
A Translocated Effector Required for Dissemination from Derma to Blood Safeguards Migratory Host Cells from Damage by Co-translocated Effectors
Cell migration, a fundamental feature of eukaryotic cells, plays a crucial role in mounting an effective immune response. However, several pathogens subvert the migratory properties of infected host cells to their benefit, such as using them as Trojan horses to disseminate within the host. Bartonella effector proteins (Beps) are bona fide virulence factors indispensable for the colonization of mammalian target cells. However, their multiple interferences with host cellular signaling processes might culminate in deleterious secondary effects that require additional effectors to maintain the host cell integrity. A striking example is BepE, which is shown here to preserve endothelial cells (ECs) from fragmentation and to inhibit the defects of dendritic cell (DCs) migration caused by BepC and possibly other Beps. Moreover, BepE is essential for Bartonella dissemination from the dermal site of inoculation to the blood stream where bacteria establish long-lasting intraerythrocytic bacteremia as a hallmark of infection in the mammalian reservoir host. Migration of Bartonella-infected DCs through a monolayer of lymphatic ECs was also found to be dependent of BepE, suggesting that BepE is required to preserve the migratory capability of DCs, a candidate cell type for systemic dissemination from the dermal site of inoculation.
Published in the journal:
. PLoS Pathog 10(6): e32767. doi:10.1371/journal.ppat.1004187
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004187
Summary
Cell migration, a fundamental feature of eukaryotic cells, plays a crucial role in mounting an effective immune response. However, several pathogens subvert the migratory properties of infected host cells to their benefit, such as using them as Trojan horses to disseminate within the host. Bartonella effector proteins (Beps) are bona fide virulence factors indispensable for the colonization of mammalian target cells. However, their multiple interferences with host cellular signaling processes might culminate in deleterious secondary effects that require additional effectors to maintain the host cell integrity. A striking example is BepE, which is shown here to preserve endothelial cells (ECs) from fragmentation and to inhibit the defects of dendritic cell (DCs) migration caused by BepC and possibly other Beps. Moreover, BepE is essential for Bartonella dissemination from the dermal site of inoculation to the blood stream where bacteria establish long-lasting intraerythrocytic bacteremia as a hallmark of infection in the mammalian reservoir host. Migration of Bartonella-infected DCs through a monolayer of lymphatic ECs was also found to be dependent of BepE, suggesting that BepE is required to preserve the migratory capability of DCs, a candidate cell type for systemic dissemination from the dermal site of inoculation.
Introduction
Pathogenic bacteria have evolved a multitude of virulence factors in order to manipulate the host to evade immune responses and to reach their replicative niche - a safe compartment to proliferate that is also a prerequisite for transmissibility [1]. Translocation of bacterial effector proteins into host cells is one of the mechanisms to manipulate the host by interfering with its signaling pathways. A prominent example is CagA, a multifunctional effector protein of the Helicobacter pylori (Hpy) type IV secretion system (T4SS). CagA modulates both innate and adaptive immune responses of the host and assists Hpy to infect the gastric mucosa in about half of the world population for their lifetime [2], [3]. Numerous effector proteins of Salmonella type III secretion systems (T3SS) SPI1 and SPI2 [1] and Shigella T3SS play a critical role in invasion of non-phagocytic intestinal cells, for further dissemination and modulation of the host inflammatory responses [4], [5]. In addition to targeting the host cellular components, some bacteria have evolved effectors that regulate an activity of each other at a specific stage of the host invasion; like Legionella Dot/Icm “metaeffector” LubX mediates the degradation of SidH. Or this interplay may happen in an indirect fashion as for many cases of T4SS/T3SS effectors [6].
Bartonella species are fastidious, Gram-negative, facultative intracellular bacteria that are highly adapted to a distinct mammalian reservoir host [7], [8], [9], [10]. Infections in the reservoir host range from asymptomatic or sub-clinical (for most animal-specific species) to clinical manifestations with low morbidity and limited mortality, such as human-specific B. quintana (Bqu) infections, or even to life-threatening disease, such as human infection by B. bacilliformis (Bba) [9], [11]. Bartonellae transmission is mediated by blood-sucking arthropod vectors. The strategy involves replication of bacteria in the gut of the arthropod vector and excretion in the feces, with subsequent survival in the environment for several days [12]. The arthropods usually defecate when feeding on mammals and provide a source of local irritation that results in itching, followed by scratching and inoculation of Bartonella-containing feces into the derma of the skin [13]. Later, Bartonella is known to appear in the blood of the reservoir host, invades erythrocytes as immune-privileged niche and develops long-lasting persistent infections for more than a year for some species [11], [14].
Bartonellae evolved two T4SSs (Trw and VirB) while adapting to a wide range of mammalian hosts [15]. Both of them are essential for the interaction with the host but at different stages of the infection cycle [14], [15], [16], [17], [18]. The Trw system seems to mediate host-specific adhesion of Bartonella to erythrocytes by binding to the cell surface with its manifold variants of pilus subunits [19], [20], while the VirB system translocates a cocktail of evolutionarily related Bartonella effector proteins (Beps) into nucleated host cells. The Beps (named BepA-G in Bhe) evolved by duplication of an ancestral bep gene followed by their functional diversification and conservation of certain domains and motifs [15]. All Beps have at least one Bartonella intracellular delivery (BID) domain and a positively-charged tail in the C-terminus as a signal for translocation through the T4SS [21].
BepE is one of the effector proteins that is conserved within lineage 4, the largest clade within the Bartonella genus comprising 11 species, e.g. B. henselae, B. quintana, B. tribocorum and B. grahamii [15]. Bhe BepE (BepEBhe) consists of 464 amino acids (aa). The N-terminus of BepEBhe, similarly to BepDBhe and BepFBhe, contains short repeated peptide sequences (EPLYA) with conserved putative tyrosine phosphorylation sites, similar to the EPIYA motif of Hpy effector CagA. BepEBhe has two BID domains in the C-terminal part [21]. The domains and the motifs but not the spacing in between is well preserved in BepE homologues [15]. Mass spectrometric analysis of BepEBhe pull-downs revealed several SH2 domain-containing eukaryotic signaling proteins that interact either with an individual phosphotyrosine of BepEBhe within a Csk-like binding motif or two ITIM/ITSM (immunotyrosine inhibitory motif/immunotyrosine switch motif) tandems [22]. Conservation of these specific motifs of BepEBhe and the described interaction partners suggest a molecular mimicry of ITIM-containing receptors by bacterial proteins and a potency to interfere with host signaling pathways.
In this study, we identified BepE as an essential bacterial factor for Bartonella reservoir host infection via the intradermal route resembling natural infection by arthropod vectors, but not for the intravenous route. This specific function was assigned to the C-terminal part of BepEBhe including the two BID domains. The same BID domains were interfering with a prominent cell fragmentation phenotype in migrating endothelial cell induced by BepC and possibly other Beps as a secondary effect. Further, we show that Bartonella translocates an effector-fusion protein (Bla-BID) into dendritic cells and affects cell migration in the absence of BepE, suggesting that DCs may be involved in the BepE-dependent dissemination of Bartonella to the blood.
Results
BepEBhe is sufficient to abolish a cell fragmentation phenotype induced by the Bhe ΔbepDEF mutant
A previous study on the interactome of Bartonella effector protein BepEBhe revealed several SH2 domain-containing signaling proteins that interact with BepEBhe upon phosphorylation of specific tyrosines within the motifs [22]. Based on these data, we hypothesized that BepE might be a factor impacting multiple cellular signaling pathways to promote the establishment of a successful Bartonella infection. To acquire first insights into the cellular phenotypes and uncover the molecular bases of BepE we used the well-established human umbilical vein endothelial cell (HUVEC) infection model for Bhe [23], [24], [25], [26], [27].
