
Growth Factor and Th2 Cytokine Signaling Pathways Converge at STAT6 to Promote Arginase Expression in Progressive Experimental Visceral Leishmaniasis
Visceral leishmaniasis (VL), caused by the intracellular protozoan Leishmania donovani, is a progressive infection that is particularly common in impoverished populations of the world. People die from this disease unless it is treated. We used an experimental infection model that mimics the clinical and pathological features of human VL to study how the parasite causes this severe disease. We found that host macrophages infected with Leishmania donovani are activated in a way that leads to the expression of arginase, an enzyme that counteracts the cell's mechanisms that control the infection. This disease-promoting activation pathway was driven by the convergence of growth factor and cytokine signaling pathways and activation of the transcription factor STAT6. Chemical inhibition of signaling through the fibroblast growth factor receptor-1 (FGFR-1) or insulin-like growth factor-1 receptor (IGF-IR), or genetic knockdown of STAT6 led to reduced expression of arginase and enhanced control of the infection by macrophages. This indicates that the growth factor signaling pathways together with the cytokine pathways promote this disease. Interventions designed to disrupt this signaling could help in the treatment of VL.
Published in the journal:
. PLoS Pathog 10(6): e32767. doi:10.1371/journal.ppat.1004165
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004165
Summary
Visceral leishmaniasis (VL), caused by the intracellular protozoan Leishmania donovani, is a progressive infection that is particularly common in impoverished populations of the world. People die from this disease unless it is treated. We used an experimental infection model that mimics the clinical and pathological features of human VL to study how the parasite causes this severe disease. We found that host macrophages infected with Leishmania donovani are activated in a way that leads to the expression of arginase, an enzyme that counteracts the cell's mechanisms that control the infection. This disease-promoting activation pathway was driven by the convergence of growth factor and cytokine signaling pathways and activation of the transcription factor STAT6. Chemical inhibition of signaling through the fibroblast growth factor receptor-1 (FGFR-1) or insulin-like growth factor-1 receptor (IGF-IR), or genetic knockdown of STAT6 led to reduced expression of arginase and enhanced control of the infection by macrophages. This indicates that the growth factor signaling pathways together with the cytokine pathways promote this disease. Interventions designed to disrupt this signaling could help in the treatment of VL.
Introduction
Visceral leishmaniasis (VL), caused by the intracellular protozoan Leishmania donovani or L. infantum, is one of the “Neglected Tropical Diseases” that impacts the poor of the world. Active VL is characterized by a relentlessly progressive infection with cachexia, massive splenomegaly, pancytopenia and ultimately death. VL ranks second to malaria in deaths caused by a protozoal pathogen; mortality is reported in up to 10–20% of patients, even with treatment [1]. The determinants of susceptibility and progressive disease are incompletely defined. However, it is clear that ineffective cellular immune function, dictated by the nature of cytokine response and polarization of macrophages [2], plays a critical role. Macrophages, the primary target of intracellular Leishmania infection, may take on distinct phenotypes in response to parasite signals and inflammatory stimuli within the infected microenvironment. Classically activated (M1) macrophages respond to IFN-γ and microbial products by generating antimicrobial molecules that effectively kill Leishmania and other intracellular pathogens [3], [4]. Central to the killing of intracellular parasites is the production of nitric oxide by the action of inducible nitric oxide synthase 2 (NOS2) on the substrate L-arginine. In contrast, alternatively activated or M2 macrophages, which are typically generated by exposure to type 2 cytokines (IL-4, IL-13), fail to produce antimicrobial effector molecules to kill intracellular pathogens and serve to dampen inflammation and promote wound healing [5], [6].
The activation status of macrophages in human VL has not been directly investigated. However, the progressive nature of the infection in the face of strong expression of IFN-γ [7]–[10], suggests that there is ineffective classical activation. The concomitant production of IL-4/IL-13 and IL-10 [7], [8], [11]–[14], which are known to impair macrophage leishmanicidal activity, may polarize macrophages toward a disease-promoting M2 phenotype. Neutralization of IL-10 in ex vivo splenocyte cultures from patients with VL promoted parasite clearance [15], but the importance of IL-4 and/or IL-13 in the pathogenesis of human VL is not clear. Additionally, Leishmania-driven subterfuge of a number of signaling pathways can render the macrophage less responsive to activating stimuli and more permissive to infection [16].
We have used the hamster model of VL, which closely mimics the clinicopathological features of human VL, to dissect the mechanisms by which L. donovani causes progressive disease. We demonstrated, similar to human VL, that progressive, lethal disease occurred in the face of what would be considered a protective type 1 cytokine response [17], [18]. Despite high expression of IFN-γ, it was ineffective in mediating classical activation of M1 macrophages and control of Leishmania infection. In fact we found that splenic macrophages from hamsters with VL were polarized to a M2-like phenotype with dominant expression of host arginase 1 (arg1) [2]. L. donovani triggered arg1 expression through a STAT6-dependent mechanism, but surprisingly it did not require type 2 cytokines [2]. Arginase contributes to intracellular Leishmania replication by competing with NOS2 for the substrate arginine (thereby reducing NO production), and by driving the generation of polyamines, which promote parasite growth [2], [19], [20]. M2-like macrophages and arginase have also been implicated in the pathogenesis of experimental cutaneous leishmaniasis [19]–[23] and infections with other intracellular pathogens [24]–[27]. Furthermore, there is accumulating evidence that arginase has a role in the pathogenesis of human disease. Although, polarization of isolated human macrophages by exposure to IL-4 in vitro did not lead to upregulation of arginase activity or arg1 expression [28], the presence of M2-like monocytes/macrophages and arginase expression has been found in cancer [29], [30], filariasis [25], tuberculosis [31], [32], and traumatic tissue injury [33]. Elevated arginase activity was also recently reported in the lesions of patients with chronic cutaneous leishmaniasis [34] and arginase expression in peripheral blood leukocytes was found to be a marker of active VL [35].
In this work we have investigated the mechanisms of the pathological upregulation of arg1 in the hamster model of progressive VL. We discovered that the expression of arg1 in L. donovani infected macrophages is driven by activation of fibroblast growth factor receptor (FGFR) and insulin-like growth factor-1 receptor (IGF-IR). Inhibition of these growth factor signaling pathways led to reduced arg1 expression and enhanced control of parasite replication. Furthermore, signaling molecules downstream of the growth factor receptors converged with IL-4 signaling to promote STAT6 activation and arg1 expression in VL. The intersection of these pathways leads to subversion of macrophage effector function and impaired host defense against VL.
Results
Receptor tyrosine kinases (RTK) are involved in parasite-induced arginase expression
We previously determined that L. donovani induced STAT6-dependent, host arg1 expression. Host arginase expression promoted parasite replication, so we sought to understand the mechanisms by which it was expressed in VL. Arg1 transcription required the de novo synthesis of protein [2] suggesting that transcription of arg1 involved signaling pathway(s) other than just direct phosphorylation of STAT6. We postulated that the newly synthesized protein could mediate its effect through RTK signaling pathways, which regulate inflammation and wound repair [36], [37]. Both of these processes are important functions of M2 macrophages. Therefore, we screened a library of 80 RTK inhibitors for inhibition of L. donovani-induced arginase transcription in an ex vivo model of infected splenocytes isolated from hamsters with VL [38]. Inhibitors of the Epidermal Growth Factor Receptor and Platelet-derived Growth Factor Receptor signaling pathways reduced arg1 transcription by >50% (Table 1). Because the RTK signaling pathways are overlapping and broad, and inhibitors of some growth factor receptors were not included in the inhibitor library, we used a RTK antibody array to further define the participation of specific RTKs in VL. We found that Fibroblast Growth Factor Receptor (FGFR) 1 and 2 and other molecules known to participate in growth factor signaling (Insulin receptor substrate 1 (IRS-1), v-akt murine thymoma viral oncogene homolog 1 and 2 (AKT 1/2), Mitogen-activated protein kinase (MAPK)-3, and Signal transducer and activator of transcription (STAT)-1, and STAT-3 were activated in splenic macrophages from hamsters infected with L. donovani (Table 2). Collectively, these data indicated that signaling through growth factor receptor pathways could contribute to the parasite-induced expression of host arg1.


Growth factors induce arg1 in L. donovani infected hamster macrophages
A significant increase in arg1 mRNA expression was observed in L. donovani infected hamster bone marrow-derived macrophages (BMDM) exposed to the recombinant growth factors FGF-2, IGF-1, and PDGF (Fig. 1A). Growth factor-induced arg1 was particularly evident in infected compared to uninfected macrophages, and it was equivalent to, or greater than, IL-4-induced arg1. Arginase protein activity was also significantly increased in L. donovani infected BMDM exposed to FGF-2, IGF-1, and PDGF (Fig. 1B). EGF did not consistently induce a significant increase arg1 mRNA or protein. Together, these data suggested that L. donovani infection of macrophages led to enhanced arg1 transcriptional responsiveness to multiple growth factors.

