
Profilin Promotes Recruitment of Ly6C CCR2 Inflammatory Monocytes That Can Confer Resistance to Bacterial Infection
Toxoplasma gondii is an apicomplexan parasite that can infect all warm blooded animals, but rodent species are considered the primary reservoirs. Mice that are infected with T. gondii become more resistant to lethal infection with other pathogens. Ly6C+ inflammatory monocytes are innate immune cells that are critical for defense against T. gondii and other infections. Mice with defects in the ability to recruit inflammatory monocytes fail to control T. gondii replication and succumb to overwhelming inflammation. In this study we used a co-infection model to explain why T. gondii-infected mice are more resistant to the bacterium Listeria monocytogenes. We show that stimulation of the rodent specific Toll-like receptor TLR11 by the T. gondii ligand profilin can recruit inflammatory monocytes, and that these monocytes can protect the host against L. monocytogenes. These findings make profilin an important tool for the study of monocyte biology during T. gondii infection of rodents and are especially interesting given that TLR11 is nonfunctional in humans and other vertebrates.
Published in the journal:
. PLoS Pathog 10(6): e32767. doi:10.1371/journal.ppat.1004203
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004203
Summary
Toxoplasma gondii is an apicomplexan parasite that can infect all warm blooded animals, but rodent species are considered the primary reservoirs. Mice that are infected with T. gondii become more resistant to lethal infection with other pathogens. Ly6C+ inflammatory monocytes are innate immune cells that are critical for defense against T. gondii and other infections. Mice with defects in the ability to recruit inflammatory monocytes fail to control T. gondii replication and succumb to overwhelming inflammation. In this study we used a co-infection model to explain why T. gondii-infected mice are more resistant to the bacterium Listeria monocytogenes. We show that stimulation of the rodent specific Toll-like receptor TLR11 by the T. gondii ligand profilin can recruit inflammatory monocytes, and that these monocytes can protect the host against L. monocytogenes. These findings make profilin an important tool for the study of monocyte biology during T. gondii infection of rodents and are especially interesting given that TLR11 is nonfunctional in humans and other vertebrates.
Introduction
Toxoplasma gondii is an obligate intracellular Apicomplexan parasite that can infect nearly any nucleated cell of all warm blooded animals. Within warm blooded hosts, T. gondii replicates as a fast growing tachyzoite form, which disseminates throughout the body during acute infection. Over time and under immune pressure, the parasite differentiates into an encysted bradyzoite form within the central nervous system and muscle tissue, which establishes a life-long chronic infection. Approximately 30% of humans are infected with T. gondii but the infection may be asymptomatic in immunocompetent hosts.
T. gondii infection is characterized by a highly polarized Th1 type immune response associated with production of IL-12 by dendritic cells (DCs), neutrophils, and macrophages which drives T and NK cell production of IFN-γ, long regarded as the main mediator of acute and chronic defenses against the parasite [1], [2], [3]. One of the T. gondii proteins known to stimulate IL-12 production is T. gondii profilin (TgPRF), which is required for parasite actin remodeling during host cell invasion and egress, and is also a ligand for TLR11 and TLR12 [4], [5], [6], [7]. Another critical factor for innate defenses are a class of Gr-1+ Ly6C+ monocytes that produce nitric oxide (NO) and TNF-α, and are recruited in a CCR2 dependent manner in response to both oral and parenteral T. gondii infections [8], [9], [10], [11]. MCP-1−/− and CCR2−/− mice do not recruit Ly6C+ monocytes to the lamina propria in response to oral infection, leading to a higher influx of neutrophils and death from intestinal necrosis and inflammation [8], [9]. Similarly, MCP-1−/− and CCR2−/− mice fail to recruit inflammatory monocytes to the peritoneal cavity following i.p. inoculation leading to increased mortality and parasite burdens [10]. Thus, Ly6C+ monocytes are necessary for early control of T. gondii replication and to prevent immune pathology. However, the specific parasite factors that elicit Ly6C+ monocytes during T. gondii infection have not been identified. Ly6Chi monocytes are also recruited during infections with other protozoan and bacterial pathogens, including Listeria monocytogenes [12], [13], [14], [15], [16].
T. gondii sexual reproduction occurs exclusively in the intestines of the feline definitive hosts, making the rodents they prey on key intermediate hosts in the T. gondii lifecycle. T. gondii infection has been shown to alter rodent aversion to cat urine and fear avoidance behaviors in ways that increase the odds of predation and thus parasite reproductive success [17], [18]. Previous studies have also reported that mice infected with T. gondii are more resistant to secondary infections with unrelated pathogens, including L. monocytogenes, Salmonella typhimurium, mengo virus, Cryptococcus neoformans, Besnoita jejuni, Moloney leukemia virus and Schistosoma monsoni [19], [20], [21], [22], [23], [24], which may also serve to increase predation. We have recently shown that stimulation with soluble T. gondii antigens (STAg) reduced viral titers and conferred a survival advantage in mice infected with highly pathogenic H5N1 avian influenza virus [25], demonstrating that treatment with STAg can stimulate immunity against unrelated pathogens. In order to further investigate the mechanisms conferring this immunological benefit, we used a highly tractable L. monocytogenes infection model.
L. monocytogenes is a Gram positive facultative intracellular bacteria commonly associated with outbreaks of the foodborne illness listeriosis. In mice, intravenous inoculation with L. monocytogenes causes highly predictable infection, involving both innate and adaptive immune responses that ultimately clear the bacteria [26], [27]. Before the onset of adaptive immunity, bacteria replicate primarily in infectious foci within cells of the spleen and liver where innate immune responses are critical for controlling early bacterial growth to prevent dissemination and lethal systemic infection. Increased early bacterial burdens in the spleen and liver correlate with the severity and outcome of infection.
Ly6Chi CCR2+ inflammatory monocytes mediate critical innate control of early bacterial replication. During L. monocytogenes infection, Ly6Chi CCR2+ cells emigrate from the bone marrow in a CCR2-dependent manor, and traffic to sites of bacterial infection to differentiate into CD11C+ TNF-α and inducible nitric oxide synthase (iNOS) producing DCs (TipDCs) that enhance bacterial clearance [12], [15], [28]. Emigration of Ly6Chi CCR2+ cells from the bone marrow is directed by MCP-1 and MCP-3, which is mainly produced by non-hematopoietic cells during infection and can be produced by bone marrow mesenchymal stem cells (BMSCs) in response to circulating TLR ligands [12], [16], [29], [30]. Accordingly, CCR2−/− mice have reduced numbers of circulating Ly6Chi monocytes, reduced numbers of TipDCs in the spleen and liver, reduced TNF-α production and are more susceptible to L. monocytogenes infection [12], [15], [16], [28]. IFN-γ and TNF-α are essential to the innate response as mice lacking either cytokine rapidly succumb to L. monocytogenes infection [31], [32], [33].
In this study we show that chronic T. gondii infection or stimulation with STAg provides resistance against L. monocytogenes bacterial infection by reducing bacterial burdens in the major sites of bacterial replication, the spleen and liver. We also show that stimulation with the TgPRF is sufficient to induce this resistance independent of IL-12, T and NK1.1+ cells but cannot completely overcome the requirement for IFN-γ mediated defenses. Most importantly, we show that TgPRF induces production of MCP-1, which results in the trafficking of Ly6Chi CCR2+ inflammatory monocytes into the blood and spleen, and that CCR2-dependent recruitment of these cells is essential to the TgPRF-induced anti-bacterial response. These results demonstrate that stimulation of TLR11 by TgPRF is sufficient to promote recruitment of Ly6Chi CCR2+ inflammatory monocytes, and that these monocytes can provide and immunological benefit against other infections.