BepE, together with BepD and BepF, belongs to the class of the Beps harboring tyrosine-containing motifs in their N-termini. In order to reduce the complexity of potential redundant functional effects by any of these three tyrosinephosphorylated effectors we infected HUVECs with a Bhe mutant carrying an in-frame deletion of the chromosomal region encoding bepD, bepE and bepF (Bhe ΔbepDEF). The infection was studied by means of microscopy of either fixed samples or by time-lapse imaging over 72 h. Starting about 20–24 hours post infection (hpi) the Bhe ΔbepDEF mutant showed a drastic phenotype of cell fragmentation (Fig. 1A and Movie S1) which was hardly detected in cells infected with wild-type bacteria at this early stage of infection (Fig. 1B, C), but became more apparent at later time points, beyond 48 hpi (Movie S2). Fragmenting cells were apparently normally moving forward but displayed pronounced difficulties in rear end retraction - such cells became more and more elongated and at some point the thin connection between a given cell body and the attached rear broke (Fig. 1A). The fragment left behind migrated on the substrate for a few hours and then came to a halt, while the cell body containing the nucleus got smaller with each fragmentation. Eventually, this process led also to a decreased number of cells within the sample (Fig. 1A and B, Movie S1).

Infections with single effector-complemented Bhe ΔbepDEF revealed that the cell fragmentation phenotype was inhibited only by expression of BepEBhe, while neither BepDBhe nor BepFBhe displayed a similar cytoprotective effect (Fig. 1B, D).
A single BID domain of BepEBhe is sufficient to interfere with the fragmentation of HUVECs
Provided that BepD and BepF could not abolish the cell fragmentation phenotype of the ΔbepDEF mutant we were interested to test whether single deletions of bepD, bepE or bepF would be sufficient to trigger cell fragmentation. Bhe ΔbepE-infected HUVECs showed indeed very similar morphological changes but the effect was less pronounced. Compared to wild-type infection, none of the other mutants could induce elevated fragmentation in a significant manner (Fig. 2A). As expected BepE could also rescue the cell fragmentation led by Bhe ΔbepE (Fig. 2B and C, Movie S3).

We next delineated the functional domain responsible for the abrogation of cell fragmentation in BepEBhe. As indicated earlier, BepEBhe has two BID domains at its C-terminus plus a positively charged C-tail. These domains (BID1.EBhe and BID2.EBhe) show high similarity (pair-wise aa identity of 53.3%) (Fig. S1A) and thus seem to be originated from a duplication event [15]. To understand which part of BepEBhe was responsible for the effect observed during the infection of HUVECs, BIDs.EBhe and BID2.EBhe were expressed in Bhe ΔbepE as they both contain an intact C-term required for translocation (Fig. 2B–E) and then used to infect HUVECs. The two BID domains (BIDs.EBhe) do overcome the Bhe ΔbepE-induced cell fragmentation phenotype. Even more, BID2.EBhe is able to complement with almost the same efficiency as full length BepE.
In summary, these data indicate that the inhibition of cell fragmentation by BepEBhe was mediated by the BID domains of BepEBhe and more specifically, the BID2.EBhe was sufficient.
Cell fragmentation induced by Bhe ΔbepDEF is inhibited by heterologous complementation with BepEBhe homologues
BepE homologues from the Bartonella lineage 4 species display significant similarity in domain and motif composition (Fig. S1B). We thus tested whether BepE homologues from different Bartonella species, i.e. BepDBtr and BepEBtr from B. tribocorum, BepEBqu from B. quintana and BepHBgr from B. grahamii, can interfere with the cell fragmentation phenotype of the Bhe ΔbepDEF. BepEBtr, BepEBqu, and BepHBgr were indeed able to functionally replace BepEBhe in Bhe ΔbepDEF background. Amongst all the homologues, BepDBtr has the least amino similarity to BepEBhe (Fig. S1B) and was also the least potent in suppressing the cell fragmentation phenotype (Fig. 3A and B). Interestingly, BepEBqu from the human-specific Bqu was the most efficient in abrogating cell fragmentation. Considering that HUVECs are primary human endothelial cells this observation is suggestive for some level of host specificity in the activity of the BepE effector.
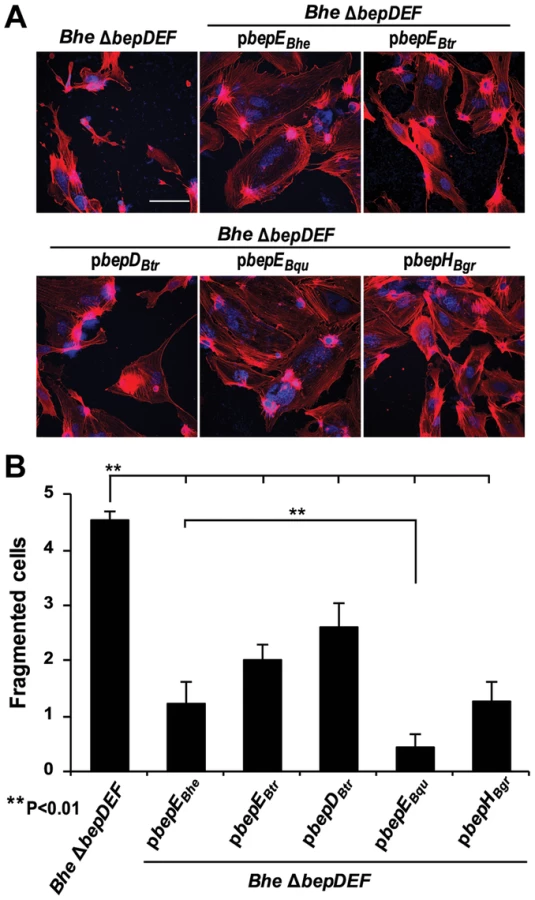
Deletion of BepC in the Bhe ΔbepE mutant background is sufficient to abolish the cell fragmentation phenotype
The cell fragmentation phenotype is triggered by the mutant strains Bhe ΔbepE and Bhe ΔbepDEF. In order to find the factor from Bhe leading to this severe phenotype we tested a set of available Bhe mutants. The effector-free mutant Bhe ΔbepA-G did not display cell fragmentation, indicating that the factor that triggers cell fragmentation in the absence of BepE must be another Bep (Fig. 4A and B). Given that the Bhe ΔbepDEF mutant strain displays severe cell fragmentation indicates that that the cell fragmentation activity is confined to either BepA, BepB, BepC or BepG or a combination thereof. A double mutant Bhe ΔbepCE did not display cell fragmentation, indicating that BepC is essential for this activity. Although we cannot exclude that either BepA, BepB, or BepG may contribute to this phenotype, ectopic expression of mCherry-BepC in HUVEC resulted in cell fragmentation that could be reduced by co-expression of GFP-BepE (Movie S4), indicating that BepC can trigger cell fragmentation on its own.

BepE translocation and interference with cell fragmentation is T4SS-dependent
To confirm that inhibition of cell fragmentation observed upon infection with strains over-expressing BepE from plasmid was fully attributable to BepE effector function within the host cell and not to competitive inhibition with translocation of other Beps, we first confirmed VirB T4SS-dependency of BepEBhe translocation into host cells. To this end we expressed Myc-BepE in Bhe wild-type or the VirB T4SS-translocation deficient Bhe ΔvirB4 mutant strain. Immunocytochemical staining with anti-Myc antibodies showed clearly that BepEBhe is translocated in a VirB T4SS-dependent manner (Fig. 5A.) as previously demonstrated for BepDBhe [21].

In order to avoid that BepE translocation interferes with BepC and possibly other Beps contributing to the cell fragmentation phenotype, we performed a mixed infection experiment with the strain Bhe ΔbepE (Fig. 5B) that triggers cell fragmentation and strain expressing BepE in an effector-free mutant background (Bhe ΔbepA-G-pBepE). The results showed that BepE translocated by one strain can suppress the cell fragmentation phenotype mediated by the effectors translocated by the other strain. As a negative control we showed that expression of BepE in a VirB T4SS-deficient background (Bhe ΔvirB4/bepA-G-pbepE) did not lead to suppression of the cell fragmentation phenotype triggered by the other strain (Fig. 5B, for Bhe ΔbepDEF see Fig. S2). These data demonstrate that BepE is translocated by the VirB T4SS system into the host cell and upon translocation shows a cytoprotective effect by interfering with the cell fragmentation.
