
ChIP-Seq and RNA-Seq Reveal an AmrZ-Mediated Mechanism for Cyclic di-GMP Synthesis and Biofilm Development by
Pathogenic bacteria such as Pseudomonas aeruginosa utilize a wide variety of systems to sense and respond to the changing conditions during an infection. When a stress is sensed, signals are transmitted to impact expression of many genes that allow the bacterium to adapt to the changing conditions. AmrZ is a protein that regulates production of several virulence-associated gene products, though we predicted that its role in virulence was more expansive than previously described. Transcription factors such as AmrZ often affect the expression of a gene by binding and promoting or inhibiting expression of the target gene. Two global techniques were utilized to determine where AmrZ binds in the genome, and what effect AmrZ has once bound. This approach revealed that AmrZ represses the production of a signaling molecule called cyclic diguanylate, which is known to induce the formation of difficult to treat communities of bacteria called biofilms. This study also identified many novel targets of AmrZ to promote future studies of this regulator. Collectively, these data can be utilized to develop treatments to inhibit biofilm formation during devastating chronic infections.
Published in the journal:
. PLoS Pathog 10(3): e32767. doi:10.1371/journal.ppat.1003984
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003984
Summary
Pathogenic bacteria such as Pseudomonas aeruginosa utilize a wide variety of systems to sense and respond to the changing conditions during an infection. When a stress is sensed, signals are transmitted to impact expression of many genes that allow the bacterium to adapt to the changing conditions. AmrZ is a protein that regulates production of several virulence-associated gene products, though we predicted that its role in virulence was more expansive than previously described. Transcription factors such as AmrZ often affect the expression of a gene by binding and promoting or inhibiting expression of the target gene. Two global techniques were utilized to determine where AmrZ binds in the genome, and what effect AmrZ has once bound. This approach revealed that AmrZ represses the production of a signaling molecule called cyclic diguanylate, which is known to induce the formation of difficult to treat communities of bacteria called biofilms. This study also identified many novel targets of AmrZ to promote future studies of this regulator. Collectively, these data can be utilized to develop treatments to inhibit biofilm formation during devastating chronic infections.
Introduction
Pseudomonas aeruginosa is a Gram-negative opportunistic pathogen that is a major burden on the health care industry. Up to 10% of all nosocomial infections are attributed to P. aeruginosa, with mortality rates approaching 40% in patients with bacteremia [1], [2]. This bacterium is often a causative agent of sepsis, as well as acute and chronic infections of the airway, burn wounds, skin, and medical devices such as catheters [1], [3].
Additionally, P. aeruginosa forms biofilms that contribute significantly to disease [4]. The formation of a biofilm by P. aeruginosa confers resistance to antibiotic treatment and immune cells [5]–[7]. The classical definition of a biofilm involves a community of bacteria adhered to a surface encased in a self-produced matrix [3], [8]–[11]. P. aeruginosa forms these biofilms in the environment, on implanted devices such as catheters, and in wound infections [12]. In addition, P. aeruginosa forms biofilms suspended in the dehydrated pulmonary mucus plugs of cystic fibrosis patients [13], [14]. Biofilms are often recalcitrant to antibiotics, have anti-phagocytic properties, and are difficult to treat, commonly accounting for the persistence of chronic infections [7], [15]–[17].
Our laboratory has identified the ribbon-helix-helix transcription factor AmrZ (alginate and motility regulator Z) as a modulator of P. aeruginosa biofilm development and virulence [18], [19]. Five AmrZ-regulated virulence factors have been identified through targeted molecular approaches; however, the global effect of AmrZ on expression of P. aeruginosa genes is unknown. AmrZ directly represses transcription of fleQ and thus motility [20], [21], and its own transcription in a feedback loop [22], [23]. Additionally, AmrZ inhibits production of the extracellular polysaccharide Psl by repressing transcription of the psl operon [19]. In contrast, AmrZ activates alginate production by binding the algD promoter [24], [25] and is essential for twitching motility and formation of a type IV pilus [26]. Each of these AmrZ-regulated genes have been linked to biofilms and P. aeruginosa pathogenicity. The major limitation of the previous approaches is that they are biased towards genes that produce an easily observed phenotype, potentially overlooking many AmrZ-regulated genes that are important in infection. Here, we present a systems-level analysis of the AmrZ regulon utilizing ChIP-Seq and RNA-Seq [27], [28]. By combining these two high-throughput techniques, the genome can be scanned for functional AmrZ binding sites. Additionally, these data allow classification of members of the AmrZ regulon into activated or repressed promoters, as well as direct vs. indirect regulation. Herein, we identified 398 regions of the genome bound by AmrZ (≥3-fold enrichment). The RNA-Seq identified 333 genes that were differentially expressed when comparing a ΔamrZ mutant to a complemented strain (≥2-fold difference). Comparison of AmrZ-bound and AmrZ-regulated genes identified 9 genes directly activated by AmrZ and 49 genes that were directly repressed. Many of these genes have been implicated in pathogenesis, highlighting the importance of AmrZ in P. aeruginosa virulence. Finally, these data allow comparisons of the sequence specificity of AmrZ bound promoters, further defining the consensus AmrZ binding site and lending insight into the mechanism of regulation by AmrZ.
One AmrZ-dependent pathway was investigated in detail since it provided important insights into earlier findings that ΔamrZ mutants form hyper biofilms compared with the parental strain, PAO1 [19]. The present study provides a molecular basis for this finding since we discovered that AmrZ directly represses a predicted diguanylate cyclase-encoding gene (PA4843), which we named adcA (AmrZ dependent cyclase A,). Repression of adcA led to reduced amounts of the second messenger c-di-GMP. This regulation event explains the hyper - aggregative and -biofilm phenotype of a ΔamrZ strain, as elevated c-di-GMP is often associated with the rugose small colony variant phenotype that shares these characteristics [29]–[31]. Recent reports indicate that reducing c-di-GMP in P. aeruginosa biofilm infections leads to biofilm dissolution [32], [33]. Regulation of c-di-GMP by AmrZ could lend insights into the establishment and persistence of chronic P. aeruginosa infections and open novel avenues of treatment.
Results
amrZ mutants have an RSCV phenotype
Upon observation of overnight growth on VBMM, the wild-type strain PAO1 formed a smooth colony, while the ΔamrZ mutant formed an aggregated rugose small colony variant (RSCV) morphology. (Figure 1A). Prevention of rugosity was dependent on AmrZ binding DNA, as the DNA binding deficient R22A AmrZ mutant is also an RSCV (Figure 1A). Chromosomal complementation of the ΔamrZ mutant relieves the rugose phenotype and returns the colony morphology to that of the smooth parental strain (Figure 1A). We have included the well-defined RSCV ΔwspF for comparison [29], [34], [35]. The RSCV phenotype of the ΔwspF mutant has been attributed to the loss of repression of the diguanylate cyclase WspR, leading to elevated intracellular c-di-GMP [32], [36]. Cyclic di-GMP modulates the activity of the transcriptional regulator FleQ at the pel locus, switching FleQ from a repressor to an activator [37], [38]. Psl and Pel polysaccharide overproduction in these strains is responsible for the hyper aggregative phenotype and rugose colony morphology observed [35]. We therefore hypothesized that the ΔamrZ mutant displayed a RSCV phenotype due to elevated intracellular c-di-GMP. To test this, we purified nucleotide pools from plate-grown cells and measured the c-di-GMP via LC-MS/MS (Figure 1B) [39]. We observed that the ΔamrZ mutant accumulated nearly double the intracellular c-di-GMP compared to parental wild type PAO1 (p≤0.01). A two-fold change in c-di-GMP levels can have drastic effects on cell physiology and biofilm formation [39]–[42]. These data are consistent with our classification of ΔamrZ mutants as RSCV. Additionally, we observed that the DNA binding deficient R22A amrZ mutant had similar intracellular levels of c-di-GMP as the ΔamrZ strain (data not shown), indicating that the AmrZ contribution to low c-di-GMP is DNA binding dependent. This observation, in combination with elevated c-di-GMP in the amrZ mutants suggests that AmrZ-mediated modulation of c-di-GMP is either through transcriptional repression of a diguanylate cyclase or activation of a phosphodiesterase. Since AmrZ is a bifunctional transcriptional regulator [22], [24], [25], either of these mechanisms is possible. Therefore, to provide a comprehensive analysis of the AmrZ regulon and to define the mechanistic basis for c-di-GMP accumulation in the ΔamrZ mutant, RNA-Seq and ChIP-Seq strategies were undertaken.

ChIP-Seq provides an unbiased analysis of AmrZ binding to genomic DNA
Previous studies identified four AmrZ-bound promoters utilizing standard molecular methods such as DNA footprinting and Electrophoretic Mobility Shift Assays (EMSA) [18], [19], [22]–[26], [43]. Though these methods are recognized as the standard for DNA binding analysis, we wished to perform a genome-wide screen for AmrZ binding sites. Chromatin immunoprecipitation (ChIP) allows us to purify DNA bound AmrZ directly from cells [27], [28], [44], [45]. In this assay, chromatin bound AmrZ was cross-linked, the DNA sheared, nonspecific proteins and nucleic acids removed, and the DNA was purified and quantified using high-throughput parallel DNA sequencing. The resulting ChIP-Seq tags were analyzed using HOMER (Hypergeometric Optimization of Motif EnRichment) a suite of tools for ChIP-Seq analysis and motif discovery [46]. This generated a complete map of genomic areas to which AmrZ binds (Table S1). Conditions were optimized by using previously studied positive control DNA (algD, amrZ) and a negative control region (algB) [22], [24], [25]. Consistent with the literature, algD and amrZ promoters were significantly enriched over input DNA (6.68 and 4.80 fold, respectively), while the algB promoter demonstrated no significant enrichment compared to input DNA. The previously published AmrZ interaction at the fleQ and pslA promoters was also confirmed with this data set (Table 1), indicating the stringency of the analysis. The relatively low enrichment of these two previously described promoters by AmrZ provides a reference to which other interactions can be compared, suggesting that the interactions described here (≥3-fold enrichment) are biologically significant in the cell. In total, we identified 398 regions of the genome that were bound by AmrZ (≥3-fold enrichment over input) (Table S1).

