
Rhadinovirus Host Entry by Co-operative Infection
All viral infections start with host entry. Entry into cells is studied widely in isolated cultures; entry into live hosts is more complicated and less well understood: our tissues have specific anatomical structures and our cells differ markedly from most cultured cells in size, shape and behaviour. The respiratory tract is a common site of virus infection. Size dictates where inhaled particles come to rest, and virus-sized particles can reach the lungs. Rhadinoviruses chronically infect both humans and economically important animals, and cause lung disease. We used a well-characterized murine example to determine how a rhadinovirus enters the lungs. At its peak, infection was prominent in epithelial cells lining the lung air spaces. However it started in macrophages, which normally clear the lungs of inhaled debris. Only epithelial cells expressed the molecules required for virus binding, but only macrophages internalized virus particles after binding; infection involved interaction between these different cell types. Blocking epithelial infection with an antibody did not stop host entry because attached antibodies increase virus uptake by lung macrophages; but an antibody that blocks macrophage infection was effective. Thus, understanding how rhadinovirus infections work in normal tissues provided important information for their control.
Published in the journal:
. PLoS Pathog 11(3): e32767. doi:10.1371/journal.ppat.1004761
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004761
Summary
All viral infections start with host entry. Entry into cells is studied widely in isolated cultures; entry into live hosts is more complicated and less well understood: our tissues have specific anatomical structures and our cells differ markedly from most cultured cells in size, shape and behaviour. The respiratory tract is a common site of virus infection. Size dictates where inhaled particles come to rest, and virus-sized particles can reach the lungs. Rhadinoviruses chronically infect both humans and economically important animals, and cause lung disease. We used a well-characterized murine example to determine how a rhadinovirus enters the lungs. At its peak, infection was prominent in epithelial cells lining the lung air spaces. However it started in macrophages, which normally clear the lungs of inhaled debris. Only epithelial cells expressed the molecules required for virus binding, but only macrophages internalized virus particles after binding; infection involved interaction between these different cell types. Blocking epithelial infection with an antibody did not stop host entry because attached antibodies increase virus uptake by lung macrophages; but an antibody that blocks macrophage infection was effective. Thus, understanding how rhadinovirus infections work in normal tissues provided important information for their control.
Introduction
The γ-herpesviruses chronically infect most mammals and cause disease in humans [1] and economically important ungulates [2]. Vaccines have failed to prevent infection. In part this reflects that host entry is ill-understood. Oral entry is often assumed, because the archetypal Epstein-Barr virus (EBV) causes acute tonsillitis. However clinical presentation occurs at least 1 month after EBV acquisition [3] and correlates better with peak salivary shedding. Thus, it may reflect host exit rather than entry, and because virus shedding involves reactivation from circulating B cells [4] entry and exit routes need not coincide.
Lymphocryptoviruses such as EBV are known only in primates; Rhadinoviruses such as the Kaposi’s Sarcoma-associated Herpesvirus (KSHV) are more widespread [5]. KSHV in nasal secretions [6], and pulmonary Kaposi’s Sarcoma complicating AIDS [7], suggest respiratory infection. Consistent with this idea, Ovine herpesvirus-2 and Alcelaphine herpesvirus-1 show salivary and nasal shedding, and infect in nebulized form [8]; Equid γHV-5 causes respiratory disease [9] and can be isolated from the respiratory tract [10]; and Bovine herpesvirus-4 colonizes the respiratory tract [11], and persists in calves after intranasal (i.n.) inoculation [12].
MuHV-4 (strain MHV-68) [13, 14] allows rhadinovirus infection to be analysed in mice. Like EBV and KSHV, it is B cell tropic. MuHV-4 infects readily mice i.n., and infects orally only if it spills into the respiratory tract [15]. When given i.n. under anesthesia it infects the lungs [16]; without anesthesia it infects the upper respiratory tract [17]; and it transmits sexually from female mice to co-caged males [18]. How MuHV-4 transmits in its natural host of yellow-necked mice [19] is unknown. Analysis of wood mice identified DNA from MuHV-4 or a related virus in lungs [20], and lung inoculation is used widely for experimental infections. Therefore MuHV-4 is suited to analyzing this mode of host entry.
A key unknown is the first infected cell type. Lungs contain type 1 AECs, which comprise 95% of the alveolar surface area; type 2 AECs, which secrete surfactant; macrophages, which phagocytose inhaled debris [21]; and some lymphocytes. By 5 days after i.n. inoculation, MuHV-4 antigens are detectable in type 1 AECs and large mononuclear cells [16]. DNA of i.n.-inoculated, replication-deficient MuHV-4 has been detected by PCR of B cells from lung homogenates [22, 23], suggesting that they are a primary infection target. However MuHV-4 normally binds to heparan, which B cells express poorly [24, 25], and shows a post-binding block to B cell infection that is not overcome until virions pass through myeloid cells [26]. PCR detects replication-deficient MuHV-4 DNA also in association with B cells recovered from splenic homogenates after intraperitoneal (i.p.) inoculation [22, 23], when in situ analysis shows no evidence of B cell infection [27]. Therefore the PCR signals associated with lung B cells might reflect adsorbed inoculum debris rather than infection.
Any understanding of MuHV-4 infection must encompass its dependence on heparan for cell binding [24], a characteristic common to many rhadinoviruses [28–30]. The MuHV-4 gp70 and gH/gL bind to heparan [31, 32], and gp150 further inhibits heparan-independent binding until heparan is engaged [24]. Thus, gL-gp70- MuHV-4 infects poorly both in vitro and in vivo, unless gp150 is disrupted also [33]. The Bovine Herpesvirus-4 gp150 homolog, gp180, is functionally similar [34]. While most transformed epithelial cells express abundant heparan, most in vivo epithelia do so only basolaterally [35]. Thus, heparan-dependent host entry by incoming, apical virions is not necessarily straightforward. The olfactory neuroepithelium, which we have analysed previously [17], unusually expresses both basolateral and apical heparan. The genital epithelium lacks apical heparan [36]; entry here may depend on epithelial trauma, as MuHV-4-infected cells are seen underneath rather than in the stratified squamous epithelium [18]. We asked which cell type MuHV-4 targets in the lungs and how this relates to heparan binding. Our aim was to understand better rhadinovirus entry into new hosts and so establish a rational basis for infection control.
Results
Viral lytic antigens appear in lung myeloid cells before epithelial cells
Four days after i.n. MuHV-4 inoculation, viral antigens were expressed in morphologically typical type 1 AECs, whose extensive cytoplasmic projections line the lung air spaces (Fig. 1A). This tropism was confirmed by co-staining lungs for viral antigens and the type 1 AEC marker podoplanin (PDP) [37] (Fig. 1B)—at 4 days post-inoculation >50% of viral antigen+ cells were PDP+ (Fig. 1C). The morphologically distinct remainder expressed the macrophage / granulocyte marker CD68 [38–40]. However at 1 day post-inoculation viral antigens were confined to CD68+ cells. Therefore while infection spread to type 1 AECs, it appeared to start in macrophages or granulocytes.

