
NEDDylation Is Essential for Kaposi’s Sarcoma-Associated Herpesvirus Latency and Lytic Reactivation and Represents a Novel Anti-KSHV Target
Kaposi’s sarcoma-associated herpesvirus (KSHV) causes Kaposi’s sarcoma (KS) and primary effusion lymphoma (PEL), often fatal malignancies afflicting HIV-infected patients. Previous research has shown that blockade of the ubiquitin proteasome system (UPS, a normal quality control pathway that degrades cellular proteins) is able to kill KSHV-infected lymphoma cells. A large component of the UPS is made up by the protein family known as the cullin-RING ubiquitin ligases (CRLs), which are activated by NEDD8 (a process known as NEDDylation). Recently, an inhibitor of NEDDylation (MLN4924) was developed and is currently in clinical trials as an anti-cancer drug. As NEDDylation has not been investigated for many viruses, we used this to compound examine its importance in KSHV biology. Firstly we show that NEDDylation is essential for the viability of KSHV-infected lymphoma cells, and MLN4924 treatment killed these cells by blocking NF-κB activity (required for KSHV latency gene expression and KSHV-associated cancer). Furthermore, we show that NEDDylation is required for KSHV to replicate its genome, a critical step in the production of new virus particles. Therefore, this research has identified a novel molecular mechanism that governs KSHV replication. Furthermore, it demonstrates that NEDDylation is a viable target for the treatment of KSHV-associated malignancies.
Published in the journal:
. PLoS Pathog 11(3): e32767. doi:10.1371/journal.ppat.1004771
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004771
Summary
Kaposi’s sarcoma-associated herpesvirus (KSHV) causes Kaposi’s sarcoma (KS) and primary effusion lymphoma (PEL), often fatal malignancies afflicting HIV-infected patients. Previous research has shown that blockade of the ubiquitin proteasome system (UPS, a normal quality control pathway that degrades cellular proteins) is able to kill KSHV-infected lymphoma cells. A large component of the UPS is made up by the protein family known as the cullin-RING ubiquitin ligases (CRLs), which are activated by NEDD8 (a process known as NEDDylation). Recently, an inhibitor of NEDDylation (MLN4924) was developed and is currently in clinical trials as an anti-cancer drug. As NEDDylation has not been investigated for many viruses, we used this to compound examine its importance in KSHV biology. Firstly we show that NEDDylation is essential for the viability of KSHV-infected lymphoma cells, and MLN4924 treatment killed these cells by blocking NF-κB activity (required for KSHV latency gene expression and KSHV-associated cancer). Furthermore, we show that NEDDylation is required for KSHV to replicate its genome, a critical step in the production of new virus particles. Therefore, this research has identified a novel molecular mechanism that governs KSHV replication. Furthermore, it demonstrates that NEDDylation is a viable target for the treatment of KSHV-associated malignancies.
Introduction
The ubiquitin-proteasome system (UPS) and associated pathways are rapidly becoming accepted as major therapeutic targets for the treatment of malignancy [1], which potentially include those associated with oncogenic viruses. Additionally, small molecule inhibitors have been successfully used for dissecting the biological roles of these intriguing pathways, which is critical for our understanding of their mechanisms of cytotoxicity. Indeed, inhibition of the UPS is cytotoxic to Kaposi’s sarcoma-associated herpesvirus (KSHV, also referred to as human herpesvirus 8 [HHV8]) infected cells [2–5]. Infection with KSHV is commonly associated with fatal malignancies, is the causative agent of primary effusion lymphoma (PEL) and Kaposi’s sarcoma (KS) and is frequently associated with multicentric Castleman’s disease (MCD) [6,7]. Like all herpesviruses, KSHV infection is lifelong and has two distinct phases to its lifecycle; latent and lytic. During latency, viral gene expression is highly restricted and, in the tumor setting, involves the expression of the latency associated nuclear antigen (LANA), the viral FLICE inhibitory protein (vFLIP), viral cyclin, kaposin and various virally encoded miRNAs. Together these promote tumorigenesis in all known KSHV-associated malignancies. Nevertheless, at least for KS, the lytic phase of KSHV, which results in the expression of the complete viral genome and the production of infectious virions, is necessary for sarcomagenesis. For this reason, the molecular mechanisms governing the switch from latency to lytic reactivation have received much attention as they may provide excellent targets for therapeutic intervention.
Current treatments of KSHV-associated malignancies have limited efficacy. PEL is treated using a combination of cyclophosphamide, doxorubicin, vincristine and prednisone (similar to CHOP therapy) and/or highly active retroviral therapy (HAART) [8,9]. For AIDS-related KS, HAART is also favored, and due to the requirement of KSHV lytic infection for the pathogenesis of KS, anti-herpesviral drugs have also been used [10]. More recently, preclinical models have demonstrated that inhibition of the UPS using bortezamib [2–5], or bortezamib in combination with a histone deacetylase (HDAC) inhibitor (vorinostat) may provide a promising new avenue [11]. Given the success of bortezamib (marketed as Velcade) for the treatment of multiple myeloma and mantle cell lymphoma, there are now various additional small molecule inhibitors of the UPS at various stages of development [1], as well as drugs that target ubiquitin-like pathways such as the NEDDylation cascade [12].