Output for one significantly enriched region is included for reference (Figure 2A). In this example, AmrZ binds upstream of the PA4843 gene in the immunoprecipitated sample (green histogram), however, this enrichment is not present in the input sample (grey histogram). Other regions of the genome that were bound by AmrZ appeared similar. Specific AmrZ binding to the PA4843 promoter region was confirmed using an Electrophoretic Mobility Shift Assay (Figure 2B).
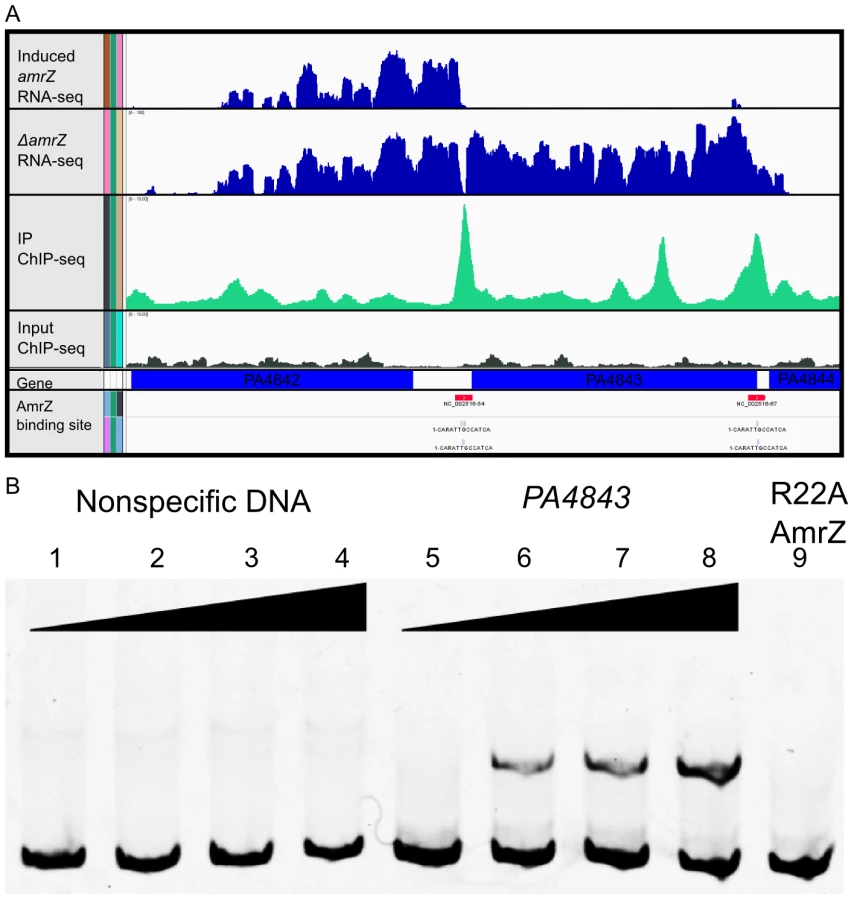
AmrZ consensus binding site is defined
The ChIP-Seq analysis allows one to predict consensus-binding sites based on identification of common sequences within enriched DNA. Based on these analyses we defined a consensus AmrZ binding site (Figure 3). The 13 nt motif was present in 54.7% of all enriched DNA fragments, but in only 10.93% of background reads, producing a significant enrichment of this sequence (p = 1e–120). This motif resembles that reported elsewhere for AmrZ binding using DNA binding and mutagenesis studies [18], [23]–[26], [43]. This motif is also contained in the crystal structure of AmrZ bound to the amrZ1 binding site identified by Pryor et al. [23]. Putative AmrZ binding sites were assigned to a selection of the AmrZ-enriched DNA fragments based on these consensus sequences and analysis of ChIP-Seq reads.

Transcriptional profiling via RNA-Seq defines the AmrZ regulon
Previous work demonstrated that AmrZ regulates genes in a variety of pathways, many of which are implicated in virulence [19], [21], [25], [26]. However, the extent of the AmrZ regulon is unknown. RNA-Seq allows comparison of sequences of the total mRNA from a ΔamrZ mutant to a complemented strain, elucidating the effect of AmrZ on all genes in the cell, both positive and negative. Total RNA was isolated from a mid-exponential culture (OD600 0.5±0.1) of a ΔamrZ mutant containing the empty pHERD20T vector and a complemented strain containing the arabinose inducible AmrZ expression vector pCJ3. These growth conditions were chosen to match those utilized in the ChIP-Seq experiment. cDNA was synthesized and the resulting product was tagged and quantified using high-throughput parallel DNA sequencing. mRNA expression levels and differential expression analysis was performed using the Bioconductor package DEseq [47]. Three hundred and thirty eight genes were significantly regulated at least 2-fold (Benjamini-Hochberg adjusted p value <0.05), with 89 genes activated - and 249 genes repressed - by AmrZ (Table S2). Several of the AmrZ-regulated genes described in the literature were identified in this analysis, including algD (activated by AmrZ 19.74 fold) and fleQ (repressed by AmrZ 8.05 fold).
The RNA-Seq data indicate that AmrZ strongly, though indirectly, represses many genes involved in iron acquisition, suggesting a novel mechanism for AmrZ mediated control of virulence (Table 1). AmrZ significantly repressed many genes in the pyochelin and pyoverdine synthesis operons, including ppyR. In addition, the Fe(III)-pyochelin receptor fptA and ferripyoverdine receptors fpvA and fpvB were all significantly repressed by AmrZ, (5.44, 2.01, and 1.45 fold, respectively), suggesting that reliance on the iron acquisition systems is reduced in strains where AmrZ is highly expressed, such as in mucoid isolates from the CF lung. Previous reports indicate that the iron concentration in the CF sputum and lung is elevated [48]-[50], supporting the hypothesis that there is sufficient iron in the CF lung for bacterial growth with reduced dependence on the high-affinity iron acquisition systems. Many virulence factors are iron-regulated, so the impact of AmrZ-mediated siderophore repression may contribute significantly to the establishment of chronic infections [51], [52]. There was no alteration of the transcription of the iron-dependent master regulator Fur in the ΔamrZ mutant, implying that AmrZ regulates these iron acquisition genes independent of Fur, perhaps through small RNAs or downstream members of the Fur regulon that have yet to be identified. Future studies will explore the relationship of AmrZ and iron acquisition during infection.
AmrZ directly regulates many genes associated with virulence
The results from the RNA-Seq and ChIP-Seq were further evaluated to determine genes potentially directly regulated by AmrZ. To accomplish this, the list of AmrZ-bound genomic regions (at least 3-fold enrichment) was filtered using the list of target genes regulated by AmrZ as determined by the RNA-Seq (at least 2-fold regulation). This approach allows classification of genes based on AmrZ binding status and AmrZ-mediated regulation. Interestingly, only 9 of the AmrZ-activated and 49 of the AmrZ-repressed genes were identified in the ChIP-Seq as also containing an AmrZ binding site within 500 base pairs of the start of the coding region of the gene (Table 2), suggesting that there are 80 activated and 200 repressed genes with promoters that were not directly bound by AmrZ, suggesting indirect regulation. One AmrZ directly activated gene is algD, a known AmrZ-dependent gene [24]. Other AmrZ-activated genes in Table 2A include a putative alginate lyase and members of the pel operon. These two genes, in combination with activation of the algD operon, suggest that when expressed, AmrZ affects the P. aeruginosa polysaccharide profile. Additionally, AmrZ directly activates the cyclic di-GMP response gene cdrA, which is correlated with polysaccharide overexpression [30]. Table 2B depicts genes directly repressed by AmrZ. In addition to the previously described fleQ, this list includes many genes that are known or predicted to be involved in virulence including: pyochelin synthesis (pchG), aggregation (siaA), flagellum synthesis (fleQ, flgG, flgE, fliF), alternative type IV pili production (flp), chemotaxis (PA2867, pctC, PA4844), multidrug transport (PA3401), and rhamnolipid production and quorum sensing (rhlR). Several of the directly AmrZ-repressed genes are predicted to be involved in Type VI secretion: PA1657, PA1664, PA1668. Type VI secretion is a recently-described system that is involved in P. aeruginosa pathogenesis and fratricide [53]–[56]. Specifically, the Type VI genes repressed by AmrZ belong to the HSI-II locus, which is involved in P. aeruginosa pathogenicity. HSI-II mutant strains exhibit a delay in mortality in both murine lung and burn wound infections [54]. This regulation may contribute to the role of AmrZ during infection.

Another group of AmrZ directly repressed genes are those predicted to be involved in cyclic diguanylate signaling. These include a predicted diguanylate cyclase (PA4843), a predicted phosphodiesterase (PA2567), and hypothetical proteins that are proposed c-di-GMP effector proteins containing PilZ domains (PA4324, PA3353). PA4324 does not appear to be part of an operon, while PA3353 is in the flgM operon and may have a function in flagella motility [57]. Dysregulation of c-di-GMP signaling could account for the hyper-aggregative phenotype of a ΔamrZ mutant. We explore this system further in this study.
