
Structure of the Trehalose-6-phosphate Phosphatase from Reveals Key Design Principles for Anthelmintic Drugs
Here, we describe the structure of trehalose-6-phosphate phosphatase (T6PP) from Brugia malayi. This enzyme is essential to the organism; deletion of the gene encoding T6PP results in toxic accumulation of trehalose 6-phosphate. Structure-guided mutagenesis coupled with kinetic analyses revealed residues important for binding and catalysis. The model for substrate binding suggests a binding mode in which shape complementarity plays a major role. Conservation of binding residues among T6PP orthologs present in pathogenic nematodes and bacteria favors T6PP as a suitable target for broad-range anthelmintic and antibacterial drug design.
Published in the journal:
. PLoS Pathog 10(7): e32767. doi:10.1371/journal.ppat.1004245
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004245
Summary
Here, we describe the structure of trehalose-6-phosphate phosphatase (T6PP) from Brugia malayi. This enzyme is essential to the organism; deletion of the gene encoding T6PP results in toxic accumulation of trehalose 6-phosphate. Structure-guided mutagenesis coupled with kinetic analyses revealed residues important for binding and catalysis. The model for substrate binding suggests a binding mode in which shape complementarity plays a major role. Conservation of binding residues among T6PP orthologs present in pathogenic nematodes and bacteria favors T6PP as a suitable target for broad-range anthelmintic and antibacterial drug design.
Introduction
Parasitic nematodes are responsible for devastating diseases that plague the tropical, low-income areas of Africa, Asia, and the Americas. In 2011, the World Health Organization estimated that 41% of the population worldwide was affected by these organisms [1]. Among the diseases caused by parasitic nematodes is the mosquito-transmitted lymphatic filariasis. It is projected that as many as 120 million people suffer from this disease with nearly 1.2 billion people at risk [2]. Lymphatic filariasis occurs upon infection of the lymphatic system by Wuchereria bancrofti, Brugia malayi, or Brugia timori and clinically manifests as lymphedema, hydrocele, and in the most extreme cases, elephantiasis. Related filarial nematodes inhabit other tissues; such infections may result in equally severe diseases, as exemplified by onchocerciasis or “river blindness” caused by Onchocerca volvulus infection. Filarial infections are responsible for extreme infirmity, distress, and social stigma. In fact, lymphatic filariasis is a major cause of permanent and long-term disability in people worldwide [3].
Due to the nature of infection and the impact on people suffering from this disease, the Global Program for the Elimination of Lymphatic Filariasis (GPELF) was established in 1999 with the major objective of ending the transmission of the disease by 2020. Currently the recommended regimen for treatment is the administration of albendazole together with either ivermectin (where onchocerciasis is endemic) or diethylcarbamazine citrate (where onchocerciasis is not present). Though community-wide treatment programs utilizing albendazole, ivermectin and/or diethylcarbamazine citrate have been effective, they are not without drawbacks. Side effects of albendazole and ivermectin are common, and although less frequent, they have also been observed with diethylcarbamazine citrate. Furthermore, these drugs only kill microfilariae, leaving the adult worms intact. Consequently, the drugs must be administered for the entire reproductive life span of the adult worm (approximately 5 years) [4]. In addition, the drug combination administered must be tailored to the specific parasite population in a given area because ivermectin administration can lead to encephalopathy in individuals with high microfilarial loads caused by Loa loa [5]. The development of new drugs that are not subject to these limitations is needed, particularly in the face of increasing drug resistance [6]. Indeed, drug resistance in veterinary nematodes is already widespread [7]–[9] and there are indications that in Ghana O. volvulus has developed a resistance to ivermectin [10].
To facilitate drug-discovery programs, the sequence determination of genomes of parasitic nematodes having human, domestic animal or plant hosts has been initiated [11]–[18]. The complicated life cycle of nematodes increases the difficulty of laboratory-based investigation. In fact, culturing W. bancrofti for in vivo studies has to date been unsuccessful. Fortunately, B. malayi can be maintained in a jird host [19] and is amenable to in vitro studies at different stages of its life-cycle [20]. Consequently, B. malayi now serves as a plausible model for research on lymphatic filarial nematodes alongside C. elegans, which for decades, has served as a model system for research on free-living nematodes. The similarities between C. elegans and parasitic nematodes with regard to genome sequences, and the phenotypes resulting from RNAi gene knockdown [21]–[23], indicate that C. elegans, being by far the most easily maintained organism, is an important resource in the development of broad-spectrum anthelminthic drugs.
Recently, top drug target candidates were identified in B. malayi using a ranking system [24], and among the highest-ranking targets is trehalose-6-phosphate phosphatase (T6PP) (UniProt: A8NS89), an enzyme that is required for the synthesis of trehalose [25]. T6PP is present in bacteria, fungi, plants, and invertebrate animals, but not in mammals. Trehalose is used by these organisms as an energy reserve, and it can also protect against environmental insults such as oxidative and osmotic stress, anoxia, heat, cold, freezing, desiccation, and anhydrobiosis [26], [27].
Trehalose is synthesized by a two-step pathway that involves T6PP and trehalose-6-phosphate synthase (genes gob-1, tps-1 and tps-2 in C. elegans, Figure 1). The synthase catalyzes the condensation of glucose 6-phosphate and UDP-glucose, forming trehalose 6-phosphate, and T6PP catalyzes phosphoryl transfer to water, forming trehalose. RNAi knockdown of the C. elegans T6PP-encoding gene gob-1 (gut-obstructed 1) gives rise to larval lethality due to intestinal blockage and subsequent starvation [28]. Importantly, this phenotype is reversed by RNAi knockdown of the tps-1 gene, suggesting that the lethality is due to a toxic accumulation of trehalose 6-phosphate [28]. A T6PP inhibitor might therefore bring about the same result, and thus we have targeted nematode T6PP for the development of small molecule anthelmintics.

As a first step toward inhibitor design the structure determination of T6PP was undertaken. The crystal structure of a putative T6PP has been reported from Thermoplasma acidophilum (PDB: 1U02) (29). Although this ortholog has low sequence identity, its structure identified it as a HAD superfamily (HADSF) phosphatase. All HADSF phosphatases possess a conserved Rossmann-fold catalytic domain, which contains the catalytic residues and the Mg2+ cofactor binding residues that together, constitute the substrate phosphoryl-group binding site. Most HADSF phosphatases, including the T6PP, also possess a cap domain (designated type C0, C1, C2A or C2B). During catalytic turnover the cap domain moves over the catalytic site through binding interactions with the substrate-leaving group, thereby forming an encapsulated active site. The size, shape and electrostatic properties of the active site are unique to each individual phosphatase. Although the sequences of the nematode T6P phosphatases are quite similar to one another, they share little identity with the T. acidophilum T6PP (12.7%). Moreover, sequence alignments revealed that the nematode orthologs possess a ∼140 amino-acid segment preceding the predicted N-terminus of the HAD phosphatase fold that is long enough to constitute a structural domain. It was thus both out of necessity for inhibitor design, and the intrigue for discovery of the fold and function of the novel N-terminal domain, that we pursued structure determination of the T6PP from B. malayi.
Herein, we report the X-ray structure of B. malayi T6PP and the findings from structure-activity analysis aimed at elucidating the role of the N-terminal domain and the active-site residues in substrate binding and catalysis.