The attachment of ubiquitin-like (Ubl) proteins to substrates represents one of the most vital posttranslational modifications in the cell and, so far, twelve Ubls have been identified including ubiquitin (Ub), NEDD8 and SUMO. The covalent attachment of Ubls to substrates requires an enzyme cascade involving an E1 activating enzyme, an E2 conjugating enzyme and an E3 ligase that provides substrate specificity, all of which are potential ‘drugable’ targets. Ubiquitylation is the principal mechanism by which proteins are targeted for proteasomal degradation, and the largest class of E3 ligases are the Cullin-RING ligases (CRLs) [13]. These modular proteins are centered on one of several Cullin scaffolds (Cul1, 2, 3, 4A, 4B, 5 and 7) and consist of a RING-binding protein (RBX1 or RBX2) that interacts with the Ub-loaded E2 and a substrate adapter protein that brings the target into close proximity with the Ub-E2. Many substrate adapters have been identified, and the various combinations of Cul-adapter complexes means there are several hundred possible CRLs, with several-fold more substrates [14]. Intriguingly, the activity of CRLs is dependent on themselves being modified by the Ubl NEDD8, which requires the activity of the NEDD8 activating enzyme, NAE1 [15]. Recently, a specific NAE1 inhibitor has been developed (MLN4924) and is currently in clinical trials for various malignancies. MLN4924-mediated inhibition of NEDDylation blocks CRL activity resulting in the global stabilization of CRL targets [12].
While the ubiquitin and SUMO pathways have been extensively studied in infectious diseases [16,17], much less attention has been given to the role of NEDD8. Recent work has shown that the large tegument protein of gammaherpesviruses has deNEDDylase activity and this was important for EBV infection [18]. Another study demonstrated that inhibition of NEDDylation blocked the ability of HIV to degrade the cellular restriction factor APOBEC3G via Vif-mediated hijack of Cul5 [19]. There are indications in the literature that NEDDylation is likely to play a central role in viral pathogenesis. For example, HIV is known to modulate the activity of at least three cullins [20,21], the KSHV LANA protein forms a CRL complex with Cul5 and targets p53 [22], and it was recently shown that human cytomegalovirus (HCMV) utilizes the nucleotide excision repair (NER) pathway, which is regulated predominantly by Cul4A/B [23], to repair its genomes during viral replication [24].
Here we asked if NEDDylation is important for the KSHV lifecycle, and to begin to dissect the functional consequences of its inhibition. Treatment of cancer cells with MLN4924 leads to dramatic cytotoxicity, and some of the best characterized mechanisms include the induction of DNA re-replication by blocking the degradation of Cdt-1 [25,26], or by inhibiting NF-κB signaling via stabilization of the inhibitor of NF-κB protein IκBα [27], both of which eventually lead to apoptosis. Given the essential role of NF-κB for the maintenance of viral latency and the survival of PEL [28], we reasoned that these cells would be highly sensitive to MLN4924. We found that, indeed, MLN4924 led to significant PEL cytotoxicity and this was mediated via inhibition of NF-κB signaling. Intriguingly, we also showed that NEDDylation was essential for amplification of the KSHV genome during reactivation of the lytic cycle and that treatment with MLN4924 prevented the recruitment of RTA to the origin of lytic replication (OriLyt). This work has highlighted the essential role of NEDD8 and CRL-mediated ubiquitylation during the life cycle of KSHV and suggests that NEDDylation may provide a novel therapeutic target for the treatment of KSHV-associated malignancies.
Results
Inhibition of NEDDylation is cytotoxic to PEL cells and leads to lytic cycle-associated gene expression
Firstly, it was important to confirm that NEDDylation still occurred in KSHV-infected cells. Transfection of rKSHV-219 cells with FLAG-NEDD8 and the indicated Myc-tagged Cullin, followed by immunoprecipitation of NEDD8 and immunoblot analysis confirmed that Cullin proteins were modified in infected cells (S1 Fig). As MLN4924 has demonstrated anti-proliferative activity in various cancer cell lines, we assessed its cytotoxic effects in PEL cells. We treated PEL cells (BCBL-1, TREx-BCBL-1-RTA and BC-3) with varying concentrations of MLN4924 and determined cell viability after 96 h using a luminescence ATP detection assay. These data demonstrated that MLN4924 was indeed cytotoxic to latently infected PEL cells with approximate EC50 values of 1.10 μM and 0.15 μM, for TREx-BCBL-1-RTA and BC-3 respectively (Fig. 1A). However, it was interesting to note that a low level of ORF57 expression was observed in TREx-BCBL-1-RTA (see 10 μM lane) and low levels of endogenous RTA in BC-3 cells treated with MLN4924, suggesting that drug treatment alone induced lytic gene expression (Fig. 1B & C). We also noted that MLN4924 led to apoptosis in BC-3 cells as shown by the cleavage of PARP (Fig. 1C). To investigate MLN4924-induced cytotoxicity, cell cycle analysis of TREx-BCBL-1-RTA cells was performed 24 h after treatment with 1 μM MLN4924. Compared to untreated TREX-BCBL-1-RTA cells (Fig. 1D (i)), MLN4924 treatment led to a reduction in S-phase and an accumulation of cells in G2/M (Fig. 1D (ii)). Clearly, inhibition of NEDDylation was toxic to PEL cells, and as shown for BC-3 this led to apoptosis. To further investigate this, we tested the activation of caspases in TREx-BCBL-1-RTA cells. Similar to BC-3, treatment of TREx-BCBL1-RTA led to the cleavage of PARP, indicative of Caspase 3/7 activation, as did the reactivation of the KSHV lytic cycle after the addition of Dox. (Fig. 1E). It has been shown previously that induction of the lytic cycle activates initiator Caspase 8, but not Caspase 9 in order to activate effector caspases such as Caspase 3 and 7 [29]. In agreement with this, we also showed that lytic reactivation did not activate Caspase 9; nevertheless, MLN4924 did, resulting in the activation of Caspase 3 (Fig. 1E). Importantly however, caspase inhibition (using the pan-caspase inhibitor, z-VAD-FMK) did not prevent PEL cell death after treatment with MLN4924 (Fig. 1F). This result showed that PEL cytotoxicity was not solely a result of apoptosis and suggests other factors, such as the inhibition latency-associated gene expression may contribute to cell death.