A common mechanism of AmrZ-mediated repression
Transcriptional start sites were obtained from RNA-Seq data by observing where the sequence reads begin upstream of a coding region [58]. By performing this analysis to a selection of directly AmrZ-regulated promoters, the proximity of the AmrZ binding site was observed relative to the transcription start site. Promoters with strong AmrZ binding (≥4-fold enrichment) and regulation (≥4-fold regulation) were chosen for an alignment of the AmrZ binding site to the start of transcription. The two strongly activated promoters did not suggest a common mechanism (Figure 4A). However, with the exception of PA3235, each of the directly AmrZ-repressed promoters observed contained an AmrZ binding site from −100 to +15 relative to the transcription start site (Figure 4B). This implies that during repression, AmrZ interferes with the binding of RNA polymerase to the promoter, a common mechanism of bacterial transcriptional repression.

Previous publications have identified two AmrZ binding sites in the amrZ promoter, amrZ1 and amrZ2 [22], [23]. The amrZ1 binding site was identified by the ChIP-Seq (Figure 4B, red binding site). The previously identified amrZ2-binding site was not specifically identified by ChIP-Seq, however, this is likely due to the reduced AmrZ affinity for the amrZ2 binding site [18], [22]. Analysis of the read alignment of the immunoprecipitated sample reveals a biphasic peak including both the amrZ1 and amrZ2 binding sites. One gene (PA3235) that was repressed by AmrZ lacked a binding site in the promoter. However, AmrZ did bind 70 bp downstream of the observed PA3235 start of transcription. This may indicate a second mechanism of AmrZ repression, where bound AmrZ interferes with the elongation of the transcript.
Analysis of the proximal promoter regions of AmrZ-regulated genes indicates that AmrZ may affect RNA polymerase assembly directed by several sigma factors. For example, the −10 and −35 boxes of siaA appear to indicate that this promoter is RpoD-dependent (−35TTGaCc/−10TAtAAT), while the promoter of PA4843 appears to match the consensus sequence for a σN dependent promoter (−24GG/−12GC) [59]. There was no discernable pattern in the relation of the AmrZ binding site to the start of transcription in the AmrZ-activated genes, indicating that there may be several mechanisms of AmrZ-mediated direct activation.
adcA (PA4843) encodes a diguanylate cyclase
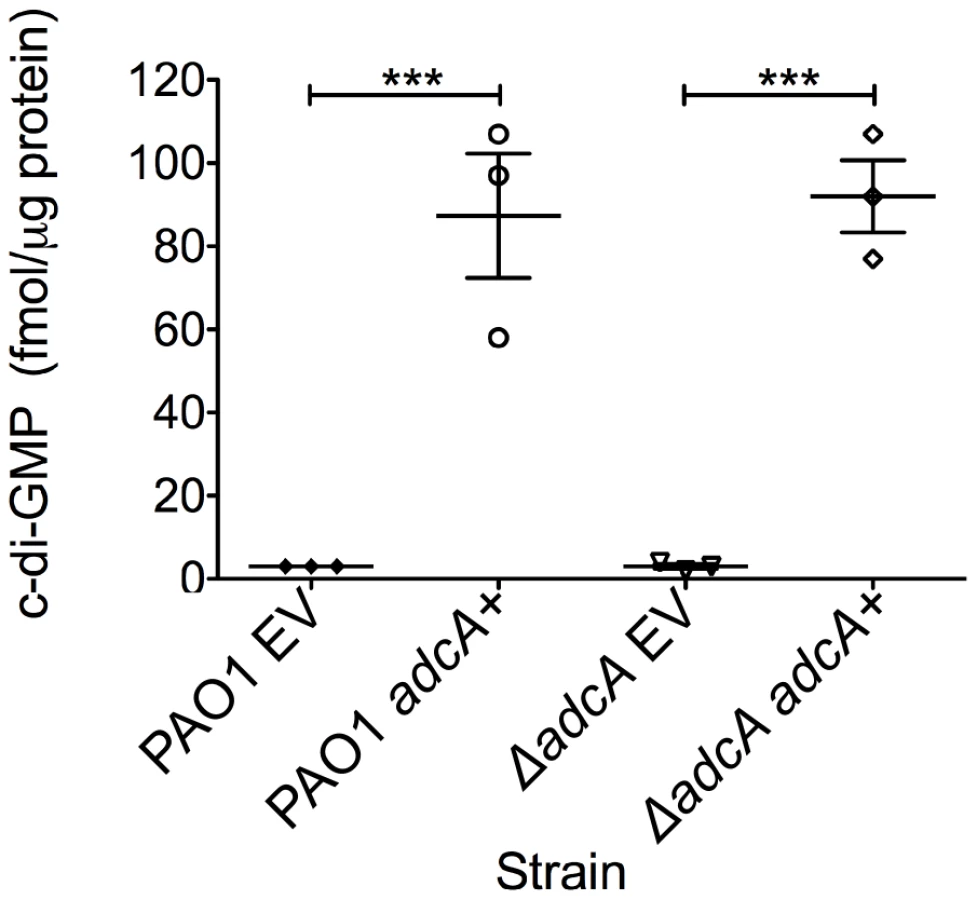
The gene most highly repressed by AmrZ was PA4843 (40-fold) (Table 2B). Predictions based on the structure and function of PleD from Caulobacter crescentus indicates that PA4843 contains two component receiver domains (Rec), an I-site, and a GGEEF cyclase domain (Figure S1). Previously, PA4843 was described as a putative diguanylate cyclase [60] since it contains a conserved cyclase domain; however, no reports demonstrate functional cyclase activity for the PA4843-encoding gene. Additionally, deletion of this gene in strain PA14 did not impact attachment or host cell cytotoxicity [60]. Because PA4843 was the most highly repressed AmrZ target gene and ΔamrZ mutants have an RSCV phenotype and elevated levels of c-di-GMP (Figure 1B), we hypothesized that PA4843 encoded a diguanylate cyclase that is de-repressed in ΔamrZ mutants. To address this, PA4843 was cloned into the arabinose inducible vector pHERD20T [61] and the plasmids transferred to wild type PAO1 or a strain lacking PA4843. c-di-GMP levels in both PAO1 or ΔPA4843 containing the induced vector control exhibited low levels of c-di-GMP (∼3 fmol/µg total protein) (Figure 5). However, expression of PA4843 in these strains generated nearly thirty fold more c-di-GMP (87 fmol/µg total protein for PAO1, and 92 fmol/µg total protein for ΔPA4843; Figure 5), supporting the hypothesis that PA4843 is a functional diguanylate cyclase. Based on these results and others below, we named PA4843 adcA, for AmrZ-dependent cyclase A. Additionally, a deletion of adcA in a ΔamrZ mutant returns the c-di-GMP to wild-type levels, (ΔamrZ mutant 7.33 fmol/µg total protein, ΔamrZ ΔadcA double mutant 2.33 fmol/µg total protein) indicating that the elevated c-di-GMP in a ΔamrZ mutant is dependent on AdcA.
The ΔamrZ mutant hyper biofilm phenotype is adcA - and c-di-GMP-dependent
As previously reported, a ΔamrZ mutant forms robust biofilms with more biomass and taller microcolonies than the parental strain, PAO1 [19]. This report demonstrated that direct repression of the psl operon by AmrZ could abrogate the hyper biofilm phenotype of the ΔamrZ mutant [19]. Here, we present data that AmrZ also regulates c-di-GMP concentrations in the cell, thus providing an additional level of control. We hypothesized that the ΔamrZ mutant hyper biofilm phenotype is due to adcA derepression and c-di-GMP accumulation in this strain. To test this hypothesis, we grew 24-hour flow cell biofilms in a PAO1 and ΔamrZ mutant background while modulating the amount of adcA expression in the cells (Figure 6A). We reasoned if the hyper biofilm phenotype is dependent on derepression of adcA and accumulation of c-di-GMP in the ΔamrZ mutant, biofilm cells formed by a ΔamrZ ΔadcA double mutant should have less intracellular c-di-GMP and biofilms with less biomass and microcolony height. Consistent with this hypothesis, the ΔamrZ ΔadcA double mutant produces biofilms with significantly less biomass than the ΔamrZ mutant (Figure 6A, Figure S3). Additionally, we observed that the ΔamrZ ΔadcA double mutant produced significantly lower c-di-GMP compared to the ΔamrZ mutant (2.33 vs 5.98 fmol/µg total protein, respectively) while the adcA overexpressing ΔamrZ mutant had significantly higher c-di-GMP (67.00 fmol/µg total protein). These data indicate that the hyper biofilm phenotype of the ΔamrZ mutant is due to loss of repression of adcA and elevated intracellular c-di-GMP. This mechanism, in addition to the previously reported direct repression of the psl-encoded biofilm polysaccharide [19], indicates that AmrZ-dependent regulation of the psl operon at multiple levels may amplify the effect on Psl production, with significant changes in the biofilm phenotype.

We also reasoned that if dysregulation of c-di-GMP production is responsible for the hyper biofilm phenotype of ΔamrZ mutants, then reducing intracellular c-di-GMP in these strains by overexpressing a phosphodiesterase (PDE) should ablate biofilm formation. For this, a plasmid encoding the arabinose inducible PDE PA2133 (pJN2133) or the empty vector pHERD20T was transformed into the ΔamrZ mutant and 16 hour flow cell biofilms were grown in the presence of inducer [32]. CLSM analysis demonstrates that PDE overexpression significantly reduces biofilm biomass and microcolony height in these biofilms (Figure. 6B). These data further support the hypothesis that the hyper biofilm phenotype of ΔamrZ mutants is dependent on elevated c-di-GMP.