Results and Discussion
Overall structure
The structure of B. malayi T6PP bound to Mg2+ was determined at 3.0 Å resolution (residues 63–491, Figure 2A, Figures S1 and S2). Electron density was not observed for residues 1–62 and SDS-PAGE analysis of dissolved crystals revealed that this region was not lost during crystallization. These residues are not conserved among the nematode T6PP enzymes, and in the case of several Caenorhabditis species, they are absent (Figure S3). In addition, the B. malayi T6PP truncation mutant Δ59 (residues 1–59 deleted) retains the native fold (Figure S4) and the same catalytic activity as the wild type enzyme (Table 1). Thus, we conclude that residues 1–62 are probably disordered and are not required for stability or catalytic activity.


The overall T6PP structure consists of a three-helix bundle N-terminal domain (residues 63–157) connected via a short linker (residues 158–205) to the Rossmann-fold catalytic core domain (residues 206–292; 398–492) and the α,β-fold cap domain (residues 293–397), which together comprise the HADSF phosphatase structure (Figure 2A). Six parallel β-strands flanked by five α-helices form the core domain. A short β-hairpin (residues 282–292) precedes the cap domain (residues 293–397) and extends the central six-stranded β-sheet of the Rossmann-fold by two strands. The C2B-type cap domain is formed by an anti-parallel β-sheet topped by two α-helices (Figure 2A). Two short loops in the cap (residues 320–322; 367–370) were not well ordered and were not modeled.
The closest related structure is the T6PP-like enzyme from T. acidophilum (which does not possess the N-terminal domain (residues 1–205; Figure 2B) [29]. Superposition of the B. malayi and T. acidophilum structures (Figure 2C) gave a three-dimensional alignment with an overall rmsd of 2.6 Å compared to the rmsd of 2.1 Å obtained for the overlay of the individual core domains (14% sequence identity) and 2.2 Å rmsd for the overlay of the cap domains (9% sequence identity). Although the tertiary structures of the paired domains are nearly identical, the difference in global conformation resulting from distinct cap-core domain orientations observed for the two crystalline states, prevented structure determination by molecular replacement, thus necessitating phasing by single wavelength anomalous diffraction of the selenomethionine-substituted B. malayi T6PP.
The MIT-like domain of T6PP
The N-terminal domain (residues 63–157) consists of a three-helix bundle which is similar in topology to the Microtubule Interacting and Transport (MIT) domains of the Vps4-like ATPases from Sulfolobus acidocaldarius (PDB ID: 2W2U, rmsd 2.4 Å, Figure 3A) and Sulfolobus solfataricus (PDB ID: 2V6Y, rmsd 2.4 Å) [30], [31]. The MIT-like domain is tethered to the core domain by a helical-connector (residues 158–206) that is comprised of a broken α-helix (helices 4 and 5), which interacts with both domains and a second, longer α-helix (helix 6), which selectively interacts with the core domain (Figures 2A,3B). The presence of the MIT-like domain is striking as no other HADSF members are known to incorporate this domain. Sequence alignment analysis revealed that that the MIT-like domain is unique to the T6PP of nematodes and in organisms from the genus Mycobacterium (Figure S5 and S6).

MIT domains are protein-interacting domains typically associated with multivesicular body formation, cytokinetic abscission, or viral budding [32]. The best-characterized MIT domains are found in the essential AAA-ATPase Vps4. Vps4 is vital for endosomal trafficking to lysosomes, where it acts to dissociate ESCRT (endosomal sorting complexes required for transport) from membranes [30]. The MIT domain of Vps4 recognizes a conserved sequence in ESCRT-III termed the MIT-interacting motif (MIM). This sequence forms a protein-protein interaction site between the second and third α-helices of the Vps4 MIT-domain. However, MIT domains can interact with MIM motifs at each helical interface in the three-helix bundle, and can interact with multiple MIM motifs at once [33]. In T6PP, an intramolecular interaction occurs at the interface between the first and third α-helices of the MIT-like domain and the C1-loop of the HAD core domain (Figure 3B), leaving the solvent-exposed interface between the second and third helices free to make intermolecular interactions with other proteins. Although the protein-protein interface is the same as that seen in other MIM-MIT domain interfaces [33] the sequence of the C1-loop of T6PP does not share significant identity with any known MIM motif. The availability of a potential protein-protein interaction interface on the MIT-like domain raised the question of whether T6PP has a “moonlighting” [34] function in addition to or in conjunction with its enzymatic phosphatase activity.
The T6PP gene was originally identified in C. elegans and was named gob-1 owing to the phenotype (gut obstruction) that occurs when the gene is knocked down. Knockdown of gob-1 as well as the upstream T6P synthase genes tps-1 and tps-2 results in a normal phenotype [28]. This suggests that the gob-1 lethality results from the buildup of the intermediate trehalose 6-phosphate, rather than the absence of trehalose (Figure 1). The presence of the MIT-like domain in the structure of T6PP led us to carry out further investigation. To test if the phenotype results from the absence of a protein-protein interaction between the T6PP MIT-like domain and another unknown protein, we repeated the RNAi experiments. Studies were carried out with the RNAi hypersensitive strain of C. elegans eri-1(mg366); lin-15B (n744). We observed that feeding dsRNA of the T6PP gob-1 gene results in arrest at the early larvae stage. In contrast, RNAi-hypersensitive worms which lack the T6P synthase gene tps-1 and which are fed gob-1 dsRNA showed a wild-type phenotype (Table S1). This observation is consistent with the earlier finding in wild-type C. elegans that the accumulation of T6P, rather than the lack of trehalose, is likely responsible for the observed lethality [28]. These results reinforce the suggestion that worm death is due to a metabolic effect, and that the gut obstruction phenotype is secondary to the accumulation of trehalose 6-phosphate. Moreover, the gene knock down lethality cannot be attributed to the removal of a hypothetical protein-protein interaction (through the MIT domain) because it occurs only if T6P synthase is present.
Deletion or mutation of the MIT-like domain is highly destabilizing to the enzyme and attempts to express the protein with the domain deleted (Δ179) or with the domain deleted and the potential hydrophobic patch “repaired” with the corresponding residues from the T. acidophilum T6PP (Δ179+L229Y/V232S/V236S/A243K) resulted in an unstable, insoluble form of the enzyme. The MIT domain itself was soluble and stable (T6PP:1-178-MIT Figure S7D). Notably, the loop region (C1-loop) that flanks the conserved active site forms extensive contacts with the MIT-like domain in the B. malayi protein, but the same C1-loop conformation is retained in the T. acidophilum ortholog lacking the MIT-like domain (Figure 2B). Thus, the interaction with the MIT-like domain is not necessary to retain the C1-loop conformation, although it is possible that in MIT-domain containing orthologs there is interdependence between the two structural units. Thermal unfolding observed by CD revealed one transition, suggesting that both domains unfold concurrently (Figure 3C). Additional proteolytic analysis confirms this observation (Figure S7) Together these findings suggest that the MIT-like domain and core domain are structurally co-dependent, and that any attempt to remove this interaction is detrimental to the overall stability of the protein. At this point it is uncertain whether the MIT-like domain plays a role in binding and catalysis of trehalose 6-phosphate hydrolysis.
The active site of T6PP
The active site of HADSF members lies at the interface of the cap and core domains, with the core domain comprising the phosphoryl-transfer catalytic machinery. Indeed, the B. malayi T6PP core holds the four conserved active-site motifs that, in the HADSF [35], coordinate both the magnesium cofactor and phosphoryl group of substrate for catalysis. A single magnesium ion is observed to be coordinated in the active site by two waters and four residues (Motif I: Asp213, Asp215 (mainchain C = O oxygen), Motif IV: Asp424 and Asp428). Thr253 (Motif II) and Lys398 (Motif III) presumably form a hydrogen bond with the phosphate during catalysis.