Incoming virions infect alveolar macrophages
MuHV-4-immune sera recognize mainly lytic antigens. As much early lung infection is latent [41], we tracked host entry more comprehensively via lytic cycle-independent [42] eGFP expression from a viral HCMV IE1 promoter (Fig. 2A). At 1 day post-inoculation, 238/238 eGFP+ cells on lung sections of 3 mice were CD68+. None was PDP+. MuHV-4 rendered incapable of uncomplemented lytic spread by disruption of its ORF50 transactivator (ORF50-) showed a similar eGFP distribution: 78/78 eGFP+ cells on lung sections from 5 mice expressed CD68, and none expressed PDP (Fig. 2B). Therefore CD68+ cells were direct infection targets. More than 80% of eGFP+ cells expressed also the tissue macrophage marker F4/80 [39] and the alveolar macrophage / dendritic cell marker CD11c [38, 40] (Fig. 2C). Macrophage populations are heterogeneous and often not clearly demarcated [38], but combined CD11c, CD68 and F4/80 expression argued that most infected cells were alveolar macrophages.
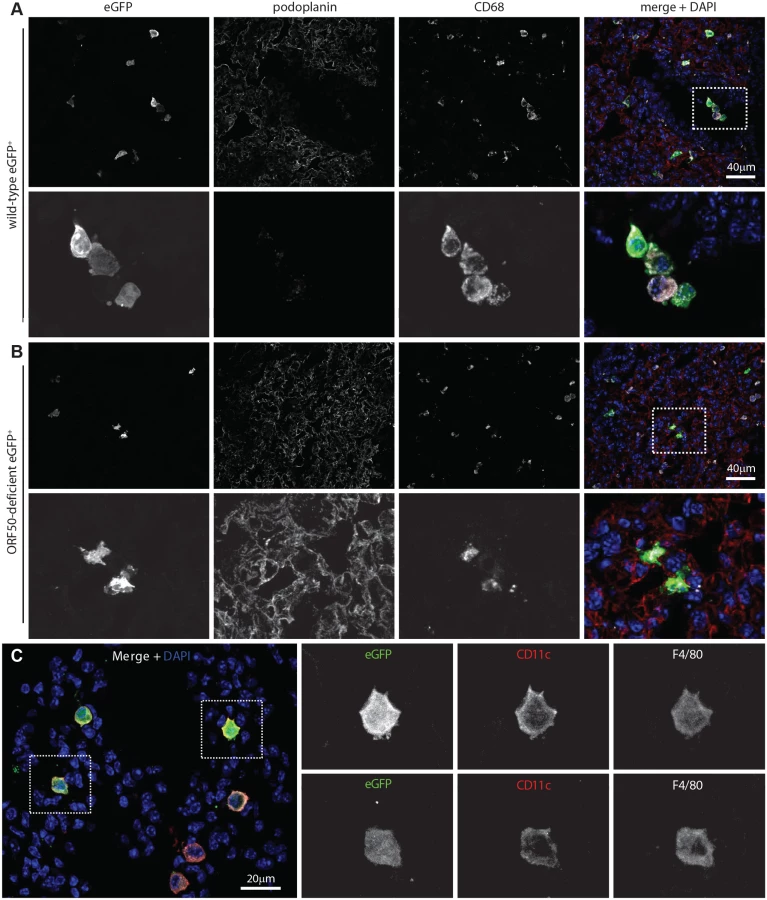
The lack of HCMV IE1-driven eGFP expression in type 1 AECs at day 1 post-inoculation established that they were uninfected rather than just slow to initiate lytic infection. We saw also no eGFP expression in B cells at day 1 by either eGFP+ wild-type (WT, >100 eGFP+ cells counted from 3 mice) or eGFP+ORF50- MuHV-4 (>50 eGFP+ cells counted from 3 mice) (S1 Fig). Nor did MuHV-4 with EF1α promoter-driven eGFP expression show a day 1 infection of B220+ lung B cells or type 1 AECs (S1 Fig, >100 eGFP+ cells counted from 3 mice). Therefore incoming virions targeted selectively alveolar macrophages.
Genetic tagging shows early virus replication in myeloid cells
Viral lytic gene expression suggests but does not prove lytic replication. To determine how alveolar macrophage infection contributed to virus production we infected lysM-cre mice with MuHV-4 in which cre-mediated recombination switches fluorochrome expression from mCherry+ (red) to eGFP+ (green) (MHV-RG) [26]. LysM-cre mice express cre from the myeloid lysozyme locus [43]. To identify cre+ lung cells we crossed them with a reporter strain in which cre activates Zsgreen expression from a widely expressed promoter [44]. PDP+ cells lacked Zsgreen (S2 Fig). CD68+ cells expressed Zsgreen in an endosomal distribution. Type 2 AECs (surfactant protein C precursor+, CD68-, MAC-2-) also expressed Zsgreen—in a uniform rather than endosomal distribution. However in C57BL/6 mice given HCMV-IE1-eGFP+ MuHV-4 they remained eGFP- (S3 Fig) and so were not infected. Thioglycollate-induced Gr-1+ peritoneal exudate neutrophils of lysM-cre x Zsgreen mice were Zsgreen+, consistent with published data [43], but lung cells expressing neutrophil markers (Gr-1 or myeloperoxidase) were not (S2 Fig). They also lacked eGFP expression by HCMV-IE1-eGFP+ MuHV-4. Therefore we interpreted MHV-RG switching in lysM-cre mice as an infection of alveolar macrophages. Some switching in lung dendritic cells was possible, but they express little lysM [43] or CD68 [39], so there was no other indication that dendritic cells were a significant target for incoming virions.
The MHV-RG recovered from lysM-cre lungs at 1 day post-inoculation was >96% eGFP+, indicating that essentially all of it had passed through an alveolar macrophage (Fig. 3A). Surprisingly the MHV-RG recovered after 4 days showed only 85% switching. This reduction in % switched virus with time was consistent and statistically significant (Fig. 3B), suggesting that virions could also enter the lungs without infecting a macrophage, but that virus production was then delayed. We confirmed myeloid infection as the predominant route by infecting CD11c-cre mice (Fig. 3C), as alveolar macrophages (and dendritic cells) are CD11c+ [38]. Again the virus recovered from lungs after 1 day was >96% eGFP+, and that recovered after 4 days was >85% eGFP+.