Discussion
Understanding how bacteria respond to varying conditions in the environment and during infection is clearly of importance. Here, we present a comprehensive analysis of a bacterial transcription factor regulon obtained by combining ChIP-Seq and RNA-Seq. The power of these techniques stems from the unbiased and genome-wide production of the entire regulon, but also the activity of the transcription factor at these binding sites. These techniques have been established in eukaryotes [62], [63], however they have recently been adapted as powerful tools to investigate the activity of bacterial transcription factors [27], [28], [58], [64]–[67]. We were able to identify 398 regions bound by AmrZ in the P. aeruginosa genome. Additionally, we developed a transcriptional profile of both the ΔamrZ mutant and its complemented strain. This allowed us to combine the results of ChIP-Seq and RNA-Seq and divide loci into several categories, either activated, repressed, or unaffected by AmrZ. Each of these groups were then further categorized into directly or indirectly regulated.
Our prior studies revealed that wild type bacteria have a competitive advantage over ΔamrZ mutant bacteria in a mixed acute pulmonary model of infection [18]. By combining ChIP-Seq and RNA-Seq analysis, we identified many genes that are AmrZ-regulated and may be important for colonization and disease progression. One of the directly AmrZ-repressed genes, a diguanylate cyclase we named adcA (PA4843), emerged as the most highly regulated AmrZ target. Deletion of adcA in a ΔamrZ mutant eliminated the accumulation of c-di-GMP and the hyper biofilm phenotype. The modulation of c-di-GMP by AmrZ is a novel observation and enhances the molecular explanation for the earlier studies regarding the role of AmrZ in biofilm phenotypes [19]. c-di-GMP has diverse functions in P. aeruginosa, regulating polysaccharide production, motility, virulence factor production, and biofilm formation [60], [68]. When competed against the wild type PAO1 in an acute pulmonary infection model, both a ΔadcA mutant and a ΔamrZ ΔadcA double mutant retained the virulence defect observed for the ΔamrZ mutant (Figure S2). We propose that AmrZ-dependent gene regulation is most important in the establishment of chronic infections, as in the cystic fibrosis lung. Therefore, lack of a phenotype in an acute model of infection does not negate a role for AmrZ in chronic infections and future studies are geared towards this line of investigation. It should be noted that suitable chronic lung infection models that faithfully reproduce CF pathology are limited, though there are several very promising developments in this area [69].
Regulation of the numerous DGC and PDE enzymes in P. aeruginosa presents a complex network of integrated stimuli sensation and physiological response. Work in other systems has demonstrated that c-di-GMP is freely diffusible in the cytoplasm and is detected by many sensors [31], [70], [71]. This work highlights the regulation of one DGC, however, deciphering the regulation of c-di-GMP production and cellular response to diverse signals is currently an area of great interest.
In addition to the DGC activity described here, AdcA contains a predicted N-terminal two-component receiver domain. This combination of receiver domain and DGC is also observed in the well-characterized PleD of C. crescentus [72], [73]. Previous studies have revealed PleC-dependent activation of the PleD receiver domain by phosphorylation, leading to dimerization and c-di-GMP production [72]. The end result of this signaling cascade is the loss of flagellum and development of the stalk leading to a sessile lifestyle. Another example of a hybrid response regulator/diguanylate cyclase with biofilm effects is WspR of P. aeruginosa [32]. Surface growth leads to phosphorylation of WspR, inducing clustering of the protein and activation of cyclase activity [74], [75]. This model of clustered cyclases suggests that such subcellular foci can lead to regional increases of c-di-GMP, which may be an explanation for why subtle changes in whole-cell c-di-GMP pools can have drastic and varied effects on biofilm and motility phenotypes [73]–[76]. Analysis of AdcA for conserved domains indicates that the aspartate at residue 300 is a probable phosphorylation site. Activation of AdcA in P. aeruginosa leads to a hyper biofilm phenotype, suggesting that AdcA, PleD, and WspR have similar cellular effects. Based on the homology between these proteins, future studies will identify the partner sensor kinase and evaluate the effects of AdcA phosphorylation.
AmrZ activates alginate transcription and twitching motility, but represses Psl, flagella, and c-di-GMP production (Figure 7). Each of these pathways have been implicated in biofilm formation and disease chronicity [77]–[85]. The complete analysis of the AmrZ regulon indicates that AmrZ may serve as a molecular switch that triggers biofilm maturation in P. aeruginosa. We have observed that nonmucoid, environmental strains produce a low amount of AmrZ, allowing for high production of the adherent and aggregative polysaccharide Psl [19]. Additionally, low AmrZ in these strains allows expression of fleQ and flagellum production, further enhancing the attachment phenotypes [21], [80]. We present here that low AmrZ also permits expression of the diguanylate cyclase adcA, producing elevated c-di-GMP in the cell. This signaling molecule can affect the production of all of the above pathways in addition to the direct regulation by AmrZ [29], [32], [37], [38]. Cumulatively, the result of de-repression of these genes results in a motile strain that is primed to colonize and form biofilms by expressing the adhesive polysaccharide Psl. We observe a hyper aggregative and hyper biofilm phenotype in the ΔamrZ mutant, supporting this hypothesis. A similar phenomenon is observed in a ΔretS mutant, where elevated c-di-GMP leads to hyper biofilm formation [40], [86]. The GacS/RetS sensor systems are involved in the transition from acute to chronic infections by regulating polysaccharide production, motility, and secretion systems [40], [86], [87]. These systems regulate virulence genes through RsmA, which has a vast regulon [41], [88]. Though AmrZ was not identified as regulating any of the members of the Gac/Rsm signaling cascade, the ultimate effects of the two pathways are strikingly similar. Further work will investigate how AmrZ is interacting or overlapping with these well-established regulators of acute to chronic transition. Identification of the signal activating AdcA will enhance the understanding of the interactions of these two functionally similar pathways.

Strains of P. aeruginosa that infect patients are Psl-producing, nonmucoid, and form biofilms more readily than mucoid strains [79], [89]. Once a cystic fibrosis patient is infected with a nonmucoid strain, there is an aggressive neutrophil influx into the lungs [90]. These neutrophils produce many antimicrobial products, including reactive oxygen species, antimicrobial peptides, and neutrophil nets [91], [92]. Additionally, CF patients with active infections are treated with high doses of antibiotics. These factors, coupled with the high salinity, low oxygen, and high viscosity of the mucus in the CF lung, provide an environment that is highly selective for bacterial variants able to persist [93]. One clear phenotype that emerges in this environment is the production of alginate (mucoidy), which provides resistance to phagocytosis and protection against antibiotics and reactive oxygen species [7], [16], [94]–[96]. Mucoid strains express AmrZ at levels much higher than those observed in nonmucoid counterparts [24], [25], [43]. We propose that AmrZ acts as a molecular switch that transitions P. aeruginosa from a motile, adherent, colonizing strain causing acute virulence and tissue damage to a nonmotile, mucoid, chronic strain that is more adept at persistence and immune evasion. We suggest that the enhanced virulence of the wild type is due to the expression of various virulence factors such as the type III secretion system regulator ExsC and iron sequestration proteins such PchC and FptA. AmrZ represses these genes (−3.4, −9.01, and −5.45-fold, respectively), though their promoters were not identified as bound by AmrZ in the ChIP-Seq analysis, suggesting that this repression is indirect. When AlgT/U is active, as in mucoid strains, the amount of AmrZ rises. This rise in AmrZ could reduce production of these proteins and limit the acute virulence of the strains, allowing for the establishment of a chronic infection. Additionally, we demonstrate that AmrZ activates expression of cdrA, encoding a biofilm matrix protein and the pel polysaccharide operon. Previous reports indicate that AmrZ can directly repress the psl operon, leading to multifactorial control of this polysaccharide [19]. Combined with the published knowledge of the effect of c-di-GMP on the psl operon through FleQ, these data further reinforce the potential for additive effects of AmrZ at multiple points of polysaccharide and matrix protein regulation. Cumulatively, these experiments suggest that the high production of AmrZ in mucoid strains during chronic infections could lead to a polysaccharide transition from expressing Psl to alginate and Pel. Additionally, CdrA has been reported to stabilize biofilm structure [30]. The overlap of these regulatory networks with the inclusion of c-di-GMP signaling could provide insight to the complexity of the contributions of polysaccharides to virulence during different stages of infection. Future work will delve into virulence contribution by the AmrZ-regulated genes to identify the molecular basis for the acute virulence defect in the ΔamrZ mutant.
Materials and Methods
Ethics statement
All animals were maintained in the OSU College of Medicine IACUC-approved vivarium located in the Biomedical Research Tower. The University has many veterinarians and trained animal caretakers available for consultation on the studies. The protocol for these studies has been approved by the OSU IACUC committee (Protocol # 2009A0177). There is adequate space for the animals to be housed in the vivarium. Animals are monitored frequently during the infection. Animals that meet the criteria for removal from study will be euthanized via CO2 inhalation. Each room contains sentinel mice that are sacrificed at regular time points for examination for infectious agents by vivarium staff. During infection, mice were lightly sedated with isoflurane and inoculated intranasally with bacteria suspended in sterile PBS. Thirty µL of the PBS solution is pipetted onto the nares of the mouse as soon as the anesthetic administration is discontinued. The animal rapidly recovers under supervision from the researcher. The mice are not in discomfort or distress during this procedure. There are no restraining devices utilized during this study. Mice were sacrificed via CO2 inhalation. This method of euthanasia causes minimal discomfort to the animals. Cardiac puncture was used as a second method of euthanasia. These methods are consistent with the recommendations of the American Veterinary Medical Association Guidelines on Euthanasia.