Results
Chronic infection with T. gondii confers resistance against L. monocytogenes infection
Previous research has shown that mice with a chronic T. gondii infection had greater survival or delayed time to death when challenged with a lethal L. monocytogenes infection [19]. Further experiments showed that this protective effect was not transferrable in the serum, and thus was likely a cell mediated response [34]. Although the specific bacterial burdens were not determined for the animals in these studies, early innate control of L. monocytogenes replication correlates well with severity of infection in mice: animals that maintain low bacterial numbers generally go on to clear the infection, whereas failure of innate immunity is associated with high numbers of bacteria, overwhelming sepsis and inevitable death. We hypothesized that the enhanced survival of T. gondii infected mice was due to innate control of L. monocytogenes replication.
To test this hypothesis, we infected naïve and T. gondii chronically infected mice with a lethal inoculum of L. monocytogenes, and then we determined the number of viable bacteria in the spleens and livers 72 hours later. T. gondii-infected mice had significant ∼3.6 log reductions in bacterial burdens in the spleen ∼4.5 log reductions in the liver compared to uninfected controls (Fig. 1A). In our experience, mice with bacterial burden less than 6 log10 CFU/g in the spleen and liver at 72 hours post infection typically remain asymptomatic and survive L. monocytogenes infection; whereas those with higher bacterial burdens usually succumb to infection. As the bacterial burdens in T. gondii infected mice were consistently less than 6 log10 CFU/g in both organs (Fig. 1A), these results suggest that survival of T. gondii infected mice reported previously [19] was due to early reductions in the numbers or replication of L. monocytogenes bacteria.

Stimulation with STAg reduces bacterial burden following L. monocytogenes infection
Our previous work with influenza virus [25] had shown that the protective effects of T. gondii infection could be replicated by treating mice with STAg, a non-infectious lysate of soluble antigens from sonicated T. gondii tachyzoites. STAg contains many T. gondii proteins, including profilin [5], and previous work has shown that STAg can stimulate immune responses similar to those induced by live parasites, including induction of IL-12, TNF-α, IFN-γ, IL-1β, IL-10 and MCP-1 in vivo or in vitro [35], [36], [37]. Consistent with these data, we observed increased levels of IL-12, TNF-α, IFN-γ and MCP-1 in the serum of STAg-stimulated mice within 24 hours (data not shown). We hypothesized that STAg treatment would reduce the bacterial burdens of L. monocytogenes infected mice as well as chronic T. gondii infection. Mice treated with 1 µl of STAg (approximately 1 µg total protein) 24 hours prior to infection with L. monocytogenes had ∼2.5 log reductions in bacterial burden in the spleens and ∼3.8 log reductions in the liver compared to PBS-treated controls (Fig. 1B). These effects were similar to the reduction in bacterial burdens we observed in T. gondii infected mice (Fig. 1A). STAg stimulated mice also experienced significantly less weight loss than PBS treated controls at 72 hours post infection (Fig. 1B). STAg stimulation was effective for reducing bacterial burdens and weight loss when given 2 or 6 hours post L. monocytogenes infection, although the reduction in bacterial burdens began to decline at 6 hours (data not shown).
To determine if the protective components in STAg were protein or other molecules such as RNA or DNA, we subjected STAg to proteinase K digestion. Proteinase K-digested STAg did not reduce bacterial burdens in L. monocytogenes infected mice (Fig. S1A), which suggested that the protective component(s) were protein. To identify the specific protein(s), we subjected STAg to ammonium sulfate (AS) precipitation and assayed the fractions for their ability to reduce the bacterial burdens. The AS precipitation fraction containing the proteins that remained soluble at AS concentrations >60% reduced the bacterial burdens similar to STAg (Fig. S1B). When we subjected these fractions to western blotting with antibodies against several T. gondii proteins, we saw TgPRF was present in the AS >60% fraction (Fig. S1C).
Recombinant TgPRF is sufficient to reduce bacterial burden and enhance survival following L. monocytogenes infection
TgPRF is an actin-binding protein involved in parasite gliding motility, host cell invasion and egress, and is known for inducing IL-12 production through stimulation of TLR11 and TLR12 expressed on DCs and macrophages [4], [5], [6]. In order to determine if TgPRF was sufficient to confer protection against L. monocytogenes we stimulated mice with purified recombinant N-terminal his-tagged TgPRF (rPRF) (Fig. 2A). Mice stimulated with 100 ng rPRF 4 hours prior to L. monocytogenes infection had a significant ∼3.4 log reduction in bacterial burdens in the spleen and ∼4 log reduction in the liver compared to PBS-treated animals (Fig. 2A). rPRF-treated mice did not exhibit weight loss in contrast to PBS-treated controls which lost 17% of their starting weight by 72 hours post infection (Fig. 2A).
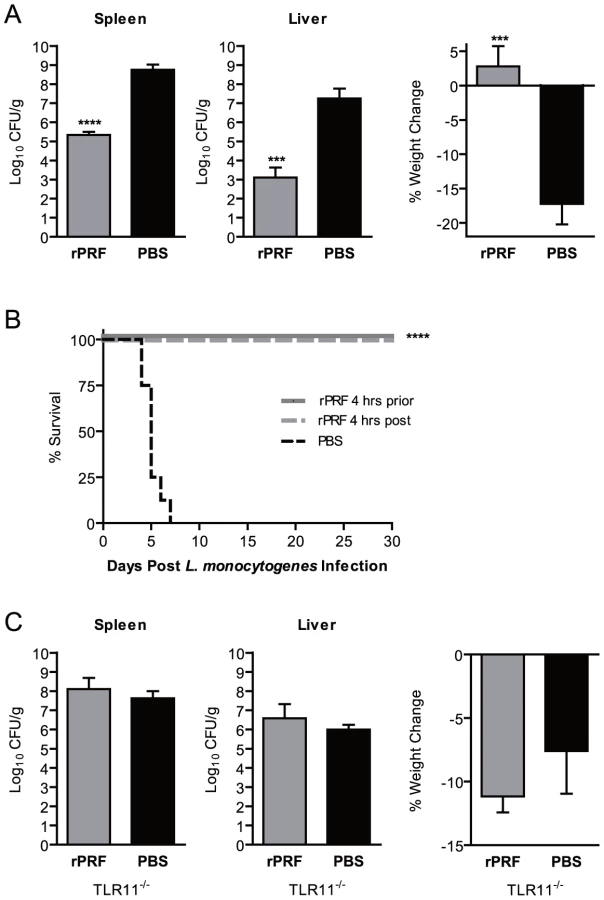
Because stimulation with rPRF was sufficient to reduce bacterial burdens similar to T. gondii infection (Fig. 1A), we expected rPRF to enhance survival of L. monocytogenes infected mice in our model (Fig. 2B). All (8/8) PBS-treated mice rapidly succumbed to L. monocytogenes infection within 7 days, with the majority of mice succumbing by day 5. In contrast, 100% (8/8 for each group) of mice stimulated with rPRF 4 hours prior to, or 4 hours after, L. monocytogenes infection survived for 30 days, at which point the experiment was terminated. These results demonstrate that rPRF-stimulation is sufficient to reduce bacterial burdens and confer a long-term survival advantage during L. monocytogenes infection.