The prominent viral lytic antigen expression in type 1 AECs at day 4 post-inoculation (Fig. 1) but not day 1 implied that they receive and amplify virus from alveolar macrophages. Their lack of HCMV IE1-driven or EF1α-driven eGFP expression at day 1 (Figs. 2, S1) excluded acute infection. Day 1 MHV-RG-infected lung sections (Fig. 3D) confirmed this: >95% of fluorescent cells were morphologically typical alveolar macrophages and PDP-. Of these 92.6 ± 4.3% were eGFP+ (mean ± SD of counts from 8 mice), confirming efficient viral fluorochrome switching in infected myeloid cells. However at day 4, when 63.7% of fluorescent cells were PDP+, 48.5 ± 10.6% of these were eGFP-mCherry+ (mean ± SD of counts from 8 mice). Thus, not all type 1 AEC infection was down-stream of myeloid cell infection. And while no eGFP+ cells were PDP+ 1 day after i.n. eGFP+ORF50- MuHV-4 inoculation of C57BL/6 mice (Fig. 2), 35.5 ± 29.7% of eGFP+ cells were PDP+ after 5 days (mean ± SD, 6 sections from 3 mice) (S4 Fig). Therefore a delayed type 1 AEC infection without prior macrophage infection seemed to account for the reduction in % virus switching between days 1 and 4 in Fig. 3A-3C. Either virions eventually penetrated AECs, or macrophages licenced AEC infection without becoming infected themselves, for example by recycling virions from early endosomes to exosomes and releasing them in a licensed form [45].
Alveolar macrophage depletion compromises host entry
Most lung macrophages express CD169 [46], so we used CD169-DTR mice [47] to test their importance for host entry (Fig. 4). Mice were given diphtheria toxin i.p. for 2 days, then given eGFP+ ORF50- MuHV-4 i.n. for 1 day. Staining lungs for CD169, CD68, F4/80 and CD206 (Fig. 4A) showed that CD169 expression was ablated completely. CD206 and F4/80 expression were ablated >90%, consistent with most lung macrophages expressing CD169. EGFP+ cell numbers in toxin-treated mice were <10% those of untreated controls (Fig. 4B). CD68+ cells were still seen, but they were larger than in undepleted mice and lacked other alveolar macrophage markers. Such cells were not evident in undepleted infected mice, and so seemed likely to be bone marrow-derived monocytes or granulocytes, recruited in response to alveolar macrophage ablation. They were clearly poor infection targets. Therefore alveolar macrophages played a crucial role in virus acquisition, for which lineage-distinct bone marrow-derived myeloid cells [48] did not substitute.

Alveolar and other macrophages are infected poorly ex vivo
Host entry via lung macrophages was unexpected, as MuHV-4 infects the lungs efficiently [49] and myeloid cells poorly [26]. The cell-free virions used for in vivo infection poorly infected ex vivo lung macrophages, peritoneal macrophages or the monocyte line RAW-264 (Fig. 5A). Virus stocks containing cell debris infected macrophages with reasonable efficiency. However cell-associated virus infections would occur only after host entry. Efficient alveolar macrophage infection by cell-free virions evidently depended on their normal tissue context.

MuHV-4 that lacks heparan binding due to gp70 and gL disruption infects mice i.n. 100-fold less well than the WT, unless its gp150, which inhibits heparan-independent binding, is disrupted too [33]. This result implies that host entry requires heparan engagement. We confirmed it by pre-incubating WT virions with soluble heparin (Fig. 5B). Seroconversion by virus-specific serum IgG ELISA—the the most sensitive measure of low dose infection in immunocompetent mice [33]—was reduced 10–100-fold by the heparin treatment. Pre-incubating virions with heparin also reduced NMuMG epithelial cell infection 100-fold (Fig. 5C). However, while low dose heparin inhibited RAW-264 monocyte infection better than NMuMG cell infection—presumably because RAW-264 cells have little heparan for virion binding to start with [26]—there was also a heparin-resistant component of infection, such that the maximal inhibition was only 4-fold. Therefore the efficiency of lung infection and its susceptibility to inhibition by heparin both suggested an initial interaction with epithelial cells rather than macrophages.
Alveolar epithelial cells express heparan; alveolar macrophages do not
To identify heparan+ cells in the lungs we stained sections with mAb 10E4, which recognizes sulfated heparan [50] (Fig. 6A), and with mAb NAH46, which recognizes non-sulfated heparan [51] (Fig. 6B). Staining by both mAbs co-localized with PDP (type 1 AECs) and not with CD68 (alveolar macrophages). To distinguish apical from basolateral heparan, we co-stained sections for the alveolar basement membrane component collagen IV (Fig. 6C). Sulfated heparan (mAb 10E4) co-localized completely with collagen IV and so was just basolateral. Unsulfated heparan (mAb NAH46) also co-localized largely with collagen IV. However there were also NAH46+ surfaces abutting the air spaces that lacked collagen IV. Therefore specifically unsulfated heparan was accessible to incoming virions on the apical AEC surface.

Incoming virions bind to type 1 AECs but accumulate in macrophages
One day after WT MuHV-4 inoculation, viral antigens were evident as isolated dots—presumably virions. Of these 94.8 ± 3.9% associated with PDP+ type 1 AECs (>300 dots counted on 6 sections from 3 mice). By contrast strong cellular staining—presumably lytic infection—was confined (78/78 cells) to CD68+ macrophages (Fig. 7A). Therefore incoming virions bound to type 1 AECs but infected macrophages. Similarly 94.0 ± 5.1% of inhaled replication-deficient MuHV-4 virions associated with PDP+ cells (ORF50-, Fig. 7B, >300 dots on 6 sections from 6 mice).

In mice given WT eGFP+ MuHV-4, 109/113 lytically infected cells (96.5%) were eGFP+ and 109/140 eGFP+ cells (77.9%) were strongly lytic antigen+ (5 sections from 3 mice) (Fig. 7C). Therefore eGFP expression identified most lytically infected cells, plus eGFP+lytic antigen- cells that were presumably latently infected. Only 8.7 ± 5.9% of antigen+ dots (virions) were associated with eGFP+ cells. Therefore most of the cells binding input virions remained uninfected.
In mice given eGFP+ORF50- MuHV-4, the lack of new lytic infection allowed us to identify diffusely antigen+ cells as those accumulating input virions (Fig. 7D). 32/35 antigen+ cells (91.4%) were eGFP+, and 32/46 eGFP+ cells (69.6%) were antigen+ (8 sections from 4 mice). The greater number of eGFP+ cells reflected presumably that detectable eGFP expression could come from a single infecting virion, but only cells accumulating multiple virions would be detectably antigen+. Nonetheless antigen accumulation and eGFP expression showed a significant correlation (p<0.0001 by Fisher's exact test). By contrast only 7.1 ± 3.8% of extracellular virions (isolated antigen+ dots) were associated with eGFP+ cells. Therefore type 1 AECs bound most input virions, but alveolar macrophages then accumulated the virions and became infected, consistent with their function of collecting particulate alveolar antigens [52].