Ectopic expression of BepE in HUVECs abrogates cell fragmentation
Next we expressed BepE ectopically in HUVECs in order to complement the data obtained for inhibition of cell fragmentation by VirB-translocated BepE. To this end we used a lentiviral transduction system to generate GFP, GFP-BepEBhe, GFP-BIDs.EBhe or GFP-BID2.EBhe-expressing HUVECs. Lentiviral transduction resulted in mixed cultures of transduced HUVECs expressing the GFP-fusion protein and GFP-negative non-transduced cells. Such mixed cultures were infected with Bhe wild-type, Bhe ΔbepE (Fig. S3) and Bhe ΔbepDEF (Fig. 6A, B) or left uninfected. Microscopic analysis showed that non-transduced and GFP-positive cells, expressing plain GFP as control, resulted in much higher fraction of HUVECs displaying cell fragmentation than cells expressing GFP-BepEBhe, GFP-BIDs.EBhe or GFP-BID2.EBhe (Fig. 6A and Fig. S3). Expression of the full-length GFP-BepEBhe fusion protein and its truncated derivatives in HUVECs were validated by anti-GFP western blot (Fig. S4). Considering that the cell fragmentation at late time points of the infection (48 h) eventually leads to the decrease of the cell number within the sample (Fig. 1B), we used flow cytometry for quantification of this phenotype. To do so, we monitored the ratio of transduced (GFP-positive) vs. non-transduced (GFP-negative) in both infected and uninfected HUVEC populations (Fig. 6B). We considered an increase in the ratio of GFP-positive cells in the infected sample to be indicative of a protective phenotype mediated by the GFP-fusion protein, knowing that cells undergoing fragmentation would be lost during the experiment. These analyses confirmed that the cells expressing GFP-BepEBhe, GFP-BID2.EBhe and to a lesser extent GFP-BIDs.EBhe were protected from fragmentation and toxic effects induced by infection with the BepE-deficient strains, Bhe ΔbepE and Bhe ΔbepDEF.

BepEBhe is recruited to the plasma membrane, localizes to cell-to-cell contacts and is enriched in the rear edge of migrating HUVECs
In the previous section we showed by anti-Myc immunocytochemistry in combination with confocal microscopy the VirB T4SS-dependent translocation of Myc-BepEBhe into HUVECs. Myc-BepEBhe localized in a punctuate staining pattern to the plasma membrane, with some enrichment to cell contact areas (Fig. 5A). Due to that latter we performed a co-staining for Myc-BepE or Myc-BIDs.EBhe additionally with VE-cadherin, a marker for adherence junctions. The stainings demonstrated co-localization of full length BepE or the BID domains with VE-cadherin (Fig. 7A).

To test for the subcellular localization of GFP-BepEBhe ectopically expressed in HUVECs, samples were analyzed by confocal microscopy and images were taken in the xy - and the xz-planes. GFP-BepEBhe clearly localized to plasma membrane with a clear exclusion of signal in the nucleus, while GFP alone localized to the cytoplasm and the nucleus (Fig. 7B). Similarly to the immunostaining of Myc-BepEBhe, confocal analysis of ectopically expressed GFP-BepEBhe revealed accumulation of BepE at cell-to-cell contacts (Fig. S4, Movie S5).
Further, when GFP-BepEBhe-expressing HUVECs were investigated in time-lapse, GFP signal was transiently accumulating and localizing to the rear edge of the cell. As displayed in Movie S6 or the series of images taken at 15 min intervals in Fig. 7C, the GFP signal was increasing at the rear sites of cell detachment, making it conceivable to assume that this transient accumulation of BepEBhe may have a role in restoring rear end retraction during cell migration when the process is distorted in cells infected with Bhe mutants deficient of BepE.
Ectopic expression of BepEBhe interferes with the disruption of stress fibers by Rho inhibitor I
RhoA is known to be involved in cell body contraction and rear release [28], [29], [30], [31]. Since the fragmentation of HUVECs by infection with Bhe ΔbepDEF or ΔbepE mutant bacteria was associated with a distortion of rear edge detachment during cell migration, and because GFP-BepEBhe transiently localizes to this rear edge during detachment, we became interested in testing the role of the RhoA signaling pathway for BepE activity.
The mixed population of GFP-BepEBhe-expressing and non-expressing HUVECs was treated with Rho inhibitor I (CTO4), which is known to inactivate RhoA, B and C by ADP-ribosylation. In response to intoxication by this inhibitor cells initially lose their stress fibers, then round up and eventually die [32], [33], [34]. Interestingly, HUVECs expressing GFP-BepEBhe showed resistance to the Rho inhibitor I as shown by the selective preservation of stress fibers, while neighboring non-transduced cells identified by the lack of GFP signal displayed complete resolution of their stress fibers (Fig. 8A). These phenotypes were quantified using HUVECs expressing plain GFP as control sample (Fig. 8B). Interference of GFP-BepEBhe with the activity of Rho inhibitor I was dose-dependent (Fig. 8B). Treatment of HUVECs expressing GFP-BepEBhe mutants with the same Rho Inhibitor I showed that the second BID domain (BID2.EBhe) of BepE is sufficient to interfere with the activity of Rho inhibition (Fig. 8D).

These results support the interaction of BepE with the Rho signaling pathway. To address the question whether BepE acts downstream of ROCK, an effector of RhoA, GFP-BepEBhe-transduced HUVECs were exposed to the inhibitor Y27632 that is known to inhibit both ROCK1 and ROCK2 [35]. In contrast to the effect mediated by the Rho inhibitor I, Y27632-treated GFP-BepEBhe HUVECs lost the stress fibers similar to the plain GFP-expressing control cells (Fig. 8C). Thus, BepE may have a direct or indirect effect on RhoA or even on ROCK, but not further downstream in this signaling pathway.
Btr ΔbepDE loses the ability to colonize rat blood in intradermal infection
Observing that BepE interferes with deleterious secondary effects of other Beps on migrating cells during Bartonella infection in vitro we became interested to examine the role of BepE in the establishment of infection in vivo. Bhe causes intraerythrocytic bacteremia as hallmark of infection in its feline natural reservoir host only, which for ethical reasons is not an accessible experimental model. However, as we have noted functional conservation among BepE homologues (including Btr) in protecting the Bhe ΔbepDEF-mediated fragmentation of HUVECs we reasoned that we can switch to Btr as appropriate species for experimental infection of rats as its natural host. Initially we employed the well-described intravenous (i.v.) infection by Btr that leads to the onset of intraerythrocytic bacteremia at 5 days post infection (dpi) following colonization of the so called “primary niche” that is considered to include vascular endothelial cells [14], [17]. Btr encodes the BepEBhe homologues BepDBtr and BepEBtr (Fig. 9A) that both cause partial protection of HUVECs from Bhe ΔbepDEF–mediated cell fragmentation (see Fig. 3). We thus constructed an in-frame deletion of bepD and bepE (Btr ΔbepDE) and compared its course of infection to wild-type bacteria. Rats were injected with Btr wild-type or Btr ΔbepDE through the i.v. route and bacteremia was monitored over time as colony forming units (CFUs) per ml of blood. In this model, the Btr ΔbepDE strain did not show any significant difference in its capacity to cause long-term bacteremia with high bacterial burden (Fig. S5, Fig. 9C). Recently, an intradermal (i.d.) infection model of B. birtlesii (Bbi) has been introduced by Marignac et al [36], where the bacteria are inoculated in the derma on the ear pinnae of mice. This model more likely reflects the natural route of infection, mimicking the animal scratching an area bitten by the arthropod vector, thereby inoculating derma with infectious bacteria from the arthropod feces. We thus adapted i.d. inoculation to our rat model to test for a possible role of BepE at the very early dermal stage of natural infection (Fig. 9B, Fig. S6A). To this end we injected rats with Btr wild-type or Btr ΔbepDE bacteria through the i.d. route and monitored the development of bacteremia of infected animals. Btr wild-type-infected animals developed blood colonization between 7–9 days post infection (dpi), which occurs with 3–4 day delay as compared to the i.v. model (Fig. S6A). In sharp contrast, 12 out of 13 Btr ΔbepDE - i.d. infected rats (from 3 different experiments) stayed abacteremic over 70 days (Fig. 9B). Only one Btr ΔbepDE-infected animal developed a delayed bacteremia starting on day 16 post infection (Fig. 9B, animal i.d. Btr ΔbepDE #4), which might result from the fact that the i.d. infection protocol can damage capillaries and small vessels in the dermis and thus inoculate bacteria directly into circulating blood similar to the i.v. inoculation protocol, yet at much smaller bacterial numbers.