In B. malayi T6PP, the cap was found to be in an open orientation relative to the core, though the crystal contacts were consistent with stabilization of the protein in this conformation and the protein may exist in alternate conformations in solution. Notably, the B. malayi T6PP has a slightly more open conformation than the orthologous T. acidophilum enzyme (Figure 2C). Because the cap domain mediates interactions with the substrate leaving group, but only the closed conformation positions substrate-binding residues for binding and catalysis, initial analysis of the structure did not reveal obvious residues involved in substrate recognition. Moreover, attempts to obtain a liganded structure or closed conformer structure have been unsuccessful thus far.
To identify residues important for ligand binding, structure-guided single site mutagenesis coupled with enzyme kinetics was performed. Several residues near the active site or in the cap region were selected for mutagenesis (Table 1, Figure 4). These residues are present on the face of the β-sheet that is oriented toward the active site and are highly conserved among T6PP enzymes from nematodes, but were not as conserved among T6PP enzymes from other phyla including Mycobacterium, other prokaryotes and Saccharomyces (Figure S6). As expected, replacement of the catalytic Asp213 or Asp215 resulted in the loss of all detectable activity (Table 1). None of the other replacements resulted in a dramatic change in Km for T6P. However, mutations in Tyr221, Gln332, Lys334, Asp336, or Arg337 resulted in large decreases in kcat (24-, 110-, 400-, 255 - or 85-fold, respectively) suggesting a role in catalysis. Each of these residues except for Tyr221 are found in the cap domain (Figure 4) and may interact with the sugar moiety in order to orient the substrate for catalysis. Since the observed changes in kcat/Km stemmed from changes in kcat, we moved to inhibitor binding studies to test if any of the protein variants differed in their affinity for substrate.
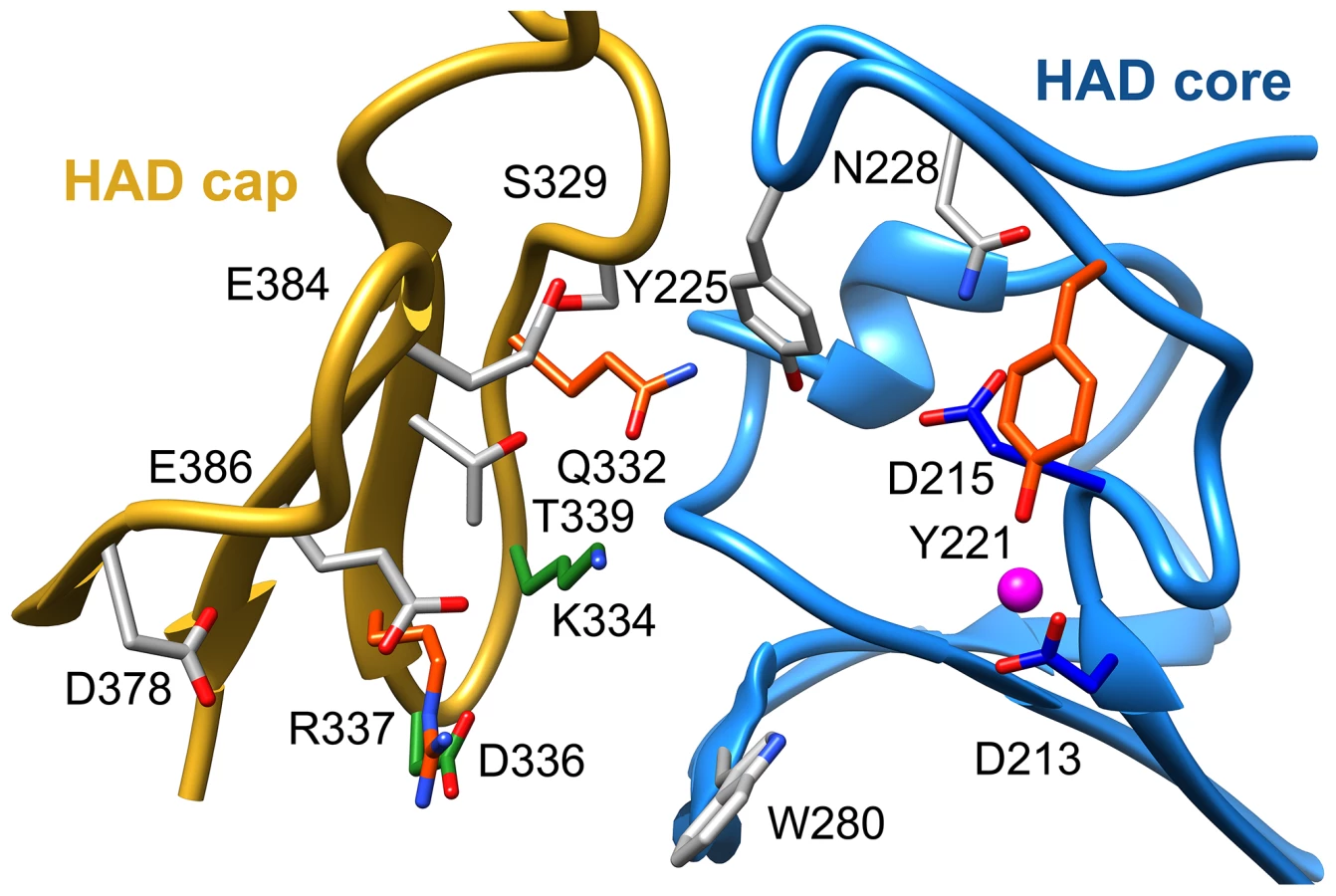
To assess substrate affinity, the substrate analogue trehalose 6-sulfate (Materials and Methods) was used to measure KI for all of the T6PP variants (Table 1). Trehalose 6-sulfate was shown by steady-state inhibition kinetics to be a competitive inhibitor of wild type T6PP with a KI of 82±7 µM against trehalose 6-phosphate as substrate. Three of the variants that showed a decrease in kcat also showed dramatic increases in KI for trehalose 6-sulfate: Tyr221, Gln332 and Arg337 (Table 1, Figure 4). From these experiments, Tyr 221, Gln332, and Arg337 are likely to be involved in binding trehalose 6-phosphate, while Lys334 and Asp336 may play other roles in catalysis. These roles may include the exclusion of solvent from the active site, steric restraint of the substrate for catalysis, or the positioning of other residues required for enzyme activity.
To determine whether homologous residues could play a role in T6PP orthologs, homology models of the enzymes from E. coli, S. cerevisiae, and M. tuberculosis were generated [36] and compared to the structure from B. malayi and T. acidophylum (because sequence identity was low, a homology model which utilizes additional constraints was the most reliable way to ensure correct alignment). The residues that affected both catalysis and KI were found to be conservatively replaced in each of these orthologs (the bulky residue Tyr221 is replaced by Ile; Gln332 is replaced by Glu/Tyr; Arg337 is replaced by Lys) (Figure S8), reinforcing the importance of the roles of the residues identified here. The presence of these residues in all T6PP enzymes examined suggests that design of broad-spectrum inhibitors may be possible.