MuHV-4 infection is endocytic [53] and virion antigens did not accumulate inside PDP+ cells. Therefore poor virion endocytosis seemed a likely explanation for type 1 AEC non-infection. Consistent with this idea, i.n. Herpes simplex virus type 1, which binds to heparan but penetrates most cells at the plasma membrane [54], showed abundant early type 1 AEC infection without marked macrophage involvement (S5 Fig).
Neutralizing virions for host entry
The γ-herpesviruses transmit from immune carriers and elicit salivary antibody responses [55]. Therefore shed virions may carry glycoprotein-specific antibodies. Also virions entering immune hosts or vaccinees could acquire antibodies before reaching mucosal surfaces. Therefore how bound antibodies affect host entry is important to understand. In vitro, immune sera inhibit MuHV-4 epithelial and fibroblast infections, but fail to inhibit and even enhance myeloid infection due to productive IgG Fc receptor (FcR)-mediated uptake [56]. This reflects that sera neutralize in vitro mainly by blocking cell binding, for which FcR binding can substitute. We tested 2 MuHV-4 glycoprotein-specific mAbs for their effect on lung infection: 8F10, which binds to gH/gL and blocks its interaction with heparan [57], and SC-9E8, which binds near the gB fusion loops and blocks membrane fusion [58] (Fig. 8).

In vitro both mAbs inhibited fibroblast infection, but while SC-9E8 inhibited RAW-264 monocyte infection 8F10 enhanced it (Fig. 8A). Enhancement reflects that FcR binding improves significantly cell-free MuHV-4 attachment to RAW-264 cells [56]. In vivo virion capture via type 1 AECs was efficient already, so here mAb 8F10 simply failed to neutralize (Fig. 8B, 8C). By contrast mAb SC-9E8, which neutralizes independent of cell binding [58], was effective at blocking host entry. Most transformed epithelial cell lines display both sulfated and unsulfated heparan and are bound by both gp70 and gH/gL [33]. However only gH/gL binds well to unsulfated heparan (10E4-NAH46+) [17], as displayed by the apical alveolar epithelium (Fig. 6). Accordingly, an additional block of gp70 heparan binding by mAb LT-6E8 [33] did not affect host entry (Fig. 8C).
To understand better the in vivo fate of 8F10-opsonized virions, we exposed WT and ORF50- eGFP+ MuHV-4 to mAb 8F10 or not, gave them i.n., and 1 day later stained lung sections for eGFP expression (Fig. 8D). Pre-incubation with 8F10 increased significantly the number of eGFP+ lung macrophages. Epithelial infection was unaffected, consistent with this occurring via macrophages rather than directly. Pre-incubating WT MuHV-4 with mAb 8F10 or not and staining lungs for virion antigens 5h after i.n. inoculation (Fig. 9) showed that 8F10 reduced significantly the virion binding to PDP+ cells without compromising their accumulation in CD68+ cells. Therefore blocking cell binding with antibody was insufficient to block host entry, because it merely diverted virions to where they were already going: macrophages.

Discussion
How rhadinoviruses first infect new hosts has been little explored. Even for lymphocryptoviruses there is considerable uncertainty. In vitro analyses have focussed on epithelial cell and lymphocyte infections. However MuHV-4, which enters the upper respiratory tract via olfactory neurons, entered the lungs via alveolar macrophages. Most entry models envisage a target cell with a specific binding receptor that is infected readily ex vivo. Alveolar macrophages lacked the key initiator of MuHV-4 binding—heparan—and were infected poorly ex vivo. These data suggest a new paradigm, in which virions with a sophisticated fusion machinery can exploit normal host pathways for entry. One such pathway appeared to be particle scavenging by alveolar macrophages; another was opsonization-dependent endocytosis. Thus, in vivo neutralization had to act post-binding. As immune sera neutralize MuHV-4 mainly by blocking cell binding [56], this explains their limited capacity to block host entry [59].
In vivo cell diversity and interaction provide a dimension lacking from most cell lines. Co-operative infections are one reflection of this and are a recurring theme in γ-herpesvirus host colonization: EBV can infect epithelial cells by transfer from B cells [60], and MuHV-4 infects olfactory sustentacular cells via binding to adjacent neurons [17]. The homeostatic interactions of macrophage with other cell types [61] make them prime candidates to participate in co-operative infections. Here, epithelial heparan binding licensed virions for macrophage infection, and interaction with macrophages in turn licensed epithelial infection.
How might epithelial cell binding license macrophage infection? The MuHV-4 gp150 promotes virion release from heparan-deficient surfaces by inhibiting heparan-independent binding [24]. (Inhibition of EBV epithelial cell infection by gp350 [60] may analogously promote virion release.) Thus, initiating MuHV-4 infection requires heparan for binding and to engage gp150. A lack of heparan normally limits macrophage infection by non-opsonized, cell-free virions. If heparan is provided, or if the need for heparan is bypassed by gp150 disruption or by virion opsonization, then infection works well [26]. Therefore type 1 AECs licensed macrophage infection by their apical heparan capturing virions and engaging gp150.
How might virion interaction with myeloid cells license epithelial infection? The next step in MuHV-4 entry after binding is endocytosis. Virions lacking gL show an endocytic defect that can be reproduced by gH/gL-directed neutralization, implying that gH/gL engages a pro-endocytic ligand [57]. (This ligand is not heparan as distinct mAbs block gH/gL heparan binding and virion endocytosis.) Thus, a paucity of pro-endocytic ligands may strand heparan-engaged virions on type 1 AECs. (By contrast the olfactory epithelium shows no barrier to virion uptake after binding [17, 26].) The MuHV-4 gB and gH change conformation between endocytosis and fusion; fusion itself involves further changes [53, 62]. Myeloid-derived virions display constitutively the intermediate, post-endocytic forms of gB and gH and overcome a post-binding block to B cell infection [26]. We envisage that they overcome the post-binding block to type 1 AEC infection in the same way. The tropism-associated gB and gH conformation changes occur in early endosomes, whereas fusion occurs in late endosomes [63]. Therefore virions sorted from myeloid early endosomes to exosomes could be released in a tropism-switched form without a need for myeloid infection. This would explain how follicular dendritic cells transfer MuHV-4 from marginal zone to follicular B cells without becoming infected [27]. A similar cycling through alveolar macrophage endosomes would explain delayed type 1 AEC infection without prior macrophage infection.
Myeloid cells have roles in host colonization by several lymphotropic viruses [64, 65] including the human MuHV-4 ortholog, KSHV [66, 67]. Although MuHV-4 myeloid infection is sparse at steady state [68], inefficient ex vivo and not prominent in disease states, it is important for host entry, lymph node colonization [69] and systemic spread [27]. Myeloid cells engulf environmental antigens at many mucosal surfaces [70–72] and this can be extensive [73]. Therefore myeloid contributions to human γ-herpesvirus infections may be under-estimated.
