
An Endogenous Foamy-like Viral Element in the Coelacanth Genome
Little is known about the origin and long-term evolutionary mode of retroviruses. Retroviruses can integrate into their hosts' genomes, providing a molecular fossil record for studying their deep history. Here we report the discovery of an endogenous foamy virus-like element, which we designate ‘coelacanth endogenous foamy-like virus’ (CoeEFV), within the genome of the coelacanth (Latimeria chalumnae). Phylogenetic analyses place CoeEFV basal to all known foamy viruses, strongly suggesting an ancient ocean origin of this major retroviral lineage, which had previously been known to infect only land mammals. The discovery of CoeEFV reveals the presence of foamy-like viruses in species outside the Mammalia. We show that foamy-like viruses have likely codiverged with their vertebrate hosts for more than 407 million years and underwent an evolutionary transition from water to land with their vertebrate hosts. These findings suggest an ancient marine origin of retroviruses and have important implications in understanding foamy virus biology.
Published in the journal:
. PLoS Pathog 8(6): e32767. doi:10.1371/journal.ppat.1002790
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002790
Summary
Little is known about the origin and long-term evolutionary mode of retroviruses. Retroviruses can integrate into their hosts' genomes, providing a molecular fossil record for studying their deep history. Here we report the discovery of an endogenous foamy virus-like element, which we designate ‘coelacanth endogenous foamy-like virus’ (CoeEFV), within the genome of the coelacanth (Latimeria chalumnae). Phylogenetic analyses place CoeEFV basal to all known foamy viruses, strongly suggesting an ancient ocean origin of this major retroviral lineage, which had previously been known to infect only land mammals. The discovery of CoeEFV reveals the presence of foamy-like viruses in species outside the Mammalia. We show that foamy-like viruses have likely codiverged with their vertebrate hosts for more than 407 million years and underwent an evolutionary transition from water to land with their vertebrate hosts. These findings suggest an ancient marine origin of retroviruses and have important implications in understanding foamy virus biology.
Introduction
Foamy viruses are complex retroviruses thought exclusively to infect mammalian species, including cats, cows, horses, and non-human primates [1]. Although human-specific foamy viruses have not been found, humans can be naturally infected by foamy viruses of non-human primate origin [2]–[4]. Comparing the phylogenies of simian foamy viruses (SFVs) and Old World primates suggests they co-speciated with each other for more than 30 million years [5]. Retroviruses can invade their hosts' genomes in the form of endogenous retroviral elements (ERVs), providing ‘molecular fossils’ for studying the deep history of retroviruses and the long-term arms races between retroviruses and their hosts [6], [7]. Although ERVs are common components of vertebrate genomes (for example, ERVs constitute around 8% of the human genome) [8], germline invasion by foamy virus seems to be very rare [9], [10]. To date, endogenous foamy virus-like elements have been discovered only within the genomes of sloths (SloEFV) [9] and the aye-aye (PSFVaye) [10]. The discovery of SloEFV extended the co-evolutionary history between foamy viruses and their mammal hosts at least to the origin of placental mammals [9]. However, the ultimate origin of foamy virus and other retroviruses remains elusive.
The continual increase in eukaryotic genome-scale sequence data is facilitating the discovery of additional ERVs, providing important insights into the origin and long-term evolution of this important lineage of viruses. In this study, we report the discovery and analysis of an endogenous foamy virus-like element in the genome of the coelacanth (Latimeria chalumnae), which we designate ‘coelacanth endogenous foamy-like virus’ (CoeEFV). The discovery CoeEFV offers unique insights into the origin and evolution of foamy viruses and the retroviruses as a whole.