Bacterial strains and growth conditions
The bacterial strains used along with genotypes are provided in Table S3. P. aeruginosa strains were inoculated in LBNS (10 g l−1 tryptone, 5 g l−1 yeast extract, pH 7.5) at 37°C for overnight cultures under shaking conditions unless otherwise noted. Strains were grown at 37°C on LANS (LBNS with 1.5% agar) or Pseudomonas Isolation Agar (Difco, Detroit, MI) agar plates. E. coli was routinely cultured at 37°C in lysogeny broth (LB, 10 g L−1 tryptone, 5 g L−1 yeast extract, 5 g L−1 NaCl). Semi-solid media was prepared by adding 1.5% Bacto agar to LB. Colony morphology was imaged on modified Vogel-Bonner minimal medium (VBMM) plates (0.2 g L−1 MgSO4 7H2O, 2.0 g L−1 citric acid, 3.5 g L−1 NaNH4HPO4 4H2O, and 10 g L−1 K2HPO4, 1 g L−1 casamino acids, and 5 mM CaCl2. Congo Red (40 µg/mL) and Brilliant Blue R (15 µg/mL) were added to VBMM to aid in visualization of morphology. Antibiotics were added to maintain or select for plasmids in P. aeruginosa as follows: gentamicin (Gm) at 100 µg/mL, Rifampicin (Rif) at 100 µg/mL and carbenicillin (Cb) at 300 µg/mL. Antibiotics were added to maintain or select for plasmids in E. coli as follows: gentamicin (Gm) at 10 µg/mL and spectinomycin (Sp) at 50 µg/mL.
Plasmid construction
Plasmids and primers used in genetic manipulations are listed in Tables S4 and S5, respectively.
Primers AmrZF2 and AmrZR2 amplified the 324 bp DNA sequence of amrZ. NEB Q5 High Fidelity DNA Polymerase was used in PCR following manufacturer's instructions. The PCR product of amrZ was inserted into pET29a (Novogen) through NdeI and NotI restriction sites. The 432 bp DNA sequence of the amrZ gene, ribosome binding site, and C-terminal 6x His tag were amplified from the resulting plasmid using primers AmrZF3 and AmrZR3. The PCR product was inserted into pHERD20T [61] through XbaI and HindIII restriction sites. The resulting construct (pCJ3) was verified by DNA sequencing.
A deletion allele for PA4843 was assembled by removing an in-frame, 1593 bp fragment of coding sequence from the PA4843 open reading frame (ORF), leaving a scar ORF encoding a 10-amino acid peptide. In a first step, two PCR products were amplified using primers that targeted the adjacent upstream and downstream regions of the chromosome flanking PA4843. Subsequently, these PCR products were joined by splicing by overlapping extension (SOE) PCR [97] to create the ΔPA4843 allele. The upstream forward and downstream reverse primers used to generate this deletion allele were tailed with attB1 or attB2 sequences as described in the Gateway Cloning Technology Manual (Invitrogen). Using Gateway technology, the ΔPA4843 allele was first recombined with pDONR223 using BP Clonase II (Invitrogen) to create pJJH125, which was sequenced using M13F and M13R primers. Finally, the ΔPA4843 allele from pJJH125 was recombined with pEX18GmGW using LR Clonase II (Invitrogen) to create the allelic exchange vector pJJH129.
The adcA overexpression plasmid pBX22 was constructed by inserting adcA coding sequence into the arabinose-inducible vector pHERD20T [61]. The 1659 bp DNA sequence of the adcA gene was amplified by primers PA4843_F and PA4843_R. NEB Q5 High Fidelity DNA Polymerase was used in PCR following manufacturer's instructions. The PCR product of adcA was inserted into pHERD20T through XbaI and HindIII restriction sites. The adcA coding sequence in pBX22 was verified by Sanger-based DNA sequencing.
Quantification of c-di-GMP by LC/MS
c-di-GMP was extracted and quantified as described previously with minor modifications [39]. Cells were cultured overnight on LANS plates. An isolated colony was transferred to a fresh LANS plate and incubated at 37°C for 24 hrs before harvesting. Colonies were scraped from agar plates and resuspended in 990 µL of LC/MS grade water (Optima). 2-chloro-adenosine-5′-O-monophosphate (2Cl-AMP, 10 µL of 10 µM, Biolog), was added as an internal standard. Nucleotides were extracted from cells by the addition of 94 µl of 70% perchloric acid and incubated for 30 min on ice. Cell debris were removed by centrifugation and reserved for subsequent protein quantification. The supernatant containing c-di-GMP was neutralized by the addition of 219 µL of 2.5 M KHCO3. The resulting precipitate was removed by centrifugation. The supernatant was stored at -80°C until LC/MS analysis. Pure c-di-GMP standards (Biolog) were extracted in parallel and treated identically to samples.
Compounds were separated on an Acuity UPLC equipped with a C18 Guard Cartridge (Phenomenex) and Synergi 4 µ Hydro RP 80A column (50×2 mm, Phenomenex). The injection volume was 20–30 µL. A gradient was established starting with 98% aqueous (10 mM formic acid in water) and 2% organic (acetonitrile). The aqueous concentration was adjusted to 70% at 2 min, 20% at 2.5 min, 100% at 3 min, and finally held at 98% from 5–7.5 min. Compounds were detected using multiple reaction monitoring on a Premier XL triple-quadrupole electrospray mass spectrometer (Waters) in positive-ionization mode. The m/z 691>152 transition was used for the identification of c-di-GMP and 382>170 for 2Cl-AMP. The cone voltages and collision energies were 40 V/30 eV and 35 V/20 eV, respectively. The capillary voltage used was 3.5 kV. The desolvation temperature was 350°C and source temperature was 120°C. Nitrogen was used as a drying gas with a flow rate of 800 L/hr. The concentrations of c-di-GMP were calculated by comparison of the peak area ratio of c-di-GMP to 2Cl-AMP to a standard curve. Moles of c-di-GMP were normalized to total protein determined from Pierce protein assay. Data represent averages of three independent cultures.
For protein quantification, cell pellets were resuspended in 220 µL of 10 mM Tris-Cl buffer (pH 8.5). The remaining acid in the pellets was neutralized by the addition of 30 µl of 1 M NaOH. Cells were lysed by the addition of 250 µl of 2X concentrated Laemilli Buffer and boiled for 30-90 min at 100°C or until the pellet had dissolved. Protein concentration was determined using Pierce 660 nm Protein Reagent with Ionic Detergent Compatibility Reagent (IDCR) as recommended by the manufacturer.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was modified from existing protocols [44], [45]. Cultures were induced with 0.5% arabinose at an OD600 of 0.1 and allowed to grow for two hours at 37°C in a roller. Protein-DNA complexes were cross-linked by addition of formaldehyde - to a final concentration of 1.0% and incubated at room temperature for ten minutes. Cross-linking was quenched by addition of glycine (final concentration 250 mM). The final OD600 was recorded and cells were collected from 1 OD600 of culture via centrifugation and washed once in LBNS. The supernatant was removed and pellets were stored for further processing at -80°C.
Cell pellets were resuspended in 1.0 mL of lysis buffer (20 mM HEPES, pH 7.9; 50 mM KCl; 0.5 mM DTT; 500 mM NaCl; 10 mM imidazole; 1% BSA; 1 µg/mL leupeptin/pepstatin; and 400 µM PMSF) per 1 OD600 of culture. Samples were sonicated on Covaris with the following conditions: Duty Cycle 20%, Intensity 8, Cycles per burst 200, with frequency sweeping 20 min total shearing time (60 sec cycles, 20 cycles). Lysate was cleared via centrifugation (20,000× g, 30 minutes, 4°C) and the supernatant was transferred to a fresh tube as the input sample. Magne-HIS beads (Promega V8560) were blocked at room temperature in lysis buffer for 30 minutes, and then 500 µl of the input sample was added to the beads. After 30 minutes of binding at room temperature with agitation, the supernatant was removed from the beads via magnetic separation. Beads were washed five times in wash buffer (100 mM HEPES, pH 7.5, 10 mM imidazole, 500 mM NaCl, and 1% BSA). Elution buffer (100 mM HEPES, pH 7.5; and 500 mM imidazole) was added to the beads and incubated at room temperature for 30 minutes. Supernatant was collected after magnetic separation and combined with SDS (1.25% final concentration), then heated to 70°C for 30 minutes to reverse cross-links. DNA was purified via phenol:chloroform extraction and ethanol precipitation [98].
ChIP-Seq library construction and sequencing
The chip DNA was quantified with Qubit 2 flurometer (Life Technologies) using Qubit dsDNA BR Assay. 10 ng of DNA was used to construct each Chip sequencing library, following NEXTflex ChIP-Seq kit (Bioo Scientific) instruction. NEXTflex ChIP-Seq Barcodes (Bioo Scientific) were used to index the library. The final DNA libraries were validated with Agilent 2100 Bioanalyzer using Agilent High Sensitivity DNA Kit. And the library concentrations were determined by Q-PCR using KAPA SYBR Fast qPCR kit. The libraries were then run on Single End flowcell on HiSeq2000.
ChIP-Seq data analysis
HiSeq2000 sequencing was performed, resulting in approximately 255 million total single-end 52 bp reads from the six control and eight treatment samples. Reads were aligned using bwa (0.5.10) to the Pseudomonas aeruginosa PAO1 reference genome [99]. Approximately 220 million reads aligned uniquely to the reference (86.3%). A TDF file was created for each sample for visualization in IGV, which was scaled to reads per 10 million data using bedtools (2.17.0) and igvtools (2.3.3). ChIP-Seq analysis was performed using HOMER (4.2). First, aligned data was transformed into a platform-independent data structure for further HOMER analyses using the makeTagDirectory function. Secondly, HOMER's findPeaks-style factor was utilized to identify peaks, or regions of the genome where more reads are present than random. Lastly, HOMER's findMotifsGenome.pl was used to analyze genomic positions for de novo enriched motif regions of length 50 or 200 and identified peaks were annotated with the motifs using the annotatePeaks.pl function.