The amount of antibody attached to shed virions is unknown, but salivary antibody has clear potential to affect tropism [74, 75]. Membrane fusion blocks or strong IgA responses might block host entry, because all infection requires fusion and mucosal IgA has a strong outward flux. However MuHV-4 elicits little post-binding neutralization [76] and little IgA [77]; antibody protects by effector recruitment [78] and poorly blocks host entry [59]. MuHV-4 is adapted to B cell physiology like EBV [79] but also to myeloid physiology, which encompasses the capture, transport and transfer of environmental antigens to B cells. Such adaptation is unlikely to be unique. Therefore γ-herpesvirus infection control strategies must consider not just the defensive functions of myeloid cells, but also their potential vulnerabilities.
Materials and Methods
Mice
C57BL/6J, BALB/c (Harlan UK or Animal Resources Centre, WA), LysM-cre [43] (Jackson Laboratories), Ai6-ZSgreen1 [44], CD11c-cre [80] and CD169-DTR mice [47] were bred in the Department of Pathology, Cambridge or the Herston Medical Research Centre, Queensland. For infection, 6–12 week old mice were anesthetized with isoflurane and virus (30μl) pipetted onto the nares, from where it was inhaled. For luciferase imaging, mice were injected i.p. with 2mg D-luciferin, anesthetised with isoflurane and monitored for light emission with a charge-coupled device camera (IVIS lumina, Caliper Life Sciences). CD169-DTR mice (CD169+/DTR) were depleted of CD169+ cells by i.p. diphtheria toxin (2μg / mouse) (Sigma-Aldrich).
Ethics statement
Animal experiments were approved by the Cambridge University ethical review board, the UK Home Office (Project Licence 80/2538), and the University of Queensland animal ethics committee.
Cells
BHK-21 cells (ATCC CCL-10), RAW-264 (ATCC TIB-71), 3T3–50 [15], NMuMG (ATCC CRL-1636) and 3T3-cre cells [24] were grown in Dulbecco’s Modified Eagle’s Medium with 2mM glutamine, 100IU/ml penicillin, 100μg/ml streptomycin, and 10% fetal calf serum (PAA laboratories). Macrophages were recovered from mice by post-mortem bronchial or peritoneal lavage, then adhered to tissue culture plastic (1h, 37°C) and washed in medium to remove contaminating B cells. The remaining cells were >95% CD68+.
Viruses
All viruses were derived from a BAC-cloned MuHV-4 genome [81] with an HCMV IE1 promoter-driven eGFP expression cassette. In some experiments this loxP-flanked cassette was retained to identify infected cells by eGFP expression; for fluorochrome-switching [26], luciferase-expressing [15] and EF1α promoter driven eGFP viruses [82] it was removed by passage in 3T3-cre cells. ORF50- MuHV-4 was grown and titered in 3T3-ORF50 cells, with ORF50 expression induced by doxycycline (1μg/ml). Herpes simplex virus type-1 (HSV-1) expressing eGFP from an HCMV IE1 promoter was grown and titered on BHK-21 cells [83]. MuHV-4 was recovered from infected cell supernatants by ultracentrifugation (13,000 x g, 2h). Any cell debris was removed by low speed centrifugation (500 x g, 5min) and filtration (0.45μm). HSV-1 was recovered from infected cells by low speed centrifugation (500 x g, 10min) then sonicated (3 x 1min). Cell-associated MuHV-4 was prepared as for HSV-1 by centrifugation and sonication of infected BHK-21 cells. All virus stocks were titered by plaque assay and stored at-70°C.
Infectivity assays
Infectious virus was measured by plaque assay [24]: dilutions of virus stocks or organ homogenates were incubated with BHK-21 cells (2h, 37°C), overlaid with 0.3% carboxymethylcellulose, cultured for 2 (HSV-1) or 4 days (MuHV-4), then fixed (4% formaldehyde) and stained (0.1% toluidine blue) for plaque counting. To assay fluorochrome switching by MHV-RG, plaque assays were performed at limiting dilution in 96 well plates (12–24 wells per dilution), and plaques scored as red or green fluorescent under ultraviolet illumination after 4 days. To assay infection by eGFP expression, cells were infected with eGFP+ virus (2h, 37°C), cultured overnight (18h, 37°C) in the presence of phosphonoacetic acid (100μg/ml) to prevent further spread, trypsinized, washed x2 in PBS, and analysed for eGFP expression with a FACS Calibur (BD Biosciences). To assay neutralization, viruses were incubated (1h, 37°C) with dilutions of mAbs 8F10 (anti-gH/gL, IgG2a), SC-9E8 (anti-gB, IgG2a), or as a negative control 28.14.8 (anti-H2Db, IgG2a, American Type Culture Collection HB-27), then assayed for infectivity as above.
Immunofluorescence
Organs were fixed in 1% formaldehyde / 10mM sodium periodate / 75mM L-lysine (24h, 4°C), equilibrated in 30% sucrose (18h, 4°C), then frozen in OCT. 9μm sections were air dried (1h, 23°C), blocked with 0.3% Triton X-100 / 5% normal goat serum (1h, 23°C), then incubated (18h, 4°C) with combinations of primary antibodies to eGFP (rabbit pAb, Abcam), mCherry (rabbit pAb, Badrilla), B220 (rat mAb RA3–6B2, Abcam), CD11c (hamster mAb N418, Abcam), CD169 (rat mAb 3D6.112, Abcam), CD206 (rat mAb MR5D3, Santa Cruz), unsulfated heparan (mouse mAb NAH46, Seikagaku Corporation), sulfated heparan (mouse mAb 10E4, Seikagaku Corporation), CD68 (rat mAb FA-11, Biolegend), PDP (goat pAb, R&D Systems), surfactant protein C precursor (goat pAb, Santa Cruz Biotechnology), MAC-2 (rat mAb M3/38, eBioscience), GR-1 (rat mAb RB6–8C5, R&D Systems), myeloperoxidase (goat pAb, R&D Systems), F4/80 (rat mAb CI:A3–1, Serotec), collagen IV (rabbit pAb, Abcam), HSV-1 (rabbit pAb, Abcam), and MuHV-4 (rabbit pAb). After incubation with primary antibodies (16h, 23°C), sections were washed x3 in PBS, incubated (1h, 23°C) with Alexa633-conjugated goat anti-rat IgG pAb, streptavidin-conjugated Alexa568 and Alexa488 - or 568-conjugated goat anti-rabbit IgG pAb (Invitrogen), washed x3 in PBS, and mounted in Prolong Gold + DAPI. Fluorescence was visualised with a Leica TCS SP5 or Zeiss LSM 510 confocal microscope or a Nikon epifluorescence microscope, and analysed with ImageJ.