Results/Discussion
Discovery of foamy virus-like elements within the genome of coelacanth
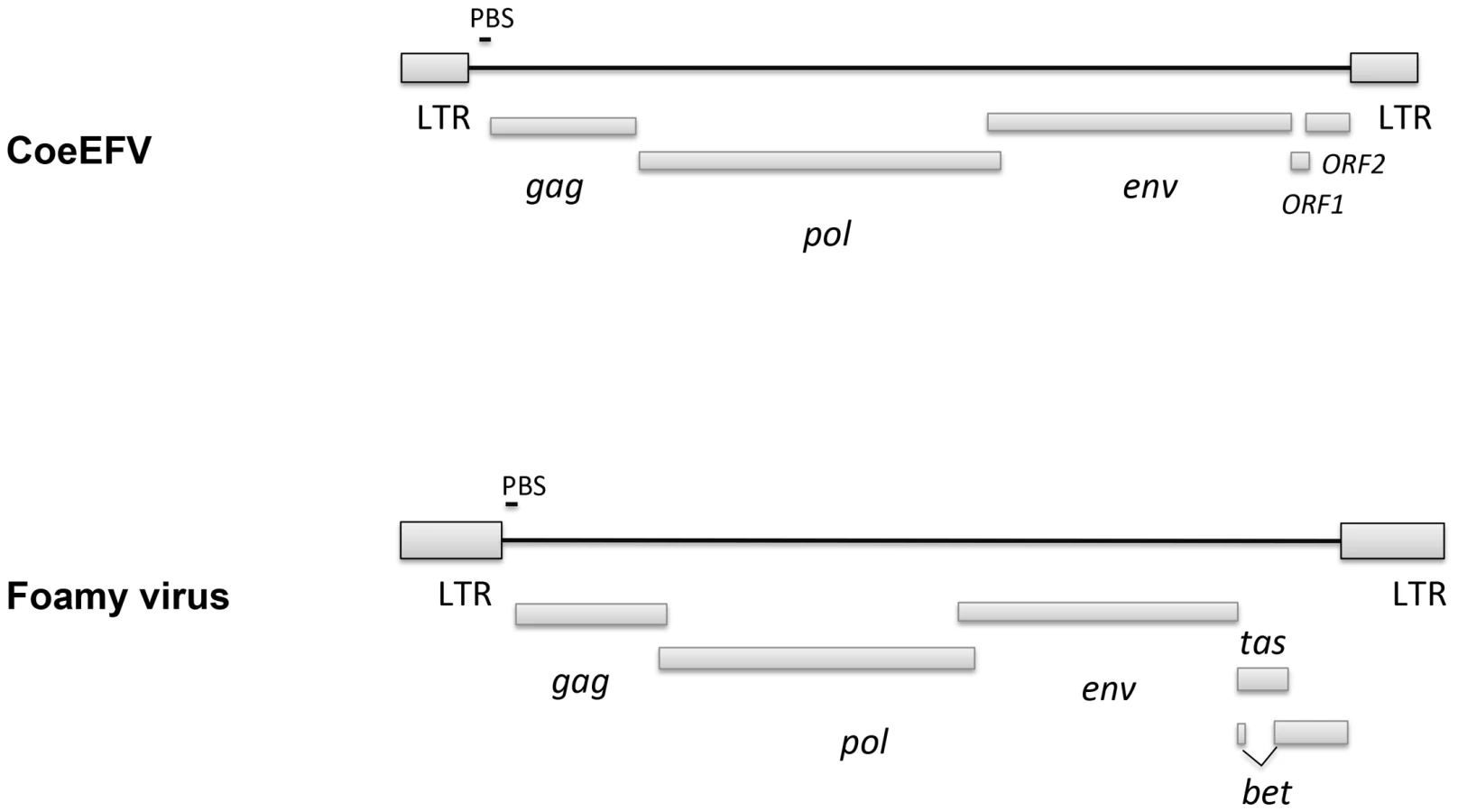
We screened all available animal whole genome shotgun (WGS) sequences using the tBLASTn algorithm using the protein sequences of representative foamy viruses (Table S1) and identified several foamy virus-like insertions (Table S2 and Fig. S1) within the genome of L. chalumnae, one of only two surviving species of an ancient Devonian lineage of lobe-finned fishes that branched off near the root of all tetrapods [11]–[15]. There are numerous in-frame stop codons and frame-shift mutations present in these CoeEFV elements, suggesting that the CoeEFV elements might be functionally defective. Although more than 230 vertebrate genome scale sequences are currently available, endogenous foamy virus elements have been only found in the aye-aye, sloths, and coelacanth, indicating that germline invasion of foamy virus is a rare process [9], [10]. We extracted all contigs containing significant matches and reconstructed a consensus CoeEFV genomic sequence (Fig. S2). The resulting consensus genome shows recognizable and typical foamy virus characteristics (Fig. 1). Its genome has long terminal repeat (LTR) sequences at both 5′ and 3′ ends and encodes the three main open reading frames (ORFs), gag, pol, and env, in positions similar to those of exogenous foamy viruses (Fig. 1). Two additional putative ORFs were found at positions similar to known foamy virus accessory genes but exhibit no significant similarity (Fig. 1). Notably, we found that the Env protein is conserved among foamy viruses and the coelacanth virus-like element (Fig. 2). A Conserved Domain search [16] identified a conserved foamy virus envelope protein domain (pfam03408) spanning most (887 of 1016 residues) of the CoeEFV Env protein, with an E-value of 1.3×10−69 (Fig. 2). The CoeEFV Env protein shares no detectable similarity with other (non-foamy virus) retroviral Env proteins or with retroviral elements within available genomic sequences of other fishes, such as the zebrafish (Danio rerio). Hence, it provides decisive evidence that CoeEFV originated from a foamy-like virus.