Taken together, the data obtained in the rat infection model suggest that either one of the missing effectors in Btr ΔbepDE, BepDBtr or BepEBtr, or both of them, are of pivotal importance for Bartonella to colonize the reservoir host when inoculated in the derma reflecting natural arthropod-borne infection, while the mutant is as infectious as wild-type bacteria via the artificial i.v. infection route.
Expression of BepE in Btr ΔbepDE restores bacteremia in i.d. infected rats
Next we complemented the Btr ΔbepDE mutant strain with various BepE homologues to test for restoration of bacteremia following i.d. inoculation in rats. To this end we expressed in trans from a plasmid under control of their native promoter(s) either both BepDBtr and BepEBtr, or BepEBtr alone. Moreover, to test for heterologous complementation we expressed BepEBhe in the Btr ΔbepDE mutant. Following inoculation by the i.d. route all complemented mutant strains had developed bacteremia on 16 dpi (Fig. 9C, Fig. S6B).
Taken together these data demonstrate for the i.d. rat model that complementation of the abacteremic Btr ΔbepDE mutant with either BepEBtr or its homologue BepEBhe is sufficient to restore bacteremia. This finding strongly supports a functional conservation of BepE between different species of the genus Bartonella that is in line with our in vitro studies on the protection from cell fragmentation (see Fig. 3B).
The BID domains of BepEBhe are sufficient to enable Btr to reach the blood
Since BepEBhe functionally complemented the ΔbepDE mutant for bacteremia establishment in the i.d. inoculation model we next tested to which domain this activity is confined and whether this might coincide to the BID domains as determined for the protection against cell fragmentation in vitro (Fig. 2 and 6). To this end, mutant variants of BepEBhe were ectopically expressed in the Btr ΔbepDE background and tested in the i.d. infection model. Two mutants of BepEBhe were chosen, one with all five tyrosines exchanged to phenylalanine (Y37,64,91,106,129→F) and the second, a truncated version of BepEBhe, with only the two BID domains and positively charged C-tail (BIDs.EBhe). The expression of the BepEBhe mutant proteins was confirmed by western blotting (Fig. S6C). At 16 dpi both complemented Btr ΔbepDE mutants triggered bacteremia at similar levels as the mutant complemented with full-length pbepEBhe or as Btr wild-type bacteria (Fig. 9D). Thus, the tyrosine-containing motifs of BepEBhe, including its N-terminal end, are not required for Bartonella to reach its replicative niche in the reservoir host. The functional part of BepE for this infection phenotype can therefore be assigned to the BID domains.
Bartonella is able to translocate the effector protein within dendritic cells and affects their migration
In vivo experimental data from rat i.d. infections suggests a role for BepE in the establishment of infection at very early stages, prior to the invasion of the blood system. In addition, the in vitro model demonstrates that BepE is essential to inhibit fragmentation of infected HUVECs by interfering with the effect of other Beps. To better understand the in vivo relevance of BepE and based on our in vitro data, we decided to further explore the impact of BepE on cell migration for a cell type potentially relevant to the very early infection process. Assuming that the initial site of Bartonella infection is in derma, we speculate that innate immune cells represent the yet unidentified primary infection niche and consider resident dendritic cells (DCs) as a candidate cell type. These cells have high potency of phagocytosis and the property to migrate towards lymph nodes [37], [38], [39].
First we assessed the effector translocation by Bhe into the BMDCs (bone marrow-derived dendritic cells). The strain expressing Bla-BID, a beta-lactamase fusion protein with a bipartite translocation signal from Bhe BepD [21], was used to infect mouse BMDCs. β-lactamase activity was measured using CCF2, a FRET substrate that emits green fluorescence but emits blue fluorescence after cleavage by β-lactamase [40]. Starting at 12 hpi the Bla-BID translocation was observed within infected BMDCs (Fig. 10A).

To investigate the migration of Bartonella-infected DC towards the draining lymph nodes, we decided to use a trans-well migration assay [41], a tissue culture model system incorporating a 3D extracellular matrix and interstitial flow, where the infected BMDCs have to migrate first through the 3D extracellular collagen matrix and then a layer of iLECs (immortalized lymphatic endothelial cells), thus mimicking the entry of lymphatic system by an infected DC.
As shown in Fig. 10B, BMDCs infected prior with Bhe ΔbepDEF revealed less efficiency in 3D trans-well migration compared to uninfected or Bhe wild-type infection conditions. Similarly to HUVEC cells, the BMDC migration is also affected by Bhe ΔbepDEF and rescued by BepE. Thus, in vitro effector translocation and modulation of cell migration, together with the general relevance of DCs for infection processes, prioritizes these cells for future investigations of the in vivo functions of Bartonella effector proteins.
Discussion
Various bacterial pathogens secrete multiple effectors that act in concert to modulate different host cellular functions during the course of infection. Often these effectors may interfere with the activity of each other – either directly or indirectly – in order to orchestrate their multi-pronged interactions with the host in a spatially and temporally controlled manner. We have discovered a particularly striking example in Bartonella. We found that BepE acts via BID2.EBhe on the RhoA signaling pathway, thereby alleviating deleterious secondary effects of BepC and possibly other Beps. These Beps have distinct functions within the host cell; however, in the absence of BepE, they result in an impaired cell migration and subsequent fragmentation of the infected ECs. Moreover, the rat model of Btr i.d. infection, which recapitulates the natural way of Bartonella infection by an arthropod, revealed the role of BepE and its BID domains on the dermal stage of infection, thus showing its essential role in establishing reservoir host infection.
Bartonella effectors are known to exhibit functional redundancy, i.e. the ability of structurally different elements to perform a similar function [42]. As an example, the F-actin-dependent uptake of Bhe via the invasome structure is triggered by distinct Bep-dependent pathways, either by BepCBhe and BepFBhe, or by BepGBhe alone [27]. All Beps harbor at their C-terminus a bipartite T4SS signal composed of a BID domain and a positively-charged tail sequence [21], while only BepDBhe, BepEBhe and BepFBhe contain multiple tyrosinephosphorylation motifs (ITIM/ITSM) in their N-terminal domains [22]. Assuming functional redundancy among this class of effectors we initially focused on the Bhe ΔbepDEF mutant in infection experiments in HUVECs, the latter representing a well-established in vitro model for Bhe infection [23], [24], [25], [26], [27]. HUVEC infection with Bhe ΔbepDEF resulted in a prominently impaired cell migration phenotype. Cell migration is a complex process that requires a series of repetitive but highly coordinated processes [43], [44], [45], [46], [47], [48], [49]. The first step of forward protrusion of lamellipodia and filopodia involves extensive remodeling of the F-actin cytoskeleton. Next, attachment at the leading edge occurs via focal adhesion complexes. After the formation of new adhesions, cells undergo actomyosin-dependent contraction to pull the cell body forward. Coincidently, cell adhesions at the rear of the cell are released such that the cell can move forward [43], [44], [45], [46], [47], [48], [49]. Bhe ΔbepDEF-infected cells were deficient in rear end detachment and thus underwent fragmentation. Despite the similarity in domain architecture of BepDBhe, BepEBhe and BepFBhe, only BepEBhe and its homologues in other bartonellae were able to suppress cell fragmentation, which was shown both by bacterial mutant complementation and by ectopic expression of GFP-BepEBhe within HUVECs.
The cell fragmentation phenotype is T4SS-dependent and strictly requires translocation of BepCBhe, while we have not tested whether other Beps not encoded in the cell fragmentation-inducing strain Bhe ΔbepDEF (i.e. BepA, BepB or BepG) may also contribute to the cell fragmentation phenotype. As already mentioned, BepCBhe, BepFBhe and BepGBhe, are involved in invasome formation via massive F-actin rearrangements [24], [27]. While this process enables Bartonella to be taken up efficiently as large aggregates it is tempting to speculate that the action of BepC and possibly BepG on cell cytoskeleton could affect rear end detachment during cell migration and thereby trigger the cell fragmentation phenotype. Although, by ectopic co-expression with BepC in HUVECs we demonstrated that BepE is reducing the stark influence on a cell cytoskeleton, future studies should address whether BepE acts as a “metaeffector” [50], which acts directly on BepC to modulate its function inside host cells, or rather indirectly tempering its activity at a different level.