RNA isolation
Cultures were induced with 0.5% arabinose at an OD600 of 0.1 and allowed to grow for two hours at 37°C in a roller. The final OD600 was recorded and 0.1 OD600 was centrifuged at 10,000-x g for 3 minutes. The supernatant was removed and pellets were resuspended in 1 mL of TRIzol (Invitrogen). Following a 5-minute incubation at room temperature, 0.2 mL of chloroform was added and the samples were shaken for 15 minutes. Phases were separated by centrifugation (12,000× g, 5 minutes, 4°C) and the aqueous phase was combined with 0.6 mL of 70% ethanol and transferred to an RNeasy mini column (Qiagen). After centrifugation, 0.7 mL of buffer RW1 (Qiagen) was added to the column and centrifuged. Samples were washed twice with 0.5 mL of Buffer RPE (Qiagen) and eluted in 50 µL of water.
RNA-Seq library construction and sequencing
Following assessment of the quality of total RNA using Agilent 2100 bioanalyzer and RNA Nano Chip kit (Agilent Technologies, CA), rRNA was removed from 2.5 µg of RNA with Ribo-Zero rRNA removal kit for Gram-negative bacteria (Epicentre Biotechnologies, WI). To generate directional signal in RNA seq data, libraries were constructed from first strand cDNA using ScriptSeq v2 RNA-Seq library preparation kit (Epicentre Biotechnologies, WI). Briefly, 50 ng of rRNA-depleted RNA was fragmented and reverse transcribed using random primers containing a 5′ tagging sequence, followed by 3′end tagging with a terminal-tagging oligo to yield di-tagged, single-stranded cDNA. Following purification by a magnetic-bead based approach, the di-tagged cDNA was amplified by limit-cycle PCR using primer pairs that anneal to tagging sequences and add adaptor sequences required for sequencing cluster generation. Amplified RNA-seq libraries were purified using AMPure XP System (Beckman Coulter). Quality of libraries were determined via Agilent 2100 Bioanalyzer using DNA High Sensitivity Chip kit, and quantified using Kappa SYBRFast qPCR kit (KAPA Biosystems, Inc, MA). 50 bp sequence reads were generated using the Illumina HiSeq 2000 platform.
RNA-Seq data analysis
HiSeq 2000 sequencing was performed, resulting in approximately 165 million total single-end 52-bp reads from the six total control and treatment samples. Reads were aligned using bwa (0.5.10) to the P. aeruginosa PAO1 reference genome [99]. Approximately 143 million reads aligned uniquely to non-ribosomal regions of the reference (86.9%). A TDF file was created for each sample for visualization in IGV, which was scaled to reads per million data using bedtools (2.17.0) and igvtools (2.3.3). A coverage file, describing the coverage for each feature in the PAO1 genome, was created using bedtools. These coverage's were normalized and the means of the control and treatment groups were tested for significant differences using the binomial test in the R package DESeq (1.10.1), producing fold changes and adjusted p-values for each feature. Resulting p-values were adjusted for multiple testing with the Benjamin-Hochberg procedure, which controls false discovery rate (FDR).
EMSA
6FAM labeled DNA used for EMSA was amplified using Quick-load Taq 2X Mastermix (New England Biolabs), FAM-labeled forward primer and non-labeled reverse primer, and PAO1 genomic DNA as the template. The EMSA procedure is similar to that previously reported [18]. Each EMSA reaction contains 4 mM Tris-HCl (pH8.0), 40 mM NaCl, 4 mM MgCl2, 4% glycerol, 150 ng/ul Poly d[(I-C)] (non-specific DNA control), 100 µg/mL BSA (non-specific protein control), 5 nM FAM labeled DNA, and a defined concentrations of AmrZ or AmrZR22A. Protein-DNA binding was equilibrated at room temperature (25°C) for 20 min after adding all reagents to each reaction. 10 µL of each reaction was loaded onto a 4% non-denaturing acrylamide gel. Electrophoresis was conducted for 22 min at 200 V in 0.5% TBE. 6FAM fluorescence was detected with a Typhoon scanner (GE Lifescience). A similar length DNA sequence within the algD coding sequence but lacking an AmrZ binding site was selected as the specificity control.
Protein structure and function prediction
Protein sequence was submitted to the Phyre2 server for analysis of homology [100]. Predicted structure was imaged in Jmol (http://www.jmol.org).
Flow cell biofilm study
Inoculation of flow cells was done by normalizing overnight cultures to an optical density of 0.5 and injecting into an Ibidi μ-Slide VI0.4 (Ibidi 80601). To seed the flow cell surface, the media flow was suspended and the bacteria allowed to adhere at room temperature for 3 hours. Flow of 5% v/v LBNS with 0.5% arabinose was initiated at a rate of 0.15 mL*min−1 and continued for 24 h. Following the biofilm growth period, the flow was terminated and the biofilms were fixed with 4% paraformaldehyde. Confocal images were taken at the Ohio State University Campus Microscopy and Imaging Facility on an Olympus Fluoview 1000 Laser Scanning Confocal microscope. Images were obtained with a 20X oil immersion objective. Images were processed using the Olympus FV1000 Viewer software. Quantitative analyses were performed using the COMSTAT software package [101] Total biomass was determined from Z-stack images using the BIOMASS command with the threshold set to 15. Three independent biofilms were imaged and analyzed. Statistical significance was determined using a Student's t-test.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. WisplinghoffH, BischoffT, TallentSM, SeifertH, WenzelRP, et al. (2004) Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39 : 309–317 doi:10.1086/421946
2. RichardsMJ, EdwardsJR, CulverDH, GaynesRP (1999) Nosocomial infections in medical intensive care units in the United States. Crit Care Med 27 : 887.
3. Hall-StoodleyL, CostertonJW, StoodleyP (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2 : 95–108 doi:10.1038/nrmicro821
4. TartAH, WozniakDJ (2008) Shifting paradigms in Pseudomonas aeruginosa biofilm research. Curr Top Microbiol Immunol 322 : 193–206.
5. EvansDJ, BrownMRW, AllisonDG, GilbertP (1990) Susceptibility of bacterial biofilms to tobramycin: role of specific growth rate and phase in the division cycle. J Antimicrob Chemother 25 : 585–591 doi:10.1093/jac/25.4.585
6. HentzerM, TeitzelGM, BalzerGJ, HeydornA, MolinS, et al. (2001) Alginate overproduction affects Pseudomonas aeruginosa biofilm structure and function. J Bacteriol 183 : 5395–5401.
7. AlkawashMA, SoothillJS, SchillerNL (2006) Alginate lyase enhances antibiotic killing of mucoid Pseudomonas aeruginosa in biofilms. APMIS 114 : 131–138 doi:_10.1111/j.1600-0463.2006.apm_356.x
8. DaviesDG, ParsekMR, PearsonJP, IglewskiBH, CostertonJW, et al. (1998) The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280 : 295–298.
9. O'TooleGA, KolterR (1998) Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol Microbiol 28 : 449–461.
10. WebbJS, ThompsonLS, JamesS, CharltonT, Tolker-NielsenT, et al. (2003) Cell death in Pseudomonas aeruginosa biofilm development. J Bacteriol 185 : 4585–4592.
11. ShroutJD, ChoppDL, JustCL, HentzerM, GivskovM, et al. (2006) The impact of quorum sensing and swarming motility on Pseudomonas aeruginosa biofilm formation is nutritionally conditional. Mol Microbiol 62 : 1264–1277 doi:10.1111/j.1365-2958.2006.05421.x
12. O'TooleGA, KolterR (1998) Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol 30 : 295–304.
13. WorlitzschD, TarranR, UlrichM, SchwabU, CekiciA, et al. (2002) Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest 109 : 317–325 doi:10.1172/JCI13870
14. HassettDJ, SuttonMD, SchurrMJ, HerrAB, CaldwellCC, et al. (2009) Pseudomonas aeruginosa hypoxic or anaerobic biofilm infections within cystic fibrosis airways. Trends Microbiol 17 : 130–138 doi:10.1016/j.tim.2008.12.003
15. DrenkardE, AusubelFM (2002) Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416 : 740–743 doi:10.1038/416740a
16. LeidJG, WillsonCJ, ShirtliffME, HassettDJ, ParsekMR, et al. (2005) The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J Immunol 175 : 7512–7518.
17. NguyenD, Joshi-DatarA, LepineF, BauerleE, OlakanmiO, et al. (2011) Active Starvation Responses Mediate Antibiotic Tolerance in Biofilms and Nutrient-Limited Bacteria. Science 334 : 982–986 doi:10.1126/science.1211037
18. WaligoraEA, RamseyDM, PryorEE, LuH, HollisT, et al. (2010) AmrZ beta-sheet residues are essential for DNA binding and transcriptional control of Pseudomonas aeruginosa virulence genes. J Bacteriol 192 : 5390–5401 doi:10.1128/JB.00711-10
19. JonesCJ, RyderCR, MannEE, WozniakDJ (2013) AmrZ modulates Pseudomonas aeruginosa biofilm architecture by directly repressing transcription of the psl operon. J Bacteriol 195 : 1637–1644 doi:10.1128/JB.02190-12
20. TartAH, WolfgangMC, WozniakDJ (2005) The alternative sigma factor AlgT represses Pseudomonas aeruginosa flagellum biosynthesis by inhibiting expression of fleQ. J Bacteriol 187 : 7955–7962 doi:10.1128/JB.187.23.7955-7962.2005
21. TartAH, BlanksMJ, WozniakDJ (2006) The AlgT-dependent transcriptional regulator AmrZ (AlgZ) inhibits flagellum biosynthesis in mucoid, nonmotile Pseudomonas aeruginosa cystic fibrosis isolates. J Bacteriol 188 : 6483–6489 doi:10.1128/JB.00636-06
22. RamseyDM, BaynhamPJ, WozniakDJ (2005) Binding of Pseudomonas aeruginosa AlgZ to sites upstream of the algZ promoter leads to repression of transcription. J Bacteriol 187 : 4430–4443 doi:10.1128/JB.187.13.4430-4443.2005
23. PryorEE, WaligoraEA, XuB, Dellos-NolanS, WozniakDJ, et al. (2012) The transcription factor AmrZ utilizes multiple DNA binding modes to recognize activator and repressor sequences of Pseudomonas aeruginosa virulence genes. PLoS Pathog 8: e1002648 doi:10.1371/journal.ppat.1002648
24. BaynhamPJ, WozniakDJ (1996) Identification and characterization of AlgZ, an AlgT-dependent DNA-binding protein required for Pseudomonas aeruginosa algD transcription. Mol Microbiol 22 : 97–108.