The FGF signaling pathway is activated in the spleens of hamsters infected with Leishmania donovani
Analysis of the FGF and IGF-1 signaling pathways in splenic macrophages from hamsters with VL by immunoblotting confirmed the finding of the antibody screening array (Figs. 2 and 3). There was no evidence for activation of other growth factor signaling pathways in VL (see Fig. S1 and S2). Our finding that inhibition of EGFR reduced arg1 mRNA expression (Table 1), when neither increased ligand expression nor receptor activation could be demonstrated, suggested that basal activity of EGF/EGFR modulated arg1 expression through an effect on downstream signaling. As we demonstrated previously [2], arg1 protein expression was increased in macrophages isolated from the spleens of hamsters with VL starting at 14 days post-infection (Fig. 2A). Of the growth factor receptor ligands, only FGF-2 expression was increased in splenic macrophages (Fig. 2B, Fig. S1, and Fig. S2) and it was accompanied by increased phosphorylation of Tyr653/654 of the FGFR-1 (Fig. 2C) relative to overall receptor protein expression (Fig. 2D). The increase in both FGF-2 and its phosphorylated receptor paralleled the expression of arg1 in the splenic macrophages. Multiple molecules involved in the signaling cascade downstream of FGFR (shown in the diagram in Fig. 2M) were activated, including members of the PI3K/AKT pathway [GAB (Fig. 2E), PI3K (Fig. 2F)] and the MAPK/ERK pathway [c-RAF (Fig. 2G), ERK1/2 (Fig. 2H)]. Activation of p38 MAPK (Fig. 2I), that leads to activation of the transcription factor ATF-2 (Fig. 2J) and the cyclic AMP response element-binding protein (CREB) (Fig. 2K) was observed at 14 days post-infection but was then down-modulated at 28 days post-infection. This suggested that sustained activation of these signaling molecules was not required for the expression of arg1 throughout the course of VL (Fig. 2A). The mechanism(s) through which these molecules are down regulated is unknown. Activation of STAT3, which was evident throughout the course of VL (Fig. 2L), may be a consequence of increased IL-10 production (Fig. S4A and reference [2]) or growth factor signaling (Fig. S4D) [39].

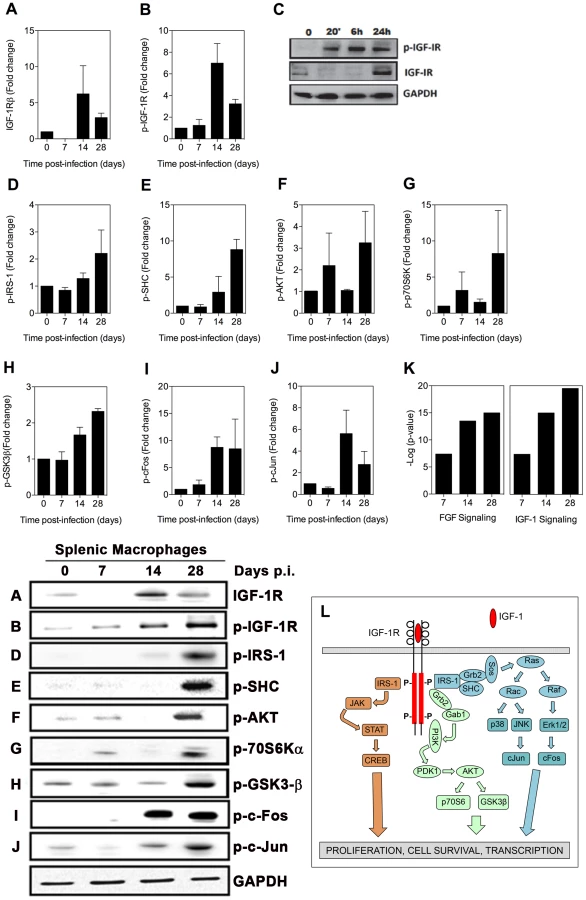
The IGF-1 signaling pathway is activated in the spleens of hamsters with VL
We were unable to detect increased expression of IGF-1 or IGF-2 in the spleen or plasma of hamsters with VL (Fig. S1; data not shown). However, by immunoblot we found increased expression of the IGF-1R after 14 days post-infection (Fig. 3A), and somewhat unexpectedly the beta (cytoplasmic) domain of the IGF-1 receptor, which mediates intracellular signaling, was phosphorylated at these time points (Fig. 3B). We confirmed these findings in BMDM exposed in vitro to L. donovani where parasite-induced IGF-1R phosphorylation was evident between 20 minutes and 24 hrs of exposure, and enhanced expression of IGF-1R protein was present at 24 hrs after infection (Fig. 3C). A number of the activated signaling molecules downstream of FGFR overlap with the canonical IGF-1R signaling pathway (compare data in Fig. 2 with schematic in Fig. 3L). Additionally, other pathway members, including IRS-1 (Fig. 3D), SHC (Fig. 3E), AKT (Fig. 3F), p70S6K (Fig. 3G), and GSK3β (Fig. 3H) were activated, as were the downstream transcription factors c-FOS (Fig. 3I) and c-Jun (Fig. 3J). When all of the activated signaling molecules were subjected to network analysis (Ingenuity Pathway Analysis) both the FGFR and IGF-1R pathways were found to be significantly upregulated in splenic macrophages during the course of VL (p<10−7; Fig. 3K).
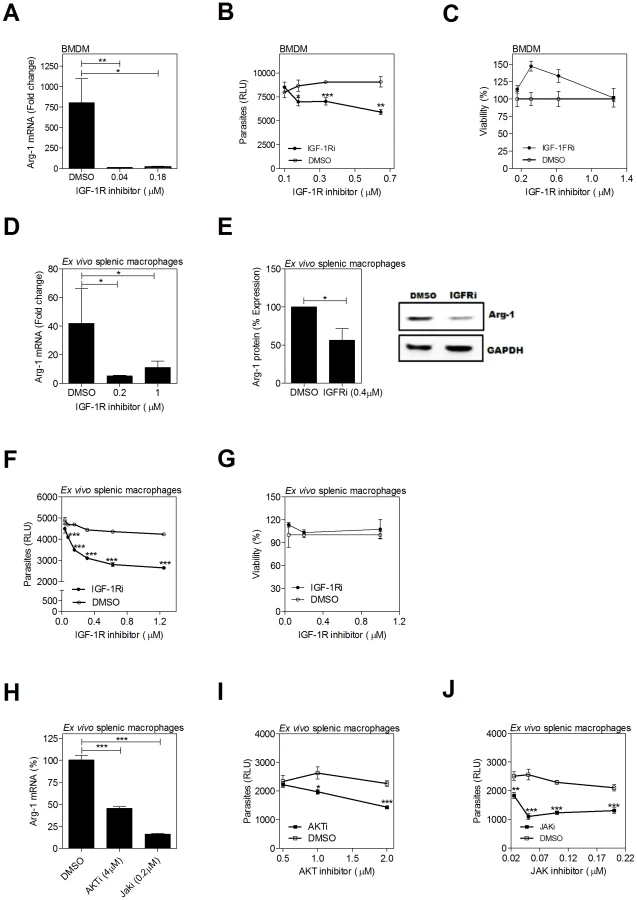
Inhibition of the FGFR and IGFR-1 and downstream signaling molecules decreases arg1 expression and parasite burden in infected macrophages
Treatment of L. donovani-infected hamster BMDMs over 24 hrs of infection with an inhibitor of FGFR-1 resulted in a significant dose-dependent reduction of arg1 mRNA expression (Fig. 4A) and parasite burden (Fig. 4B) without affecting cell viability (Fig. 4C). Notably the concentration of FGFR inhibitor required to inhibit parasite replication was higher than the concentration that reduced arg1 expression. This suggests that growth factor signaling supported parasite growth/survival through additional arg1-independent mechanisms, or that residual arginase activity at the lower inhibitor concentration is enough to support parasite growth. The latter possibility is consistent with our previous finding that >90% arg1 knockdown led to approximately 50% reduction of parasite load [2]. The FGFR inhibitor also blocked the expression of arg1 mRNA (Fig. 4D) and protein (Fig. 4E), and reduced the parasite burden (Fig. 4F) without affecting cell viability (Fig. 4G) in ex vivo cultured spleen cells from infected hamsters. Similar effects were found by inhibition of IGF-1R. In the in vitro infection model, IGF-1R inhibition reduced parasite-induced expression of host arg1 mRNA (Fig. 5A) and the intracellular parasite load (Fig. 5B), without decreasing cell viability (Fig. 5C). Similarly, the inhibitor reduced arg1 mRNA (Fig. 5D) and protein (Fig. 5E), and reduced the parasite burden (Fig. 5F) without affecting cell viability (Fig. 5G) in ex vivo cultured spleen cells from infected hamsters. The FGFR and IGF-1R inhibitors did not have a direct effect on the viability of L. donovani cultured promastigotes (Fig. S3), suggesting that the effect of receptor inhibition was through modulation of the host cell. Inhibition of JAK, which plays a key role in the phosphorylation of STAT proteins following cytokine and growth factor signaling, dramatically reduced arg1 transcription in ex vivo cultured splenocytes from infected hamsters (Fig. 5H). To a lesser degree, inhibition of the protein AKT, which is involved in signal transduction downstream of the IGF-1 and FGF receptors, also decreased Arg-1 expression (Fig. 5H). Both the AKT and JAK inhibitors significantly reduced parasite load (Figs. 5I and 5J).