To exclude the possibility that these CoeEFV elements result from laboratory contamination, we obtained a tissue sample of L. chalumnae and succeeded in amplifying CoeEFV insertions within the genome of L. chalumnae via PCR with degenerate primers designed for conserved regions of foamy virus pol and env genes.
To establish the position of CoeEFV on the retrovirus phylogeny, conserved regions of the Pol protein sequences of CoeEFV and various representative endogenous and exogenous retroviruses were used to reconstruct a phylogenetic tree with a Bayesian approach. The phylogenetic tree shows that CoeEFV groups with the foamy viruses with strong support (posterior probability = 1.00; Figs. 3 and S3), confirming that CoeEFV is indeed an endogenous form of a close relative of extant foamy viruses. The discovery of CoeEFV establishes that a distinct lineage of exogenous foamy-like viruses existed (and may still exist) in species outside the Mammalia.

CoeEFV likely invaded the coelacanth genome more than 19 million years ago
Endogenous retroviruses are likely to undergo a gradual accumulation of neutral mutations with host genome replication after endogenization [17]. To date the invasion of CoeEFV into coelacanth genome, we identified two sets of sequences, each of which arose by segmental duplication because each set of sequences shares nearly identical flanking regions (Fig. S4). The two sets contain five and two sequences, respectively. Because the divergence time of the two extant coelacanth species (L. chalumnae and L. menadoensis) is uncertain [11], it is impossible to obtain a reliable neutral evolutionary rate of coelacanth species. Nevertheless, even using the mammalian neutral evolutionary rate [18] as a proxy for the coelacanth rate, the invasion dates were conservatively estimated at 19.3 (95% highest posterior density [HPD]: 15.3–23.6) million years ago for the dataset of five sequences. For the dataset containing two sequences, the divergence between the pair is estimated to be 4.1% and the invasion time is estimated to be approximately 9.3 million years ago. Because the CoeEFV invasion almost certainly occurred earlier than the duplication events within the host genome and because the evolutionary rate of coelacanth species is thought to be lower than other vertebrate species [19], [20], the time of CoeEFV integration might much more than 19 million years. Additional phylogenetic evidence (see below) suggests that its exogenous progenitors likely infected coelacanths for hundreds of millions of years prior to the event that fossilized CoeEFV within its host's genome.
Foamy-like viruses have likely codiverged with their vertebrate hosts for at least 407 million years
To further evaluate the relationship of foamy viruses, we reconstructed phylogenetic trees based on the conserved region of Pol proteins of foamy viruses and Class III retroviruses, the conserved region of foamy virus Pol and Env protein concatenated alignment, and the conserved region of foamy virus Env protein alignment, respectively. The three phylogenies have the same topology in terms of foamy viruses (Figs. 4, S5, and S6). CoeEFV was positioned basal to the known foamy viruses (Fig. 4), suggesting a remarkably ancient ocean origin of foamy-like viruses: the most parsimonious explanation of this phylogenetic pattern is that foamy viruses infecting land mammals originated ultimately from a prehistoric virus circulating in lobe-finned fishes. The branching order of the three foamy virus phylogenies (Fig. 4, S5, and S6) is completely congruent with the known relationships of their hosts, and each node on the three virus trees is supported by a posterior probability of 1.0 (except the node leading to equine, bovine, and feline foamy viruses on the Env phylogeny, which is supported by a posterior probability of 0.94; Fig. S6). The common ancestor of coelacanths and tetrapods must have existed prior to the earliest known coelacanth fossil, which is 407–409 million years old [21]. The completely congruent virus topology, therefore, strongly indicates that an ancestral foamy-like virus infected this ancient animal. Crucially, the foamy viral branch lengths of the three phylogenies are highly significantly correlated with host divergence times (R2 = 0.7115, p = 1.10×10−5, Fig. 5; R2 = 0.7024, p = 1.41×10−5, Fig. S5; and R2 = 0.7429, p = 4.26×10−6, Fig. S6), a pattern that can reasonably be expected only if the viruses and hosts codiverged. It is worth emphasizing that we used a consensus sequence to represent CoeEFV in these analyses, so its branch length should correspond roughly to that of the exogenous virus that integrated >19 million years ago, rather than within-host mutations since that time.