ELISA
MuHV-4 virions were disrupted with 0.05% Triton-X100 in 50mM sodium carbonate pH = 8.5, and coated (18h, 4°C) onto Maxisorp ELISA plates (Nalge Nunc). The plates were washed x3 in PBS / 0.1% Tween-20, blocked with 2% bovine serum albumin in PBS / 0.1% Tween-20, incubated with 3-fold dilutions of serum from MuHV-4-exposed mice (1h, 23°C), washed x4 in PBS / 0.1% Tween-20, incubated (1h, 23°C) with alkaline phosphatase-conjugated goat anti-mouse IgG-Fc pAb (Sigma Chemical Co.), washed x5, and developed with nitrophenylphosphate substrate (Sigma). Absorbance was read at 405nm (Biorad), and the presence of a MuHV-4-specific response determined by comparison with sera from age-matched, uninfected controls.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Ambinder RF, Cesarman E (2007) Clinical and Pathological aspects of EBV and KSHV infection. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, et al, editors. Human Herpesviruses: Biology, Therapy and Immunoprophylaxis. Cambridge University Press. Chapter 50.
2. O'Toole D, Li H (2014) The pathology of malignant catarrhal fever, with an emphasis on ovine herpesvirus 2. Vet Pathol 51 : 437–452. doi: 10.1177/0300985813520435 24503439
3. Hoagland RJ (1964) The incubation period of Infectious Mononucleosis. Am. J. Public Health Nations Health 54 : 1699–1705. 14240492
4. Rickinson AB, Yao QY, Wallace LE (1985) The Epstein-Barr virus as a model of virus-host interactions. Br Med Bull 41 : 75–79. 2982449
5. Ehlers B, Dural G, Yasmum N, Lembo T, de Thoisy B, et al (2008) Novel mammalian herpesviruses and lineages within the Gammaherpesvirinae: cospeciation and interspecies transfer. J Virol 82 : 3509–3516. doi: 10.1128/JVI.02646-07 18216123
6. Blackbourn DJ, Lennette ET, Ambroziak J, Mourich DV, Levy JA (1998) Human herpesvirus 8 detection in nasal secretions and saliva. J Infect Dis 177 : 213–216. 9419191
7. Garay SM, Belenko M, Fazzini E, Schinella R (1987) Pulmonary manifestations of Kaposi's sarcoma. Chest 91 : 39–43. 3792084
8. Li H, Cunha CW, Davies CJ, Gailbreath KL, Knowles DP, et al (2008) Ovine herpesvirus 2 replicates initially in the lung of experimentally infected sheep. J Gen Virol 89 : 1699–1708. doi: 10.1099/vir.0.2008/000554-0 18559941
9. Hartley CA, Dynon KJ, Mekuria ZH, El-Hage CM, Holloway SA, et al (2013) Equine gammaherpesviruses: perfect parasites? Vet Microbiol 167 : 86–92. doi: 10.1016/j.vetmic.2013.05.031 23845734
10. Bell SA, Balasuriya UB, Gardner IA, Barry PA, Wilson WD, et al (2006) Temporal detection of equine herpesvirus infections of a cohort of mares and their foals. Vet Microbiol 116 : 249–257. 16774810
11. Bartha A, Juhász M, Liebermann H (1966) Isolation of a bovine herpesvirus from calves with respiratory disease and keratoconjunctivitis. A preliminary report. Acta Vet Acad Sci Hung 16 : 357–358. 6005954
12. Castrucci G, Frigeri F, Ferrari M, Ranucci S, Aldrovandi V, et al (1987) Experimental infection of calves with strains of Bovid herpesvirus-4. Comp Immunol Microbiol Infect Dis 10 : 41–49. 3034501
13. Stevenson PG, Simas JP, Efstathiou S (2009) Immune control of mammalian gamma-herpesviruses: lessons from murid herpesvirus-4. J Gen Virol 90 : 2317–2330. doi: 10.1099/vir.0.013300-0 19605591
14. Barton E, Mandal P, Speck SH (2011) Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu Rev Immunol 29 : 351–397. doi: 10.1146/annurev-immunol-072710-081639 21219186
15. Milho R, Smith CM, Marques S, Alenquer M, May JS, et al (2009) In vivo imaging of murid herpesvirus-4 infection. J Gen Virol 90 : 21–32. doi: 10.1099/vir.0.006569-0 19088269
16. Sunil-Chandra NP, Efstathiou S, Arno J, Nash AA (1992) Virological and pathological features of mice infected with murine gamma-herpesvirus 68. J Gen Virol 73 : 2347–2356. 1328491
17. Milho R, Frederico B, Efstathiou S, Stevenson PG (2012) A heparan-dependent herpesvirus targets the olfactory neuroepithelium for host entry. PLoS Pathog 8: e1002986. doi: 10.1371/journal.ppat.1002986 23133384
18. François S, Vidick S, Sarlet M, Desmecht D, Drion P, et al (2013) Illumination of murine gammaherpesvirus-68 cycle reveals a sexual transmission route from females to males in laboratory mice. PLoS Pathog 9: e1003292. doi: 10.1371/journal.ppat.1003292 23593002
19. Kozuch O, Reichel M, Lesso J, Remenová A, Labuda M, et al (1993) Further isolation of murine herpesviruses from small mammals in southwestern Slovakia. Acta Virol 37 : 101–105. 8105644
20. Blasdell K, McCracken C, Morris A, Nash AA, Begon M, et al (2003) The wood mouse is a natural host for Murid herpesvirus 4. J Gen Virol 84 : 111–113. 12533706
21. Stone KC, Mercer RR, Freeman BA, Chang LY, Crapo JD (1992) Distribution of lung cell numbers and volumes between alveolar and nonalveolar tissue. Am Rev Respir Dis 146 : 454–456. 1489139
22. Moser JM, Farrell ML, Krug LT, Upton JW, Speck SH (2006) A gammaherpesvirus 68 gene 50 null mutant establishes long-term latency in the lung but fails to vaccinate against a wild-type virus challenge. J Virol 80 : 1592–1598. 16415035
23. Kayhan B, Yager EJ, Lanzer K, Cookenham T, Jia Q, et al (2007) A replication-deficient murine gamma-herpesvirus blocked in late viral gene expression can establish latency and elicit protective cellular immunity. J Immunol 179 : 8392–8402. 18056385
24. de Lima BD, May JS, Stevenson PG (2004) Murine gammaherpesvirus 68 lacking gp150 shows defective virion release but establishes normal latency in vivo. J Virol 78 : 5103–5112. 15113892
25. Jarousse N, Chandran B, Coscoy L (2008) Lack of heparan sulfate expression in B-cell lines: implications for Kaposi's sarcoma-associated herpesvirus and murine gammaherpesvirus 68 infections. J Virol 82 : 12591–12597. doi: 10.1128/JVI.01167-08 18842731