Therefore, we next measured the effect of treatment on viral gene expression. Latently infected TREx-BCBL-1-RTA were treated with 1 μM MLN4924 and harvested 24 h later. As shown in Fig. 2A, the expression of all lytic-cycle associated genes tested were induced after inhibition of NEDDylation. In contrast, the expression of latency-associated transcripts ORF73 (latency associated nuclear antigen [LANA]) and ORF71 (vFLIP) were reduced in the presence of drug. Interestingly, the expression of K12 followed that of latency-associated genes. While K12 is dramatically induced upon activation of the KSHV lytic cycle [30], its expression during latency is initiated from the LANA promoter [31]. This suggested that MLN4924 treatment modulated latency-associated viral gene expression, rather than inducing the full KSHV lytic cycle. To further investigate the potential of MLN4924 to reactivate lytic cycle gene expression, rKSHV.219 cells were cultured for 36 h in the presence of 0.1 μM MLN4924, a concentration that was tolerated while in culture. These HEK293-based cells are latently infected with recombinant KSHV that contains a red fluorescent protein (RFP) reporter gene under the control of an RTA-responsive promoter (the PAN promoter); hence expression of RTA (and thus reactivation of the lytic cycle) leads to RFP expression. As shown in Fig. 2B, RFP expression was observed in numerous cells demonstrating that MLN4924 was indeed able to induce the expression of RTA. MLN4924 was also capable of initiating expression of ORF57 (activated by RTA during reactivation) (Fig. 2C) and enhancing lytic cycle gene expression in cells reactivated with TPA alone (Fig. 2D). Taken together, these results confirmed that inhibition of NEDDylation was clearly essential for PEL viability and that its inhibition was able to induce low level, lytic cycle-associated viral gene expression. We next investigated the potential mechanisms of MLN4924-mediated cytotoxicity.

MLN4924-induced cytotoxicity is due to the inhibition of NF-κB
The reported mechanisms of MLN4924-induced cytotoxicity is the induction of DNA re-replication (by blocking the CRL-mediated degradation of Cdt-1) [26], or by inhibiting NF-κB signaling as CRL-mediated degradation of IκBα is prevented, thus precluding NF-κB’s transcriptional activity [27]. It is well established that PEL is dependent on NF-κB signaling, and that KSHV vFLIP drives the constitutive activation of NF-κB in order to maintain viability. NF-κB activity also inhibits RTA-mediated transactivation of lytic genes, and thus it is important for maintaining latency. As the viral gene expression profiles following MLN4924 treatment (Fig. 2A) are consistent with an inhibition of NF-κB, we tested whether it was inhibited after MLN4924 treatment. The NF-κB transcription factor is tightly regulated via its interaction with IκBα which maintains it in the cytoplasm and away from its transcriptional targets. Upon stimulation, the Iκκ complex phosphorylates IκBα at ser32 and 36, which signals for the recruitment of the Cul1-containing βTrCP E3 ligase leading to the subsequent degradation of IκBα. This releases NF-κB allowing to translocate to the nucleus and drive transcription [32]. Therefore, MLN4924 inhibition of CRL1 function should stabilize phosphorylated IκBα (pIκBα) leading to an inhibition in NF-κB function.
To investigate the most likely mechanism of MLN4924-induced cytotoxicity we performed a timecourse experiment (Fig. 3A). Here we show that as little as 1 h treatment of TREX-BCBL-1-RTA cells with MLN4924 led to an inhibition of NEDDylation (as shown by the lack of NEDDylated Cul2 compared to DMSO-treated cells). We also observed the stabilization of pIκBα as early as 1 h post-treatment. By contrast, the accumulation of Cdt1 did not occur until 4 h with significant levels not observed until 6–8 h later. Furthermore, cell cycle analysis of TREx-BCBL-1-RTA cells revealed an accumulation of cells in G2/M after 24 h, indicative of a block at the DNA damage check point. Concomitant with this, we observed the phosphorylation of p53 at 24 h post treatment, but not at earlier time points. Likewise, we did not observe significant alterations in the levels of phosphorylated H2Ax (γH2Ax)—a marker of DNA strand breaks—although γH2Ax is known to be present in latently infected cells due to its association with LANA expression [33]. Importantly, the MLN4924-associated reduction of ORF73 mRNA expression occurs prior to any indication of DNA damage (Fig. 3B). Along with cell cycle analysis (Fig. 1D) showing that treatment of TREx-BCBL-1-RTA cells led to a reduction of cells in S-Phase (which is inconsistent with Cdt1-associated DNA-rereplication), and the rapid stabilization of pIκBα suggests that inhibition of NF-κB signaling was responsible for cytotoxicity. Additionally, pIκBα levels increased in an MLN4924-dose-dependent manner in latently-infected TREx-BCBL-1-RTA (Fig. 3C) and BC-3 cells (Fig. 3D) treated for 24 h, showing that, as expected, treatment did stabilize pIκBα. In accordance with this, the cytoplasmic levels of the p65/RelA subunit of NF-κB were increased in a dose-dependent manner (Fig. 3E-F—enrichment of the cytoplasmic compartment was verified by the lack of nuclear Lamin B—compared to the nuclear fraction control which maintained Lamin B but lacked GAPDH). Previous reports have shown that upon lytic reactivation, IκBα is posttranscriptionally downregulated [34] and consistent with this, cytoplasmic p65/RelA decreased in Dox-treated (reactivated) cells further highlighting the involvement of IκBα for MLN4924-induced inhibition of NF-κB.

Additionally, we did not observe pIκBα expression in reactivated cells (those with RTA expression) regardless of MLN4924 treatment (Fig. 3C-D). This was also highlighted in our confocal analysis of p65/RelA localization showing it had translocated to the nucleus in reactivated cells even when treated with MLN4924 (S2 Fig). We therefore surmised that inhibition of NF-κB, via stabilization of IκBα was responsible for cytotoxicity and demonstrated that inhibition of NEDDylation may provide a therapeutic option for the treatment of NF-κB-dependent PEL.