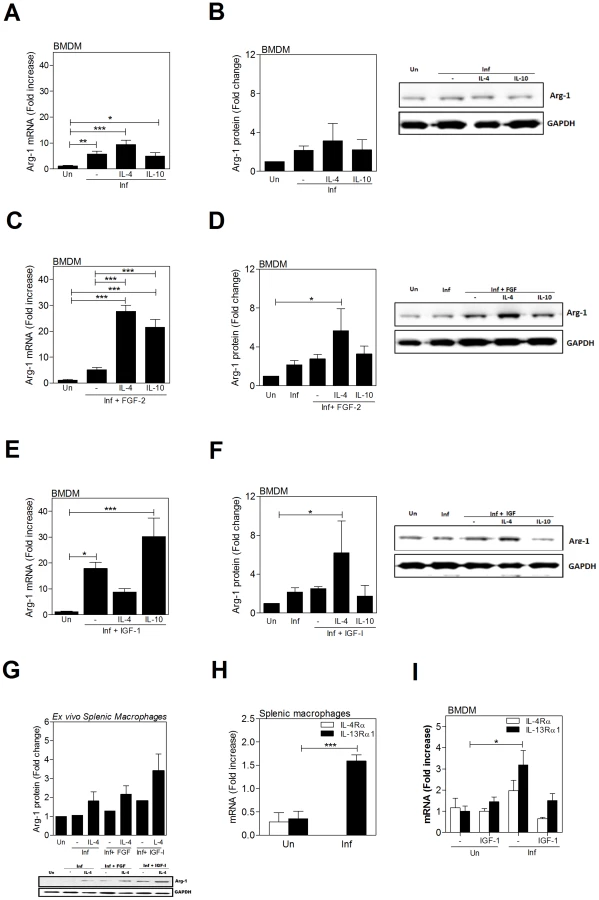
Cytokines amplify the L. donovani - and growth factor-induced expression of arginase 1
Since cytokines (IL-4 and IL-10) are known to stimulate the expression of arginase [5], [6], and we demonstrated that growth factors also induced arginase (Fig. 1), we investigated the potential for amplification of arg1 expression in macrophages by simultaneous exposure to these stimuli (all of which are expressed in the spleen during VL (reference [2] and Figs. 2 and 3). The L. donovani-induced expression of arg1 in BMDM was modestly amplified by IL-4 but not IL-10 at the mRNA level (Fig. 6A), but neither significantly amplified the arg1 protein (Fig. 6B). However, IL-4 and IL-10 dramatically enhanced the FGF-2-induced arg1 mRNA (Fig. 6C), and IL-4 (but not IL-10) enhanced FGF-2-induced arg1 protein (Fig. 6D) expression in infected macrophages. IL-4 did not amplify IGF-1-induced arg1 mRNA expression in infected BMDMs (Fig. 6E) but augmented arg1 protein expression (Fig. 6F). Similar to IL-10 and FGF-2, IL-10 enhanced IGF-1-induced arg1 mRNA but not protein expression. A trend of an additive effect of IL-4 and the growth factors was also found in splenic macrophages from infected animals exposed to the cytokine and growth factors ex vivo (Fig. 6G). The additive effect of IL-4 and growth factors in the induction of arg1 expression prompted us to consider that there may be cross-regulation of receptor expression. We found that the expression of IL-13Rα1, but not IL-4Rα, was upregulated in splenic macrophage from hamsters with VL (Fig. 6H) and in BMDMs infected with L. donovani (Fig. 6I). Addition of FGF-2 or IGF-1 to infected macrophages did not further increase the expression of either of these receptor components (data not shown and Fig. 6I). IL-10Rα expression (along with IL-10) was also increased in splenic macrophages from infected hamsters (Fig. S4A) and in in vitro infected BMDMs (Fig. S4B), but FGF-2 or IGF-1 did not augment IL-10 or IL-10Rα expression (Fig. S4B). These data, coupled with the data shown in Figs. 2 and 3, suggest that the cytokine-mediated amplification of growth factor driven arg1 could occur by either increased IL-4-mediated signaling through upregulated type II receptor (IL-13Rα1) expression [40] or through activation of signaling proteins (e.g. Jak-1, STAT6, IRS-1, PI3K, AKT) common to the two pathways.
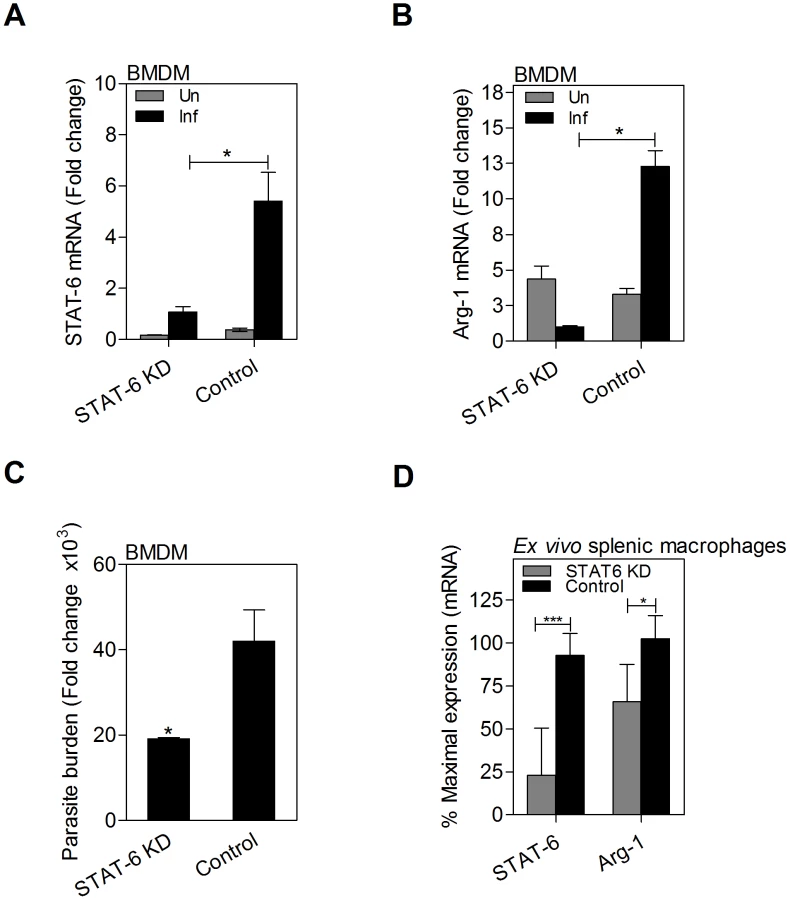
STAT6 is required for L. donovani induced expression of arg1 in macrophages
We previously demonstrated that STAT6 was required for L. donovani-induced arg1 expression in fibroblasts [2]. Here we confirmed that siRNA-mediated knockdown of STAT6 mRNA (Fig. 7A) and protein (see Fig. 8F and 8I) in in vitro infected macrophages led to reduced arg1 mRNA (Fig. 7B) expression, and improved control of parasite replication (Fig. 7C). Similarly, knockdown of STAT6 (75% reduction) in ex vivo cultured splenic macrophages from infected hamsters led to significantly reduced arg1 mRNA expression (Fig. 7D). These data confirm the critical importance of STAT6 in the parasite-driven expression of arg1 in macrophages in VL.