25. BaynhamPJ, BrownAL, HallLL, WozniakDJ (1999) Pseudomonas aeruginosa AlgZ, a ribbon-helix-helix DNA-binding protein, is essential for alginate synthesis and algD transcriptional activation. Mol Microbiol 33 : 1069–1080.
26. BaynhamPJ, RamseyDM, GvozdyevBV, CordonnierEM, WozniakDJ (2006) The Pseudomonas aeruginosa ribbon-helix-helix DNA-binding protein AlgZ (AmrZ) controls twitching motility and biogenesis of type IV pili. J Bacteriol 188 : 132–140 doi:10.1128/JB.188.1.132-140.2006
27. DaviesBW, BogardRW, MekalanosJJ (2011) Mapping the regulon of Vibrio cholerae ferric uptake regulator expands its known network of gene regulation. Proc Natl Acad Sci U S A 108 : 12467–12472 doi:10.1073/pnas.1107894108
28. GalaganJ, LyubetskayaA, GomesA (2013) ChIP-Seq and the complexity of bacterial transcriptional regulation. Curr Top Microbiol Immunol 363 : 43–68 doi:__10.1007/82_2012_257
29. StarkeyM, HickmanJH, MaL, ZhangN, De LongS, et al. (2009) Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol 191 : 3492–3503 doi:10.1128/JB.00119-09
30. BorleeBR, GoldmanAD, MurakamiK, SamudralaR, WozniakDJ, et al. (2010) Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol Microbiol 75 : 827–842 doi:10.1111/j.1365-2958.2009.06991.x
31. BoydCD, O'TooleGA (2012) Second messenger regulation of biofilm formation: breakthroughs in understanding c-di-GMP effector systems. Annu Rev Cell Dev Biol 28 : 439–462 doi:10.1146/annurev-cellbio-101011-155705
32. HickmanJW, TifreaDF, HarwoodCS (2005) A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci U S A 102 : 14422–14427 doi:10.1073/pnas.0507170102
33. Christensen LD, van Gennip M, Rybtke MT, Wu H, Chiang WC, et al. (2013) Clearance of Pseudomonas aeruginosa foreign-body biofilm infections through reduction of the c-di-GMP level in the bacteria. Infect Immun [epub ahead of print]. doi:10.1128/IAI.00332-13.
34. HausslerS (2003) Highly adherent small-colony variants of I in cystic fibrosis lung infection. J Med Microbiol 52 : 295–301 doi:10.1099/jmm.0.05069-0
35. KirisitsMJ, ProstL, StarkeyM, ParsekMR (2005) Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl Environ Microbiol 71 : 4809–4821 doi:10.1128/AEM.71.8.4809-4821.2005
36. MaloneJG, WilliamsR, ChristenM, JenalU, SpiersAJ, et al. (2007) The structure-function relationship of WspR, a Pseudomonas fluorescens response regulator with a GGDEF output domain. Microbiology 153 : 980–994 doi:10.1099/mic.0.2006/002824-0
37. HickmanJW, HarwoodCS (2008) Identification of FleQ from Pseudomonas aeruginosa as a c-di-GMP-responsive transcription factor. Mol Microbiol 69 : 376–389 doi:10.1111/j.1365-2958.2008.06281.x
38. BaraquetC, MurakamiK, ParsekMR, HarwoodCS (2012) The FleQ protein from Pseudomonas aeruginosa functions as both a repressor and an activator to control gene expression from the pel operon promoter in response to c-di-GMP. Nucleic Acids Res 40 : 7207–7218 doi:10.1093/nar/gks384
39. IrieY, BorleeBR, O'ConnorJR, HillPJ, HarwoodCS, et al. (2012) Self-produced exopolysaccharide is a signal that stimulates biofilm formation in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 109 : 20632–20636 doi:10.1073/pnas.1217993109
40. MoscosoJA, MikkelsenH, HeebS, WilliamsP, FillouxA (2011) The Pseudomonas aeruginosa sensor RetS switches type III and type VI secretion via c-di-GMP signalling. Environ Microbiol 13 : 3128–3138 doi:10.1111/j.1462-2920.2011.02595.x
41. Frangipani E, Visaggio D, Heeb S, Kaever V, Cámara M, et al. (2013) The Gac/Rsm and cyclic-di-GMP signalling networks coordinately regulate iron uptake in Pseudomonas aeruginosa. Environ Microbiol [epub ahead of print] doi:10.1111/1462-2920.12164.
42. RömlingU, GalperinMY, GomelskyM (2013) Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev 77 : 1–52 doi:10.1128/MMBR.00043-12
43. YuH, MuddM, BoucherJC, SchurrMJ, DereticV (1997) Identification of the algZ gene upstream of the response regulator algR and its participation in control of alginate production in Pseudomonas aeruginosa. J Bacteriol 179 : 187–193.
44. CastangS, McManusHR, TurnerKH, DoveSL (2008) H-NS family members function coordinately in an opportunistic pathogen. Proc Natl Acad Sci U S A 105 : 18947–18952 doi:10.1073/pnas.0808215105
45. GilbertKB, KimTH, GuptaR, GreenbergEP, SchusterM (2009) Global position analysis of the Pseudomonas aeruginosa quorum-sensing transcription factor LasR. Mol Microbiol 73 : 1072–1085 doi:10.1111/j.1365-2958.2009.06832.x
46. HeinzS, BennerC, SpannN, BertolinoE, LinYC, et al. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. MolCell 38 : 576–589 doi:10.1016/j.molcel.2010.05.004
47. AndersS, HuberW (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106 doi:10.1186/gb-2010-11-10-r106
48. StitesSW, WaltersB, O'Brien-LadnerAR, BaileyK, WesseliusLJ (1998) Increased iron and ferritin content of sputum from patients with cystic fibrosis or chronic bronchitis. Chest 114 : 814–819.
49. StitesSW, PlautzMW, BaileyK, O'Brien-LadnerAR, WesseliusLJ (1999) Increased concentrations of iron and isoferritins in the lower respiratory tract of patients with stable cystic fibrosis. Am J Respir Crit Care Med 160 : 796–801 doi:10.1164/ajrccm.160.3.9811018
50. PalmerKL, AyeLM, WhiteleyM (2007) Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J Bacteriol 189 : 8079–8087 doi:10.1128/JB.01138-07
51. OglesbyAG, FarrowJM, LeeJH, TomarasAP, GreenbergEP, et al. (2008) The Influence of Iron on Pseudomonas aeruginosa Physiology: A REGULATORY LINK BETWEEN IRON AND QUORUM SENSING. J Biol Chem 283 : 15558–15567 doi:10.1074/jbc.M707840200
52. JimenezPN, KochG, ThompsonJA, XavierKB, CoolRH, et al. (2012) The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol Mol Biol Rev 76 : 46–65 doi:10.1128/MMBR.05007-11
53. MougousJD, CuffME, RaunserS, ShenA, ZhouM, et al. (2006) A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 312 : 1526–1530 doi:10.1126/science.1128393
54. LesicB, StarkeyM, HeJ, HazanR, RahmeLG (2009) Quorum sensing differentially regulates Pseudomonas aeruginosa type VI secretion locus I and homologous loci II and III, which are required for pathogenesis. Microbiology 155 : 2845–2855 doi:10.1099/mic.0.029082-0
55. HoodRD, SinghP, HsuF, GüvenerT, CarlMA, et al. (2010) A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 7 : 25–37 doi:10.1016/j.chom.2009.12.007
56. RussellAB, HoodRD, BuiNK, LeRouxM, VollmerW, et al. (2011) Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475 : 343–347 doi:10.1038/nature10244
57. FriskA, JyotJ, AroraSK, RamphalR (2002) Identification and Functional Characterization of flgM, a Gene Encoding the Anti-Sigma 28 Factor in Pseudomonas aeruginosa. J Bacteriol 184 : 1514–1521 doi:10.1128/JB.184.6.1514-1521.2002
58. DötschA, EckweilerD, SchniederjansM, ZimmermannA, JensenV, et al. (2012) The Pseudomonas aeruginosa transcriptome in planktonic cultures and static biofilms using RNA sequencing. PLoS ONE 7: e31092 doi:10.1371/journal.pone.0031092.s006
59. PotvinE, SanschagrinF, LevesqueRC (2008) Sigma factors in Pseudomonas aeruginosa. FEMS Microbiol Rev 32 : 38–55 doi:10.1111/j.1574-6976.2007.00092.x
60. KulasakaraH, LeeV, BrencicA, LiberatiN, UrbachJ, et al. (2006) Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3“-5”)-cyclic-GMP in virulence. Proc Natl Acad Sci U S A 103 : 2839–2844 doi:10.1073/pnas.0511090103
61. QiuD, DamronFH, MimaT, SchweizerHP, YuHD (2008) PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl Environ Microbiol 74 : 7422–7426 doi:10.1128/AEM.01369-08
62. JohnsonDS, MortazaviA, MyersRM, WoldB (2007) Genome-wide mapping of in vivo protein-DNA interactions. Science 316 : 1497–1502 doi:10.1126/science.1141319
63. NagalakshmiU, WangZ, WaernK, ShouC, RahaD, et al. (2008) The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320 : 1344–1349 doi:10.1126/science.1158441
64. PerkinsTT, DaviesMR, KlemmEJ, RowleyG, WilemanT, et al. (2012) ChIP-seq and transcriptome analysis of the OmpR regulon of Salmonella enterica serovars Typhi and Typhimurium reveals accessory genes implicated in host colonization. Mol Microbiol 87 : 526–538 doi:10.1111/mmi.12111
65. LiaoJ, SchurrMJ, SauerK (2013) The MerR-like regulator BrlR confers biofilm tolerance by activating multidrug-efflux pumps in Pseudomonas aeruginosa biofilms. J Bacteriol 195(15): 3352–63 doi:10.1128/JB.00318-13
66. BalasubramanianD, KumariH, JaricM, FernandezM, TurnerKH, et al. (2013) Deep sequencing analyses expands the Pseudomonas aeruginosa AmpR regulon to include small RNA-mediated regulation of iron acquisition, heat shock and oxidative stress response. Nucleic Acids Res 42(2): 979–98 doi:10.1093/nar/gkt942
67. GalaganJE, MinchK, PetersonM, LyubetskayaA, AziziE, et al. (2013) The Mycobacterium tuberculosis regulatory network and hypoxia. Nature 499 : 178–183 doi:10.1038/nature12337
68. LeeVT, MatewishJM, KesslerJL, HyodoM, HayakawaY, et al. (2007) A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol Microbiol 65 : 1474–1484 doi:10.1111/j.1365-2958.2007.05879.x
69. StoltzDA, MeyerholzDK, PezzuloAA, RamachandranS, RoganMP, et al. (2010) Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med 2 : 29ra31 doi:10.1126/scitranslmed.3000928
70. HenggeR (2009) Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol 7 : 263–273 doi:10.1038/nrmicro2109
71. SondermannH, ShikumaNJ, YildizFH (2012) You've come a long way: c-di-GMP signaling. Curr Opin Microbiol 15 : 140–146 doi:10.1016/j.mib.2011.12.008
72. HechtGB, NewtonA (1995) Identification of a novel response regulator required for the swarmer-to-stalked-cell transition in Caulobacter crescentus. J Bacteriol 177 : 6223–6229.