GFP-tagged BepEBhe was found to be transiently enriched at the rear edge of migrating cells. This localization is consistent with a potential role of BepE in modulating focal adhesion turnover at the rear edge of the cell as a possible mechanism to counteract the cell fragmentation phenotype triggered by other Bep(s). Focal adhesion kinase (FAK), GEFs for Rho and the Rho effectors ROCK and mDia are crucial for the regulation of the adhesion turnover and rear edge retraction [28], [29]. mDia is a formin that produces F-actin filaments by nucleation and polymerization, while ROCK activates myosin to cross-link to F-actin for induction of actomyosin bundles and contractility [51], [52]. Functional interferences with any of these components would disturb normal cell migration. Small GTPases are targeted by numerous bacterial effectors and several examples are known for pairs of effectors secreted by a given pathogen that have opposing effects on their regulation. Salmonella SopE and SptP have adopted GEF and GAP functions for CDC42 and RAC1, respectively, that allow to temporally switch between their active and inactive forms; first to induce membrane ruffle-mediated uptake of bacteria, which is followed by stabilization of the remodeled F-actin cytoskeleton [53]. Another example is the Legionella pneumophila effectors Drra/SidM and LepB that are known to regulate the Rab1 activity and dynamic membrane remodeling of the Legionella-containing phagosome [54]. The Bep-dependent cell fragmentation phenotype, observed in the absence of BepE, might be related to the inhibition of RhoA. Inhibition of RhoA activity by its inhibitor, C3 (C. botulinum exoenzyme), leads to marked morphological changes of migrating monocytes, with frequent polarization and long tails trailed behind the cell body [30] - a phenotype reminiscent to an early stage of cell fragmentation. A counteraction of BepE on the RhoA signaling pathway is supported by the fact that ectopic expression of GFP-BepEBhe interferes with the inhibitory effect of the C3-based Rho inhibitor 1, as shown by stress fiber preservation in GFP-BepEBhe-expressing cells. However, BepE was not able to potentiate any stress fibers when using the ROCK inhibitor Y27632. Based on these observations, BepE could be a factor that directly (or indirectly) activates RhoA independently of Rho inhibitor 1-mediated ADP-ribosylation at asparagine 41 [33].
The VirB T4SSs of bartonellae and its multiple Bep effectors evolved as a toolbox facilitating the adaptation to specific mammalian reservoir hosts [15], [55]. The prominent activity of BepE homologues in restoring cell migration raised our interest to decipher the in vivo function of this effector protein. Bartonella-reservoir host interaction has been studied in a rat model developed by Schulein et al [14]. In this model, intravenously (i.v.) injected Btr wild-type are rapidly cleared from the blood stream, which remains sterile for at least three days. On day four, the bacteria re-appear in the blood where they adhere to and invade erythrocytes. Approximately every five days, a new wave of bacteria is seeded into the blood stream and invades erythrocytes, sustaining the bacteremia for about ten weeks. This is in accordance with other animal models, such as B. grahamii (Bgr) i.v. infection of mice [56]. The intimate interaction of bartonellae with ECs in vitro and the clinical descriptions of Bhe association to human vasculature, together with the disappearance of bacteria from blood for several days following i.v. infection lead to the proposition that vascular ECs represent, at least in part, the “primary niche” that is colonized initially and from where bacteria are seeded to the blood stream to invade erythrocytes. Upon i.v. infection the course of bacteremia was similar in cases of wild-type and Btr ΔbepDE mutant bacteria. In an i.d. infection model that reflects much better the initial dermal stage of Bartonella infection, wild-type bacteria still caused bacteremia, yet with a 4–5 day time delay compared to the i.v. model. In contrast, animals infected with the Btr ΔbepDE mutant remained abacteremic, while complementation with BepE restored bacteremia. This data suggests (i) that Bartonella presumably initially infects some cell type(s) in the derma, and (ii) that spreading the infection to the blood stream (possibly via the lymphatic system) is BepE-dependent. Previous studies based on the i.v. infection model describe components of Btr VirB T4SS to be essential for colonization of the “primary niche”, while those were dispensable for the subsequent stage of intraerythrocytic infection [17], [19]. Addressing an earlier stage of vector-borne infection, our results in the i.d. model demonstrate that the VirB T4SS and specifically BepE are already required at the dermal stage of infection. This revisits our understanding of intracellular niches of Bartonella. We suggest replacing the old term “primary niche” by “blood seeding niche” and introducing “dermal niche” as a new term describing the preceding dermal stage of arthropod-borne infection.
The BepE-dependent phenotypes in vivo (progression from dermal to blood stage infection in the i.d. model) and in vitro (suppression of cell fragmentation and stress fiber disassembly by Rho inhibitor 1 activity) were consistently mediated by the BID domains of BepEBhe. In fact, most phenotypes mediated by Beps of Bhe are attributed to a BID domain – further to the essential role of the most C-terminal BID domain as part of the bi-partite secretion signal for T4SS-dependent effector translocation [21] this domain has thus adopted diverse effector functions within host cells [15]. The anti-apoptotic activity of BepABhe is confined to its single BID domain [27], and the F-actin remodeling activity of BepF is mediated by its multiple BID domains. Surprisingly, the phosphotyrosine-containing motifs in the N-terminal part of BepE that were shown to recruit multiple SH2-domain containing signaling proteins of the host cell [22] revealed no essential role in the here described BepE-dependent in vitro and in vivo phenotypes. However, we suggest that investigation of these motifs may be relevant in cells infected in the “dermal niche”, e.g. to modulate immune responses. DCs represent one of the candidate cell types for Bartonella infection in this “dermal niche”. In steady state skin DCs include immature epidermal DCs, Langerhans cells (LCs) and immature dermal DCs [57]. Those have a high phagocytic activity and constantly sample the environment as sentinels for foreign antigens (Ag) [58]. Ag uptake induces the activation/maturation of DCs by starting a directional migration towards the draining lymph nodes and up-regulation of MHC class II/co-stimulatory molecules. Finally, in the lymph nodes DCs form an immune synapse with naive T cells and present Ags, and as a consequence initiate effective adaptive immune responses [59], [60]. Considering that BepE is recruited to cell-to-cell contacts, the phosphotyrosine motifs and their proposed mimickry of immune inhibitory receptors [22] could be a valid tool to modulate signaling pathways within the immune synapse and thus altering the initiation of an adaptive immunity. On the way to lymph nodes DCs may serve as transporters of bacterial cargo. This strategy is considered to be used by other bacterial pathogens, such as Brucella abortus [61] or Bordetella bronchiseptica [39], to disseminate within their mammalian hosts. When Btr ΔbepDE infects the rat reservoir host we think that at the dermal stage fragmentation or impaired migration of yet unknown infected cells may occur that would prevent further dissemination of bacteria and colonization of the blood stream. This is further supported by our finding that migration of BMDCs in a trans-well migration assay is blocked by infection with Bhe ΔbepDEF, while expression of BepEBhe in this mutant background restores it. Albeit it is difficult to compare directly the phenotypic changes in DC (altered cell migration) and HUVECs (fragmentation) upon Bartonella infection, yet considering dermal DCs as “dermal niche” for Bartonella represents a valid hypothesis to be investigated in vivo. Future work should also address how exactly the complex in vitro phenotype of cell fragmentation translates into the striking in vivo phenotype - lack of spreading from derma to blood.
In summary, this study builds on extending the “primary niche” of Bartonella infection that precedes blood stage infection in the mammalian reservoir host to a dermal stage reflecting natural infection by blood-sucking arthropods. Using this i.d. model we describe for the first time a specific role for a Bartonella effector protein in vivo, i.e. BepE is required to spread infection from the dermal site of inoculation to the blood, possibly via promoting normal migration of infected DCs by alleviating deleterious secondary effects mediated by other Beps targeting the RhoA pathway. As a result, this functional crosstalk between Beps could facilitate that Bartonella reaches its replicative niche in the blood, where it multiplies and persists within the immune-privileged intracellular compartment of erythrocytes, from where it is picked up by blood-sucking arthropods for transmission to naive hosts.
Materials and Methods
Ethics statement
Animals were handled in strict accordance with good animal practice as defined by the regulations from the Federal Veterinary Office (FVO) of Switzerland: Animal Welfare Act (TSchG;45), Animal Welfare Ordinance (TSchV;455.1) and Animal Experimentation Ordinance (Tierversuchs-V;455.163). Animal work was approved by the Veterinary Office of the Canton Basel City, Kantonales Veterinäramt, on January 2011 (licence no. 1741). Analyses of human umbilical cord endothelial cells (HUVECs) were performed with written informed consent of patients from Bruderholzspital Basel under approval from the Local Research Ethics Committee, Ethikkommission beider Basel EKBB (protocol no. 102/01). All patient samples were anonymized.