Identification of Cul4B as a novel regulator of KSHV latency
Treatment of cells with MLN4924 blocks total NEDDylation, which undoubtedly has far-reaching implications on cellular function. To determine if inhibition of CRL activity was responsible for the phenotypes we have observed, and to identify which CRLs are implicated, we expressed dominant-negative versions of the different cullin family members (DNCul) in cells latently infected with KSHV (rKSHV.219) and screened for reactivation of the lytic cycle by expression of ORF57. DNCuls are truncated forms of cullin proteins that are still able to engage their respective substrates, but are unable to bind to the Ub-loaded E2 enzyme (and are unable to be NEDDylated), thus preventing substrate degradation [35]. Using this approach, we showed that CRL1 was able to reactivate ORF57 expression, albeit at a low level (Fig. 4A-B). That CRL1 inhibition reactivated lytic expression is not surprising as it is known to regulate NF-κB signaling, corroborating our earlier analyses (Fig. 2 & 3) [36,37]. However, we were surprised to note that CRL4B inhibition was able to reactivate ORF57 expression (Fig. 4A-B). Cul4A and 4B both regulate chromatin-associated functions, and they share ca. 80% amino acid identity [38]. However, Cul4B has an extended N-terminus that contains a nuclear localization signal (NLS) and its expression is confined to the nucleus, whereas Cul4A is recruited to the nucleus following its interaction with DDB1 in response to genotoxic stress. Due to the high degree of similarity, they are both capable of binding the same CRL4 components (e.g. DNA damage binding proteins 1 and 2 [DDB1, DDB2] and the various DDB and Cul4-associated factors [DCAFs]). Interestingly, when DNCul4A and 4B were expressed together, the activation of ORF57 was reduced. We believe that this was due to the fact that these two proteins compete for the same binding partners meaning that the degree of nuclear inhibition of CRL4B was reduced, further implicating the importance of CRL4B in the regulation of KSHV latency. Moreover, as CRL4B resided in the nucleus, this particular E3 ligase is the most likely one to be involved in KSHV biology. To confirm this, independent shRNA knockdown of Cul4B in TREx-BCBL-1-RTA cells was performed which led to an ca. 50% reduction in Cul4B expression (Fig. 4C) resulting in the expression of various lytic cycle-associated genes (Fig. 4D). Interestingly, expression of the latency-associated gene LANA did not alter suggesting that Cul4B specifically regulated the expression of lytic genes. Here, we also investigated the expression of K12, which in contrast to the reduced expression we observed in the presence of MLN4924, was increased by Cul4B knockdown (Fig. 4D). Although a thorough investigation of individual CRLs is required (for example, using RNAi experiments to inhibit the various CRL components), this data suggests that MLN4924’s effect on KSHV latency is through its inhibition of CRL activity. In addition, we have identified CRL4B as a novel regulator of KSHV latency.

NEDDylation is essential for replication of the KSHV genome
The KSHV lytic cycle is intimately linked to the pathogenesis of KSHV malignancies and the role of NEDDylation or the importance of CRL-mediated ubiquitylation during this process is currently unknown. To investigate if ubiquitylation is indeed a feature of KSHV reactivation, we stained reactivated cells with anti-Ub FK2 (which only recognizes ubiquitylated proteins and not free ubiquitin) and to increase the stringency of this assay, we removed soluble nuclear material prior to fixation (see Materials and Methods). As shown in Fig. 5A, reactivation is associated with significant levels of ubiquitylation which appeared to localize to the edges of the replication compartments (Fig. 5A inset). However, in the presence of low concentration MLN4924 (1 μM), the degree of replication compartment ubiquitylation was reduced. There are two potential reasons that explain this observation; that CRLs are the predominant E3 ligases that ubiquitylate replication compartment factors, or that NEDDylation is required within replication compartments, and this is important for recruiting ubiquitin E3 ligases (as recently demonstrated during the DNA double-strand break repair mechanism [39]). As NEDDylation-dependent ubiquitylation occurred around replication compartments, we asked whether this modification was required for KSHV lytic reactivation. For these experiments, we employed TREx-BCBL-1-RTA cells, where the lytic cycle is induced by the addition of doxycycline (Dox.) by virtue of Dox-inducible expression of exogenous RTA-Myc. The advantage of using TREx-BCBL-1-RTA cells is that the lytic cycle is robustly induced (more so than with TPA/NaB) and any effect is a result of viral gene expression and not due to the general effects of TPA/NaB. Cells were treated with varying concentrations of MLN4924, the lytic cycle was induced and 24 h later cells were harvested and viral protein expression was assessed by immunoblotting. As shown in Fig. 5B, MLN4924 was able to inhibit the expression of RTA-Myc and its downstream target ORF57 in a dose-responsive manner, corresponding to the levels of Cul2 NEDDylation. As RTA-Myc represents exogenous protein expression (i.e. not from the KSHV genome) in TREx-BCBL-1-RTA cells, these results showed that MLN4924 inhibited all transcription in these cells. Using qPCR analysis of viral genomes, we also confirmed that MLN4924 was able to inhibit KSHV DNA replication (Fig. 5C). However, KSHV genome replication was inhibited at lower concentrations than was required to inhibit viral protein expression (compare protein expression and genome replication levels in cells treated with 1 μM) highlighting a potentially novel mechanism, mediated by NEDDylation, that uncouples these two features of KSHV biology. These data show for the first time that NEDDylation (or NEDDylation-dependent ubiquitylation) plays a significant role during the KSHV lytic cycle.