Mutagenesis-guided model of the T6PP-trehalose 6-phosphate complex
Attempts to visualize the closed-cap form of the B. malayi enzyme using molecular dynamics simulations were unsuccessful (see Materials and Methods). To provide a platform for inhibitor design, a model of T6PP with trehalose 6-phosphate bound in the active site was constructed. To accomplish this, trehalose 6-phosphate was manually placed in the active site of T6PP such that it was positioned for “in-line” attack by Asp213 (the Asp nucleophile) and coordinated to the Mg2+ cation as has been observed in all HADSF enzyme-substrate or enzyme transition-state analogue complex structures [37]–[40]. In addition, the side chain of Asp215 (the general acid/base catalyst conserved in phosphatase members of the HAD superfamily [35]) was positioned to promote leaving-group protonation. To position the trehalose moiety, the FTMap server [41], [42] was used to identify putative hot spots in the cap and core domains of both the B. malayi and T. acidophilum T6PPs. Intriguingly, FTMap analysis of the two enzymes revealed several hot spots forming a pocket extending from the phosphate-binding site to the C1 loop (Figures 5A,B). The identified binding pocket was juxtaposed to the important residues identified by mutagenesis in B. malayi in the cap (Figure 5B). The trehalose moiety was manually rotated to fit within the hot-spot binding sites and the model was then minimized using NAMD [43]. Tyr221, Lys 334, Gln332 and Arg337 are positioned near the substrate and, consistent with the mutagenesis results, may play important roles in binding (Figure 5C). Overall, the interface between the cap and core domains forms a substrate-binding pocket for trehalose 6-phosphate that may exclude solvent, provide steric constraint through van der Waals contacts, and may form only a few specific hydrogen bonds.

Although the T6P docked in the crystal structure of B. malayi T6PP brings the proposed binding residues proximal to the substrate, the model is not in a closed conformation. FTMap analysis suggests that the T. acidophilum T6PP enzyme is in a more closed conformation compared to that of B. malayi. Superimposition of the B. malayi cap with the T. acidophilum T6PP cap and subsequent analysis by DynDom [44]–[46] predicts that the cap rotates 45.6° with respect to the core (Figure S9A). Placement of the B. malayi cap in this orientation positions the residues identified by mutagenesis within contact distance of the predicted T6P model position (Figure S9B).
Comparison to other HAD phosphatases
To compare our model of the T6PP-T6P complex to other phosphatases the DALI server [47] was used to identify HAD members with similar structures. Because the orientation between cap and core varies and the cap is the most variable portion of HAD family members, the cap domain from T6PP was used to find similar structures with C2B-type caps. Besides the T6PP-like enzyme from T. acidophilum, one of the highest scoring structures was that of sucrose-6-phosphate phosphatase (S6PP) from the cyanobacteria Synechocystis sp. PCC 6803 [48]. Sucrose plays a similar role in cells to that of trehalose, and is synthesized in response to osmotic stress in a two-step pathway involving the dephosphorylation of a sucrose 6-phosphate (S6P) intermediate [49], [50]. The structure of the S6PP-S6P complex (PDB: 1U2T) reveals a closed cap conformation with an extensive hydrogen-bonding network providing substrate stabilization (Figure 6A). The ability of these enzymes to discriminate between T6P and S6P may be due to the different glycosidic-linkages of sucrose and trehalose (sucrose: α(1→2)β vs. trehalose: α(1→1)α). According to our model, T6P packs against Tyr221 in the C1-Loop and may be stabilized for catalysis via hydrogen bonds from Gln332 and Arg337. S6P is oriented in the opposite direction and is stabilized by several hydrogen bonds from cap-residues (Figure 6A). The C1-loop in S6PP is much shorter and less pronounced, emphasizing its importance in T6P recognition by T6PP.

Overlay of S6P in the active site of T6PP reveals a steric clash with a loop in T6PP (Figure 6B). This is consistent with the fact that T6PP does not accept S6P as a substrate as well as the inability of S6P to bind to or act as an inhibitor of T6PP. In addition, the use of hydrogen bonds as a mode of sugar recognition in S6PP versus van der Waals interactions in T6PP explains the inhibition of S6PP by the monosaccharide glucose moiety comparable to that of sucrose itself [48]. In contrast in T6PP, we found that there is no inhibition by the trehalose or glucose 6-phosphate substituents of trehalose 6-phosphate (KI>10 mM for these compounds, Figures 7C, D). Thus, it is expected that a critical issue in designing inhibitory ligands for T6PP will be the occupancy of the phosphoryl binding site in conjunction with shape recognition of the leaving group (Figures 7A, B). Indeed, T6PP acts as an excellent system for the exploration of inhibitory ligands of phosphoryl binding sites as the majority of inhibitors to phosphoryl transfer enzymes [51] take advantage of adjacent binding sub-sites (e.g. the nucleotide or peptide/protein binding subsites in kinases [52], [53] rather than the transferring phosphate site.

Concluding remarks
In order to improve our understanding of the structure/function relationship in T6PP enzymes from parasitic nematodes we determined the structure of T6PP from B. malayi. Although the cap-open, unliganded form was observed in the structure, site-directed mutagenesis coupled with substrate and inhibition kinetics identified five residues (Table 1, Figure 4) important for catalysis and/or substrate affinity. Notably, no single residue is critical to binding, consistent with a mode of binding of the trehalose moiety by van der Waals contacts and shape complementarity between the sugar and enzyme. Moreover, the trehalose binding site is not dominated by hydrophobic residues that could form ring-stacking interactions like those found in other carbohydrate binding proteins [54]–[57]. The conservation of several key binding site residues among T6P phosphatases from parasitic nematodes and pathogenic bacteria indicates that a common strategy for T6PP inhibitor design might be used in the development of antibiotics as well as anthelmintics.
An inhibitor design strategy is now afforded by the availability of the T6PP structure. Based on inspection of the structure and the inhibition kinetic analysis of wild-type and mutant B. malayi T6PP, there are two independent regions that can be simultaneously targeted for inhibitor design, namely the phosphoryl group binding site and the trehalose leaving group binding site (Figure 7). The lack of inhibition of the B. malayi enzyme by trehalose (KI>10 mM) compared to the observed tight binding of the inert substrate analog trehalose 6-sulfate (KI = 82 µM) shows that electrostatic interaction with the phosphoryl group binding site provides a significant amount of binding energy that can be captured by a strategically positioned inhibitor sulfate group. Although the T6P trehalose moiety is essential for properly orienting the substrate in the catalytic site for catalytic turnover, it does not appear to provide the amount of binding energy needed for a lead inhibitor. Thus, we envision the design of a bi-dentate inhibitor comprised of the high affinity sulfate group (or similar) for interaction with the catalytic site of the core domain with an organic moiety that maximizes the intrinsic binding energy derived from hydrophobic and electrostatic interaction with the binding region made available by the interfaced cap domain (not necessarily mimicking those made by the substrate trehalose moiety).