Growth factors activate STAT6 and increase STAT6-dependent arg1 expression
Since STAT6 had a critical role in parasite-induced arg1 transcription, activation of growth factor signaling was evident in L. donovani infection, and there was an additive effect of IL-4 and growth factors in the induction of arg1 expression, we wanted to know if the FGF-2 - and/or IGF-1-induced arg1 expression was dependent on the activation of STAT6. In a STAT6 reporter assay (hamster fibroblast cell line; reference [2]), we found that recombinant FGF-2 and IGF-1 induced STAT6 activation, which was blocked when cells were pre-treated with an inhibitor of the corresponding growth factor receptor (Figure 8A). In the fibroblast cell line, exposure to parasites had a relatively weak effect on STAT6 activation, probably because at this parasite dose the cells are infected at a very low level. The growth factor-induced activation of STAT6 in macrophages was confirmed by detection of phosphorylated STAT6 in immunoprecipitated lysates of splenic macrophages from L. donovani infected hamsters exposed ex vivo to recombinant FGF-2 or IGF-1 (Fig. 8B). Parasite-induced STAT6 activation was abrogated completely by an IGF-1R inhibitor and partially by an FGFR inhibitor (Fig. 8C). Conversely, siRNA-mediated knockdown of STAT6 mRNA in infected, FGF-2-treated BMDM (Fig. 8D) identified the requirement for STAT6 in the FGF-2-induced expression of arg1 mRNA (Fig. 8E) and protein (Fig. 8F). Similarly, siRNA-mediated knockdown of STAT6 in infected IGF-1-treated BMDM (Fig. 8G) identified the contribution of, but not absolute requirement for, STAT6 in the IGF-1-induced expression of arg1 mRNA (Fig. 8H) and protein (Fig. 8I). Collectively these data identify the critical importance of growth factor signaling in the parasite-induced activation of STAT6, and of STAT6 in the IGF-1 and FGF-2 driven expression of arg1 in L. donovani infected macrophages.
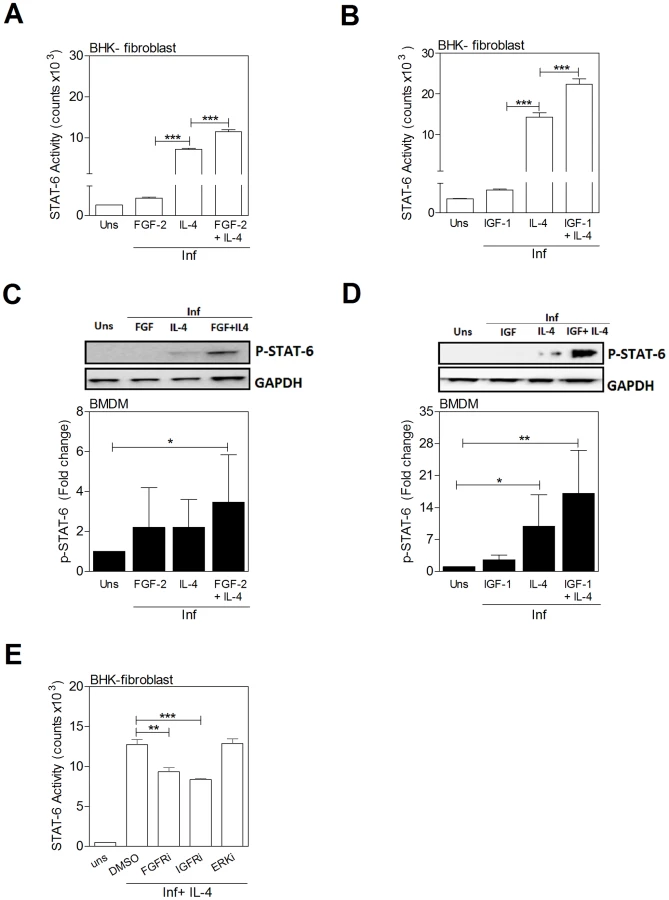
IL-4 and growth factors have an additive effect in the activation of STAT6
Since simultaneous exposure of infected macrophages to IL-4 and FGF-2 or IGF-1 led to enhanced arginase expression, and the growth factor - and cytokine-induced expression of arg1 was dependent on STAT6, we reasoned that there might be enhanced activation of STAT6 in cells exposed to both IL-4 and growth factors. Stimulation of the reporter cells with either growth factors (see also Fig. 8A) or IL-4 activated STAT6. There was evidence of an additive effect when the growth factor and cytokine were combined (Figs. 9A and 9B). By immunoblotting, STAT6 phosphorylation was amplified when IL-4 was combined with the growth factors (Figs. 9C and 9D). Inhibition of FGFR and IGF-1R activation led to decreased IL-4-induced STAT6 activation (Fig. 9E). Taken together, these data indicate that bi-directional crosstalk between the growth factor and IL-4 signaling pathways converges at STAT6 to drive arg1 expression in VL.
Discussion
In an experimental model of progressive VL, we demonstrated previously that parasitized macrophages were polarized to an M2-like phenotype [2], characteristic of macrophages at a site of chronic injury and wound healing [5], [6], and were massively expanded in the spleen [2], [38]. These macrophages had dominant expression of arg1, which promoted parasite growth. The L. donovani-induced macrophage arg1 expression did not require, but was amplified by, type 2 cytokines [2]. In this work we focused our attention on the mechanisms through which pathological arg1 expression occurs in VL. We discovered that FGF-2 and IGF-1 signaling pathways were activated in splenic macrophages from animals with progressive VL. These growth factors, which may be produced by macrophages, fibroblasts, or endothelial cells [41]–[43], induced macrophage arg1 expression. Inhibition of FGFR1 and IGF-1R signaling led to both reduced arg1 expression and improved control of intracellular L. donovani infection. Parasite-induced FGFR and IGF-1R signaling converged with the canonical type 2 cytokine signaling pathway through STAT6 activation to induce arg1 expression. Simultaneous exposure of macrophages to growth factors and IL-4, as would occur in the spleen during VL, enhanced the activation of STAT6 and expression of arg1. The interplay of STAT6 and growth factor signaling was confirmed by demonstrating that FGF-2 - and IGF-1-induced arg1 expression was abrogated by knockdown of STAT6, and conversely, that inhibition of growth factor signaling reduced parasite - and IL-4-mediated STAT6 activation and arg1 expression.
Arginase expression contributes to the pathogenesis of cutaneous L. major infection in mice [19]–[23] and progressive experimental VL caused by L. donovani [2]. Its expression in blood leukocytes was also found to be a marker of active VL in patients from Ethiopia [35]. In that study the blood leukocytes that produced arginase were found in the mononuclear cell fraction but expressed CD15 so were identified as low-density granulocytes. Those cells were not further characterized, and we have not evaluated expression of arg1 in granulocytes in our model of experimental VL. Therefore, it remains to be determined if there is a fundamental difference in the source of arg1 in experimental and human VL, or if further characterization of the cell populations will resolve the apparent difference. The disease-promoting effect of arg1 may be mediated through several mechanisms. First, arg1 metabolizes arginine such that this substrate is not available for the generation of the antimicrobial effector molecule, nitric oxide, by the action of inducible nitric oxide synthase. Second, arg1 expression leads to the production of polyamines, which promote intracellular Leishmania growth [2], [19], [20]. Lastly, local depletion of arginine leads to impaired anti-leishmanial T cell responses [44]. The relative contributions of each of these effects on the pathogenesis of VL remain to be determined.
The role of growth factors in modulation of arg1 expression and macrophage function in response to Leishmania or other pathogens has received little attention. The induction of arginase expression is classically a type 2 cytokine (IL-4/IL-13) - and STAT6-driven process [5], although some parasites or parasite products have been shown to directly induce an M2-like macrophage phenotype [2], [20], [45]. Since growth factors modulate inflammation and tissue repair [46]–[50], processes in which M2 macrophages have an integral part, it is not surprising that there would be interconnections between growth factors, type 2 cytokines, and M2 polarization. The tissue remodeling [51], [52], accumulation of macrophages [38], [52]–[55] and collagen deposition/fibrosis [38], [54] observed in the spleens in experimental and human VL are processes that suggest growth factors may contribute to VL pathology. Cytosolic IGF-1 was found increased in L. major infected murine macrophages [56], and IGF-1 induced parasite arginase in L. amazonensis infected macrophages [57]. Although we cannot exclude the potential contribution of parasite arginase in the IGF-1 and FGF-2-mediated effects on macrophages, we found previously that L. donovani arginase contributed little to the overall arginase expression at the site of infection in this model of progressive VL [2]. The increased expression of FGF-2 and evidence of signaling through the IGF-1 and FGF receptors to our knowledge had not been described previously in VL. Surprisingly, robust IGF-1R phosphorylation was evident in the infected spleen in the absence of increased IGF-1, suggesting cross-activation by FGF-2 [58] or by an unknown host or parasite-derived factor. We think cross-activation by FGF-2 is unlikely in the case of VL since we did not find IGF-1R phosphorylation in BMDMs infected with L. donovani and treated with FGF-2 for 20 minutes to 48 hours post-infection (data not shown). Of note, it was reported previously that Leishmania expressed an ortholog of FGF-2 [59] so conceivably other parasite-produced growth factor orthologs could be driving the activation of IGF-1R in the absence of host IGF-1. Insulin-like growth factor binding proteins (IGFBPs) or IGFBP proteases [60] could also be modulating the local availability and activity of IGF-1 during the infection.