There are two alternative explanations for these phylogenetic patterns. One is that the exogenous progenitor of CoeEFV is not truly the sister taxon to the mammalian foamy viruses, but a more distant relative. The robust posterior probability (1.00) placing them in the same clade and the absence of evidence for viruses or virus-like elements from other species disrupting this clade argue against this view, as does the significant similarity between the Env proteins of CoeEFV and the foamy viruses (Fig. 2). Moreover, its branch length would be difficult to explain under such a scenario. If the coelacanth foamy-like virus lineage and the mammalian foamy virus lineage did not share a most recent common ancestor in their ancestral host, why is CoeEFV neither more nor less divergent from the mammalian foamy viruses than one might expect if they did?
The other alternative to the hypothesis that these viruses have co-diverged over more than 407 million years is that they somehow moved, in more recent times, from terrestrial hosts to sarcopterygian hosts that inhabited the deep sea, and that the similarity of the coelacanth virus to the mammalian viruses is due to cross-species (in fact cross-class) transmission, rather than shared history. However, as illustrated by the significant correlation between host divergence times and viral distances (Figs. 5, S5, and S6), the long branches leading to CoeEFV and the clade of mammal foamy viruses suggest the virus had already circulated in vertebrates for an extremely long time before the origin of mammal foamy virus. Given that there is strong evidence that placental mammals were already being infected with foamy viruses by about 100 million years ago [9], the distinctness of the coelacanth virus suggests that it would have to have crossed from some other unidentified host, one whose foamy-like virus was already hundreds of millions of years divergent from the mammalian viruses. This seems highly unlikely. Although cross-species transmission of SFVs has been observed [2]–[5], [22], foamy viruses seem to mainly follow a pattern of co-diversification with their hosts [5], [9]. If one accepts that the endogenous foamy viruses within the genomes sloths indicate more than 100 million years of host-virus co-divergence, it seems plausible that CoeEFV extends that timeline by an additional 300 million years.
Moreover, the habitat isolation of the coelacanth and terrestrial vertebrates would have provided limited opportunities for direct transfer of foamy viruses to coelacanths. Taken together, these lines of evidence strongly suggest that foamy viruses and their vertebrate hosts have codiverged for more than 407 million years, and that foamy viruses underwent a remarkable evolutionary transition from water to land simultaneously with the conquest of land by their vertebrate hosts.
Our analyses provide compelling evidence for the existence of retroviruses going back at least to the Early Devonian. This is the oldest estimate, to our knowledge, for any group of viruses, significantly older than the previous estimates for hepadnaviruses (19 million years) [23] and large dsDNA viruses of insects (310 million years) [24]. Although highly cytopathic in tissue culture, foamy viruses do not seem to cause any recognizable disease in their natural hosts [1], [25], [26]. Such long-term virus-host coevolution may help explain the low pathogenicity of foamy viruses. The fact that the Env is well conserved between CoeEFV and foamy viruses is consistent with the fact that these viruses are asymptomatic and mainly co-evolve with their hosts in a relatively conflict-free relationship. It is easy to imagine that previously overlooked examples of such a non-pathogenic virus may yet be found in hosts that fill in some of the gaps in the phylogeny, namely amphibians, reptiles, and birds. It will be of interest to screen these hosts, but also various fish species, for evidence of exogenous and/or endogenous foamy-like viruses.