Materials and Methods
Bioinformatic analysis of the T6PP enzyme family
Sequences of T6PP enzymes were downloaded from UniProt, aligned using T-Coffee [58] and visualized using ESPript [59]. A sequence alignment of all T6PP enzymes from nematodes revealed a high degree of conservation with the exception of the N-termini which vary in length (1–60 amino acids) and sequence (Figure S3). In addition, an extra N-terminal domain consisting of approximately 150 residues was found in T6PP enzymes from nematodes and in organisms from the genus Mycobacterium (Figs. S5 and S6)
Protein preparation and crystallization
The gene Bm1_08695 encoding T6PP from B. malayi was cloned from a cDNA library by PCR using primers with the restriction sites for NdeI and BamHI embedded. Standard cloning procedures were used to place the gene into a modified pET-15b vector (thrombin site replaced with a TEV site; pET-15(TEV)). The individual domains were also separately cloned into a pETDUET-1 vector by placing the HAD core (180–492) into MCS1 using XbaI and BamHI and placing the MIT domain (1–179) into MCS2 using NdeI and KpnI. To create variants for probing residues important to activity (Table S2), the Q5 Site-Directed Mutagenesis Kit (New England Biolabs, NEB) was used following the manufacturer's protocol. NEB 10-beta Competent E. coli (NEB 3019) were used for plasmid propagation and purification, and all plasmids were sequenced to confirm accuracy before use. For expression, T6PP, T6PP-Δ59, T6PP-Δ179, T6PP:1-178 (MIT) or the T6PP variants were prepared with T7 Express lysY/Iq Competent E. coli (NEB C3013) that were transformed with appropriate expression vectors. Flask cultures were grown in LB medium containing 0.1% glucose. Fermentation medium was as follows: 2% soytone, 1% yeast extract, 171 mM NaCl, 1.87 mM KH2PO4, 5.97 mM Na2HPO4, 13.1 mM NH4Cl, 0.5 mM K2SO4, 8% glycerol, 5 mM MgSO4, 10 mM betaine, 1× Korz trace metals [60], and 0.01% antifoam 204 (Sigma). For flask cultures 100 µg/ml of ampicillin was used and 150 µg/ml ampicillin was used in fermenters. One colony was inoculated into 7 ml of LB and 0.1 ml of the suspension was inoculated into 500 ml of LB for seed cultures. The seed cultures were incubated overnight at 30°C to an OD600 of 2. Ten liter Fermentations were done in 19.7 L Bioflo 510 fermenters (Eppendorf). The pH was controlled at 7.0 with automatic addition of 30% NH4OH and 46 N H3PO4. The dissolved oxygen was controlled at 20% of air saturation using constant gas flow at 0.5 vvm and an agitation-oxygen cascade. The culture was grown at 30°C to an OD600 of 4. IPTG was then added to 0.5 mM and the temperature was dropped to 18°C for induction. The culture was induced for 18 h before harvesting. Cells containing pET-15b (TEV):T6PP-Δ179 were induced with 1 mM IPTG for 3 hours at 37°C once the OD600 reached 0.6–0.8. Cells were pelleted and frozen at −20°C until further processing. For lysis, cell pellets were thawed and resuspended (3 ml buffer per g pellet) in a solution consisting of 50 mM Tris pH 7.5, 500 mM NaCl, 20 mM imidazole, 1 mM EDTA, 1 mM PMSF, 30 µg/ml DNase I (Sigma) and 1 protease inhibitor cocktail tablet (Sigma S8830) per every 100 g of cell pellet. Cells were lysed via a single pass through a microfluidizer (Microfluidics Model #M110P) at 20,000 PSI. Lysate was clarified at high speed (100,000× g), bound to a HisTrap FF column using an ÄKTA FPLC (GE Biosciences) and washed extensively before elution using a gradient with a buffer consisting of 50 mM Tris pH 7.5, 500 mM NaCl, 500 mM imidazole. The resulting protein fraction was then diluted 20-fold in a low-salt buffer consisting of 20 mM Tris pH 7.5 and 10 mM NaCl and bound to a HiTrap Q column (GE Biosciences). The protein was eluted using a gradient with a buffer comprised of 25 mM Tris pH 7.5 and 1 M NaCl. The N-terminal His-tag was removed by TEV protease (Blommel and Fox, 2007) using a 1∶50 ratio (mg∶mg) of TEV:T6PP prior to dialysis into a storage buffer consisting of 25 mM Tris pH 7.5, 10 mM NaCl, 5 mM MgCl2. Purified T6PP was concentrated to 15 mg/ml using an Amicon Ultra concentrator (10K MWCO, Millipore), aliquoted in small volumes and stored at −80°C.
To prepare selenomethionine-incorporated protein, the same procedure was used with two minor changes. Protein was expressed in the T7 Express Crystal Competent E. coli methionine-auxotroph cell strain (NEB C3022), following the manufacturer's protocol. Second, all buffers used contained 5 mM DTT to prevent oxidation of selenium.
Initial crystal screens were set up with either full-length T6PP or T6PP-Δ59 at 15 mg/ml in 96-well microplates (Corning 3552) using the Crystal Screen HT, PegRx HT and Index HT sparse matrix crystallization screens (Hampton Research) and a final drop size of 1.5 µl at 290 K. The full-length protein yielded hexagonal crystals in several low molecular weight polyethylene glycol (PEG) solutions buffered within a pH range of 5.0–8.5 and were optimized using the Additive HT and Detergent HT screens (Hampton Research). For data collection, optimized crystals of native T6PP were grown at 290 K in hanging drops (concentration of 15 mg/ml) in 33% PEG 300, 0.1 M sodium citrate pH 5.0, 13% ethylene glycol (EG) and 10 mM CoCl2. For cryoprotection, the concentration of EG was slowly increased to 25% by adding a concentrated solution directly to the crystal drop. Crystals were harvested using CryoLoops (Hampton Research), looped through LV CryoOil (MiTeGen), frozen in liquid nitrogen and stored until data collection. Crystals of selenomethionine-incorporated T6PP were grown at 290 K in hanging drops (concentration 10 mg/ml) in 30% PEG 300, 0.1 M sodium citrate pH 5.0 and 20 mM NDSB-256. For cryoprotection, 2-methyl-2,4-pentanediol (MPD) was slowly added to crystallization drops (final concentration of 25%). Crystals were harvested and stored in the same way as the native crystals.
X-ray data collection and structure determination
Native and derivative datasets were collected at either the National Synchrotron Light Source (NSLS, beamline ×25) or the Stanford Synchrotron Radiation Lightsource (SSRL, beamlines 7-1, 12-2) using automounter systems. Data was collected under nitrogen gas flow at 100 K in 1° oscillations using either a Dectris Pilatus 6M detector (NSLS ×25; SSRL 12-2) or an ADSC Quantum 315r detector (SSRL 7-1) and processed with HKL-3000 [61]. Initial electron density maps were calculated via single-wavelength anomalous dispersion (SAD) with Phenix AutoSol using a 3.4 Å selenomethionine derivative dataset. This revealed 15 anomalous selenium peaks corresponding to 15 (out of 16) selenomethionine residues. Phases were improved and extended to 3.0 Å with Phenix AutoBuild using a native dataset [62]. The model was built iteratively with Coot [63] and refined in Phenix Refine [62] with 10% of the reflections excluded for the calculation of R-free. The final model converged to an R-value of 21.5% and an R-free of 25.9% with 93.2% of residues in the favored region and 6.6% of residues in the allowed region of the Ramachandran plot (Lovell et al., 2003). Full data collection and refinement statistics are summarized in Table S3.
The quality of the completed model was assessed by MolProbity [64]. Structural similarity to known folds was determined using either the PDBeFold (http://www.ebi.ac.uk/msd-srv/ssm) [65] or DALI (http://ekhidna.biocenter.helsinki.fi/dali_server/start) [47] servers.
Generation of tps-1 knockout C. elegans for RNAi studies
The eri-1(mg366); lin-15B(n744) tps-1(ok373) worms were constructed by crossing tps-1(ok373) worms with eri-1(mg366); lin-15B(n744) worms. The genes lin-15B and tps-1 are closely linked on the X chromosome. Animals (n = 600) were genotyped to identify a recombination event that occurred between lin-15B and tps-1. Progeny (n = 600) of worms heterozygous for tps-1(ok373) and lin-15B (n744) were picked individually to a petri plate. After the worms generated progeny on the new plate the genomic DNA from each parent worm was isolated as previously described [66]. The genomic DNA of each worm was screened by PCR for homozygosity of the tps-1(ok373) deletion allele and heterozygosity of the lin-15B (n744) allele. Homozygosity for tps-1(ok373) was determined by the presence of the deletion mutation and absence of the wild-type gene. Additionally, the worms were tested for the presence of the lin-15B (n744) mutation by performing PCR of the lin-15B locus and digesting the product with BccI, which only cuts the mutant n744 mutant lin-15B allele. Only animals that were homozygous for the tps-1(ok373) deletion allele and heterozygous for lin-15B(n744) would had the recombination event between the two genes that was necessary for the eri-1(mg366); lin-15B(n744) tps-1(ok373) strain construction. Finally, the homozygosity for the eri-1(mg366) mutation was established by PCR and sequencing. L4 worms (n = 45) of each genotype were picked to individual plates with HT115 bacteria expressing gob-1 dsRNA [67]. The progeny of each animal was examined for defects.