From this work we have begun to understand the mechanistic basis for the interplay of L. donovani, IL-4 and growth factors in the induction of arg1. IL-4, which is increased in the spleen during VL in humans and hamsters [2], [12], amplifies the parasite - and growth factor-induced expression of arg1. IL-10 appears to have a more limited role in that it upregulates arg1 mRNA, but not protein expression, in infected macrophages, and does not amplify the growth factor effect. Similarly, in L. donovani-infected mice, IL-10 does not directly induce macrophage arginase, but contributes indirectly to its expression by upregulating the type I IL-4 receptor [61]. FGF-2 - and IGF-1 enhance expression of arg1 in L. donovani infected macrophages, but have a more modest effect on uninfected macrophages. Thus, the concomitant expression of IL-4 and growth factors in the infected spleen provide an environment highly suited for arg1 expression. IL-4 and IL-13 were shown previously to induce the expression of macrophage IGF-1 [62] and coincident expression of type 2 cytokines and IGF-1 was demonstrated in experimental helminth infection [63]. The amplification of growth factor-induced arg1 by IL-4 in experimental VL is not associated with growth factor-mediated upregulation of the type 1 IL-4 receptor (IL-4rα). Although we found that L. donovani infection increased expression of IL-13Rα1, which partners with IL-4rα to transduce a signal via IL-4 or IL-13 [40], this receptor is thought to be a less-potent driver of M2 macrophage activation than is IL-4 signaling through the type I receptor. Furthermore, IRS-1/2, which we found strongly activated in VL, is activated primarily via IL-4 signaling through the type I rather than the type II receptor [64], [65]. Collectively, these data suggest that the growth factor/IL-4-mediated amplification of arg1 expression results from an effect downstream of the IL-4 receptor. Since IL-13 [2] and its receptor are also increased in the spleen during VL, they may also contribute to the induction of arg1.
The body of work presented here supports the conclusion that the signaling pathways downstream of the growth factor and IL-4 receptors converge at STAT6 to drive pathological arg1 expression. Figure 10 illustrates our current working model for the expression of arg1 in VL. IGF-1 is known to activate STAT6 through an IRS-1/2-dependent pathway. IL-4, which is also an activator of IRS-1/2 [64], can amplify this effect [66], [67]. To our knowledge FGF-2 had not been shown previously to activate STAT6. The pathway through which the growth factors activate STAT6-dependent arg1 transcription in VL remains to be fully elucidated, but for the reasons noted above, IRS-1/2 and JAKs, which are activated in splenic macrophages during VL, are likely key intermediates (see Fig. 10). The downstream activation of other transcription factors (CREB, STAT-3) and signaling molecules, including PI3K/AKT, ERK, p38 MAPK, and GSK3β, are also likely to directly or indirectly contribute to the growth factor induced macrophage polarization and arg1 expression. Notably, p38 MAPK and downstream transcription factors (CREB and ATF-2) are only transiently upregulated so do not account for the sustained increase in arg1 expression over time (reference [2] and this work). Down-modulation of the p38 pathway, however, may contribute to the survival and local expansion of splenic arginase-expressing macrophages [68]. Leishmania infection of macrophages was shown previously to activate the PI3K/AKT pathway, which is a critical regulator of the IL-10 and IL-12 response [69]–[71]. L. donovani-induced production of IL-10 by macrophages involved activation of the PI3K/AKT pathway and downstream phosphorylation-mediated inactivation of GSK-3beta and phosphorylation of CREB [72]. Our data suggest that parasite-induced arg1 is driven at least in part through activation of the same pathway that mediates production of IL-10 by macrophages. However, arg1 and IL-10 expression appear to result from parallel rather than interdependent processes because IL-10 was not a strong inducer of arg1 and did not amplify growth factor-induced arg1 as did IL-4. Also the inhibition of the AKT pathway, which drives IL-10 production, had a less dramatic effect in the down regulation of arg1 than the inhibition of JAKs with the consequent block in STAT activation. Taken together, these data indicate that the expression of arg1 downstream of the growth factor/PI3K/AKT pathway, which is enhanced by IL-4/STAT6 signaling, is an additional mechanism of parasite-mediated subversion of macrophage effector function. Further work is needed to definitively determine the role of IL-10 and STAT3 in this process.

The pathological signaling through the IGF-1R and FGFR that leads to arginase expression in progressive VL is a potential target for adjunctive chemotherapy. Therapies targeting these pathways have recently emerged for a number of proliferative diseases, in particular hematopoietic malignancies and solid tumors (reviewed in [73], [74]). Our ex vivo data suggest that inhibition of FGFR or IGF-1R signaling could have therapeutic potential. Furthermore, it was previously demonstrated in a murine model of L. donovani infection that in vivo administration of a receptor tyrosine kinase inhibitor, when combined with conventional anti-leishmanial chemotherapy had a therapeutic effect [75]. Future pre-clinical studies of FGFR and IGF-1R inhibitors, alone or in combination with current anti-leishmanial therapies, are warranted.
In summary, we determined that the convergence of FGFR/IGFR and IL-4 signaling pathways is responsible for the expression of arg1 in disease-promoting macrophages during chronic progressive VL. FGF-2 and Th2 cytokines [2] are produced in the spleen and lead to activation of the FGFR and STAT6 in infected splenic macrophages. Although the infection does not appear to increase IGF-1 production, the IGF-1R is activated on splenic macrophages through a yet to be identified host or parasite factor. Activation of the FGF and IGF-1 receptors leads to phosphorylation of downstream signaling molecules such as IRS1/2, PI3K, and AKT, which lead to expression of IL-10 [72] and converge with downstream components of the IL-4R pathway to drive arg1 expression. Activation of these pathways, along with the parallel effects of IL-10 in subverting macrophage function [16], [76], [77], plays an important role in the pathogenesis of VL. Targeted interruption of these pathological processes offers an approach to restrain this relentlessly progressive disease.
Materials and Methods
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Texas Medical Branch, Galveston, Texas (protocol number 1101004).
Hamsters
6–8 week old Syrian golden hamsters (Mesocricetus auratus) were obtained from Harlan Laboratories.
Parasites and infection
L. donovani (MHOM/SD/001S-2D) promastigotes were cultured as described previously [78]. Hamsters were infected by intracardial injection of 106 peanut agglutinin purified metacyclic promastigotes [78]. For in vitro infections, stationary phase promastigotes were washed with PBS and used immediately to infect hamster BMDMs. Cells were infected at a promastigote to macrophage ratio of 2∶1 and cultured thereafter in complete medium (CM) composed of DMEM supplemented with 1 mM sodium pyruvate (Gibco), 1× MEM amino acids solution (Sigma), 10 mM HEPES buffer (Cellgro), and 100 IU/mL penicillin/100 mg/mL streptomycin solution (Cellgro), which was supplemented with 2% heat inactivated fetal bovine serum (HIFBS). When infecting BMDMs at this ratio all parasites were internalized so that no extracellular parasites could be observed by light microscopy at 24 hrs post-infection.
Isolation of bone marrow derived macrophages
Bone marrow cells were flushed from normal hamster femurs and adjusted to 8×106/mL in RPMI with 10% HIFBS, 50 µM β-mercaptoethanol (Sigma), and supplemented with 20 ng/mL recombinant human macrophage-colony stimulating factor (M-CSF) (R&D Systems). After 3 days of culture the medium was changed and at 6–7 days of culture the cell monolayer (>95% macrophages as determined by microscopy) was washed 3 times with PBS and detached with Trypsin/EDTA (Gibco) and cell scraping. The cells were starved of M-CSF or serum in CM with 2% HIFBS overnight before the assays.
Measurement of arginase
Arg1 expression and arginase enzymatic activity in BMDM was determined at 24 hrs or 48 hrs by real-time RT-PCR or by production of urea, respectively, as described previously [2]. For Western blot a goat anti-hamster arg1 polyclonal antibody was used [2]. The antibody used for detection of hamster arg1 did not react with L. donovani parasite lysates. The cells were left unstimulated or exposed to recombinant human Epidermal Growth Factor (EGF), mouse Insulin-like Growth factor-1 (IGF-1), human Platelet-Derived Growth Factor (PDGF) (Cell Signaling), human Fibroblast Growth Factor basic (heparin stabilized) (Sigma), 0.3–2.5% recombinant hamster IL-4 conditioned medium (equivalent to 3–25 IU/mL determined by STAT6 reporter bioassay) [2], or human IL-10 (R&D Systems) and/or infected with L. donovani promastigotes at 1∶2 macrophage∶parasite ratio. The activity of human IL-10 on hamster cells was verified using hamster BMDMs transiently transfected with a STAT3 lentiviral reporter construct (Cignal Lenti-reporter, SA Biosciences).