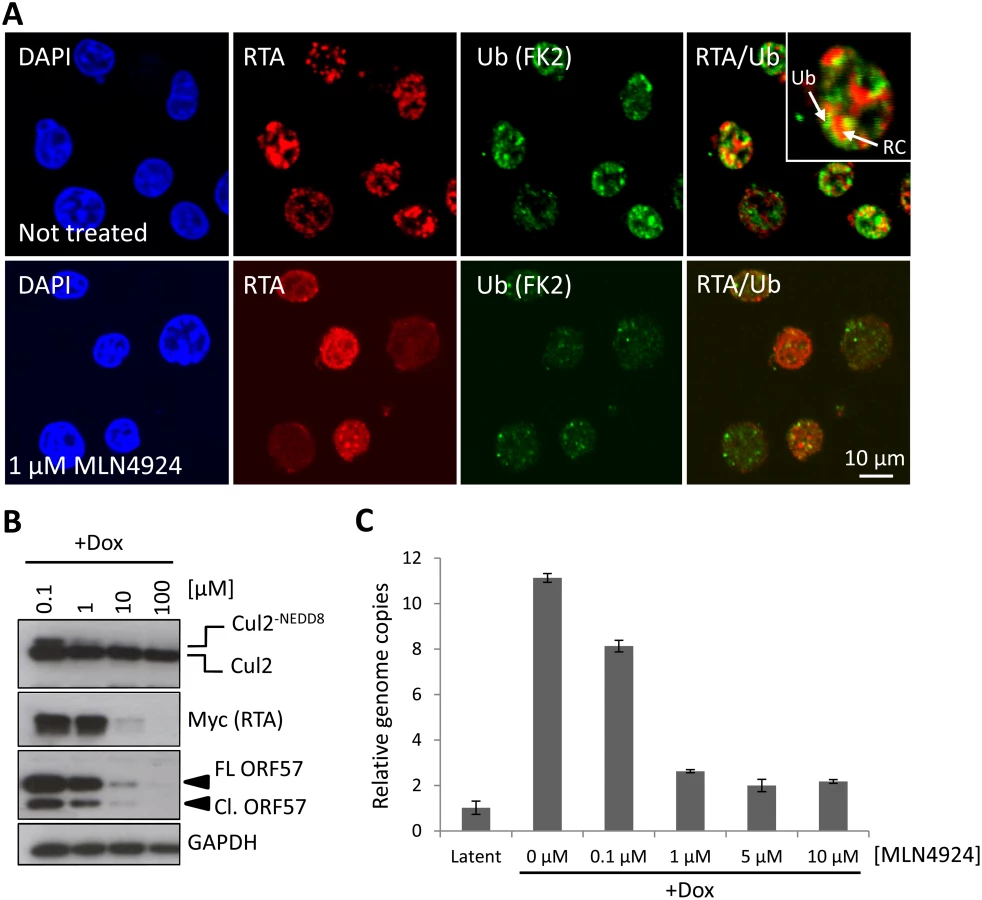
MLN4924-mediated inhibition of KSHV genome replication is not due to caspase activation
We have shown that the inhibition of NEDDylation leads to caspase activation (Fig. 3B) which may account for the ability of MLN4924 to prevent KSHV genome replication. Furthermore, caspase activation has been shown to regulate viral gene expression via Caspase 7 cleavage of ORF57 [29]. We therefore asked whether caspases were responsible for the inhibition of KSHV reactivation. Treatment of cells with z-VAD-FMK efficiently blocked caspase-mediated cleavage of PARP-1 (Cl. PARP-1) and ORF57 (Cl. ORF57) in reactivated cells; caspase inhibition also led to an increase in viral protein expression as noted by the increased levels of full length ORF57 (FL ORF57) and RTA expression in reactivated cells (lane 4, Fig. 6A). Importantly, inhibition of caspase activity restored ORF57 and RTA-Myc expression, clearly demonstrating that MLN4924-induced apoptosis was responsible for inhibiting viral protein expression (lane 6, Fig. 6A). To our surprise however, when we treated cells with z-VAD-FMK and MLN4924 and investigated viral genome copy number by qPCR, viral DNA replication was still inhibited (Fig. 6B).

An additional measure of viral genome replication involves the incorporation of EdU into DNA specifically marking replicating viral genomes in replication compartments, where proteins such as RTA are also localized (Fig. 7A). However, even in cells clearly expressing RTA, MLN4924 treatment prevented virus replication, as shown by the lack of EdU-positive replication compartments. Inhibition of caspases restored viral protein expression in MLN4924-treated cells; however, viral DNA replication was still inhibited (Fig. 6B). We therefore repeated our EdU analysis of drug-treated cells in the presence of z-VAD-FMK. Inhibition of caspases permitted the formation of replication centers and amplification of KSHV DNA in non-treated cells. Nevertheless, z-VAD-FMK was still unable to restore viral genome replication in MLN4924-treated cells (Fig. 7B). These data confirm that MLN4924-mediated activation of caspases was not responsible for the inhibition of KSHV genome replication upon MLN4924 treatment.