Although TgPRF can be recognized by TLR11 and TLR12 [5], [6], [7], the ability of rPRF to reduce bacterial burdens was strictly dependent on TLR11. In multiple experiments, TLR11-deficient (TLR11−/−) mice treated with 40-fold more protein (4 µg rPRF) 4 hours prior to L. monocytogenes infection had no reduction in bacterial burden in either the spleen or liver compared to PBS-stimulated controls (Fig. 2C). rPRF-stimulated TLR11−/− mice also showed equivalent weight loss as the PBS controls (Fig. 2C). These results demonstrate that the effects of rPRF are dependent on recognition by TLR11 and that potential contaminants such as LPS do not contribute to the effect.
To determine whether TgPRF was the major T. gondii factor in STAg responsible for the resistance to L. monocytogenes, we stimulated TLR11−/− mice with STAg. Doses of STAg up to 200 µl did not result in significant reductions in bacterial burdens or reduced weight loss during L. monocytogenes infection (Fig. S2 and data not shown). We did observe modest but statistically significant reductions in bacterial burdens in the spleen (∼20-fold) and liver (∼80-fold) with 200 µl of STAg generated from twice the normal number of parasites, or 400 µg of protein (Fig. S2). These results were in contrast to WT mice, in which 1 µg of STAg reduced bacterial burdens by up 300-fold in the spleen and 6,000-fold in the liver (Fig. 1B). It is possible that the TLR11 independent effects of STAg could be due to parasite derived TLR ligands such as nucleic acids and GPI moieties, or other parasite derived proteins. However, it is unlikely that such large doses of STAg, equivalent to material from 1.6×108 lysed parasites, are relevant during natural infection and thus TgPRF is likely to be the main factor in STAg responsible for the resistance to L. monocytogenes.
TgPRF induces production of IL-12, IFN-γ, MCP-1 and TNF-α
STAg has been shown to induce cytokines and chemokines including IL-12, TNF-α, IFN-γ, IL-1β, IL-10 and MCP-1 [35], [36], [37]. TgPRF has been shown to induce IL-12 by classes of DCs and macrophages, IFN-α by CD11c+ spleenocytes, and to promote IFN-γ production by NK1.1+ cells [6]. To determine if TgPRF could induce production of other anti-listerial cytokines, we stimulated mice with rPRF then analyzed serum 2 or 24 hours later. rPRF stimulation induced significant production of IL-12 and MCP-1 at 2 and 24 hours, and IFN-γ and TNF-α by 24 hours (Fig. 3). These results show that TgPRF can stimulate the production of multiple cytokines and chemokines in addition to IL-12.

Signaling through IL-12Rβ1 is not required for TgPRF-induction reduction in bacterial burden
IL-12 mediates defenses against T. gondii by inducing IFN-γ production from NK and T cells, which in turn helps to activate macrophage effector functions, enhancing antigen presentation, and by promoting the differentiation of Th1 cells [2]. IL-12 plays a similar and critical role in L. monocytogenes infections [26], [27]. We hypothesized that the ability of rPRF to reduce the bacterial burdens would require IL-12 signaling. However, we determined that IL-12 signaling was not required for rPRF-induced resistance to L. monocytogenes infection using IL-12Rβ1 deficient (IL-12Rβ1−/−) mice (Fig. 4A). Compared to PBS-treated controls, rPRF-treated IL-12Rβ1−/− mice had significant ∼2.6 log and ∼2.8 log reductions in bacterial burdens in the spleen and livers, respectively. rPRF-treated IL-12Rβ1−/− mice exhibited only mild weight loss of 1.5%, in contrast to PBS-treated controls which lost 14% of their weight. IL-12Rβ1 is also a component of the IL-23 receptor, so these results indicate that both IL-12 and IL-23 signaling are not required for rPRF-induced resistance to L. monocytogenes infection.

IFN-γ but not T and NK1.1+ cells are required for TgPRF induced protection
IFN-γ is a critical mediator of innate defenses against both L. monocytogenes [31], [32] and T. gondii [1], [3]. Our previous work with STAg and influenza virus found that STAg-induced IFN-γ from NK cells was required to mediate protection against influenza virus [25]. To determine the role of IFN-γ mediated defenses in rPRF-induced protection against L. monocytogenes we treated IFN-γ deficient (IFN-γ−/−) mice with rPRF (Fig. 4B) then infected them with a low but lethal dose of L. monocytogenes, 200 CFU/animal, to account for the extreme susceptibility imposed by IFN-γ deficiency [31], [32]. rPRF-treated IFN-γ−/− mice had a slight but statistically significant 6-fold reduction in bacterial burden in the spleen and 10-fold reduction in the liver compared to PBS-treated controls. Although the bacterial burdens were still high, rPRF-treated IFN-γ−/− mice experienced less weight loss than PBS-treated controls. While all rPRF-treated IFN-γ−/− mice did succumb to L. monocytogenes infection within eight days, the delay was significant relative to PBS-stimulated animals (Fig. 4C). These results suggest that IFN-γ is at least partially required for rPRF-induced protection against L. monocytogenes and that rPRF stimulation cannot overcome the requirement for IFN-γ mediated defenses even at the low infectious doses used.
The major sources of IFN-γ are T cells and NK cells. NK1.1+ cells are the critical source of IFN-γ for early defense against T. gondii [38] and for STAg-induced protection against influenza virus [25]. Similarly, the majority of IFN-γ during early L. monocytogenes infection is produced by NK1.1+ cells [39], [40]. However, T cells can also produce IFN-γ in early responses to T. gondii [41] and L. monocytogenes infection [40], [42]. To determine if either NK1.1+ or T cells were required for rPRF-induced for protection against L. monocytogenes, we created mice deficient in both T and NK cells by depleting Rag1 deficient (Rag1−/−) mice with PK136 (anti-NK1.1) monoclonal antibody. In contrast to IFN-γ−/− mice, Rag1−/− NK1.1-depleted mice had no increase in susceptibility to L. monocytogenes infection and rPRF-stimulation was highly effective in Rag1−/− NK1.1-depleted mice infected with 6×104 CFU/animal, the same dose used for experiments with WT animals (Fig. 4D). rPRF-treated mice had a ∼3.7 log reduction in bacterial burden in the spleen and ∼1.8 log reduction in the liver, compared to PBS-treated controls. rPRF-treated Rag1−/− NK1.1-depleted mice also did not show weight loss (Fig. 4D). Similar results were observed with rPRF treatment in singly deficient Rag1−/− or wild-type NK1.1-depleted mice (data not shown). These data suggest that neither T nor NK cells are required for rPRF-induced reduction in bacterial burdens and survival. However, in the absence of T and NK cells, mice may develop compensatory defense mechanisms, so it is possible the factors required in these animals are different than in WT mice.