An ancient marine origin of retroviruses
Dating analyses provide the clearest evidence for when and where retroviruses originated. There is strong evidence that foamy viruses shared a common, exogenous retroviral ancestor more than 400 million years ago (since Env was present in both terrestrial and marine lineages). The discovery of endogenous lentiviruses demonstrates that lentiviruses, a distinct retroviral lineage that includes HIV, are also millions of years old [27]–[30]. Foamy viruses and lentiviruses share a distantly related ancestor (Figs. 3, S3) and the foamy virus clade alone almost certainly accounts for more than 407 million years of retroviral evolution. It follows that the origin of at least some retroviruses is older than 407 million years ago. As with the coelacanth lineage in the foamy virus clade, we found that retroviruses of fishes occupy the most basal positions within both the Class I and Class III retroviral clades (walleye dermal sarcoma virus (WSDV) and snakehead retrovirus (SnRV), respectively, blue asterisks), (Figs. 3, S3). This pattern provides additional evidence of a marine origin and long-term coevolution of these major retroviral lineages. However, to be specific, the phylogenetic reconstruction in Fig. 3 reflects the history of only of the Pol protein, not a comprehensive history of retroviral genomic evolution. Nevertheless, our analyses support a very ancient marine origin of retroviruses.
Materials and Methods
Screening and consensus genome construction
All available animal whole genome shotgun (WGS) sequences from GenBank were screened for endogenous foamy viruses using the tBLASTn algorithm and the protein sequences of representative exogenous and endogenous foamy viruses (Table S1). Sequences highly similar to foamy virus proteins discovered within the coelacanth WGS were aligned to generate a CoeEFV consensus genome. Conserved domains were identified using CD-Search service [16].
PCR amplification and cloning of CoeEFV
Ethanol preserved Latimeria chalumnae tissue sample was obtained from Ambrose Monell Cryo Collection (AMCC) at the American Museum of Natural History, New York. Genomic DNA was extracted using the DNeasy tissue kit (QIAGEN, MD) following the manufacturer's instructions. Amplification of ∼680 bp gag gene and ∼650 bp env gene fragments was performed with the degenerate primer pairs, FVpol-F (5′-AACAGTGYCTYGACCMAACC-3′) and FVpol-R (5′-TAGTGAGCGCTGCTTTGAGA-3′), FVenv-F (5′-CTGGGGATGACAAYCAGAGT-3′) and FVenv-R (5′-CCACTCRGGAGAGAGGCAAC-3′). PCR was performed in 25 µl of final volume reactions with 0.1 µl Platinum Taq HiFi enzyme (Invitrogen, CA), 1 µl primer mix (10 µM each), 0.5 µl of 10 mM dNTP mixture, 1 µl of 50 mM MgSO4, 2.5 µl of 10× PCR buffer, and 1 µl of template DNA. The PCR reactions were cycled under the following conditions: initial denaturation at 94°C for 2 minutes, 45 cycles of (94°C for 15 seconds, 60°C for 60 seconds, and 72°C for 30 seconds), and final elongation at 72°C for 5 minutes. The PCR products were purified using QIAquick spin columns (QIAGEN, MD). Purified PCR products were cloned into the pGEM-T Easy vector (Promega, WI). Cloned products were sequenced by the University of Arizona Genetics Core with an Applied Biosystems 3730XL DNA Analyzer. The sequences have been deposited in GenBank (Accession Nos. JX006240-JX006251).
Phylogenetic analysis
All protein sequences were aligned using Clustal Omega [31]. Gblocks 0.91b was used to eliminate ambiguous regions and extract conserved regions from the alignments [32]. To determine the phylogenetic relationship between CoeEFV and other retrovirus, we reconstructed a phylogeny based on the conserved region of Pol proteins of CoeEFV and various representative exogenous and endogenous retroviruses (Table S1; Dataset S1). To further evaluate the relationship and divergence of foamy viruses, the conserved region of the foamy viruses and Class III endogenous retroviruses Pol protein (Dataset S2), the conserved region of foamy virus Pol and Env protein concatenated alignment (Dataset S3), and the conserved region of foamy virus Env protein alignment (Dataset S4) were used to infer phylogenetic trees. We were unable to discern positional homology for the first 143 residues of the Pol protein with reasonable certainty. These regions were excluded from all subsequent analyses. All the phylogenetic analyses were performed with MrBayes 3.1.2 [33] using 1,000,000 generations in four chains, sampling posterior trees every 100 generations. The rtREV amino acid substitution model [34] was used. The first 25% of the posterior trees were discarded. MCMC convergence was indicated by an effective sample size >300 as calculated in the program Tracer v1.5.