NEDDylation is required for the proper formation of KSHV replication compartments and the recruitment of RTA to OriLyt
Throughout our studies, we noticed that reactivated cells treated with MLN4924 displayed an unusual RTA expression pattern; as shown in Figs. 5A & 7A, MLN4924 treatment is associated with pan-RTA localization, rather than in discrete, EdU-positive foci. We also observed the same phenotype when we costained for RTA and a second replication compartment-associated factor, RNA Pol II (S3 Fig). We therefore hypothesized that this would have implications for RTA’s ability to mediate KSHV genome replication, and therefore provide a rationale for the MLN4924-induced block in replication. OriLyt is the cis-acting loci where proteins required for KSHV genome replication assemble. The mechanisms of this have been well characterized and RTA is central to this process. For example, it has been shown that the pre-replication complex (which involves RTA, K8, the core replication proteins and various cellular proteins) is formed prior to its loading onto OriLyt, and that RTA, through its direct DNA binding activity with RRE, recruits these factors to OriLyt [40–42]. Therefore, as MLN4924 inhibited viral genome replication and prevented the correct localization of RTA, we asked whether treatment precluded the loading of the replication complex. To do this, TREx-BCBL-1-RTA cells were treated with 1 μM MLN4924 (a concentration that permits viral protein expression but inhibits replication) and 18 h later, ChIP analysis was performed using antibodies specific for IgG (isotype control), RNA Pol II and Myc-tag (for RTA), and primers specific for the RTA responsive element (RRE) of OriLyt. Firstly, we performed this analysis on latently infected cells (Fig. 7C), where we observed an enrichment of RNA Pol II (6.45-fold over IgG), whereas RTA levels were lower than those observed for IgG control. Interestingly, when cells were treated, RNA Pol II occupancy at OriLyt approximately doubled, and RTA levels increased 1.6-fold over IgG. These results confirm that MLN4924 was able to induce transcription in latently infected cells; however, they show that only low levels of RTA are required. During lytic replication, both RNA Pol II (63-fold over IgG) and RTA (44-fold over IgG) were increased at OriLyt (Fig. 7D). However, after MLN4924 treatment, RNA Pol II occupancy reduced ca. 3-fold (to 21-fold over IgG) and RTA occupancy decreased ca. 4-fold (to 10-fold over IgG) confirming that the inhibition in KSHV genome replication was due to a block in the recruitment of the viral pre-replication complex. Consequently, by using MLN4924 to investigate the role of NEDDylation in KSHV biology, we have uncovered a novel mechanism that regulates lytic reactivation.
Discussion
KSHV infection is responsible for various malignancies, including KS, PEL and many cases of MCD. As these diseases are highly associated with compromised immune function, they represent some of the most common cancers in areas of the world where HIV infection is also high. Indeed, KS is the most prevalent cancer in many sub-Saharan countries. Therefore, understanding the molecular mechanisms that underlie KSHV biology is of the utmost importance if therapeutic targets are to be identified. Given the success of drugs such as bortezamib (UPS inhibitor) for the treatment of various malignancies, interest has grown in the development of inhibitors that target additional aspects of the UPS, or additional Ubls. An example of this is MLN4924, a small molecule inhibitor that blocks the function of NAE1, the first enzyme (E1) in the NEDDylation cascade. Inhibition of NEDDylation leads to a global stabilization of CRL targets, and this drug has proved to be potently cytotoxic in many cancer models. The principal mechanisms of MLN4924 cytotoxicity in cancer cells appear to involve blocking NF-κB signaling (via stabilization of IκBα, and the retention of the NF-κB transcription factor in the cytoplasm) or leading to unlicensed DNA replication (via stabilization Cdt-1), ultimately leading to apoptotic cells death. Given that PEL cells absolutely require NF-κB signaling for their survival, we asked whether MLN4924 was able to kill these cells. Furthermore, as the lytic cycle plays a significant role during the pathogenesis of KS, we also asked whether the NEDDylation cascade was necessary for virus reactivation, and if so, what role it plays.
The activation of NF-κB is central to KSHV infection (by modulating viral gene expression) and for the pathogenesis of KSHV-associated malignancies (via induction of inflammatory mediators and the expression of antiapoptotic genes). Inhibiting NEDDylation was clearly cytotoxic to PEL cells, and mechanistically, it appears that this was due to the inhibition of NF-κB. For example, we showed that MLN4924 led to: i) accumulation of pIκBα, ii) a KSHV gene expression profile consistent with inhibition of NF-κB and iii) the induction of apoptosis (Fig. 8). This corroborates recent work demonstrating that MLN4924 killed NF-κB-dependent ABC-DLBCL cells in a comparable fashion [27]. In fact, ABC-DLBCL cells were more sensitive than GCB-DLBCLs (that are not reliant on NF-κB) [27] suggesting that this drug may be more efficacious for cancers such as PEL and KS, where NF-κB activation is a requirement for malignancy. NF-κB is also required for transcription of the latency control locus which expresses an alternatively spliced transcript that produces ORF71 (vFLIP), ORF72 (vCyclin) and ORF73 (LANA). As LANA is essential for maintenance of the KSHV genome during mitosis, NF-κB signaling is clearly required for sustained infection. Interestingly, MLN4924 did not appear to overtly affect LANA expression (despite reducing its mRNA level) which is in line with previous reports showing that its half-life exceeds the time course of our experiments [43] and signifying that cytotoxicity was not a result of KSHV genome loss. Moreover, the histological signature of KS involves elongated, spindle-shaped endothelial cells and vFLIP expression alone is sufficient to bring about this morphological change in endothelial cell cultures [44,45]. Importantly, this phenotypic alteration has been shown to be NF-κB-dependent; therefore, it will be of particular interest to investigate if MLN4924 can inhibit this characteristic feature of KS.

In contrast to its role in latency-associated transcription, NF-κB is a negative regulator of numerous lytic cycle-associated genes, including those analyzed in this study (RTA, ORF57 and ORF47) [46]. The KSHV protein RTA is both necessary and sufficient for the induction of the lytic cycle, resulting in the expression of the entire KSHV genome and virion production. Many RTA-responsive genes contain RBP-Jκ binding sites, and the translocation of NF-κB into the nucleus allows it to bind RBP-Jκ and prevent its association with RTA. Hence, the induction of lytic cycle-associated gene expression in PEL and rKSHV.219 cells is very likely due to MLN4924-induced inhibition of NF-κB. The Cul1-containing βTrCP is the principal E3 ligase responsible for IκBα degradation [37]. In agreement the above hypothesis, we found that individual expression of dominant-negative versions of Cul1 (DNCul) was able to activate lytic cycle gene expression. Importantly, these data also suggest that MLN4924’s effect on viral gene expression was due to its inhibition of the CRL activity as opposed to a direct role of NEDDylation in KSHV gene expression or the over-arching effects a global block in NEDDylation would have on cellular function. However, an in-depth investigation of the various CRL components is required to fully elucidate the question of specificity.