26. Frederico B, Milho R, May JS, Gillet L, Stevenson PG (2012) Myeloid infection links epithelial and B cell tropisms of Murid Herpesvirus-4. PLoS Pathog 8: e1002935. doi: 10.1371/journal.ppat.1002935 23028329
27. Frederico B, Chao B, May JS, Belz GT, Stevenson PG (2014) A murid gamma-herpesviruses exploits normal splenic immune communication routes for systemic spread. Cell Host Microbe 15 : 457–470. doi: 10.1016/j.chom.2014.03.010 24721574
28. Vanderplasschen A, Bublot M, Dubuisson J, Pastoret PP, Thiry E (1993) Attachment of the gammaherpesvirus bovine herpesvirus 4 is mediated by the interaction of gp8 glycoprotein with heparinlike moieties on the cell surface. Virology 196 : 232–240. 8356795
29. Akula SM, Wang FZ, Vieira J, Chandran B (2001) Human herpesvirus 8 interaction with target cells involves heparan sulfate. Virology 282 : 245–255. 11289807
30. Means RE (2004) Characterization of the Herpesvirus saimiri Orf51 protein. Virology 326 : 67–78. 15262496
31. Gillet L, Adler H, Stevenson PG (2007) Glycosaminoglycan interactions in murine gammaherpesvirus-68 infection. PLoS ONE 2: e347. 17406671
32. Gillet L, Colaco S, Stevenson PG (2008) The Murid Herpesvirus-4 gH/gL Binds to Glycosaminoglycans. PLoS ONE 3: e1669. doi: 10.1371/journal.pone.0001669 18301747
33. Gillet L, May JS, Stevenson PG (2009) In vivo importance of heparan sulfate-binding glycoproteins for murid herpesvirus-4 infection. J Gen Virol 90 : 602–613. doi: 10.1099/vir.0.005785-0 19218205
34. Machiels B, Lété C, de Fays K, Mast J, Dewals B, et al (2011) The bovine herpesvirus 4 Bo10 gene encodes a nonessential viral envelope protein that regulates viral tropism through both positive and negative effects. J Virol 85 : 1011–1024. doi: 10.1128/JVI.01092-10 21068242
35. Hayashi K, Hayashi M, Jalkanen M, Firestone JH, Trelstad RL, et al (1987) Immunocytochemistry of cell surface heparan sulfate proteoglycan in mouse tissues. A light and electron microscopic study. J Histochem Cytochem 35 : 1079–1088. 2957423
36. Hayashi K, Hayashi M, Boutin E, Cunha GR, Bernfield M, et al (1988) Hormonal modification of epithelial differentiation and expression of cell surface heparan sulfate proteoglycan in the mouse vaginal epithelium. An immunohistochemical and electron microscopic study. Lab Invest 58 : 68–76. 2961930
37. Vanderbilt JN, Allen L, Gonzalez RF, Tigue Z, Edmondson J, et al (2008) Directed expression of transgenes to alveolar type I cells in the mouse. Am J Respir Cell Mol Biol 39 : 253–362. doi: 10.1165/rcmb.2008-0049OC 18367724
38. Hume DA (2011) Applications of myeloid-specific promoters in transgenic mice support in vivo imaging and functional genomics but do not support the concept of distinct macrophage and dendritic cell lineages or roles in immunity. J Leukoc Biol 89 : 525–538. doi: 10.1189/jlb.0810472 21169519
39. Gordon S, Plϋddemann A (2013) Tissue macrophage heterogeneity: issues and prospects. Semin Immunopathol 35 : 533–540. doi: 10.1007/s00281-013-0386-4 23783507
40. Zaynagetdinov R, Sherrill TP, Kendall PL, Segal BH, Weller KP, et al (2013) Identification of myeloid cell subsets in murine lungs using flow cytometry. Am J Respir Cell Mol Biol 49 : 180–189. doi: 10.1165/rcmb.2012-0366MA 23492192
41. Flaño E, Jia Q, Moore J, Woodland DL, Sun R, et al (2005) Early establishment of gamma-herpesvirus latency: implications for immune control. J Immunol 174 : 4972–4978. 15814726
42. Smith CM, Gill MB, May JS, Stevenson PG (2007) Murine gammaherpesvirus-68 inhibits antigen presentation by dendritic cells. PLoS One 2: e1048. 17940612
43. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8 : 265–277. 10621974
44. Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, et al (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13 : 133–140. doi: 10.1038/nn.2467 20023653
45. Braun V, Niedergang F (2006) Linking exocytosis and endocytosis during phagocytosis. Biol Cell 98 : 195–201. 16480341
46. Bilyk N, Holt PG (1991) The surface phenotypic characterization of lung macrophages in C3H/HeJ mice. Immunology 74 : 645–651. 1783423
47. Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, et al (2011) CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity 34 : 85–95. doi: 10.1016/j.immuni.2010.12.011 21194983
48. Epelman S, Lavine KJ, Randolph GJ (2014). Origin and functions of tissue macrophages. Immunity 41 : 21–35. doi: 10.1016/j.immuni.2014.06.013 25035951
49. Tibbetts SA, Loh J, Van Berkel V, McClellan JS, Jacoby MA, et al (2003) Establishment and maintenance of gammaherpesvirus latency are independent of infective dose and route of infection. J Virol 77 : 7696–7701. 12805472
50. David G, Bai XM, Van der Schueren B, Cassiman JJ, Van den Berghe H (1992) Developmental changes in heparan sulfate expression: in situ detection with mAbs. J Cell Biol 119 : 961–975. 1385449
51. Suzuki K, Yamamoto K, Kariya Y, Maeda H, Ishimaru T, et al (2008) Generation and characterization of a series of monoclonal antibodies that specifically recognize [HexA(+/−2S)-GlcNAc]n epitopes in heparan sulfate. Glycoconj J 25 : 703–712. doi: 10.1007/s10719-008-9130-z 18461440
52. Geiser M. 2002. Morphological aspects of particle uptake by lung phagocytes. Microsc Res Tech 57 : 512–522. 12112434
53. Gillet L, Colaco S, Stevenson PG (2008) Glycoprotein B switches conformation during murid herpesvirus 4 entry. J Gen Virol 89 : 1352–1363. doi: 10.1099/vir.0.83519-0 18474550
54. Minson AC (1994) Interactions of herpes simplex viruses with the host cell. Biochem Soc Trans 22 : 298–301. 7958311
55. Yao QY, Rowe M, Morgan AJ, Sam CK, Prasad U, et al (1991) Salivary and serum IgA antibodies to the Epstein-Barr virus glycoprotein gp340: incidence and potential for virus neutralization. Int J Cancer 48 : 45–50. 1850382