Synthesis of trehalose 6-sulfate
To a solution of SO3/C5H5N complex (432 mg, 2.7 mmol) in freshly distilled pyridine (5 mL) was added a solution of the benzyl-protected trehalose (1.345 g, 1.35 mmol) in freshly distilled pyridine (5 mL) was added. The mixture was stirred for 5 h at room temperature, neutralized by addition of Na2CO3(aq) (1 M, 10 mL) and concentrated under reduced pressure. The residue was titrated with anhydrous methanol and the triturate was filtered. The filtrate was concentrated in vacuo giving a residue that was subjected to silica gel column chromatography (30∶1 MeOH/EtOAc) affording the protected trehalose 6-sulfate (1.35 g, 93%) as a white powder. A solution of the white powder (0.96 g, 0.89 mmol) in MeOH/H2O (1∶1,V/V, 40 mL), 20% Pd(OH)2/C (1.26 g) was repeatedly purged with hydrogen. The resulting mixture was stirred under 1 atm of H2 at room temperature for 24 h and filtered through a Celite pad. The filtrate was concentrated in vacuo giving a residue that was subjected to silica gel column chromatography (1∶4∶4, water/isopropanol/ethyl acetate) to give trehalose 6-sulfate as a white powder (366 mg, 93%). TLC (water/isopropanol/ethyl acetate, 1∶4∶4, v/v/v): Rf = 0.40; 1H NMR (D2O): 3.39–3.52 (2H, m), 3.60–3.67 (2H, m), 3.72–3.86 (5H, m), 3.97–4.0 2(1H, m), 4.23–4.26 (2H, m), 5.16–5.18 (2H, m); 13C NMR (D2O): 60.3, 66.7, 69.1, 69.5 70.0, 70.68, 70.74, 72.0, 72.19, 72.25; HRMS(ES) m/z: [M+Na+] calculated for C12H21O14S, 421.0652; found, 421.0648.
Substrate specificity profile and steady-state kinetic characterization of T6PP
To determine the substrate specificity profile for T6PP, purified enzyme was diluted to 10 µM into a buffer consisting of 25 mM Tris pH 7.5, 50 mM NaCl, 5 mM MgCl2 and 1 mM DTT and aliquoted into 96-well plates in 5 µl volumes. Equivalent volumes of 20 mM substrate from a 167-compound in-house screen were then added and incubated for 45 minutes at room temperature. Next, 52.5 µl of BioMol Green (Enzo Life Sciences) was added to the wells to detect enzymatically released phosphate and incubated for another 30 minutes. After incubation, the absorbance of each well at 625 nm was measured using a Molecular Devices SpectraMax M5 microtiter plate reader. T6PP was found to be highly specific for trehalose 6-phosphate, eliminating the need to measure steady-state kinetic constants for other substrates.
For steady-state kinetic characterization, purified T6PP and the variants made by site-directed mutagenesis were diluted to between 25–75 nM into a buffer consisting of 25 mM Tris pH 7.5, 50 mM NaCl and 5 mM MgCl2. The steady-state kinetic parameters (Km and kcat) for trehalose 6-phosphate were determined from initial reaction velocities measured at varying concentrations (0.0625–5 mM) using the EnzCheck Phosphate Assay Kit (Invitrogen). Absorbance measurements were performed with a Beckman DU800 spectrophotometer using quartz cuvettes (Starna Cells, 18B-Q-10) and 250 µl volumes. Data were fit to the following using SigmaPlot Enzyme Kinetics Module:


The steady-state competitive inhibition constant KI was determined for trehalose 6-sulfate by fitting the initial velocity data, measured as a function of trehalose 6-phosphate (0.5 Km to 5 Km) and inhibitor (0, KI, 2 KI) concentration to the following using SigmaPlot Enzyme Kinetics Module:

Homology modeling of related T6PP enzymes
To generate homology models of related T6PP enzymes, FASTA sequences for E. coli (UniProt: E8Y507), S. cerevisiae (A6ZY39), and M. tuberculosis (H8EZ37) were uploaded to the Protein Homology/analogY Recognition Engine V 2.0 (Phyre2) server, where models were automatically generated using the best possible template model [36]. In this case, each model was based on 1U02 with a >95% confidence level. To model the putative MIT-like domain in M. tuberculosis, one-to-one threading on the Phyre2 server was used utilizing the structure of T6PP from B. malayi as the template.
Hot spot mapping using the FTMap server
To map substrate binding residues in the HAD domain, the FTMap server (http://ftmap.bu.edu) was used to identify hot spots. To map putative MIT interactions, PDB files consisting of only the MIT domains (residues 63–156 for B. malayi; 2W2U chain A; 2V6Y chain A) were uploaded and analyzed in PPI mode. To map putative substrate binding residues, PDB files (residues 63–491 for B. malayi; 1U02 chain A) were uploaded and analyzed using default settings.
Modeling of the T6PP-T6P complex
To model the T6PP-T6P complex, trehalose 6-phosphate was built (α-D-glucopyranosyl-(1→1)-α-D-glucopyranoside 6-phosphate) and manually placed in the active site of T6PP. The phosphate was placed such that it was positioned for “in-line” attack by Asp213 (the Asp nucleophile) and coordinated to the Mg2+ cation. In addition, Asp215 (Asp+2, general acid/base residue) was positioned to promote leaving-group protonation. The sugar was manually rotated to lie against the C1-loop guided by mutagenesis and FTMap hot spot results. The model was then minimized using NAMD [43] and analyzed using UCSF Chimera [68].
Molecular dynamics simulations
Hydrogen atoms were added to the docked T6PP-T6P model, using the HBUILD function in CHARMM [69] and the coordinates gently minimized in general Born implicit solvent (GBIS) [70] by gradually removing harmonic restraints on the system heavy atoms and performing 25 steps of steepest descent followed by 250 to 500 steps of advanced basis Newton-Raphson (ABNR) steps. A final, unrestrained minimization using ABNR steps was performed until the energy gradient stabilized below 10−5 kcal/mol-step. A further 200 steps of minimization was performed in NAMD [43] and rigid bonds. GBIS solvation was employed with an effective ion concentration of 150 mM and a 12 Å cutoff for calculating the Born derivatives. All other parameters were set to NAMD defaults. Surface tension terms in the solvation model were ignored. A non-bonded cutoff distance of 15 Å was employed with a switching function employed to 16 Å. Langevin dynamics was initiated at 300 K utilizing a damping coefficient of 1/ps, a 2 fs timestep and rigid bonds enforced. This configuration was integrated for a total of 1.2 ns.