Bacterial strains and growth conditions
The bacterial strains used in this study are listed in Table S1. Bartonella spp were grown on Columbia agar plates containing 5% defibrinated sheep blood (CBA plates) at 35°C and 5% CO2 for 2–3 days. Bhe RSE247 [26] and Btr RSE149 [14] spontaneous streptomycin-resistant strains served as Bhe wild-type and Btr wild-type respectively in this study. When indicated, media were supplemented with 30 µg/ml kanamycin, 100 µg/ml streptomycin, 10 µg/ml gentamycin, and/or 500 µM isopropyl-β-D-thiogalactoside (IPTG, Applichem). E. coli strains were cultivated in Luria-Bertani liquid medium (LB) or on Luria-Bertani agar on plates (LA) at 37°C overnight. When indicated, media were supplemented with 50 µg/ml kanamycin, 200 µg/ml ampicillin, 20 µg/ml gentamycin, 500 µM IPTG, and/or 1 mM diaminopimelic acid (DAP).
Conjugation of Bartonella-expression plasmids into Bartonella
Conjugation of plasmids from the E. coli dap – mutant b2150 [26] into Bartonella spp. was done by triparental mating in the presence of helper plasmid pRK2013 as described previously by Dehio et al., 1998 [62].
DNA manipulations
Plasmids used in this study are listed in Table S1, primers are listed in Table S2. Generation of plasmids for expression of Myc-Beps or lentiviral expression, plasmids used for chromosomal integrations and in-frame deletions are detailed described in Materials and Methods S1.
Western blot analysis of protein expression in Bartonella
Western blot analysis of expressed protein levels in Bartonella was performed as described previously by Schulein et al., 2005 [21].
Infection of rats
Female Wistar rats were obtained at the age of 10 weeks from Harlan, RCC-Füllinsdorf. All animal studies were approved by the authors' institutional review boards.
After two weeks of adaptation the rats were infected with Btr. Bacterial strains were grown as described above, harvested in phosphate-buffered saline (PBS), and diluted to OD595 = 1. Rats were anesthetized with a 2–3% Isoflurane/O2 mixture and infected with 10 µl or 200 µl of the bacterial suspension in the dermis of the right ear or in the tail vein respectively. Blood samples were taken at the tail vein and immediately mixed with PBS containing 3.8% sodium-citrate to avoid coagulation. After freezing to −70°C and subsequent thawing, undiluted and diluted blood samples were plated on Columbia agar plates containing 5% defibrinated sheep blood (CBA plates) at 35°C and 5%. CFUs were counted after 7–10 days of growth.
Cell culture
Human umbilical vein endothelial cells (HUVEC) were isolated as described before [26] and cultured in EGM medium (Promocell) in a humidified atmosphere at 37°C and 5% CO2.
Mouse bone marrow-derived dendritic cells (BMDCs) were differentiated in vitro using standard protocol [63]. Briefly, bone marrow cells were flushed from the tibias and femurs of Balb/c mouse with culture medium composed of DMEM medium (Invitrogen Life Technologies) supplemented with 10% fetal calf serum (FCS, Invitrogen Life Technologies). After one centrifugation, BM cells were resuspended in Tris-ammonium chloride for 2 min to lyse RBC. After one more centrifugation, BM cells were cultured for 9–10 days at 1*106 cells/ml in culture medium supplemented with 200 ng/ml recombinant human Flt3L (produced by 40E1 hybridoma cells, kind gift from prof. A. Rolink). Cultures were incubated at 37°C in 5% CO2-humidified atmosphere.
iLECs (immortalized lymphatic endothelial cells) were cultured as described by Vigl et al., 2011 [64].
Lentiviral transduction of HUVECs
Subconfluent (3*106) HEK 293T cells in 10 cm cell-culture dishes were transfected with a total of 5 µg of plasmid DNA following the FuGENE transfection protocol (FuGENE 6 Transfection Reagent, Roche). After 12 h, the cell culture media was replaced and the cells were kept in culture for virus production in the supernatant for additional 24 h. 7*104 HUVECs/well was seeded in gelatin coated 6-well plate 24 h before the viral infection. On day of infection the viral supernatant from transfected HEK 293T cells was filtered with 0.45 µm filter and transferred onto the monolayer of HUVECs, 2 ml of viral supernatant in presence of 0.5 µg/ml Polybrene (Sigma) was applied on each well. After the first harvesting of viral supernatant the HEK 293T cells were supplemented with a fresh medium. 24 h later the HUVECs were re-infected repeating the same procedure with followed replacement of the medium in 24 h.
Infection of HUVECs
HUVECs (passage 4–7) were plated on gelatin-coated glass slides in 24-well plates at 5*104/ml using EGM or alternatively 3*103/well seeded in 96-well plate. The next day cells were washed twice with M199 with Earls salts (M199, Gibco, Invitrogen Life Technologies) supplemented with 10% FCS (Invitrogen Life Technologies) and infected with a multiplicity of infection (MOI) of 200 bacteria per cell or in case of mixed infections 200 plus 200 of corresponding strains in M199/10% FCS/500 µM IPTG and incubated for 24, 36 and 48 h or 72 h for the time-lapse microscopy. Where necessary the infection was fixed with 3,7% paraformaldehyde (PFA).
Immunofluorescent labeling
Indirect immunofluorescent labeling was performed as described [26]. Standard 96-well plate assays were stained with TRITC-Phalloidin (Sigma, 100 µg/ml stock solution, final concentration 1∶400) or Dy-647-Phalloidin (Dyomics, 100 µg/ml stock solution, final concentration 1∶200) and DAPI (Roche, 0.1 mg/ml). Samples on glass slides for confocal microscopy were stained with Cy3-Phalloidin (Sigma, 100 µg/ml stock solution, final concentration 1∶100) or Dy-647-Phalloidin (Dyomics, 100 µg/ml stock solution, final concentration 1∶200), DAPI and anti-Myc antibodies (mouse anti-Myc, Invitrogen, Carlsbad, CA/USA) and/or VE-Cadherin (rabbit anti-VE-Cadherin, Bender MedSystems, Burlingame, CA/USA). Secondary antibodies for Immunofluorescence used in this study were Cy5-conjugated goat anti-rabbit Ig antibodies (Dianova, Hamburg, Germany, 1∶100).
Inhibitor treatment of transduced HUVECs
Mixed culture of non-transduced and lentivirally transduced HUVECs by pRO300, pRO301, pRO302, pRO303 or pRO304 were seeded in gelatin coated wells at 3,5*103/100 µl using endothelial complete medium (PromoCell) in 96 well-plates in the morning. Cells were starved in endothelial medium with reduced serum (containing 20× reduced SupplementMix) for overnight. The next day the wells were treated with Rho Inhibitor I (CTO4, Cytoskeleton, Denver, CO) or Y27632 (Sigma) at different concentrations for 4 h or 30 m respectively. The experiment was stopped by fixation with 3.7% PFA, stained for F-actin and nucleus as described above and subjected to automated microscopy.
Image analysis and quantification of cell fragmentation or loss of stress fibers
Experiments performed in 96-well plates were subjected to automated microscopy, using MD ImageXpress Micro automated microscopes. In every well, 6×6 sites were imaged in 2–3 different wavelengths corresponding the applied cell staining. Images were visualized using MetaXpress software (MDC) and the number of cells per image was determined automatically by MetaXpress in-build analysis modules (CountNuclei). The cells with the thin elongations or stress fiber-containing cells were defined and counted by eye. The percentage of elongated cells or stress fiber-containing cells was calculated using Microsoft Office Excel. In every condition randomly picked 10 fields of magnification 10 within the well and triplicates of the wells were analyzed.
For confocal laser microscopy, the stained samples were analyzed using a Zeis Point Scanning Confocal, LSM700 Upright microscope (Imaging Core Facility, Biozentrum, University of Basel, Switzerland). Z-stacks with 20–30 focal planes with a spacing of 0.1–0.3 µm were recorded and xz - and yz-planes were reconstructed using Zen 2010 software. Images were exported and finalized using MetaMorph, ImageJ and Adobe Photoshop.