Host-virus branch length analysis
For the phylogenetic tree based on the foamy viruses and Class III endogenous retroviruses Pol protein, Class III endogenous retroviruses were used to root the foamy viral phylogeny (Fig. 4). Because there is no obvious outgroup for foamy virus Env protein, we rooted the phylogenetic trees inferred from foamy virus Pol and Env concatenated alignment and Env alignment using midpoint method (Figs. S5 and S6). Because the topologies of the host and virus trees were identical for the foamy viruses (Figs. 4, S5, and S6), we were able to plot host branch length (in millions of years) versus virus branch length (in expected amino acid substitutions per site) for every branch (both internal and external). The vertebrate host divergence times are based on references [21], [35], and [36].
Dating analysis
The nucleotide sequences were aligned using MUSCLE [37]. To estimate the age of the CoeEFV invasion, we identified two sets of sequences, which contain five sequences (contig270160, contig184752, contig185880, contig245863, and contig236769) (Dataset S5) and two sequences (contig243355 and contig219087) (Dataset S6). Sharing the same flanking region, each set of sequences arose from segmental duplication. I) For the dataset of five sequences: the best-fitting model of nucleotide substitution was determined using jModelTest [38]. The typical mammal neutral evolutionary rate (2.2×10−9 substitutions per site per year, standard deviation = 0.1×10−9) was used as the rate prior [18]. The HKY substitution model was used. BEAST v1.6.1 (http://beast.bio.ed.ac.uk) was employed for Bayesian MCMC analysis with a strict clock model [39] and Yule model of speciation. MCMC chains were run for 100 million steps twice to achieve adequate mixing for all parameters (effective sample size >200). Tracer v1.5 was used to summarize and analyze the resulting posterior sample. II) For the dataset of two sequences: we calibrated the genetic distance between the pair based on the Kimura two-parameter model, in which transitions and transversions are treated separately.
Supporting Information
Zdroje
1. MeieringCDLinialML 2001 Historical perspective of foamy virus epidemiology and infection. Clin Microbiol Rev 14 165 176
2. WolfeNDSwitzerWMCarrJKBhullarVBShanmugamV 2004 Naturally acquired simian retrovirus infections in central African hunters. Lancet 363 932 937
3. SwitzerWMBhullarVShanmugamVCongMEParekhB 2004 Frequent simian foamy virus infection in persons occupationally exposed to nonhuman primates.) . J Virol 78 2780 2789
4. HeneineWSchweizerMSandstromPFolksT 2003 Human infection with foamy viruses. Curr Top Microbiol Immunol 277 181 196
5. SwitzerWMSalemiMShanmugamVGaoFCongME 2005 Ancient co-speciation of simian foamy viruses and primates. Nature 434 376 380
6. HolmesEC 2011 The evolution of endogenous viral elements. Cell Host Microbe 10 368 377
7. PatelMREmermanMMalikHS 2011 Paleovirology - Ghosts and gifts of viruses past. Curr Opin Virol 1 304 309
8. GriffithsDJ 2001 Endogenous retroviruses in the human genome sequence. Genome Biol 2 1017.1 1017.5
9. KatzourakisAGiffordRJTristemMGilbertMTPybusOG 2009 Macroevolution of complex retroviruses. Science 325 1512
10. HanGWorobeyM 2012 An endogenous foamy virus in the aye-aye (Daubentonia madagascariensis). J Virol (In press)
11. NikaidoMSasakiTEmersonJJAibaraMMzighaniSI 2011 Genetically distinct coelacanth population off the northern Tanzanian coast. Proc Natl Acad Sci USA 108 18009 18013
12. TakezakiNFigueroaFZaleska-RutczynskaZTakahataNKleinJ 2004 The phylogenetic relationship of tetrapod, coelacanth, and lungfish revealed by the sequences of forty-four nuclear genes. Mol Biol Evol 21 1512 1524
13. ZardoyaRCaoYHasegawaMMeyerA 1998 Searching for the closest living relative(s) of tetrapods through evolutionary analyses of mitochondrial and nuclear data. Mol Biol Evol 15 506 517