Quantitative RT-PCR
Real time RT-PCR for arg1 and STAT6 mRNA was performed as described [2].
Screening of Receptor Tyrosine Kinase (RTK) inhibitors
Spleen cells from 28-day L. donovani infected hamsters were cultured ex vivo as described previously [38] and treated for 24 hrs with each inhibitor from a library of 80 RTK inhibitors (Biomol International, Inc.) at twice the dose reported to cause 50% inhibition. Total RNA was isolated and the level of arg1 transcription determined by real time PCR as described [2].
Screening for activated RTKs
An RTK antibody array (PathScan Array, Cell Signaling), which contains antibodies against 28 phospho-RTKs and 11 key signaling nodes of the RTK pathways, was used to identify RTKs activated by L. donovani infection. The mean dot-spot chemiluminescent intensity of splenic macrophages (n = 4) from infected hamsters (28 days post-infection) was compared to that of 4 uninfected hamsters by densitometry analysis (GeneTools Analysis Software, Syngene).
Chemical inhibition of growth factor receptors
BMDMs were seeded in white clear bottom 96-well plates at 20,000 cells per well in CM and pre-treated with Fibroblast Growth Factor Receptor-1 inhibitor (PD166866; CAS 192705-79-6; Calbiochem) or Insulin-like Growth Factor Receptor inhibitor (PPP; CAS 477-47-4; Calbiochem). After 1–2 hrs the medium containing the inhibitor was discarded and the cells infected with L. donovani promastigotes for 20 min. Medium containing fresh inhibitors was then added back to the infected cells and the cells collected at 24 h post-infection for measurement of arg1 expression and parasite burden. Parasite load was determined by measurement of luciferase activity from luciferase-transfected parasites as described previously [38] or by real time RT-PCR using primers and a Taqman probe against the conserved sequence of the 18S gene of Leishmania [79] (forward primer: TTACCACCTTACGTA TCTTTTCTATTCG; reverse primer: AAAACAGAAAACGTGCTGAGG AT; Taqman probe: FAM-CT TTACCGGCCACCCACGGGA-TAMRA). Similar experiments were performed with adherent spleen cells cultured ex vivo from hamsters infected with L. donovani (21 days post-infection) as described [38]. The viability of treated cells was assessed in parallel experiments (20,000 cells/well/100 µL in 96-well white plates) by luminometric measurement of ATP (Cell Titer Glo Assay, Promega).
Measurement of growth factor receptor activation in hamsters infected with L. donovani
To confirm the results of the PathScan Array, we immunoprecipitated (IP) selected growth factor receptors from fresh lysates of splenic macrophages isolated from infected hamsters using cross-reacting anti-mouse/human/rat growth factor receptor antibodies (Table S1). Following cell lysis in RIPA buffer supplemented with protease/phosphatase inhibitors (Santa Cruz) the protein concentration of total cell lysates was adjusted to 3 µg/300 µL buffer and the IP procedure was followed according the manufacturer's instructions using protein A/G agarose (Santa Cruz). In brief, pre-cleared samples were incubated with the anti-receptor antibody overnight at 4°C on an Orbital shaker, then 20 µL protein A/G agarose was added to the Ag-antibody complex and incubated for 4 hr at 4°C. The protein A/G/antibody complex was precipitated by centrifugation, washed 3 times with PBS, suspended in 50 µL of 1× LDS running buffer (Invitrogen) and the antibodies released from the agarose beads by heat (100°C, 5 min). After resolving 20 µL of sample by SDS PAGE the separated proteins were transferred to nitrocellulose membranes, blocked with TBS-T 5% milk with 1 mM sodium orthovanadate (Na3VO4) and incubated overnight at 4°C with the anti-phospho RTK in TBS-T with 0.4% BSA or TBS-T 3% milk with 1 mM Na3VO4.
Measurement of growth factors in hamsters infected with L. donovani
Growth factor receptor ligands were measured in plasma or spleen homogenates from uninfected or infected hamsters. IGF-I and PDGF-β were measured by ELISA using anti-rat/mouse IGF-I and anti-rat/mouse PDGF-β using ELISA kits (R&D Systems). Epidermal Growth Factor, heparin-binding EGF-like growth factor (HB-EGF), Epiregulin and Amphiregulin were measured by immunoprecipitation/western blot using antibodies reactive against the mouse/rat/human proteins (Santa Cruz).
Identification of activated signaling proteins
Splenic macrophages isolated by adherence from infected or uninfected hamsters were lysed and suspended in RIPA buffer containing 1× protease/phosphatase inhibitors. Lysates were stored at −80°C and used within 2 months. Ten µg of total protein was suspended in 1× LDS sample buffer and separated by SDS-PAGE in pre-cast gels (NuPage, Bis-Tris 4–12%). The separated proteins were transferred to nitrocellulose membranes using the iBlot system (20 V, 9 min) (Invitrogen). Then membranes were incubated with primary antibody (Table S1) either in TBS-T with 0.4% BSA or TBS-T with 3% milk and 1 mM Na3VO4 followed by the secondary antibody conjugated to HRP. The reaction was detected with enhanced chemiluminescent substrate (West Pico; Thermo Scientific) and captured with a Chemi X T4 camera (G BOX, SynGene) and analyzed with Gene Tools analysis Software (SynGene). The fold change of protein expression was calculated by densitometry analysis of western blot bands of infected samples (at 7, 14 and 28 days post-infection) with reference to uninfected samples.
Transcriptional activation of STAT6
STAT6 activity was determined in the hamster BHK-21 cell line stably transfected with the luciferase reporter plasmid p(IE-IL4RE)4-LUC as described previously [2]. p-STAT6 was detected by immunoprecipitation of cell lysates (5×106 cells/300 µL RIPA with phosphatase inhibitors) with 1 µg of STAT6 capture antibody (M-20, Santa Cruz Biotechnology) at 4°C. overnight. Protein A/G immunoprecipitated complexes were washed 4 times with PBS, eluted by heat 5 min 100°C in 50 µl of 1× LDS loading buffer and detected by SDS-page using anti-p-STAT6 antibody (# 9361, Cell Signaling, 1∶1000 TBS-T, 0.4% BSA, 4°C, overnight), anti-rabbit HRP conjugate, and West Pico substrate (Thermo Scientific) as above.
Knockdown of STAT6 in hamster BMDMs
Stealth RNAi sequences were designed in silico using the BLOCK-iT RNAi Designer (Life Technologies) and chosen based on the sequences spanning 2 regions that were successfully targeted in knockdown of STAT6 previously [2] as follows: region 1: top, UGGCCACCAUCAGACAAAUACUUCA; bottom, UGAAGUAUUUGUCUGAUGGUGGCCA; region 2: top, CACAGUUCAACAAGGAGAUCCUGUU; bottom, AACAGGAUCUCCUUGUUGAACUGUG (each duplex synthetized and annealed by Life Technologies). Hamster BMDMs were differentiated for 6 days with 20 ng/mL of recombinant human M-CSF (R&D Systems) and plated overnight (250,000 cells per well in 24-well plates and 500 µL CM with 10% HIFBS). For transfection, 25 nM of each stealth duplex (239 and 1451) targeting hamster STAT6 was mixed in a volume of 100 µL Optimem with 0.9 µL of Lipofectamine RNAiMAX (Invitrogen) according the manufacturer's instruction. A non-targeting oligonucleotide (low GC, Invitrogen) was used as a control. Then the culture medium was discarded and 500 µL of Optimem (Invitrogen) with 10% HIFBS without antibiotics was added to the cell monolayer together with 100 µl of the transfection mix to achieve a final concentration of 8.3 nM of siRNAi oligos in 600 µL per well. The next day the transfection medium was changed for fresh Optimem with 10% HIFBS without antibiotics. At 48 hr post-transfection cells were serum starved in CM overnight, and stimulated with either L. donovani promastigotes or growth factors at 72 hr of transfection. Both STAT6 knockdown efficiency and arg1 transcription was measured 24 h later by real time RT-PCR and Western blot.
Statistical analysis
Comparison between groups was typically performed using ANOVA. A parametric or non-parametric test was selected according the distribution of the raw data, followed by a post-test analysis for multiple groups (e.g. Dunnett's Multiple Comparison Test) as appropriate. Paired t test and Wilcoxon signed rank test were used to identify differences between inhibitors and vehicle controls. All analyses were conducted using GraphPad InStat version 3.00 software for Windows 95 (GraphPad Software, San Diego California USA). P values of <0.05 were considered significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. AlvarJ, VelezID, BernC, HerreroM, DesjeuxP, et al. (2012) Leishmaniasis worldwide and global estimates of its incidence. PLoS One 7: e35671.