Time-lapse microscopy
Time-lapse microscopy was carried out using an MD ImagXpress Micro automated microscope from Molecular devices (inside a controlled chamber held at 37°C and 5% CO2). Images were recorded using a CoolSNAP ES digital CCD camera and processed using MetaXpress software. Briefly, HUVECs were infected with Bhe strains in 96-well format as described above. After incubating for 8 h, 2 to 4 points in each well were chosen for time-lapse microscopy. The points were imaged for 72 h with 10 min lapse between each imaging. Both, red and green, fluorescence were detected at 20× magnification, and the corresponding videos were processed in MetaXpress and compiled into QuickTime (Apple, Cupertino, CA) movies.
Quantification of cell survival by FACS analysis
Mixed population of non-transduced HUVECs with lentivirally transduced HUVECs by pRO300, pRO301, pRO302, pRO303 or pRO304 were seeded in gelatin coted 6 well-plates at 105 cell/well the evening before experiment. Next day the infection was performed with MOI 200 as described above. After 48 hpi the cells were detached from the wells by using trypsin, resuspended in medium and analyzed for quantification of GFP-expressing cells by FACS (BD Biosciences). The data were analyzed using the FlowJo software.
Infection and Bla analysis of mouse BMDCs
Day 9–10 BMDCs were harvested from the flask by vigorous pipetting followed by washing twice with room temperature PBS without Ca2+ or Mg2+ (Invitrogen Life Technologies). 4*103 BMDCs were seeded within the wells of 96-well plate. Next day supernatant was replaced with the fresh M199 with Earls salts (M199, Gibco, Invitrogen Life Technologies) supplemented with 10% FCS (Invitrogen Life Technologies) and infected with a MOI of 50, 100, 300. At 3, 6, 9, 12, 24 and 48 h post infection the wells were loaded with 1 µM CCF2 - AM (Invitrogen Life Technologies) in M199/10% FCS and incubated at room temperature for 60–120 min. The plate was imaged in two channels (blue and green) by Leica DM-IRBE inverted fluorescence microscope. Data were analyzed in MetaMorph and further processed in Microsoft Office Excel.
BMDC trans-well migration
Trans-well migration assay was performed as described previously [41]. Experiments were performed with 12 mm diameter, 8 µm pore cell-culture inserts (Millipore, Billerica, MA) in a modified Boyden chamber assay. One day before the experiment, iLECs were seeded onto the collagen-coated underside of the chamber at 105 cells/well and in parallel 2*105 mouse BMDCs were infected with Bartonella at MOI = 200. The next day, infected BMDCs were harvested and stained with DiD-Vybrant (Invitrogen Life Technologies), then seeded in 200 µl (1 mm thick) Matrigel (4.65 mg/ml, BD Biosciences, San Jose, CA) within the inserts. Basal medium was placed in the top and bottom chamber. After 15 h in a 35°C/5% CO2 incubator, Matrigel containing non-migrated cells was removed and the inserts were fixed in 3.7% PFA. The number of migrated cells was counted by microscope on the underside of the chamber.
Accession numbers for the proteins used in this study
The Uniport (http://www.uniprot.org) accession numbers for the protein sequences to BepABhe (Q6G2A9), BepCBhe (Q5QT03), BepDBhe (Q5QT02), BepEBhe (Q5QT01), BepFBhe (Q5QT00), BepGBhe (Q5QSZ9), BepDBtr (A9IWP9), BepEBtr (A9IWQ0), BepEBqu (Q6FYV7), BepHBgr (C6AES9), VirB4Bhe (Q9R2W4).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MonackDM, MuellerA, FalkowS (2004) Persistent bacterial infections: the interface of the pathogen and the host immune system. Nat Rev Microbiol 2 : 747–765.
2. LeeKS, KalantzisA, JacksonCB, O'ConnorL, Murata-KamiyaN, et al. (2012) Helicobacter pylori CagA triggers expression of the bactericidal lectin REG3gamma via gastric STAT3 activation. PLoS One 7: e30786.
3. KidoM, WatanabeN, AokiN, IwamotoS, NishiuraH, et al. (2011) Dual roles of CagA protein in Helicobacterpylori-induced chronic gastritis in mice. Biochem Biophys Res Commun 412 : 266–272.
4. SchroederGN, HilbiH (2008) Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin Microbiol Rev 21 : 134–156.
5. DiacovichL, GorvelJP (2010) Bacterial manipulation of innate immunity to promote infection. Nat Rev Microbiol 8 : 117–128.
6. ShamesSR, FinlayBB (2012) Bacterial effector interplay: a new way to view effector function. Trends Microbiol 20 : 214–219.
7. ChomelBB, BoulouisHJ, MaruyamaS, BreitschwerdtEB (2006) Bartonella spp. in pets and effect on human health. Emerg Infect Dis 12 : 389–394.
8. BoulouisHJ, ChangCC, HennJB, KastenRW, ChomelBB (2005) Factors associated with the rapid emergence of zoonotic Bartonella infections. Vet Res 36 : 383–410.
9. ChomelBB, BoulouisHJ, BreitschwerdtEB, KastenRW, Vayssier-TaussatM, et al. (2009) Ecological fitness and strategies of adaptation of Bartonella species to their hosts and vectors. Vet Res 40 : 29.
10. BreitschwerdtEB, MaggiRG, ChomelBB, LappinMR (2010) Bartonellosis: an emerging infectious disease of zoonotic importance to animals and human beings. J Vet Emerg Crit Care (San Antonio) 20 : 8–30.
11. DehioC (2004) Molecular and cellular basis of bartonella pathogenesis. Annu Rev Microbiol 58 : 365–390.
12. FoilL, AndressE, FreelandRL, RoyAF, RutledgeR, et al. (1998) Experimental infection of domestic cats with Bartonella henselae by inoculation of Ctenocephalides felis (Siphonaptera: Pulicidae) feces. J Med Entomol 35 : 625–628.
13. RaoultD, AboudharamG, CrubezyE, LarrouyG, LudesB, et al. (2000) Molecular identification by “suicide PCR” of Yersinia pestis as the agent of medieval black death. Proc Natl Acad Sci U S A 97 : 12800–12803.
14. SchuleinR, SeubertA, GilleC, LanzC, HansmannY, et al. (2001) Invasion and persistent intracellular colonization of erythrocytes. A unique parasitic strategy of the emerging pathogen Bartonella. J Exp Med 193 : 1077–1086.
15. EngelP, SalzburgerW, LieschM, ChangCC, MaruyamaS, et al. (2011) Parallel Evolution of a Type IV Secretion System in Radiating Lineages of the Host-Restricted Bacterial Pathogen Bartonella. PLoS Genet 7: e1001296.
16. SeubertA, HiestandR, de la CruzF, DehioC (2003) A bacterial conjugation machinery recruited for pathogenesis. Mol Microbiol 49 : 1253–1266.
17. SchuleinR, DehioC (2002) The VirB/VirD4 type IV secretion system of Bartonella is essential for establishing intraerythrocytic infection. Mol Microbiol 46 : 1053–1067.
18. DehioC (2008) Infection-associated type IV secretion systems of Bartonella and their diverse roles in host cell interaction. Cell Microbiol 10 : 1591–1598.
19. Vayssier-TaussatM, Le RhunD, DengHK, BivilleF, CescauS, et al. (2010) The Trw type IV secretion system of Bartonella mediates host-specific adhesion to erythrocytes. PLoS Pathog 6: e1000946.
20. NystedtB, FrankAC, ThollessonM, AnderssonSG (2008) Diversifying selection and concerted evolution of a type IV secretion system in Bartonella. Mol Biol Evol 25 : 287–300.
21. SchuleinR, GuyeP, RhombergTA, SchmidMC, SchroderG, et al. (2005) A bipartite signal mediates the transfer of type IV secretion substrates of Bartonella henselae into human cells. Proc Natl Acad Sci U S A 102 : 856–861.
22. SelbachM, PaulFE, BrandtS, GuyeP, DaumkeO, et al. (2009) Host cell interactome of tyrosine-phosphorylated bacterial proteins. Cell Host Microbe 5 : 397–403.