In addition to the ability of DNCul1 to reactivate lytic cycle associated gene expression, DNCul4B and shRNA knockdown of Cul4B were both able to induce lytic cycle gene expression. Using K12 expression as an indicator of latency versus lytic cycle-associated gene expression, the complete blockade of NEDDylation led to a reduction in its expression, whereas knockdown of Cul4B led to its increase, pointing to differing mechanisms. These data are in line with the hypothesis that complete blockade (via MLN4924) effects NF-κB expression by inhibiting CRL1 and that inhibition of Cul1 NEDDylation is more sensitive than its inhibition of Cul4B modification. It is not currently known whether differences exist in the sensitivity of individual CRLs following MLN4924 treatment. Of the known CRL functions, Cul4 activities are centered on chromatin regulation, such as chromosome condensation, heterochromatin formation and DNA replication and repair processes. Therefore, although Cul4’s potential role in the maintenance of KSHV latency is novel, it might not be surprising. The two Cul4 proteins are virtually identical apart from an extended N-terminal portion found in Cul4B that encodes a nuclear localization signal. Therefore, Cul4B predominantly resides in the nucleus, whereas Cul4A is recruited to the nucleus in response to genotoxic stress. This provides further credence to the hypothesis that Cul4B is important for the regulation of KSHV gene expression. The DNA damage response is a recurring theme in virus biology as it either aids or is a consequence of infection [24,47–49]. Cul4A and 4B play a pivotal role in the nucleotide excision repair (NER) process, and it was recently reported that human cytomegalovirus (HCMV) is dependent on NER during replication of its genome. Therefore, the role of Cul4 during KSHV infection merits further investigation.
A striking observation however, was that MLN4924 treatment blocked viral genome replication despite appreciable levels of viral gene expression. At 1 μM MLN4924, ORF57 expression was not significantly reduced, but genome replication was reduced by ca. 80%. We considered the possibility that the induction of apoptosis may be responsible for this, and so we repeated the experiments in the presence of a pan-caspase inhibitor (z-VAD-FMK). Even though this led to a recovery in KSHV protein expression even when up to 10 μM MLN4924 was used, genome replication was still not observed. Over the course of our studies, we did notice aberrant RTA localization in BCBL-1 cells treated with MLN4924, followed by reactivation (e.g. Fig. 7). Given that RTA’s function is to recruit the viral pre-replication complex, along with various cellular factors required for KSHV genome replication, its localization is intimately linked with sites of viral genome replication occurs. These sites are termed replication compartments (or replication centers) and can be observed as discrete foci at the nuclear periphery that co-stain with RTA and newly replicated viral DNA (this can be observed using BrdU or EdU that specifically marks viral DNA due to KSHV’s ability to block cellular DNA replication during reactivation). We therefore hypothesized that NEDDylation, or CRL-mediated ubiquitylation was required for KSHV genome replication (but not viral gene expression).
Firstly, we showed that reactivation of KSHV is associated with ubiquitylation within replication compartments, and that MLN4924 treatment inhibited this. We next investigated whether NEDDylation was required for RTA’s recruitment to the origins of lytic replication (OriLyt). Using ChIP analysis, we observed that treatment of latent cells led to an increase in Pol II (ca. 2-fold) and RTA (ca. 1.6-fold) occupancy, which agrees with our data showing that treatment led to activation of viral gene expression. Treatment followed by reactivation with Dox. however showed us that NEDDylation was required for RTA recruitment to OriLyt. Here we showed that 1 μM MLN4924 (a concentration that still permits viral gene expression) blocked RTA recruitment to OriLyt. Likewise, Pol II occupancy was reduced to similar levels. This confirmed to us that NEDDylation was required for the proper recruitment of the RTA (and therefore the pre-replication complex) to OriLyt, and offers and explanation for the aberrant localization of RTA and the inhibition of genome replication.
There are various hypotheses that may explain the importance of NEDDylation for the recruitment of the pre-replication complex to OriLyt (Fig. 8). It might be possible that proteins within the pre-replication complex are themselves targets for NEDD8 modification. We have addressed this for RTA and showed that RTA is not NEDDylated (S4 Fig); given that RTA is the protein responsible for recruitment, this result suggested that this hypothesis is unlikely [40]. It might also be possible that NEDDylation or CRL-mediated ubiquitylation of OriLyt-associated chromatin might be important for the recruitment of the pre-replication complex, as has recently been reported for the recruitment of repair enzymes during a DNA damage response [50]. Although it is still possible that these modifications play a role in cells, RTA binds to DNA in a sequence-specific manner, and it can do this in vitro in the absence of chromatin suggesting that this may not be the role of NEDDylation during reactivation [51]. A further hypothesis is that modification of a factor that resides at OriLyt during latency (in order to maintain latency) may be required during reactivation. This may include CRL-mediated ubiquitylation (via K48) that targets a latency-associated protein for degradation thus allowing access to OriLyt by the pre-replication complex. It may also involve Ub-modification via K63 linkage that might enhance binding in vivo. Various cellular proteins have been reported to interact with OriLyt during reactivation, but in that study, proteins that bound during latency were not investigated [42]. All of the above hypotheses merit further investigation but are unfortunately beyond the scope of this study.
In summary, we have demonstrated that NEDDylation is essential for various aspects of KSHV infection. Inhibition of NEDDylation using MLN4924 proved cytotoxic to PEL cells due to their dependence on NF-κB. Remarkably, we also show that a functioning NEDDylation cascade is essential for KSHV genome replication as it was required for the recruitment of the RTA-mediated pre-replication complex to OriLyt. These new findings have opened up new avenues of investigation regarding the regulation of herpesvirus latency and reactivation. Moreover, they demonstrate that inhibition of NEDDylation represents a novel approach for the treatment of KSHV-associated malignancies, including KS that is dependent on both lytic replication and the latency-associated activation of NF-κB.