Ly6Chi CCR2+ inflammatory monocytes and Ly6Cint Ly6G+ neutrophils are recruited in response to TgPRF
During L. monocytogenes infection, MCP-1 and MCP-3 signals promote emigration of TipDC precursors, Ly6Chi inflammatory monocytes, out of the bone marrow and into circulation in a CCR2-dependent manner [16]. Because serum levels of MCP-1 in rPRF-stimulated mice were significantly increased within 2 hours (Fig. 3) and because T. gondii infection is also known to elicit a population of Ly6C+ monocytes via CCR2 [8], [9], [10], we examined the ability of rPRF to promote emigration of Ly6Chi monocytes. Within four hours after rPRF stimulation, there was an ∼3 fold average increase in the frequency of CD11b+ Ly6Chi monocytes in both the blood and spleens of TgPRF stimulated animals (Fig. 5A and B).The Ly6Chi monocyte population expressed CCR2 (data not shown), consistent with an inflammatory monocyte and TipDC precursor populations described previously [9], [10], [11], [12], [13], [14], [15]. There was also an ∼2.7 fold average increase in the frequency of neutrophils (CD11b+ Ly6Cint Ly6G+) in the blood and a ∼2.5 fold average increase in the spleens of rPRF stimulated mice (Fig. 5A and B). To confirm that these results were specifically attributable to TgPRF, we measured monocyte and neutrophil recruitment in TLR11−/− mice. As expected, there was not an increase the percentage of Ly6Chi monocytes or neutrophils in TLR11−/− mice stimulated with 100 ng rPRF compared to PBS stimulated controls (Fig. S3), demonstrating the specificity of the TgPRF-TLR11 interaction in monocyte and neutrophil recruitment.

Ly6Chi CCR2+ inflammatory monocytes are required for TgPRF-induced defenses
Ly6Chi CCR2+ monocyte emigration from the bone marrow into circulation is CCR2-dependent [9], [12]. To determine if Ly6Chi CCR2+ cells recruited in response to rPRF were essential for the reductions in bacterial burdens, we rPRF-stimulated CCR2 deficient (CCR2−/−) mice (Fig. 6A). rPRF-stimulated CCR2−/− mice did not have large reductions in bacterial burdens compared to PBS-treated controls, 2-fold in the spleen and 10-fold in the liver. Although the reductions were statistically significant, they are not likely biologically relevant given the overall high burdens. In addition, both groups experienced equal weight loss.

Although CCR2−/− mice have diminished levels of circulating Ly6Chi monocytes, they have increased numbers in the bone marrow at rest, and large numbers of activated TNF-α producing Ly6Chi monocytes accumulate in the bone marrow during infection [12]. Thus, the small reduction in bacterial burden we saw in rPRF-stimulated CCR2−/− mice could still be dependent on Ly6Chi CCR2+ monocytes, either by activation of a limited number of cells in circulation, or via soluble cytokines such as TNF-α produced by those cells restricted to the bone marrow. To deplete Ly6Chi CCR2+ monocytes, we treated mice with the anti-Gr-1 MAb RB6-8C5, which recognizes a common epitope shared by Ly6C and Ly6G [43]. Depletion with MAb RB6-8C5 reduced neutrophils in the spleens of rPRF stimulated mice by ∼95% and inflammatory monocytes by ∼85% (data not shown). We consistently observed that rPRF-stimulation did not offer any protection in RB6-8C5 depleted mice. There were no significant difference in bacterial burdens between rPRF - and PBS-stimulated mice in either the spleens or livers (Fig. 6B), and both groups experienced equal weight loss (Fig. 6B). Because TLR11 and TLR12 are expressed on macrophages and DCs [7], which may express Ly6C and thus would be depleted by RB6-8C5, we tested the ability of RB6-8C5 depleted mice to respond to profilin by measuring serum cytokine levels 2 hours post rPRF stimulation. rPRF-stimulated RB6-8C5 depleted mice produced significant amounts of IL-12 and MCP-1 (Fig. S4) at levels similar to WT mice at the same timepoint (Fig. 3). rPRF-stimulated RB6-8C5 depleted mice also produced significant levels of TNF-α (Fig. S4).This suggests that the cell population required for recognition of profilin and production of MCP-1 is not subject to depletion by RB6-8C5 MAb.
Because RB6-8C5 significantly depletes Ly6G+ neutrophils as well as Ly6Chi monocytes, we also depleted mice with the Ly6G specific MAb 1A8 [44] to establish the relative contribution of Ly6G+ cells. In contrast to CCR2−/− and RB6-8C5-depleted mice, 1A8-depleted rPRF-stimulated animals were consistently protected against L. monocytogenes infection (Fig. 6C). rPRF-stimulation reduced bacterial burdens in the spleens of 1A8-depleted mice by ∼3 logs and in the livers by ∼2.3 logs, although bacterial burdens in the livers of all 1A8 depleted mice were highly variable. This observation along with the fact that rPRF stimulated CCR2−/− mice had a 10-fold reduction in liver bacterial burdens may indicate that neutrophils play a minor role in defense in this organ. rPRF-stimulated 1A8-depleted mice also did not show weight loss in contrast to PBS-stimulated controls which lost significantly more weight (Fig. 6C).
Together, these results indicate that although rPRF stimulates a large influx of Ly6Cint LyG+ neutrophils into the blood and spleen, these cells are largely dispensable for rPRF induced protection and reduction of bacterial burden in the spleen and liver. While Ly6G+ neutrophils may have a small contribution in the liver following rPRF-treatment, CCR2-dependent recruitment of Ly6Chi CCR2+ inflammatory monocytes plays the central and essential role in rPRF-induced clearance of L. monocytogenes.
Discussion
In this study we investigated how chronic infection with T. gondii protects the rodent host against unrelated pathogens [19], [20], [21], [22], [23], [24], [25]. Because rodents are the primary reservoir for T. gondii, elucidating the key ligand/receptor interactions is essential for understanding host defense. Our work identifies TgPRF as a T. gondii factor that recruits inflammatory monocytes and demonstrates that stimulation of TLR11 or TLR11/TLR12 heterodimers provides an immunological benefit to a T. gondii-infected host against another pathogen. Stimulation with TgPRF results in production of MCP-1 and recruitment of Ly6Chi CCR2+ inflammatory monocytes and Ly6G+ neutrophils into the blood and spleen, although only Ly6Chi CCR2+ inflammatory monocytes and CCR2-signaling are essential to reduce bacterial burdens. These data have significant implications for our understanding of the biology of T. gondii infection and the evolutionary maintenance of TLR11 in rodents.
Ly6Chi CCR2+ inflammatory monocytes were first identified in L. monocytogenes infection, where they differentiate into TipDCs at the sites of bacterial infection and are essential for early control of bacterial replication. Emigration of these cells out of the bone marrow is directed by the chemokines MCP-1 and MCP-3 and their receptor CCR2 [12], [16]. Accordingly, CCR2 mice have diminished numbers of TipDCs in the spleen and are highly susceptible to L. monocytogenes infection [15]. Ly6Chi monocytes have also been implicated in defense against many other pathogens, including T. gondii. In both oral and parenteral T. gondii inoculation, Gr-1+ Ly6C+ monocytes are recruited to sites of infection and are critical for acute survival [8], [9], [10], [11]. These cells have been shown to produce TNF-α and iNOS, and their emigration is dependent on MCP-1 and CCR2 consistent with inflammatory monocytes or TipDC precursor populations, although interestingly they do not appear to acquire CD11c [8], [9], [10], [11]. MCP-1−/− and CCR2−/− mice, which fail to recruit inflammatory monocytes, have enhanced mortality, greater parasite burdens, and die of pathological inflammation and intestinal necrosis [8], [9], [10], [11]. These studies show that Ly6C+ monocytes are essential for early control of T. gondii replication and to prevent immune pathology. However, the parasite factors that elicit Ly6C+ monocytes had not been identified. Here we identify TgPRF as a mechanism by which T. gondii can elicit emigration of a Ly6Chi CCR2+ inflammatory monocyte population and show that these cells are required for TgPRF to confer resistance to L. monocytogenes infection. In this study stimulation by TgPRF was associated with production of the CCR2 ligand MCP-1 but we did not examine production of other notable CCR2 ligands such as MCP-3. Presumably MCP-3 is also involved in CCR2 dependent inflammatory monocyte recruitment during T. gondii infection as mortality and defects in monocyte recruitment and are less severe in MCP-1−/− than CCR2−/− mice [10], although no specific studies have addressed the role of this chemokine. We also did not determine if TgPRF recruited monocytes acquire CD11c or differentiate into TipDCs during the context of L. monocytogenes infection.