73. PaulR, AbelS, WassmannP, BeckA, HeerklotzH, et al. (2007) Activation of the diguanylate cyclase PleD by phosphorylation-mediated dimerization. J Biol Chem 282 : 29170–29177 doi:10.1074/jbc.M704702200
74. GuvenerZT, HarwoodCS (2007) Subcellular location characteristics of the Pseudomonas aeruginosa GGDEF protein, WspR, indicate that it produces cyclic-di-GMP in response to growth on surfaces. Mol Microbiol 66 : 1459–1473 doi:10.1111/j.1365-2958.2007.06008.x
75. HuangyutithamV, GuvenerZT, HarwoodCS (2013) Subcellular clustering of the phosphorylated WspR response regulator protein stimulates its diguanylate cyclase activity. MBio 4: e00242–13 doi:10.1128/mBio.00242-13
76. PaulR, WeiserS, AmiotNC, ChanC, SchirmerT, et al. (2004) Cell cycle-dependent dynamic localization of a bacterial response regulator with a novel di-guanylate cyclase output domain. Genes Dev 18 : 715–727 doi:10.1101/gad.289504
77. KlausenM, HeydornA, RagasP, LambertsenL, Aaes-JørgensenA, et al. (2003) Biofilm formation by Pseudomonas aeruginosa wild type, flagella and type IV pili mutants. Mol Microbiol 48 : 1511–1524.
78. KlausenM, Aaes-JørgensenA, MolinS, Tolker-NielsenT (2003) Involvement of bacterial migration in the development of complex multicellular structures in Pseudomonas aeruginosa biofilms. Mol Microbiol 50 : 61–68.
79. OverhageJ, SchemionekM, WebbJS, RehmBHA (2005) Expression of the psl operon in Pseudomonas aeruginosa PAO1 biofilms: PslA performs an essential function in biofilm formation. Appl Environ Microbiol 71 : 4407–4413 doi:10.1128/AEM.71.8.4407-4413.2005
80. ToutainCM, CaizzaNC, ZegansME, O'TooleGA (2007) Roles for flagellar stators in biofilm formation by Pseudomonas aeruginosa. Res Microbiol 158 : 471–477 doi:10.1016/j.resmic.2007.04.001
81. Byrd MS, Pang B, Mishra M, Swords WE, Wozniak DJ (2010) The Pseudomonas aeruginosa exopolysaccharide Psl facilitates surface adherence and NF-kappaB activation in A549 cells. MBio 1 pii: e00140-10. doi:10.1128/mBio.00140-10.
82. ByrdMS, PangB, HongW, WaligoraEA, JuneauRA, et al. (2011) Direct evaluation of Pseudomonas aeruginosa biofilm mediators in a chronic infection model. Infect Immun 79 : 3087–3095 doi:10.1128/IAI.00057-11
83. YangL, HuY, LiuY, ZhangJ, UlstrupJ, et al. (2011) Distinct roles of extracellular polymeric substances in Pseudomonas aeruginosa biofilm development. Environ Microbiol 13 : 1705–1717 doi:10.1111/j.1462-2920.2011.02503.x
84. YangL, HengzhuangW, WuH, DamkiærS, JochumsenN, et al. (2012) Polysaccharides serve as scaffold of biofilms formed by mucoid Pseudomonas aeruginosa. FEMS Immunol Med Microbiol 65 : 366–376 doi:10.1111/j.1574-695X.2012.00936.x
85. WangS, ParsekMR, WozniakDJ, MaLZ (2013) A spider web strategy of type IV pili-mediated migration to build a fibre-like Psl polysaccharide matrix in Pseudomonas aeruginosa biofilms. Environ Microbiol 15(8): 2238–53 doi:10.1111/1462-2920.12095
86. GoodmanAL, KulasekaraB, RietschA, BoydD, SmithRS, et al. (2004) A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev Cell 7 : 745–754 doi:10.1016/j.devcel.2004.08.020
87. GoodmanAL, MerighiM, HyodoM, VentreI, FillouxA, et al. (2009) Direct interaction between sensor kinase proteins mediates acute and chronic disease phenotypes in a bacterial pathogen. Genes Dev 23 : 249–259 doi:10.1101/gad.1739009
88. BurrowesE, BaysseC, AdamsC, O'GaraF (2006) Influence of the regulatory protein RsmA on cellular functions in Pseudomonas aeruginosa PAO1, as revealed by transcriptome analysis. Microbiology 152 : 405–418 doi:10.1099/mic.0.28324-0
89. MannEE, WozniakDJ (2011) Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol Rev 36 : 893–916 doi:10.1111/j.1574-6976.2011.00322.x
90. WalkerTS, TomlinKL, WorthenGS, PochKR, LieberJG, et al. (2005) Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infect Immun 73 : 3693–3701 doi:10.1128/IAI.73.6.3693-3701.2005
91. MishraM, ByrdMS, SergeantS, AzadAK, ParsekMR, et al. (2012) Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement-mediated opsonization. Cell Microbiol 14 : 95–106 doi:10.1111/j.1462-5822.2011.01704.x
92. YoungRL, MalcolmKC, KretJE, CaceresSM, PochKR, et al. (2011) Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PLoS ONE 6: e23637 doi:10.1371/journal.pone.0023637
93. GovanJR, DereticV (1996) Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60 : 539–574.
94. LearnDB, BrestelEP, SeetharamaS (1987) Hypochlorite scavenging by Pseudomonas aeruginosa alginate. Infect Immun 55 : 1813–1818.
95. PedersenSS, KharazmiA, EspersenF, HøibyN (1990) Pseudomonas aeruginosa alginate in cystic fibrosis sputum and the inflammatory response. Infect Immun 58 : 3363–3368.
96. MaiGT, SeowWK, PierGB, McCormackJG, ThongYH (1993) Suppression of lymphocyte and neutrophil functions by Pseudomonas aeruginosa mucoid exopolysaccharide (alginate): reversal by physicochemical, alginase, and specific monoclonal antibody treatments. Infect Immun 61 : 559–564.
97. HoangTT, Karkhoff-SchweizerRR, KutchmaAJ, SchweizerHP (1998) A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212 : 77–86.
98. Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press. 2028 p.
99. WinsorGL, LamDKW, FlemingL, LoR, WhitesideMD, et al. (2011) Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res 39: D596–D600 doi:10.1093/nar/gkq869
100. KelleyLA, SternbergMJE (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4 : 363–371 doi:10.1038/nprot.2009.2
101. HeydornA, NielsenAT, HentzerM, SternbergC, GivskovM, et al. (2000) Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 146 (Pt 10): 2395–2407.
102. StoverCK, PhamXQ, ErwinAL, MizoguchiSD, WarrenerP, et al. (2000) Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406 : 959–964 doi:10.1038/35023079
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 3
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Cytomegalovirus m154 Hinders CD48 Cell-Surface Expression and Promotes Viral Escape from Host Natural Killer Cell Control
- Human African Trypanosomiasis and Immunological Memory: Effect on Phenotypic Lymphocyte Profiles and Humoral Immunity
- DHX36 Enhances RIG-I Signaling by Facilitating PKR-Mediated Antiviral Stress Granule Formation
- Conflicting Interests in the Pathogen–Host Tug of War: Fungal Micronutrient Scavenging Versus Mammalian Nutritional Immunity