytic choriomeningitis virus infection. Eur J Immunol 27 2626 2633
46. MatloubianMSomasundaramTKolhekarSRSelvakumarRAhmedR 1990 Genetic basis of viral persistence: single amino acid change in the viral glycoprotein affects ability of lymphocytic choriomeningitis virus to persist in adult mice. J Exp Med 172 1043 1048
47. BaconKBPremackBAGardnerPSchallTJ 1995 Activation of dual T cell signaling pathways by the chemokine RANTES. Science 269 1727 1730
48. MolonBGriGBettellaMGomez-MoutonCLanzavecchiaA 2005 T cell costimulation by chemokine receptors. Nat Immunol 6 465 471
49. MullerUSteinhoffUReisLFHemmiSPavlovicJ 1994 Functional role of type I and type II interferons in antiviral defense. Science 264 1918 1921
50. HuangSHendriksWAlthageAHemmiSBluethmannH 1993 Immune response in mice that lack the interferon-gamma receptor. Science 259 1742 1745
51. SullivanBMJuedesASzaboSJvon HerrathMGlimcherLH 2003 Antigen-driven effector CD8 T cell function regulated by T-bet. Proc Natl Acad Sci U S A 100 15818 15823
52. IntlekoferAMBanerjeeATakemotoNGordonSMDejongCS 2008 Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science 321 408 411
53. GalvaniAPSlatkinM 2003 Evaluating plague and smallpox as historical selective pressures for the CCR5-Delta 32 HIV-resistance allele. Proc Natl Acad Sci U S A 100 15276 15279
54. PulendranBMillerJQuerecTDAkondyRMoseleyN 2008 Case of yellow fever vaccine–associated viscerotropic disease with prolonged viremia, robust adaptive immune responses, and polymorphisms in CCR5 and RANTES genes. J Infect Dis 198 500 507
55. ThioCLSeabergECSkolaskyRJrPhairJVisscherB 2002 HIV-1, hepatitis B virus, and risk of liver-related mortality in the Multicenter Cohort Study (MACS). Lancet 360 1921 1926
56. ZhouJDoreGJZhangFLimPLChenYM 2007 Hepatitis B and C virus coinfection in The TREAT Asia HIV Observational Database. J Gastroenterol Hepatol 22 1510 1518
57. AlterMJ 2006 Epidemiology of viral hepatitis and HIV co-infection. J Hepatol 44 S6 9
58. ZeremskiMPetrovicLMTalalAH 2007 The role of chemokines as inflammatory mediators in chronic hepatitis C virus infection. J Viral Hepat 14 675 687
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 7
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Requires Glycerol for Maximum Fitness During The Tick Phase of the Enzootic Cycle
- Comparative Genomics Yields Insights into Niche Adaptation of Plant Vascular Wilt Pathogens
- The Role of IL-15 Deficiency in the Pathogenesis of Virus-Induced Asthma Exacerbations
- “Persisters”: Survival at the Cellular Level