Stimulation with TgPRF also results in trafficking of Ly6Cint Ly6G+ neutrophils into the blood and spleen. Early work suggested that neutrophils were the major cells responsible for controlling the early growth and dissemination of L. monocytogenes [45], [46]. These observations were based mainly on studies using an anti-granulocyte receptor-1 (Gr-1) MAb, which is now known to recognize both neutrophils (Ly6Cint Ly6G+) and non-neutrophil Ly6C+ cells, including subsets of monocytes, macrophages, DCs and lymphocytes [43]. Recent work has suggested that Ly6G+ neutrophils are largely dispensable for innate defenses [47] while others have shown that these cells contribute to significant anti-listerial defenses in the liver [48], [49]. Consistent with these findings, we observed that 1A8 depleted mice are slightly more susceptible to L. monocytogenes than WT mice (lethal dose 1×104 versus 6×104 CFU), although not as susceptible as CCR2−/− (8×103 CFU) or RB6-8C5 depleted animals (200 CFU). The fact that rPRF-stimulated 1A8 depleted mice are resistant to L. monocytogenes infection demonstrates that rPRF-recruited Ly6G+ neutrophils are dispensable for TgPRF-induced protection. Rather, Ly6Chi CCR2+ inflammatory monocytes and TipDCs play the predominant role in TgPRF-mediated defenses.
There are several mechanisms by which TgPRF recruited monocytes may contribute to early control of L. monocytogenes and that could also account for the requirement for IFN-γ. First, inflammatory monocytes may be directly bactericidal. Inflammatory monocytes recruited to the peritoneal cavity during T. gondii infection express iNOS and have enhanced parasite killing in vitro [11], so it is reasonable to infer that TgPRF recruited monocytes would display enhanced activity against L. monocytogenes as well. However, rPRF treatment effectively reduced bacterial burdens in L. monocytogenes infected iNOS deficient mice (data not shown) suggesting that NO production is unlikely to be a primary mechanism of killing. The impaired protection we observed in IFN-γ−/− mice could be due to generalized defects in antimicrobial effector mechanisms dependent on IFN-γ that stimulation with TgPRF cannot overcome or because the development of Ly6Chi inflammatory monocytes into TipDCs and inflammatory DCs during L. monocytogenes and T. gondii infections is largely dependent on NK1.1+ cell derived IFN-γ [50], [51].
Noncognate antigen driven proliferation and activation of memory T cells and innate NK cells could also mediate a degree of resistance dependent on IFN-γ and explain the IFN-γ dependence of TgPRF induced protection. Memory T cells can proliferate, produce IFN-γ and acquire effector cell functions during bacterial infection, which contributes to IFN-γ mediated defenses [52], [53], [54]. Activation is driven by IL-15 and IL-18 production by inflammatory monocytes and CD8α+ DCs, dependent on inflammasome activation, type I interferon and TLR priming [53], [54]. TgPRF could contribute to induction of noncognate memory T cell responses by increases in the number of inflammatory monocytes or serving as the TLR-based priming signal via stimulation of TLR11. Activation of transferred IFN-γ sufficient memory T cells mediated a ∼100-fold reduction in L. monocytogenes bacterial burden in IFN-γ-/ - mice, but only modest 3-fold reduction in mice with intact IFN-γ responses [52], [53], [54]. Thus, it is unclear if the 2,500 - to 30,000-fold reductions we describe in T. gondii infected or rPRF stimulated IFN-γ sufficient mice can be entirely attributed to cognate antigen independent induction of IFN-γ by memory T cells. Inflammatory monocytes can also induce IFN-γ production by NK cells [53]. Along these lines, TgPRF has been shown to stimulate IFN-γ production by NK1.1+ cells [6] and NK1.1+ derived IFN-γ is required for T. gondii induced protection against influenza [25]. In our model however, NK1.1+ cells do not appear to be essential for TgPRF mediated defenses against L. monocytogenes. The increased importance of NK cells in defense against influenza may be attributable to the comparatively increased role of NK cells in viral infections and killing of virus infected cells.
All of these mechanisms are unable to fully account for the fact that stimulation with rPRF was able to reduce L. monocytogenes bacterial burdens in Rag1−/− NK1.1 depleted mice. It is possible that in the absence of T and NK cells, alternative mechanisms leading to production of IFN-γ may be induced. Neutrophils could be an important source of IFN-γ independent of T and NK cells in our model. Recent evidence has clearly shown that IFN-γ producing neutrophils are present in the peritoneal cavity during T. gondii infection of WT and TLR11−/− mice and are a biologically relevant source of IFN-γ [55]. Neutrophil derived IFN-γ is produced independent of IL-12 [55], which is consistent with our results showing that neither T cells, NK1.1+ cells, nor IL-12 are required for TgPRF-induced resistance to L. monocytogenes. The fact that stimulation with TgPRF elicited a significant number of neutrophils suggests that IFN-γ producing neutrophils could provide a relevant source of non NK1.1+ derived IFN-γ in our model. Future studies will determine if TgPRF elicits these IFN-γ producing neutrophils during T. gondii infection of mice.
The identification of TgPRF as a T. gondii factor that elicits Ly6Chi inflammatory monocytes and neutrophils is especially important for our understanding of T. gondii infection given that humans presumably lack functional TLR11 and TLR12 receptors for TgPRF, yet inflammatory monocytes are critical for innate defenses against T. gondii. In mice, Ly6C+ and Gr-1+ cells are recruited to sites of T. gondii infection in a CCR2 dependent manner and produce TNF-α and iNOS [8], [9], [10], [11]. CCR2−/− and MCP1−/− mice fail to control parasite replication and are highly susceptible to both oral and parenteral T. gondii infection [8], [9], [10]. Lack of inflammatory monocytes is associated with severe inflammation at the sites of T. gondii infection, including increased numbers of neutrophils, intestinal necrosis and CNS pathology [8], [9], [10]. However, the beneficial versus detrimental role of TgPRF is unclear. Similar to mice lacking inflammatory monocytes, lack of TLR11 during systemic T. gondii infection is associated with inappropriate inflammation [56], which suggests a role for TgPRF recruited monocytes in the regulation of systemic immunopathological responses. In contrast, recognition of TgPRF is detrimental during oral T. gondii infection, likely because gut commensal bacteria stimulate anti-parasitic immune responses [57]. WT, but not TLR11−/−, mice develop acute ileitis and liver pathology suggesting that additional parasite or bacterial factors may be sufficient to direct recruitment of inflammatory monocytes in the absence of TLR11, but concurrent stimulation by gut microbes, TgPRF and other T. gondii molecules promotes overwhelming pathological inflammation. The detrimental effects of TgPRF recognition may also be due to TgPRF mediated recruitment of neutrophils, which lead to mucosal pathology [8], [9], [58] and contribute to parasite spread within the intestine [59]. Even so our work presented here shows that recognition of TgPRF and subsequent recruitment of inflammatory monocytes provides a host the benefit of innate defense against an unrelated pathogen. It is possible that carriage of T. gondii may have driven the maintenance of TLR11 specifically in rodent hosts due to this property, and that the interaction of TgPRF with TLR11 or TLR11/TLR12 heterodimers may be critical for this beneficial host-microbe interaction.