23. SchmidMC, SchuleinR, DehioM, DeneckerG, CarenaI, et al. (2004) The VirB type IV secretion system of Bartonella henselae mediates invasion, proinflammatory activation and antiapoptotic protection of endothelial cells. Mol Microbiol 52 : 81–92.
24. RhombergTA, TruttmannMC, GuyeP, EllnerY, DehioC (2009) A translocated protein of Bartonella henselae interferes with endocytic uptake of individual bacteria and triggers uptake of large bacterial aggregates via the invasome. Cell Microbiol 11 : 927–945.
25. SchmidMC, ScheideggerF, DehioM, Balmelle-DevauxN, SchuleinR, et al. (2006) A translocated bacterial protein protects vascular endothelial cells from apoptosis. PLoS Pathog 2: e115.
26. DehioC, MeyerM, BergerJ, SchwarzH, LanzC (1997) Interaction of Bartonella henselae with endothelial cells results in bacterial aggregation on the cell surface and the subsequent engulfment and internalisation of the bacterial aggregate by a unique structure, the invasome. J Cell Sci 110 (Pt 18) 2141–2154.
27. TruttmannMC, RhombergTA, DehioC (2011) Combined action of the type IV secretion effector proteins BepC and BepF promotes invasome formation of Bartonella henselae on endothelial and epithelial cells. Cell Microbiol 13 : 284–299.
28. IwanickiMP, VomastekT, TilghmanRW, MartinKH, BanerjeeJ, et al. (2008) FAK, PDZ-RhoGEF and ROCKII cooperate to regulate adhesion movement and trailing-edge retraction in fibroblasts. J Cell Sci 121 : 895–905.
29. RidR, SchiefermeierN, GrigorievI, SmallJV, KaverinaI (2005) The last but not the least: the origin and significance of trailing adhesions in fibroblastic cells. Cell Motil Cytoskeleton 61 : 161–171.
30. WorthylakeRA, LemoineS, WatsonJM, BurridgeK (2001) RhoA is required for monocyte tail retraction during transendothelial migration. J Cell Biol 154 : 147–160.
31. HeasmanSJ, CarlinLM, CoxS, NgT, RidleyAJ (2010) Coordinated RhoA signaling at the leading edge and uropod is required for T cell transendothelial migration. J Cell Biol 190 : 553–563.
32. FanH, HallP, SantosLL, GregoryJL, Fingerle-RowsonG, et al. (2011) Macrophage migration inhibitory factor and CD74 regulate macrophage chemotactic responses via MAPK and Rho GTPase. J Immunol 186 : 4915–4924.
33. KimMJ, KimS, KimY, JinEJ, SonnJK (2012) Inhibition of RhoA but not ROCK induces chondrogenesis of chick limb mesenchymal cells. Biochem Biophys Res Commun 418 : 500–505.
34. AktoriesK, WildeC, VogelsgesangM (2004) Rho-modifying C3-like ADP-ribosyltransferases. Rev Physiol Biochem Pharmacol 152 : 1–22.
35. GroegerG, NobesCD (2007) Co-operative Cdc42 and Rho signalling mediates ephrinB-triggered endothelial cell retraction. Biochem J 404 : 23–29.
36. MarignacG, BarratF, ChomelB, Vayssier-TaussatM, GandoinC, et al. (2010) Murine model for Bartonella birtlesii infection: New aspects. Comp Immunol Microbiol Infect Dis 33 : 95–107.
37. SavinaA, AmigorenaS (2007) Phagocytosis and antigen presentation in dendritic cells. Immunol Rev 219 : 143–156.
38. ShortmanK, NaikSH (2007) Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol 7 : 19–30.
39. SkinnerJA, PilioneMR, ShenH, HarvillET, YukMH (2005) Bordetella type III secretion modulates dendritic cell migration resulting in immunosuppression and bacterial persistence. J Immunol 175 : 4647–4652.
40. ZlokarnikG, NegulescuPA, KnappTE, MereL, BurresN, et al. (1998) Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science 279 : 84–88.
41. ShieldsJD, FleuryME, YongC, TomeiAA, RandolphGJ, et al. (2007) Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine CCR7 signaling. Cancer Cell 11 : 526–538.
42. EdelmanGM, GallyJA (2001) Degeneracy and complexity in biological systems. Proc Natl Acad Sci U S A 98 : 13763–13768.
43. LarsenM, TremblayML, YamadaKM (2003) Phosphatases in cell-matrix adhesion and migration. Nat Rev Mol Cell Biol 4 : 700–711.
44. LamaliceL, Le BoeufF, HuotJ (2007) Endothelial cell migration during angiogenesis. Circ Res 100 : 782–794.
45. InsallRH, MacheskyLM (2009) Actin dynamics at the leading edge: from simple machinery to complex networks. Dev Cell 17 : 310–322.
46. PontiA, MachacekM, GuptonSL, Waterman-StorerCM, DanuserG (2004) Two distinct actin networks drive the protrusion of migrating cells. Science 305 : 1782–1786.
47. Zaidel-BarR, BallestremC, KamZ, GeigerB (2003) Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J Cell Sci 116 : 4605–4613.
48. WoodW, MartinP (2002) Structures in focus–filopodia. Int J Biochem Cell Biol 34 : 726–730.
49. MiaoL, VanderlindeO, StewartM, RobertsTM (2003) Retraction in amoeboid cell motility powered by cytoskeletal dynamics. Science 302 : 1405–1407.
50. JermyA (2011) Bacterial pathogenesis: legionella effector under friendly fire. Nat Rev Microbiol 9 : 80.
51. ChaturvediLS, MarshHM, BassonMD (2011) Role of RhoA and its effectors ROCK and mDia1 in the modulation of deformation-induced FAK, ERK, p38, and MLC motogenic signals in human Caco-2 intestinal epithelial cells. Am J Physiol Cell Physiol 301: C1224–1238.
52. NarumiyaS, TanjiM, IshizakiT (2009) Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev 28 : 65–76.
53. KuboriT, GalanJE (2003) Temporal regulation of salmonella virulence effector function by proteasome-dependent protein degradation. Cell 115 : 333–342.
54. IngmundsonA, DelpratoA, LambrightDG, RoyCR (2007) Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature 450 : 365–369.
55. SaenzHL, EngelP, StoeckliMC, LanzC, RaddatzG, et al. (2007) Genomic analysis of Bartonella identifies type IV secretion systems as host adaptability factors. Nat Genet 39 : 1469–1476.
56. KoeslingJ, AebischerT, FalchC, SchuleinR, DehioC (2001) Cutting edge: antibody-mediated cessation of hemotropic infection by the intraerythrocytic mouse pathogen Bartonella grahamii. J Immunol 167 : 11–14.
57. Lopez-BravoM, ArdavinC (2008) In vivo induction of immune responses to pathogens by conventional dendritic cells. Immunity 29 : 343–351.
58. RandolphGJ, OchandoJ, Partida-SanchezS (2008) Migration of dendritic cell subsets and their precursors. Annu Rev Immunol 26 : 293–316.
59. AlvarezD, VollmannEH, von AndrianUH (2008) Mechanisms and consequences of dendritic cell migration. Immunity 29 : 325–342.
60. GorvelJP (2008) Brucella: a Mr “Hide” converted into Dr Jekyll. Microbes Infect 10 : 1010–1013.
61. SalcedoSP, MarchesiniMI, LelouardH, FugierE, JollyG, et al. (2008) Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog 4: e21.
62. DehioM, KnorreA, LanzC, DehioC (1998) Construction of versatile high-level expression vectors for Bartonella henselae and the use of green fluorescent protein as a new expression marker. Gene 215 : 223–229.
63. BraselK, De SmedtT, SmithJL, MaliszewskiCR (2000) Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood 96 : 3029–3039.
64. ViglB, AebischerD, NitschkeM, IolyevaM, RothlinT, et al. (2011) Tissue inflammation modulates gene expression of lymphatic endothelial cells and dendritic cell migration in a stimulus-dependent manner. Blood 118 : 205–215.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 6
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Fungal Nail Infections (Onychomycosis): A Never-Ending Story?
- Profilin Promotes Recruitment of Ly6C CCR2 Inflammatory Monocytes That Can Confer Resistance to Bacterial Infection
- IscR Is Essential for Type III Secretion and Virulence
- Contribution of Specific Residues of the β-Solenoid Fold to HET-s Prion Function, Amyloid Structure and Stability