56. Rosa GT, Gillet L, Smith CM, de Lima BD, Stevenson PG (2007) IgG fc receptors provide an alternative infection route for murine gamma-herpesvirus-68. PLoS One 2: e560. 17593961
57. Glauser DL, Gillet L, Stevenson PG (2012) Virion endocytosis is a major target for murid herpesvirus-4 neutralization. J Gen Virol 93 : 1316–1327. doi: 10.1099/vir.0.040790-0 22377583
58. Glauser DL, Kratz AS, Gillet L, Stevenson PG (2011) A mechanistic basis for potent, glycoprotein B-directed gammaherpesvirus neutralization. J Gen Virol 92 : 2020–2033. doi: 10.1099/vir.0.032177-0 21593277
59. Gillet L, May JS, Stevenson PG (2007) Post-exposure vaccination improves gammaherpesvirus neutralization. PLoS One 2: e899. 17878934
60. Shannon-Lowe CD, Neuhierl B, Baldwin G, Rickinson AB, Delecluse HJ (2006) Resting B cells as a transfer vehicle for Epstein-Barr virus infection of epithelial cells. Proc Natl Acad Sci USA 103 : 7065–7070. 16606841
61. Ginhoux F, Jung S (2014) Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 14 : 392–404. doi: 10.1038/nri3671 24854589
62. Gillet L, Colaco S, Stevenson PG (2008) The Murid Herpesvirus-4 gL regulates an entry-associated conformation change in gH. PLoS One 3: e2811. doi: 10.1371/journal.pone.0002811 18665235
63. Glauser DL, Kratz AS, Stevenson PG (2012) Herpesvirus glycoproteins undergo multiple antigenic changes before membrane fusion. PLoS One 7: e30152. doi: 10.1371/journal.pone.0030152 22253913
64. Teleshova N, Derby N, Martinelli E, Pugach P, Calenda G, et al (2013) Simian immunodeficiency virus interactions with macaque dendritic cells. Adv Exp Med Biol 762 : 155–181. doi: 10.1007/978-1-4614-4433-6_6 22975875
65. Ludlow M, Lemon K, de Vries RD, McQuaid S, Millar EL, et al (2013) Measles virus infection of epithelial cells in the macaque upper respiratory tract is mediated by subepithelial immune cells. J Virol 87 : 4033–4042. doi: 10.1128/JVI.03258-12 23365435
66. Knowlton ER, Lepone LM, Li J, Rappocciolo G, Jenkins FJ, et al (2013) Professional antigen presenting cells in human herpesvirus 8 infection. Front Immunol 3 : 427. doi: 10.3389/fimmu.2012.00427 23346088
67. Wang LX, Kang G, Kumar P, Lu W, Li Y, et al (2014) Humanized-BLT mouse model of Kaposi's sarcoma-associated herpesvirus infection. Proc Natl Acad Sci USA 111 : 3146–3151. doi: 10.1073/pnas.1318175111 24516154
68. Marques S, Efstathiou S, Smith KG, Haury M, Simas JP (2003) Selective Gene Expression of Latent Murine Gammaherpesvirus 68 in B Lymphocytes. J Virol 77 : 7308–7318. 12805429
69. Gaspar M, May JS, Sukla S, Frederico B, Gill MB, et al (2011) Murid herpesvirus-4 exploits dendritic cells to infect B cells. PLoS Pathog 7: e1002346. doi: 10.1371/journal.ppat.1002346 22102809
70. Chieppa M, Rescigno M, Huang AY, Germain RN (2006) Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med 203 : 2841–2852. 17145958
71. Jahnsen FL, Strickland DH, Thomas JA, Tobagus IT, Napoli S, et al (2006) Accelerated antigen sampling and transport by airway mucosal dendritic cells following inhalation of a bacterial stimulus. J Immunol 177 : 5861–5967. 17056510
72. Iijima N, Thompson JM, Iwasaki A (2008) Dendritic cells and macrophages in the genitourinary tract. Mucosal Immunol 1 : 451–459. doi: 10.1038/mi.2008.57 19079212
73. Katila T (2012) Post-mating inflammatory responses of the uterus. Reprod Domest Anim 47 Suppl 5 : 31–41. doi: 10.1111/j.1439-0531.2012.02120.x 22913558
74. Sixbey JW, Yao QY (1992) Immunoglobulin A-induced shift of Epstein-Barr virus tissue tropism. Science 255 : 1578–1580. 1312750
75. Turk SM, Jiang R, Chesnokova LS, Hutt-Fletcher LM (2006) Antibodies to gp350/220 enhance the ability of Epstein-Barr virus to infect epithelial cells. J Virol 80 : 9628–9633. 16973566
76. Gillet L, May JS, Colaco S, Stevenson PG (2007) The murine gammaherpesvirus-68 gp150 acts as an immunogenic decoy to limit virion neutralization. PLoS One 2: e705. 17684552
77. Tan CS, Stevenson PG (2014) B cell response to herpesvirus infection of the olfactory neuroepithelium. J Virol 88 : 14030–14039. doi: 10.1128/JVI.02345-14 25253348
78. Wright DE, Colaco S, Colaco C, Stevenson PG (2009) Antibody limits in vivo murid herpesvirus-4 replication by IgG Fc receptor-dependent functions. J Gen Virol 90 : 2592–2603. doi: 10.1099/vir.0.014266-0 19625459
79. Thorley-Lawson DA, Miyashita EM, Khan G (1996) Epstein-Barr virus and the B cell: that's all it takes. Trends Microbiol 4 : 204–208. 8727601
80. Caton ML, Smith-Raska MR, Reizis B (2007) Notch-RBP-J signaling controls the homeostasis of CD8 - dendritic cells in the spleen. J Exp Med 204 : 1653–1664. 17591855
81. Adler H, Messerle M, Wagner M, Koszinowski UH (2000) Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J Virol 74 : 6964–6974. 10888635
82. May JS, Stevenson PG (2010) Vaccination with murid herpesvirus-4 glycoprotein B reduces viral lytic replication but does not induce detectable virion neutralization. J Gen Virol 91 : 2542–2552. doi: 10.1099/vir.0.023085-0 20519454
83. Shivkumar M, Milho R, May JS, Nicoll MP, Efstathiou S, et al (2013) Herpes simplex virus 1 targets the murine olfactory neuroepithelium for host entry. J Virol 87 : 10477–10488. doi: 10.1128/JVI.01748-13 23903843
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 3
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Bacterial Immune Evasion through Manipulation of Host Inhibitory Immune Signaling
- BILBO1 Is a Scaffold Protein of the Flagellar Pocket Collar in the Pathogen
- Antimicrobial-Induced DNA Damage and Genomic Instability in Microbial Pathogens
- Attenuation of Tick-Borne Encephalitis Virus Using Large-Scale Random Codon Re-encoding