Other microbes are known to confer symbiotic-like protection against unrelated pathogens. Latent infection with the murine γ-herpesvirus MHV68 and the β-herpes virus MCMV conferred protection against the bacterial pathogens L. monocytogenes and Yersinia pestis [60]. Protection resulted in increased survival and correlated with 100-fold reductions in L. monocytogenes burdens in the spleen and liver, similar to the results we observed with chronic infection by T. gondii and stimulation with TgPRF. MHV68 infection also confers enhanced resistance to influenza A virus infection associated decreased viral titers, similar to previous results we reported for T. gondii infection [25], [61]. γHV68-induced protection against both L. monocytogenes and influenza was associated with elevated IFN-γ and increased numbers of activated macrophages with enhanced antibacterial activity [60], [61]. These results suggest that herpes virus and T. gondii exploit similar mechanisms to enhance antibacterial innate immunity.
Inflammatory monocytes and TipDCs play key roles in defense against several other pathogens. Ly6C+ monocytes are recruited in CCR2 dependent manner and help initiate protective T cell responses following infection with Mycobacterium tuberculosis, Leishmania major, and Cryptococcus neoformans [14]. The importance of Ly6C+ monocytes against C. neoformans infection may explain prior observations that chronic T. gondii infection confers a survival benefit during co-infection with this pathogen [21]. Ly6C+ monocytes have been shown to reduce Plasmodium chabaudi circulating parasitemia in a mouse model of malaria and to enhance clearance of West Nile Virus [14]. Inflammatory monocytes and TipDCs may play a more limited or even detrimental role in other infections. TNF-α and nitric oxide produced by TipDCs contribute to tissue injury and liver necrosis during infection with Trypanosoma brucei [14]. TipDCs are recruited to the bladder via CCR2 during uropathogenic E. coli infection but are dispensable for bacterial clearance [62]. Future studies will examine the role of TgPRF recruited inflammatory monocytes during T. gondii and other infections.
Materials and Methods
Ethics statement
Animals were housed under conventional, specific-pathogen-free conditions and were treated in compliance with guidelines set by the Institutional Animal Care and Use Committee of the University of Wisconsin School of Medicine and Public Health (IACUC), according to IACUC approved protocol number M01545. This protocol adheres to the regulations and guidelines set by the National Research Council. The University of Wisconsin is accredited by the International Association for Assessment and Accreditation of Laboratory Animal Care.
Mice
Unless indicated otherwise, all mice used in this study were on a C57BL/6 background and used at 6–8 weeks of age. Wild-type (WT) mice were purchased from National Cancer Institute – Harlan, Frederick, MD. IL-12Rβ1−/− (002984, B6.129S1-Il2rb1tm1Jm/J), IFN-γ−/− (002287, B6.129S7-Ifngtm1Ts/J), Rag1−/− (002216, B6.129S7-Rag1tm1Mom/J), CCR2−/− (004999, B6.129S4-Ccr2tm1Ifc/J) mice were purchased from Jackson Laboratory (Bar Harbor, ME). TLR11−/− mice were a generous gift from Felix Yarovinsky [5] and were rederived at the University of Wisconsin. A/J mice (National Cancer Institute) were used for protein purification experiments because they more susceptible to L. monocytogenes infection than C57BL/6 mice [63], which allowed us to detect subtle changes in bacterial burdens in partially active fractions. All animals were housed and bred under specific pathogen free conditions at an AALAC accredited facility at the University of Wisconsin School of Medicine and Public Health. All experiments were conducted in accordance with an IACUC approved protocol.
L. monocytogenes infections
L. monocytogenes strain EGD was a kind gift from C. Czuprynski. Mice were anesthetized with an isofluorane vaporizer connected to an IVIS 200 imaging system (Caliper Life Sciences, Hopkington, MA) then infected via retro-orbital i.v. injection with an appropriate number of bacteria to cause lethal infection as indicated. Animals were monitored daily for clinical signs of disease (ruffled fur, hunched posture, paralysis, etc.) and were euthanized if moribund. At 72 hours post infection, weight loss and bacterial burdens (CFU/g) in the spleen and liver were determined. WT mice were infected with approximately 6×104 CFU (∼6 LD50's), which consistently resulted in death or euthanasia of 100% of control animals. TLR11−/−, IL-12Rβ1−/−, IFN-γ−/−, Rag1−/− NK1.1-depleted, WT RB6-8C5 (Ly6C/Ly6G)-depleted, CCR2−/−, and WT 1A8 (Ly6G)-depleted mice were infected with approximately 4×104 CFU, 1×104 CFU, 200 CFU, 8×104 CFU, 200 CFU, 8×103 CFU and 8×103 CFU respectively. These doses were chosen because they resulted in bacterial burdens and weight loss similar to lethally infected WT mice.
T. gondii infection
10 week old WT mice were injected i.p. with 250 tachyzoites of the T. gondii strain PruΔ. In order to increase the number of animals that survived greater than 30 days into chronic infection, T. gondii-infected and control uninfected mice were all fed a diet containing sulfadiazine (1,365 ppm) and trimethoprim (275ppm) (TD.06596, Harlan Teklad, Madison, WI) from days 9 through 14 post T. gondii infection, then returned to a normal diet on day 15 through the duration of the experiment.
STAg
For experiments in WT mice, soluble T. gondii antigens (STAg) was made from sonicated tissue culture grown tachyzoites (4×108/ml) essentially as described previously [25] and typically had a protein concentration of ∼1 mg/ml. For experiments in TLR11−/− mice, double the amount of parasites (8×108/ml) were used.
Recombinant profilin
Purified recombinant his-tagged T. gondii profilin (rPRF) was a kind gift from F. Yarovinsky [5]. rPRF preparations used in this study had endotoxin levels of 6×10−4 EU or 1×10−2 EU per 100 ng dose (estimated 0.06 and 1 pg endotoxin respectively) as measured by LAL assay (Pierce, Rockford, IL).
Cytokine analysis
Blood was collected from mice via the lateral tail vein 2 or 24 hours post stimulation with rPRF as indicated. Serum was frozen in aliquots at −80°C and then analyzed using a Mouse Inflammation Cytometric Bead Array kit (BD Biosciences, San Jose, CA) according to the manufacturer's instructions.