Accession numbers
Coordinates for T6PP and structure factors have been deposited in the Protein Data Bank (PDB ID: 4OFZ).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MartinJ, AbubuckerS, HeizerE, TaylorCM, MitrevaM (2012) Nematode.net update 2011: addition of data sets and tools featuring next-generation sequencing data. Nucleic Acids Res 40: D720–8 doi:10.1093/nar/gkr1194
2. OttesenEa, HooperPJ, BradleyM, BiswasG (2008) The global programme to eliminate lymphatic filariasis: health impact after 8 years. PLoS Negl Trop Dis 2: e317 doi:10.1371/journal.pntd.0000317
3. WHO (2009) World Health Organization Global Program to Eliminate Lymphatic Filariasis. Available: http://www.who.int/lymphatic_filariasis/disease/en/. Accessed 2 June 2014.
4. GyapongJO, KumaraswamiV, BiswasG, OttesenEa (2005) Treatment strategies underpinning the global programme to eliminate lymphatic filariasis. Expert Opin Pharmacother 6 : 179–200 doi:10.1517/14656566.6.2.179
5. KamgnoJ, BoussinesqM (2008) Encephalopathy after Ivermectin Treatment in a Patient Infected with Loa Loa and Plasmodium spp. Am J Trop Med Hyg 78 : 546–551.
6. BockarieMJ, DebRM (2010) Elimination of lymphatic filariasis: do we have the drugs to complete the job? Curr Opin Infect Dis 23 : 617–620 doi:10.1097/QCO.0b013e32833fdee5
7. ColesGC, JacksonF, PomroyWE, PrichardRK, von Samson-HimmelstjernaG, et al. (2006) The detection of anthelmintic resistance in nematodes of veterinary importance. Vet Parasitol 136 : 167–185 doi:10.1016/j.vetpar.2005.11.019
8. TraversaD, von Samson-HimmelstjernaG, DemelerJ, MililloP, SchürmannS, et al. (2009) Anthelmintic resistance in cyathostomin populations from horse yards in Italy, United Kingdom and Germany. Parasit Vectors 2 Suppl 2: S2 doi:10.1186/1756-3305-2-S2-S2
9. BradyHa, NicholsWT (2009) Drug Resistance in Equine Parasites: An Emerging Global Problem. J Equine Vet Sci 29 : 285–295 doi:10.1016/j.jevs.2009.04.186
10. TaylorMJ, AwadziK, BasáñezM-G, BiritwumN, BoakyeD, et al. (2009) Onchocerciasis Control: Vision for the Future from a Ghanian perspective. Parasit Vectors 2 : 7 doi:10.1186/1756-3305-2-7
11. GhedinE, WangS, SpiroD, CalerE, ZhaoQ, et al. (2007) Draft genome of the filarial nematode parasite Brugia malayi. Science (80-) 317 : 1756–1760 doi:10.1126/science.1145406
12. DesjardinsCa, CerqueiraGC, GoldbergJM, Dunning HotoppJC, HaasBJ, et al. (2013) Genomics of Loa loa, a Wolbachia-free filarial parasite of humans. Nat Genet 45 : 495–500 doi:10.1038/ng.2585
13. JexAR, LiuS, LiB, YoungND, HallRS, et al. (2011) Ascaris suum draft genome. Nature 479 : 529–533 doi:10.1038/nature10553
14. MitrevaM, JasmerDP, ZarlengaDS, WangZ, AbubuckerS, et al. (2011) The draft genome of the parasitic nematode Trichinella spiralis. Nat Genet 43 : 228–235 doi:10.1038/ng.769
15. OppermanCH, BirdDM, WilliamsonVM, RokhsarDS, BurkeM, et al. (2008) Sequence and genetic map of Meloidogyne hapla: A compact nematode genome for plant parasitism. Proc Natl Acad Sci U S A 105 : 14802–14807 doi:10.1073/pnas.0805946105
16. DieterichC, CliftonSW, SchusterLN, ChinwallaA, DelehauntyK, et al. (2008) The Pristionchus pacificus genome provides a unique perspective on nematode lifestyle and parasitism. Nat Genet 40 : 1193–1198 doi:10.1038/ng.227
17. C. elegans Sequencing Consortium (1998) Genome Sequence of the Nematode C. elegans: A Platform for Investigating Biology. Science 282 : 2012–2018 doi:10.1126/science.282.5396.2012
18. GuptaBP, SternbergPW (2003) The draft genome sequence of the nematode Caenorhabditis briggsae, a companion to C. elegans. Genome Biol 4 : 238 doi:10.1186/gb-2003-4-12-238
19. AshLR, RileyJM (1970) Development of Subperiodic Brugia malayi in the Jird, meriones unguiculatus, with notes on infections in other rodents. J Parasitol 56 : 969–973.
20. FalconeFH, SchlaakM, HaasH (1995) In vitro cultivation of Brugia malayi, a parasitic nematode that causes human lymphatic filariasis. ALTEX 12 : 179–187.
21. AboobakerAA, BlaxterML (2003) Use of RNA interference to investigate gene function in the human filarial nematode parasite Brugia malayi. Mol Biochem Parasitol 129 : 41–51 doi:10.1016/S0166-6851(03)00092-6
22. LustigmanS, ZhangJ, LiuJ, OksovY, HashmiS (2004) RNA interference targeting cathepsin L and Z-like cysteine proteases of Onchocerca volvulus confirmed their essential function during L3 molting. Mol Biochem Parasitol 138 : 165–170 doi:10.1016/j.molbiopara.2004.08.003
23. PfarrK, HeiderU, HoeraufA (2006) RNAi mediated silencing of actin expression in adult Litomosoides sigmodontis is specific, persistent and results in a phenotype. Int J Parasitol 36 : 661–669 doi:10.1016/j.ijpara.2006.01.010
24. KumarS, ChaudharyK, FosterJM, NovelliJF, ZhangY, et al. (2007) Mining predicted essential genes of Brugia malayi for nematode drug targets. PLoS One 2: e1189 doi:10.1371/journal.pone.0001189
25. PelleroneF, ArcherS, BehmC, GrantW, LaceyM, et al. (2003) Trehalose metabolism genes in Caenorhabditis elegans and filarial nematodes. Int J Parasitol 33 : 1195–1206 doi:10.1016/S0020-7519(03)00173-5
26. BehmC (1997) The Role of Trehalose in the Physiology of Nematodes. Int J Parasitol 27 : 215–229.
27. ElbeinAD, PanYT, PastuszakI, CarrollD (2003) New insights on trehalose: a multifunctional molecule. Glycobiology 13 : 17R–27R doi:10.1093/glycob/cwg047
28. KormishJD, McGheeJD (2005) The C. elegans lethal gut-obstructed gob-1 gene is trehalose-6-phosphate phosphatase. Dev Biol 287 : 35–47 doi:10.1016/j.ydbio.2005.08.027
29. RaoK, KumaranD (2006) Crystal structure of trehalose-6-phosphate phosphatase – related protein: Biochemical and biological implications. Protein Sci 15 : 1735–1744 doi:10.1110/ps.062096606.ily
30. ObitaT, SaksenaS, Ghazi-TabatabaiS, GillDJ, PerisicO, et al. (2007) Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature 449 : 735–739 doi:10.1038/nature06171
31. SamsonRY, ObitaT, FreundSM, WilliamsRL, BellSD (2008) A Role for the ESCRT System in Cell Division in Archaea. Science 322 : 1710–1713.