2. OsorioEY, ZhaoW, EspitiaC, SaldarriagaO, HawelL, et al. (2012) Progressive visceral leishmaniasis is driven by dominant parasite-induced STAT6 activation and STAT6-dependent host arginase 1 expression. PLoS Pathog 8: e1002417.
3. GreenSJ, NacyCA, MeltzerMS (1991) Cytokine-induced synthesis of nitrogen oxides in macrophages: a protective host response to Leishmania and other intracellular pathogens. J Leukoc Biol 50 : 93–103.
4. LiewFY, LiY, MossD, ParkinsonC, RogersMV, et al. (1991) Resistance to Leishmania major infection correlates with the induction of nitric oxide synthase in murine macrophages. Eur J Immunol 21 : 3009–3014.
5. GordonS, MartinezFO (2010) Alternative activation of macrophages: mechanism and functions. Immunity 32 : 593–604.
6. MartinezFO, HelmingL, GordonS (2009) Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 27 : 451–483.
7. KenneyRT, SacksDL, GamAA, MurrayHW, SundarS (1998) Splenic cytokine responses in Indian kala-azar before and after treatment. J Infect Dis 177 : 815–818.
8. KarpCL, el-SafiSH, WynnTA, SattiMM, KordofaniAM, et al. (1993) In vivo cytokine profiles in patients with kala-azar. Marked elevation of both interleukin-10 and interferon-gamma [see comments]. J Clin Invest 91 : 1644–1648.
9. GidwaniK, JonesS, KumarR, BoelaertM, SundarS (2011) Interferon-gamma release assay (modified QuantiFERON) as a potential marker of infection for Leishmania donovani, a proof of concept study. PLoS Negl Trop Dis 5: e1042.
10. SinghOP, GidwaniK, KumarR, NylenS, JonesSL, et al. (2012) Reassessment of immune correlates in human visceral leishmaniasis as defined by cytokine release in whole blood. Clin Vaccine Immunol 19 : 961–966.
11. HailuA, van BaarleD, KnolGJ, BerheN, MiedemaF, et al. (2005) T cell subset and cytokine profiles in human visceral leishmaniasis during active and asymptomatic or sub-clinical infection with Leishmania donovani. Clin Immunol 117 : 182–191.
12. NylenS, MauryaR, EidsmoL, ManandharKD, SundarS, et al. (2007) Splenic accumulation of IL-10 mRNA in T cells distinct from CD4+CD25+ (Foxp3) regulatory T cells in human visceral leishmaniasis. J Exp Med 204 : 805–817.
13. SundarS, ReedSG, SharmaS, MehrotraA, MurrayHW (1997) Circulating T helper 1 (Th1) cell - and Th2 cell-associated cytokines in Indian patients with visceral leishmaniasis. Am J Trop Med Hyg 56 : 522–525.
14. ZwingenbergerK, HarmsG, PedrosaC, OmenaS, SandkampB, et al. (1990) Determinants of the immune response in visceral leishmaniasis: evidence for predominance of endogenous interleukin 4 over interferon-gamma production. Clin Immunol Immunopathol 57 : 242–249.
15. GautamS, KumarR, MauryaR, NylenS, AnsariN, et al. (2011) IL-10 neutralization promotes parasite clearance in splenic aspirate cells from patients with visceral leishmaniasis. J Infect Dis 204 : 1134–1137.
16. OlivierM, GregoryDJ, ForgetG (2005) Subversion mechanisms by which Leishmania parasites can escape the host immune response: a signaling point of view. Clin Microbiol Rev 18 : 293–305.
17. MelbyPC, ChandrasekarB, ZhaoW, CoeJE (2001) The hamster as a model of human visceral leishmaniasis: progressive disease and impaired generation of nitric oxide in the face of a prominent Th1-like response. J Immunol 166 : 1912–1920.
18. PerezLE, ChandrasekarB, SaldarriagaOA, ZhaoW, ArteagaLT, et al. (2006) Reduced nitric oxide synthase 2 (NOS2) promoter activity in the Syrian hamster renders the animal functionally deficient in NOS2 activity and unable to control an intracellular pathogen. J Immunol 176 : 5519–5528.
19. IniestaV, Gomez-NietoLC, CorralizaI (2001) The inhibition of arginase by N(omega)-hydroxy-l-arginine controls the growth of Leishmania inside macrophages. J Exp Med 193 : 777–784.
20. StempinCC, DulgerianLR, GarridoVV, CerbanFM (2010) Arginase in parasitic infections: macrophage activation, immunosuppression, and intracellular signals. J Biomed Biotechnol 2010 : 683485.
21. IniestaV, Carlos Gomez-NietoL, MolanoI, MohedanoA, CarcelenJ, et al. (2002) Arginase I induction in macrophages, triggered by Th2-type cytokines, supports the growth of intracellular Leishmania parasites. Parasite Immunol 24 : 113–118.
22. IniestaV, CarcelenJ, MolanoI, PeixotoPM, RedondoE, et al. (2005) Arginase I induction during Leishmania major infection mediates the development of disease. Infect Immun 73 : 6085–6090.
23. KropfP, FuentesJM, FahnrichE, ArpaL, HerathS, et al. (2005) Arginase and polyamine synthesis are key factors in the regulation of experimental leishmaniasis in vivo. Faseb J 19 : 1000–1002.
24. StempinC, GiordanengoL, GeaS, CerbanF (2002) Alternative activation and increase of Trypanosoma cruzi survival in murine macrophages stimulated by cruzipain, a parasite antigen. J Leukoc Biol 72 : 727–734.
25. BabuS, KumaraswamiV, NutmanTB (2009) Alternatively activated and immunoregulatory monocytes in human filarial infections. J Infect Dis 199 : 1827–1837.
26. BenoitM, BarbaratB, BernardA, OliveD, MegeJL (2008) Coxiella burnetii, the agent of Q fever, stimulates an atypical M2 activation program in human macrophages. Eur J Immunol 38 : 1065–1070.
27. El KasmiKC, QuallsJE, PesceJT, SmithAM, ThompsonRW, et al. (2008) Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol 9 : 1399–1406.
28. RaesG, Van den BerghR, De BaetselierP, GhassabehGH, ScottonC, et al. (2005) Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J Immunol 174 : 6561 author reply 6561–6562.
29. RodriguezPC, OchoaAC (2008) Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev 222 : 180–191.
30. RodriguezNE, ChangHK, WilsonME (2004) Novel program of macrophage gene expression induced by phagocytosis of Leishmania chagasi. Infect Immun 72 : 2111–2122.
31. MattilaJT, OjoOO, Kepka-LenhartD, MarinoS, KimJH, et al. (2013) Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol 191 : 773–784.
32. PessanhaAP, MartinsRA, Mattos-GuaraldiAL, ViannaA, MoreiraLO (2012) Arginase-1 expression in granulomas of tuberculosis patients. FEMS Immunol Med Microbiol 66 : 265–268.
33. OchoaJB, BernardAC, O'BrienWE, GriffenMM, MaleyME, et al. (2001) Arginase I expression and activity in human mononuclear cells after injury. Ann Surg 233 : 393–399.
34. AbebeT, HailuA, WoldeyesM, MekonenW, BilchaK, et al. (2012) Local increase of arginase activity in lesions of patients with cutaneous leishmaniasis in ethiopia. PLoS Negl Trop Dis 6: e1684.
35. AbebeT, TakeleY, WeldegebrealT, ClokeT, ClossE, et al. (2013) Arginase activity - a marker of disease status in patients with visceral leishmaniasis in ethiopia. PLoS Negl Trop Dis 7: e2134.
36. MullerAK, MeyerM, WernerS (2012) The roles of receptor tyrosine kinases and their ligands in the wound repair process. Semin Cell Dev Biol 23 : 963–970.
37. PortaC, LarghiP, RimoldiM, TotaroMG, AllavenaP, et al. (2009) Cellular and molecular pathways linking inflammation and cancer. Immunobiology 214 : 761–777.
38. OsorioY, TraviBL, RensloAR, PenicheAG, MelbyPC (2011) Identification of small molecule lead compounds for visceral leishmaniasis using a novel ex vivo splenic explant model system. PLoS Negl Trop Dis 5: e962.
39. ZongCS, ChanJ, LevyDE, HorvathC, SadowskiHB, et al. (2000) Mechanism of STAT3 activation by insulin-like growth factor I receptor. J Biol Chem 275 : 15099–15105.
40. JiangH, HarrisMB, RothmanP (2000) IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol 105 : 1063–1070.
41. LuH, HuangD, SaederupN, CharoIF, RansohoffRM, et al. (2011) Macrophages recruited via CCR2 produce insulin-like growth factor-1 to repair acute skeletal muscle injury. FASEB J 25 : 358–369.
42. WernerS, GroseR (2003) Regulation of wound healing by growth factors and cytokines. Physiol Rev 83 : 835–870.
43. YunYR, WonJE, JeonE, LeeS, KangW, et al. (2010) Fibroblast growth factors: biology, function, and application for tissue regeneration. J Tissue Eng 2010 : 218142.