14. BrinkmannHVenkateshBBrennerSMeyerA 2004 Nuclear protein-coding genes support lungfish and not the coelacanth as the closest living relatives of land vertebrates. Proc Natl Acad Sci USA 101 4900 4905
15. ShanYGrasR 2011 43 genes support the lungfish-coelacanth grouping related to the closest living relative of tetrapods with the Bayesian method under the coalescence model. BMC Res Notes 4 49
16. Marchler-BauerABryantSH 2004 CD-Search: protein domain annotations on the fly. Nucleic Acids Res 32 W327 331
17. JohnsonWECoffinJM 1999 Constructing primate phylogenies from ancient retrovirus sequences. Proc Natl Acad Sci USA 96 10254 10260
18. KumarSSubramanianS 2002 Mutation rates in mammalian genomes. Proc Natl Acad Sci USA 99 803 808
19. AmemiyaCTPowersTPProhaskaSJGrimwoodJSchmutzJ 2010 Complete HOX cluster characterization of the coelacanth provides further evidence for slow evolution of its genome. Proc Natl Acad Sci USA 107 3622 3627
20. NoonanJPGrimwoodJDankeJSchmutzJDicksonM 2004 Coelacanth genome sequence reveals the evolutionary history of vertebrate genes. Genome Res 14 2397 2405
21. JohansonZLongJATalentJAJanvierPWarrenJW 2006 Oldest coelacanth, from the Early Devonian of Australia. Biol Lett 2 443 446
22. LeendertzFHZirkelFCouacy-HymannEEllerbrokHMorozovVA 2008 Interspecies transmission of simian foamy virus in a natural predator-prey system. J Virol 82 7741 7744
23. GilbertCFeschotteC 2010 Genomic fossils calibrate the long-term evolution of hepadnaviruses. PLoS Biol 8 e1000495
24. ThézéJBézierAPeriquetGDrezen JMHerniou EA 2011 Paleozoic origin of insect large dsDNA viruses. Proc Natl Acad Sci U S A 108 15931 15935
25. LinialM 2000 Why aren't foamy viruses pathogenic? Trends Microbiol 8 284 289
26. MurraySMLinialML 2006 Foamy virus infection in primates. J Med Primatol 35 225 235
27. KatzourakisATristemMPybusOGGiffordRJ 2007 Discovery and analysis of the first endogenous lentivirus. Proc Natl Acad Sci U S A 104 6261 6265
28. GilbertCMaxfieldDGGoodmanSMFeschotteC 2009 Parallel germline infiltration of a lentivirus in two Malagasy lemurs. PLoS Genet 5 e1000425
29. GiffordRJKatzourakisATristemMPybusOGWintersM 2008 A transitional endogenous lentivirus from the genome of a basal primate and implications for lentivirus evolution. Proc Natl Acad Sci USA 105 20362 20367
30. HanGZWorobeyM 2012 Endogenous lentiviral elements in the weasel family (Mustelidae). Mol Biol Evol (in press)
31. SieversFWilmADineenDGibsonTJKarplusK 2011 Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7 539
32. TalaveraGCastresanaJ 2007 Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56 564 577
33. RonquistFHuelsenbeckJP 2003 Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19 1572 1574
34. DimmicMWRestJSMindellDPGoldsteinD 2002 rtREV: An amino acid substitution matrix for inference of retrovirus and reverse transcriptase phylogeny. J Mol Evol 55 65 73
35. MeredithRWJanečkaJEGatesyJRyderOAFisherCA 2011 Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science 334 521 524
36. KumarSHedgesSB 2011 TimeTree2: species divergence times on the iPhone. Bioinformatics 27 2023 2024
37. EdgarRC 2004 MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32 1792 1797
38. PosadaD 2008 jModelTest: phylogenetic model averaging. Mol Biol Evol 25 1253 1256
39. DrummondAJHoSYPhillipsMJRambautA 2006 Relaxed phylogenetics and dating with confidence. PLoS Biol 4 e88
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2012 Číslo 6
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Protecting against Pneumococcal Disease: Critical Interactions between Probiotics and the Airway Microbiome
- Manipulation of Costimulatory Molecules by Intracellular Pathogens: Veni, Vidi, Vici!!
- Highly Efficient Prion Transmission by Blood Transfusion
- A Highly Intensified ART Regimen Induces Long-Term Viral Suppression and Restriction of the Viral Reservoir in a Simian AIDS Model