32. SchmidtO, TeisD (2012) The ESCRT machinery. Curr Biol 22: R116–20 doi:10.1016/j.cub.2012.01.028
33. YangZ, VildC, JuJ, ZhangX, LiuJ, et al. (2012) Structural basis of molecular recognition between ESCRT-III-like protein Vps60 and AAA-ATPase regulator Vta1 in the multivesicular body pathway. J Biol Chem 287 : 43899–43908 doi:10.1074/jbc.M112.390724
34. CopleySD (2012) Moonlighting is mainstream: paradigm adjustment required. BioEssays 34 : 578–588 doi:10.1002/bies.201100191
35. AllenKN, Dunaway-MarianoD (2009) Markers of fitness in a successful enzyme superfamily. Curr Opin Struct Biol 19 : 658–665 doi:10.1016/j.sbi.2009.09.008
36. KelleyLa, SternbergMJE (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4 : 363–371 doi:10.1038/nprot.2009.2
37. LuZ, Dunaway-MarianoD, AllenKN (2008) The catalytic scaffold of the haloalkanoic acid dehalogenase enzyme superfamily acts as a mold for the trigonal bipyramidal transition state. Proc Natl Acad Sci U S A 105 : 5687–5692 doi:10.1073/pnas.0710800105
38. LahiriSD, ZhangG, Dunaway-MarianoD, AllenKN (2003) The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction. Science 299 : 2067–2071 doi:10.1126/science.1082710
39. DaughtryKD, HuangH, MalashkevichV, LiuW, RamagopalU, et al. (2013) Structural Basis for the Divergence of Substrate Speci fi city and Biological Function within HAD Phosphatases in Lipopolysaccharide and Sialic Acid Biosynthesis. Biochemistry 52 : 5372–5386.
40. NguyenHH, WangL, HuangH, PeisachE, Dunaway-MarianoD, et al. (2010) Structural determinants of substrate recognition in the HAD superfamily member D-glycero-D-manno-heptose-1,7-bisphosphate phosphatase (GmhB). Biochemistry 49 : 1082–1092 doi:10.1021/bi902019q
41. BrenkeR, KozakovD, ChuangG-Y, BeglovD, HallD, et al. (2009) Fragment-based identification of druggable “hot spots” of proteins using Fourier domain correlation techniques. Bioinformatics 25 : 621–627 doi:10.1093/bioinformatics/btp036
42. KozakovD, HallDR, ChuangG-Y, CencicR, BrenkeR, et al. (2011) Structural conservation of druggable hot spots in protein-protein interfaces. Proc Natl Acad Sci U S A 108 : 13528–13533 doi:10.1073/pnas.1101835108
43. PhillipsJC, BraunR, WangW, GumbartJ, TajkhorshidE, et al. (2008) Scalable Molecular Dynamics with NAMD. J Comput Chem 26 : 1781–1802.
44. LeeRa, RazazM, HaywardS (2003) The DynDom database of protein domain motions. Bioinformatics 19 : 1290–1291 doi:10.1093/bioinformatics/btg137
45. QiG, LeeR, HaywardS (2005) A comprehensive and non-redundant database of protein domain movements. Bioinformatics 21 : 2832–2838 doi:10.1093/bioinformatics/bti420
46. TaylorD, CawleyG, HaywardS (2013) Classification of Domain Movements in Proteins Using Dynamic Contact Graphs. PLoS One 8 : 1–10 doi:10.1371/journal.pone.0081224
47. HolmL, RosenströmP (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res 38: W545–9 doi:10.1093/nar/gkq366
48. FieulaineS, LunnJE, BorelF, FerrerJ (2005) The Structure of a Cyanobacterial Sucrose-Phosphatase Reveals the Sugar Tongs That Release Free Sucrose in the Cell. Plant Cell 17 : 2049–2058 doi:10.1105/tpc.105.031229.1
49. LunnJE (2002) Evolution of Sucrose Synthesis [w]. Plant Physiol 128 : 1490–1500 doi:10.1104/pp.010898.1490
50. LunnJE, PriceGD, FurbankRT (1999) Cloning and expression of a prokaryotic sucrose-phosphate synthase gene from the cyanobacterium Synechocystis sp. PCC 6803. Plant Mol Biol 40 : 297–305.
51. ZhangM, YogeshaSD, MayfieldJE, GillGN, ZhangY (2013) Viewing serine/threonine protein phosphatases through the eyes of drug designers. FEBS J 280 : 4739–4760 doi:10.1111/febs.12481
52. NobleMEM, EndicottJa, JohnsonLN (2004) Protein kinase inhibitors: insights into drug design from structure. Science 303 : 1800–1805 doi:10.1126/science.1095920
53. HanK-C, KimSY, YangEG (2012) Recent advances in designing substrate-competitive protein kinase inhibitors. Curr Pharm Des 18 : 2875–2882.
54. ChaptalV, KwonS, SawayaMR, GuanL, KabackHR, et al. (2011) Crystal structure of lactose permease in complex with an affinity inactivator yields unique insight into sugar recognition. Proc Natl Acad Sci U S A 108 : 9361–9366 doi:10.1073/pnas.1105687108
55. BorastonAB, BolamDN, GilbertHJ, DaviesGJ (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J 382 : 769–781 doi:10.1042/BJ20040892
56. MuraseT, ZhengRB, JoeM, BaiY, MarcusSL, et al. (2009) Structural insights into antibody recognition of mycobacterial polysaccharides. J Mol Biol 392 : 381–392 doi:10.1016/j.jmb.2009.06.074
57. AsensioJL, ArdáA, CañadaFJ, Jiménez-BarberoJ (2013) Carbohydrate-aromatic interactions. Acc Chem Res 46 : 946–954 doi:10.1021/ar300024d
58. NotredameC, HigginsD, HeringaJ (2000) T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 205–217 doi:10.1006/jmbi.2000.4042
59. GouetP (2003) ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res 31 : 3320–3323 doi:10.1093/nar/gkg556
60. RinasU, HellmuthK, SandersEA (1995) Simple fed-batch technique for high cell density cultivation of Escherichia coli. J Biotechnol 39 : 59–65.
61. MinorW, CymborowskiM, OtwinowskiZ, ChruszczM (2006) HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62 : 859–866 doi:10.1107/S0907444906019949
62. AdamsPD, AfoninePV, BunkócziG, ChenVB, DavisIW, et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66 : 213–221 doi:10.1107/S0907444909052925
63. EmsleyP, CowtanK (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60 : 2126–2132 doi:10.1107/S0907444904019158
64. ChenVB, ArendallWB, HeaddJJ, KeedyDa, ImmorminoRM, et al. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66 : 12–21 doi:10.1107/S0907444909042073
65. KrissinelE, HenrickK (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60 : 2256–2268 doi:10.1107/S0907444904026460
66. WilliamsBD (1995) Genetic mapping with polymorphic sequence-tagged sites. Methods Cell Biol 48 : 81–96.
67. TimmonsL, CourtDL, Firea (2001) Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263 : 103–112.
68. PettersenEF, GoddardTD, HuangCC, CouchGS, GreenblattDM, et al. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25 : 1605–1612 doi:10.1002/jcc.20084
69. BrooksBR, IiiCLB, MackerellAD, NilssonL, PetrellaRJ, et al. (2009) CHARMM: The Biomolecular Simulation Program. J Comput Chem 30 : 1545–1614 doi:10.1002/jcc
70. DominyBN, BrooksCL (1999) Development of a Generalized Born Model Parametrization for Proteins and Nucleic Acids. J Phys Chem B 103 : 3765–3773 doi:10.1021/jp984440c
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 7
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Molecular and Cellular Mechanisms of KSHV Oncogenesis of Kaposi's Sarcoma Associated with HIV/AIDS
- Holobiont–Holobiont Interactions: Redefining Host–Parasite Interactions
- BCKDH: The Missing Link in Apicomplexan Mitochondrial Metabolism Is Required for Full Virulence of and
- Helminth Infections, Type-2 Immune Response, and Metabolic Syndrome