Flow cytometry
Spleens were dissociated by mechanical disruption and digested with collagenase/dispase (20 ug/ml, Roche, Indianapolis, IN) and DNAse I (300 ug/ml, Roche) for 30 min at 37°C and passed through a 70 um cell strainer (BD Biosciences, San Jose, CA). Heparinized blood was collected via cardiac puncture and RBCs were removed by dextran sedimentation. Remaining RBCs were lysed with ammonium chloride. Cells were stained at 4°C in PBS with Live/Dead Violet Fixable Stain kit (Invitrogen, Carlsbad, CA), washed, then stained in PBS with 0.5 mM EDTA, 0.2% BSA, 0.09% azide, and 2% normal rat serum (Jackson ImmunoResearch, West Grove, PA). Anti-mouse CD45 (30-F11) APC, anti-mouse CD11b (M1/70) PerCpCy5.5 (eBioscience, San Diego, CA), anti-mouse Ly6C (AL-21) PE, anti-mouse CCR2 (475301) Fluorescein (R&D Systems, Minneapolis, MN), and anti-mouse Ly6G (1A8) PE-Cy7 antibodies were purchased from BD Biosciences except as indicated. Anti-Rat/Hamster CompBeads (BD Biosciences) were used to set compensation. Data were collected on an LSRII cytometer (BD Biosciences) and analyzed with FlowJo 7.6.1 (TreeStar, Ashland, OR).
In vivo depletions
All antibodies used for depletions were purchased from BioXCell (West Lebanon, NH). To deplete NK cells, mice were treated with the anti-NK1.1 MAb PK136 (250 µg/animal). To deplete both Ly6Chi inflammatory monocytes and Ly6Cint Ly6G+ neutrophils, mice were treated with anti-Gr-1 MAb RB6-8C5 (250 µg/animal) which recognizes a common epitope shared by Ly6C and Ly6G [43]. To deplete only neutrophils, mice were treated with anti-Ly6G MAb 1A8 (250 µg/animal) which has been shown to deplete neutrophils in the spleen, liver and blood [44], [48], [49]. All depletion treatments were administered in PBS via i.p. injection beginning 48 hours prior to stimulation with rPRF, then continued every 48 (1A8) or 96 (PK136 and RB6-8C5) hours after for the duration of the experiment.
Statistical analysis
Graphs and statistical analysis were made using Graph Pad Prism (San Diego, CA).Graphs represent means and error bars represent standard deviation except where noted otherwise. Bacterial burden data and cytokine were analyzed with the two-tailed student's t-test, and survival data were analyzed using the Log-rank (Mantel-Cox) method. p-values are represented by asterisks in figures as follows: *p<0.05, **p<0.01, ***p<0.001, and ****p<0.001. We consider all p-values <0.05 to be significant.
Supporting Information
Zdroje
1. SuzukiY, ConleyFK, RemingtonJS (1989) Importance of endogenous IFN-gamma for prevention of toxoplasmic encephalitis in mice. J Immunol 143 : 2045–2050.
2. SherA, CollazzoC, ScangaC, JankovicD, YapG, et al. (2003) Induction and regulation of IL-12-dependent host resistance to Toxoplasma gondii. Immunol Res 27 : 521–528.
3. SuzukiY, OrellanaMA, SchreiberRD, RemingtonJS (1988) Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 240 : 516–518.
4. PlattnerF, YarovinskyF, RomeroS, DidryD, CarlierMF, et al. (2008) Toxoplasma profilin is essential for host cell invasion and TLR11-dependent induction of an interleukin-12 response. Cell Host Microbe 3 : 77–87.
5. YarovinskyF, ZhangD, AndersenJF, BannenbergGL, SerhanCN, et al. (2005) TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308 : 1626–1629.
6. KoblanskyAA, JankovicD, OhH, HienyS, SungnakW, et al. (2013) Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity 38 : 119–130.
7. AndradeWA, Souza MdoC, Ramos-MartinezE, NagpalK, DutraMS, et al. (2013) Combined action of nucleic acid-sensing Toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe 13 : 42–53.
8. DunayIR, FuchsA, SibleyLD (2010) Inflammatory monocytes but not neutrophils are necessary to control infection with Toxoplasma gondii in mice. Infect Immun 78 : 1564–1570.
9. DunayIR, DamattaRA, FuxB, PrestiR, GrecoS, et al. (2008) Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity 29 : 306–317.
10. RobbenPM, LaReginaM, KuzielWA, SibleyLD (2005) Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J Exp Med 201 : 1761–1769.
11. MordueDG, SibleyLD (2003) A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. J Leukoc Biol 74 : 1015–1025.
12. SerbinaNV, PamerEG (2006) Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 7 : 311–317.
13. SerbinaNV, ShiC, PamerEG (2012) Monocyte-mediated immune defense against murine Listeria monocytogenes infection. Adv Immunol 113 : 119–134.
14. ShiC, PamerEG (2011) Monocyte recruitment during infection and inflammation. Nat Rev Immunol 11 : 762–774.
15. SerbinaNV, Salazar-MatherTP, BironCA, KuzielWA, PamerEG (2003) TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity 19 : 59–70.
16. JiaT, SerbinaNV, BrandlK, ZhongMX, LeinerIM, et al. (2008) Additive roles for MCP-1 and MCP-3 in CCR2-mediated recruitment of inflammatory monocytes during Listeria monocytogenes infection. J Immunol 180 : 6846–6853.
17. VyasA, KimSK, GiacominiN, BoothroydJC, SapolskyRM (2007) Behavioral changes induced by Toxoplasma infection of rodents are highly specific to aversion of cat odors. Proc Natl Acad Sci U S A 104 : 6442–6447.
18. AfonsoC, PaixaoVB, CostaRM (2012) Chronic Toxoplasma infection modifies the structure and the risk of host behavior. PLoS One 7: e32489.
19. RuskinJ, RemingtonJS (1968) Immunity and Intracellular Infection: Resistance to Bacteria in Mice Infected with a Protozoan. Science 160 : 72–74.
20. RemingtonJS, MeriganTC (1969) Resistance to virus challenge in mice infected with protozoa or bacteria. Proc Soc Exp Biol Med 131 : 1184–1188.
21. GentryLO, RemingtonJS (1971) Resistance against Cryptococcus conferred by intracellular bacteria and protozoa. J Infect Dis 123 : 22–31.
22. MahmoudAA, WarrenKS, StricklandGT (1976) Acquired resistance to infection with Schistosoma mansoni induced by Toxoplasma gondii. Nature 263 : 56–57.
23. McLeodR, EstesRG, CohenH (1985) Influence of Toxoplasma on manifestations of Moloney virus infections. Trans R Soc Trop Med Hyg 79 : 781–787.
24. McLeodR, RemingtonJS (1977) Studies on the specificity of killing of intracellular pathogens by macrophages. Cell Immunol 34 : 156–174.
25. O'BrienKB, Schultz-CherryS, KnollLJ (2011) Parasite-mediated upregulation of NK cell-derived gamma interferon protects against severe highly pathogenic H5N1 influenza virus infection. J Virol 85 : 8680–8688.
26. PamerEG (2004) Immune responses to Listeria monocytogenes. Nat Rev Immunol 4 : 812–823.
27. ZenewiczLA, ShenH (2007) Innate and adaptive immune responses to Listeria monocytogenes: a short overview. Microbes Infect 9 : 1208–1215.
28. ShiC, VelazquezP, HohlTM, LeinerI, DustinML, et al. (2010) Monocyte trafficking to hepatic sites of bacterial infection is chemokine independent and directed by focal intercellular adhesion molecule-1 expression. J Immunol 184 : 6266–6274.
29. SerbinaNV, KuzielW, FlavellR, AkiraS, RollinsB, et al. (2003) Sequential MyD88-independent and -dependent activation of innate immune responses to intracellular bacterial infection. Immunity 19 : 891–901.