44. ModolellM, ChoiBS, RyanRO, HancockM, TitusRG, et al. (2009) Local suppression of T cell responses by arginase-induced L-arginine depletion in nonhealing leishmaniasis. PLoS Negl Trop Dis 3: e480.
45. NoelW, RaesG, Hassanzadeh GhassabehG, De BaetselierP, BeschinA (2004) Alternatively activated macrophages during parasite infections. Trends Parasitol 20 : 126–133.
46. PuzikA, RuppJ, TrogerB, GopelW, HertingE, et al. (2012) Insulin-like growth factor-I regulates the neonatal immune response in infection and maturation by suppression of IFN-gamma. Cytokine 60 : 369–376.
47. Jimenez-SousaMA, AlmansaR, de la FuenteC, Caro-PatonA, RuizL, et al. (2010) Increased Th1, Th17 and pro-fibrotic responses in hepatitis C-infected patients are down-regulated after 12 weeks of treatment with pegylated interferon plus ribavirin. Eur Cytokine Netw 21 : 84–91.
48. MlamboNC, HylanderB, BraunerA (1999) Increased levels of transforming growth factor beta 1 and basic fibroblast growth factor in patients on CAPD: a study during non-infected steady state and peritonitis. Inflammation 23 : 131–139.
49. SkevakiCL, PsarrasS, VolonakiE, PratsinisH, SpyridakiIS, et al. (2012) Rhinovirus-induced basic fibroblast growth factor release mediates airway remodeling features. Clin Transl Allergy 2 : 14.
50. TourdotS, MathieS, HussellT, EdwardsL, WangH, et al. (2008) Respiratory syncytial virus infection provokes airway remodelling in allergen-exposed mice in absence of prior allergen sensitization. Clin Exp Allergy 38 : 1016–1024.
51. EngwerdaCR, AtoM, CotterellSE, MynottTL, TschannerlA, et al. (2002) A role for tumor necrosis factor-alpha in remodeling the splenic marginal zone during Leishmania donovani infection. Am J Pathol 161 : 429–437.
52. YurdakulP, DaltonJ, BeattieL, BrownN, ErguvenS, et al. (2011) Compartment-specific remodeling of splenic micro-architecture during experimental visceral leishmaniasis. Am J Pathol 179 : 23–29.
53. KayePM, SvenssonM, AtoM, MaroofA, PolleyR, et al. (2004) The immunopathology of experimental visceral leishmaniasis. Immunol Rev 201 : 239–253.
54. VeressB, OmerA, SatirAA, El HassanAM (1977) Morphology of the spleen and lymph nodes in fatal visceral leishmaniasis. Immunology 33 : 605–610.
55. WoodruffAW, TopleyE, KnightR, DownieCG (1972) The anaemia of kala-azar. Br J Haematol 22 : 319–329.
56. ReisLC, Ramos-SanchezEM, GotoH (2013) The interactions and essential effects of intrinsic insulin-like growth factor-I on Leishmania (Leishmania) major growth within macrophages. Parasite Immunol 35 : 239–244.
57. VendrameCM, CarvalhoMD, RiosFJ, ManuliER, Petitto-AssisF, et al. (2007) Effect of insulin-like growth factor-I on Leishmania amazonensis promastigote arginase activation and reciprocal inhibition of NOS2 pathway in macrophage in vitro. Scand J Immunol 66 : 287–296.
58. YoshinouchiM, MiuraM, GaozzaE, LiSW, BasergaR (1993) Basic fibroblast growth factor stimulates DNA synthesis in cells overexpressing the insulin-like growth factor-I receptor. Mol Endocrinol 7 : 1161–1168.
59. KardamiE, PearsonTW, BeecroftRP, FandrichRR (1992) Identification of basic fibroblast growth factor-like proteins in African trypanosomes and Leishmania. Mol Biochem Parasitol 51 : 171–181.
60. BaxterRC (2000) Insulin-like growth factor (IGF)-binding proteins: interactions with IGFs and intrinsic bioactivities. Am J Physiol Endocrinol Metab 278: E967–976.
61. BiswasA, BhattacharyaA, KarS, DasPK (2011) Expression of IL-10-triggered STAT3-dependent IL-4Ralpha is required for induction of arginase 1 in visceral leishmaniasis. Eur J Immunol 41 : 992–1003.
62. WynesMW, RichesDW (2003) Induction of macrophage insulin-like growth factor-I expression by the Th2 cytokines IL-4 and IL-13. J Immunol 171 : 3550–3559.
63. ChenF, LiuZ, WuW, RozoC, BowdridgeS, et al. (2012) An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat Med 18 : 260–266.
64. MyersMGJr, GrammerTC, WangLM, SunXJ, PierceJH, et al. (1994) Insulin receptor substrate-1 mediates phosphatidylinositol 3′-kinase and p70S6k signaling during insulin, insulin-like growth factor-1, and interleukin-4 stimulation. J Biol Chem 269 : 28783–28789.
65. HellerNM, QiX, JunttilaIS, ShireyKA, VogelSN, et al. (2008) Type I IL-4Rs selectively activate IRS-2 to induce target gene expression in macrophages. Sci Signal 1: ra17.
66. KimJH, ParkHH, LeeCE (2003) IGF-1 potentiation of IL-4-induced CD23/Fc(epsilon)RII expression in human B cells. Mol Cells 15 : 307–312.
67. PatelBK, WangLM, LeeCC, TaylorWG, PierceJH, et al. (1996) Stat6 and Jak1 are common elements in platelet-derived growth factor and interleukin-4 signal transduction pathways in NIH 3T3 fibroblasts. J Biol Chem 271 : 22175–22182.
68. ZuluagaS, Alvarez-BarrientosA, Gutierrez-UzquizaA, BenitoM, NebredaAR, et al. (2007) Negative regulation of Akt activity by p38alpha MAP kinase in cardiomyocytes involves membrane localization of PP2A through interaction with caveolin-1. Cell Signal 19 : 62–74.
69. CheekatlaSS, AggarwalA, NaikS (2012) mTOR signaling pathway regulates the IL-12/IL-10 axis in Leishmania donovani infection. Med Microbiol Immunol 201 : 37–46.
70. RuhlandA, KimaPE (2009) Activation of PI3K/Akt signaling has a dominant negative effect on IL-12 production by macrophages infected with Leishmania amazonensis promastigotes. Exp Parasitol 122 : 28–36.
71. RuhlandA, LealN, KimaPE (2007) Leishmania promastigotes activate PI3K/Akt signalling to confer host cell resistance to apoptosis. Cell Microbiol 9 : 84–96.
72. NandanD, Camargo de OliveiraC, MoeenrezakhanlouA, LopezM, SilvermanJM, et al. (2012) Myeloid cell IL-10 production in response to leishmania involves inactivation of glycogen synthase kinase-3beta downstream of phosphatidylinositol-3 kinase. J Immunol 188 : 367–378.
73. ChavesJ, SaifMW (2011) IGF system in cancer: from bench to clinic. Anticancer Drugs 22 : 206–212.
74. DanieleG, CorralJ, MolifeLR, de BonoJS (2012) FGF receptor inhibitors: role in cancer therapy. Curr Oncol Rep 14 : 111–119.
75. DaltonJE, MaroofA, OwensBM, NarangP, JohnsonK, et al. (2010) Inhibition of receptor tyrosine kinases restores immunocompetence and improves immune-dependent chemotherapy against experimental leishmaniasis in mice. J Clin Invest 120 : 1204–1216.
76. NylenS, SacksD (2007) Interleukin-10 and the pathogenesis of human visceral leishmaniasis. Trends Immunol 28 : 378–384.
77. WilsonME, JeronimoSM, PearsonRD (2005) Immunopathogenesis of infection with the visceralizing Leishmania species. Microb Pathog 38 : 147–160.
78. SacksDL, MelbyPC (2001) Animal models for the analysis of immune responses to leishmaniasis. Curr Protoc Immunol Chapter 19: Unit 19 12.
79. van der MeideW, GuerraJ, SchooneG, FarenhorstM, CoelhoL, et al. (2008) Comparison between quantitative nucleic acid sequence-based amplification, real-time reverse transcriptase PCR, and real-time PCR for quantification of Leishmania parasites. J Clin Microbiol 46 : 73–78.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 6
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Fungal Nail Infections (Onychomycosis): A Never-Ending Story?
- Profilin Promotes Recruitment of Ly6C CCR2 Inflammatory Monocytes That Can Confer Resistance to Bacterial Infection
- IscR Is Essential for Type III Secretion and Virulence
- Contribution of Specific Residues of the β-Solenoid Fold to HET-s Prion Function, Amyloid Structure and Stability