30. ShiC, JiaT, Mendez-FerrerS, HohlTM, SerbinaNV, et al. (2011) Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity 34 : 590–601.
31. BuchmeierNA, SchreiberRD (1985) Requirement of endogenous interferon-gamma production for resolution of Listeria monocytogenes infection. Proc Natl Acad Sci U S A 82 : 7404–7408.
32. HartyJT, BevanMJ (1995) Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity 3 : 109–117.
33. PasparakisM, AlexopoulouL, EpiskopouV, KolliasG (1996) Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med 184 : 1397–1411.
34. RuskinJ, McIntoshJ, RemingtonJS (1969) Studies on the mechanisms of resistance to phylogenetically diverse intracellular organisms. J Immunol 103 : 252–259.
35. Del RioL, ButcherBA, BennounaS, HienyS, SherA, et al. (2004) Toxoplasma gondii triggers myeloid differentiation factor 88-dependent IL-12 and chemokine ligand 2 (monocyte chemoattractant protein 1) responses using distinct parasite molecules and host receptors. J Immunol 172 : 6954–6960.
36. LiZY, MantheyCL, PereraPY, SherA, VogelSN (1994) Toxoplasma gondii soluble antigen induces a subset of lipopolysaccharide-inducible genes and tyrosine phosphoproteins in peritoneal macrophages. Infect Immun 62 : 3434–3440.
37. GrunvaldE, ChiaramonteM, HienyS, WysockaM, TrinchieriG, et al. (1996) Biochemical characterization and protein kinase C dependency of monokine-inducing activities of Toxoplasma gondii. Infect Immun 64 : 2010–2018.
38. JohnsonLL, VanderVegtFP, HavellEA (1993) Gamma interferon-dependent temporary resistance to acute Toxoplasma gondii infection independent of CD4+ or CD8+ lymphocytes. Infect Immun 61 : 5174–5180.
39. DunnPL, NorthRJ (1991) Early gamma interferon production by natural killer cells is important in defense against murine listeriosis. Infect Immun 59 : 2892–2900.
40. ThaleC, KiderlenAF (2005) Sources of interferon-gamma (IFN-gamma) in early immune response to Listeria monocytogenes. Immunobiology 210 : 673–683.
41. GazzinelliRT, WysockaM, HienyS, Scharton-KerstenT, CheeverA, et al. (1996) In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J Immunol 157 : 798–805.
42. CarreroJA, CalderonB, UnanueER (2006) Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J Exp Med 203 : 933–940.
43. FlemingTJ, FlemingML, MalekTR (1993) Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol 151 : 2399–2408.
44. DaleyJM, ThomayAA, ConnollyMD, ReichnerJS, AlbinaJE (2008) Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol 83 : 64–70.
45. CzuprynskiCJ, BrownJF, MaroushekN, WagnerRD, SteinbergH (1994) Administration of anti-granulocyte mAb RB6-8C5 impairs the resistance of mice to Listeria monocytogenes infection. J Immunol 152 : 1836–1846.
46. ConlanJW, NorthRJ (1994) Neutrophils are essential for early anti-Listeria defense in the liver, but not in the spleen or peritoneal cavity, as revealed by a granulocyte-depleting monoclonal antibody. J Exp Med 179 : 259–268.
47. ShiC, HohlTM, LeinerI, EquindaMJ, FanX, et al. (2011) Ly6G+ neutrophils are dispensable for defense against systemic Listeria monocytogenes infection. J Immunol 187 : 5293–5298.
48. CarrKD, SieveAN, IndramohanM, BreakTJ, LeeS, et al. (2011) Specific depletion reveals a novel role for neutrophil-mediated protection in the liver during Listeria monocytogenes infection. Eur J Immunol 41 : 2666–2676.
49. EdelsonBT, BradstreetTR, HildnerK, CarreroJA, FrederickKE, et al. (2011) CD8alpha(+) dendritic cells are an obligate cellular entry point for productive infection by Listeria monocytogenes. Immunity 35 : 236–248.
50. KangSJ, LiangHE, ReizisB, LocksleyRM (2008) Regulation of hierarchical clustering and activation of innate immune cells by dendritic cells. Immunity 29 : 819–833.
51. GoldszmidRS, CasparP, RivollierA, WhiteS, DzutsevA, et al. (2012) NK cell-derived interferon-gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity 36 : 1047–1059.
52. BergRE, CrossleyE, MurrayS, FormanJ (2003) Memory CD8+ T cells provide innate immune protection against Listeria monocytogenes in the absence of cognate antigen. J Exp Med 198 : 1583–1593.
53. SoudjaSM, RuizAL, MarieJC, LauvauG (2012) Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity 37 : 549–562.
54. KupzA, GuardaG, GebhardtT, SanderLE, ShortKR, et al. (2012) NLRC4 inflammasomes in dendritic cells regulate noncognate effector function by memory CD8(+) T cells. Nat Immunol 13 : 162–169.
55. SturgeCR, BensonA, RaetzM, WilhelmCL, MirpuriJ, et al. (2013) TLR-independent neutrophil-derived IFN-gamma is important for host resistance to intracellular pathogens. Proc Natl Acad Sci U S A 110 : 10711–10716.
56. YarovinskyF, HienyS, SherA (2008) Recognition of Toxoplasma gondii by TLR11 prevents parasite-induced immunopathology. J Immunol 181 : 8478–8484.
57. BensonA, PiferR, BehrendtCL, HooperLV, YarovinskyF (2009) Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe 6 : 187–196.
58. GraingerJR, WohlfertEA, FussIJ, BouladouxN, AskenaseMH, et al. (2012) Inflammatory monocytes regulate pathologic responses to commensals during acute gastrointestinal infection. Nat Med 19 : 713–721.
59. CoombesJL, CharsarBA, HanSJ, HalkiasJ, ChanSW, et al. (2013) Motile invaded neutrophils in the small intestine of Toxoplasma gondii-infected mice reveal a potential mechanism for parasite spread. Proc Natl Acad Sci U S A 110: E1913–1922.
60. BartonES, WhiteDW, CathelynJS, Brett-McClellanKA, EngleM, et al. (2007) Herpesvirus latency confers symbiotic protection from bacterial infection. Nature 447 : 326–329.
61. SaitoF, ItoT, ConnettJM, SchallerMA, CarsonWFt, et al. (2009) MHV68 latency modulates the host immune response to influenza A virus. Inflammation 36 : 1295–1303.
62. EngelD, DobrindtU, TittelA, PetersP, MaurerJ, et al. (2006) Tumor necrosis factor alpha - and inducible nitric oxide synthase-producing dendritic cells are rapidly recruited to the bladder in urinary tract infection but are dispensable for bacterial clearance. Infect Immun 74 : 6100–6107.
63. GervaisF, StevensonM, SkameneE (1984) Genetic control of resistance to Listeria monocytogenes: regulation of leukocyte inflammatory responses by the Hc locus. J Immunol 132 : 2078–2083.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 6
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Fungal Nail Infections (Onychomycosis): A Never-Ending Story?
- Profilin Promotes Recruitment of Ly6C CCR2 Inflammatory Monocytes That Can Confer Resistance to Bacterial Infection
- IscR Is Essential for Type III Secretion and Virulence
- Contribution of Specific Residues of the β-Solenoid Fold to HET-s Prion Function, Amyloid Structure and Stability