Materials and Methods
Cell lines and drug treatments
BCBL-1 and BC-3 are primary effusion lymphoma (PEL) B cell lines latently infected with KSHV. TREx-BCBL-1-RTA cells (a kind gift of Dr. Jae Jung, University of Southern California) are a BCBL-1-based cell line that has been engineered to inducibly express exogenous Myc-tagged RTA by the addition of doxycycline, leading to a robust reactivation of the full KSHV lytic cycle [52]. The rKSHV.219 cell line maintains KSHV as a latent infection and was generated by infecting HEK293T cells with a recombinant KSHV that contains a constitutively active puromycin resistance and GFP gene, and an RFP gene that is fused to an RTA-responsive lytic cycle (PAN) promoter; hence, expression of RFP can be used as a reporter of RTA activity [53]. HEK293T cells were also used for co-immunoprecipitation experiments. All cells were maintained at 37°C in a humidified incubator with 5% CO2. The PEL cell lines were maintained in RPMI-1640 (Lonza) supplemented with 10% FBS (Life Technologies). TREx-BCBL-1-RTA media also contained 200 μg/ml hygromycin B (Life Technologies). The rKSHV.219 and HEK293T cell lines were maintained in DMEM (Lonza) supplemented with 10% FBS (Life Technologies). The rKSHV.219 cultures also contained 1 μg/ml puromycin. KSHV reactivation was induced in BCBL-1 and rKSHV.219 by the addition of 20 ng/ml 12-O-tetradecanoylphorbol 13-acetate (TPA) and 1.5 mM sodium butyrate (NaB) or 1 μg/ml doxycycline hyclate (Sigma) in TREx-BCBL-1-RTA cells.
MLN4924 [12] (Millennium Pharmaceuticals) stock solutions (10 mM) were prepared in DMSO and diluted in media prior to its addition to cells at the indicated concentrations, and for the indicated times. Routinely, 106 cells in 12-well plates were treated. Inhibition of caspase enzymes was achieved by the addition of 50 μM z-VAD-FMK or FMK-negative control (MBL International) 30 min prior to MLN4924 and/or reactivation treatment. Inhibitors remained on the cells for the duration of the experiments.
Viability assays
Cell viability was determined using the standard trypan blue exclusion method and using the ATPlite Luminescence ATP Detection Assay (Perkin Elmer). For the ATPlite assay, 10,000 cells were seeded into white 96-well plates and allowed to settle for 16 h at 37°C. Varying concentrations of MLN4924 were added to the cells followed by 96 h incubation at 37°C. Cellular proliferation was determined according to the manufacturer’s instructions.
Cell cycle analysis
Cells (106) were treated with 1 μM MLN4924 (or left untreated) for 24 h, washed in cold PBS and fixed for 24 h in cold 70% ethanol at -20°C. Prior to analysis, cells were washed in PBS and treated with 1 ml PBS, 10 μg/ml propidium iodide (Sigma), 0.5 mg RNase A for 3 h at 37°C. Cells were then pelleted, resuspended in 1 ml PBS and analysed using a Becton Dickinson BD-LSRFortessa flow cytometer. Data were fitted using ModFit software.
Expression vectors and transfection
Most expression vectors were obtained from Addgene: pcDNA3-DN-hCul1-FLAG (plasmid 15818), pcDNA3-DN-hCul2-FLAG (plasmid 15819), pcDNA3-DN-hCul3-FLAG (plasmid 15820), pcDNA3-DN-hCul4A-FLAG (plasmid 15821), pcDNA3-DN-hCul4B-FLAG (plasmid 15822), pcDNA3-DN-hCul5-FLAG (plasmid 15823) [35], pcDNA3-HA-Cullin4A (plasmid 19907), pcDNA3-Myc-NEDD8 (plasmid 19943). pFLAG-CMV-4-NEDD8 was a kind gift from Dr Eric Stebbins (Rockefeller University) [54]. RTA expression vectors (pRTA and pRTAH145L) were gifts of Dr Gary Hayward (Johns Hopkins University). Cells were plated into 6-well plates and transfections routinely used 1 μg plasmid DNA and Lipofectamine 2000 (Life Technologies) following the manufacturer’s instructions.
Knockdown of gene expression
Knockdown of Cul4B expression was accomplished using four individual SureSilencing shRNA expression vectors (Qiagen). TREx-BCBL-1-RTA cells (3 x 106 per transfection) were transfected with 20 μg scramble shRNA (control) or 5 μg of each of four shCul4B vectors by nucleofection (Lonza; solution V and nucleofector program T-01). Four days post-transfection, RNA was extracted using Trizol (Life Technologies) and used to generate cDNA for qRT-PCR analysis (see below).
Immunoblot analysis
Cells were washed in PBS and proteins extracted in lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% NP-40 and 1x protease inhibitor cocktail (Roche) for 15 min on ice and clarified by centrifugation at 12,000 xg for 10 min, 4°C. Cells used in Nuclear/cytoplasmic enrichments assays (ca. 106) were washed in PBS, lysed in 50 μl PBS, 1% Triton X-100 and 1x protease inhibitor cocktail (Roche) for 15 min on ice and nuclei were pelleted at 2000 xg for 5 min. Cytoplasmic fractions (supernatant) were moved to new tubes and the nuclear pellets were washed three times in lysis buffer. SDS-PAGE and immunoblotting of normalized protein concentrations followed standard techniques using the following antibodies [55]: RTA, rabbit antisera (1 : 400), mouse mAb anti-Myc-tag (1 : 5000; Sigma), rabbit pAb anti-FLAG-tag (1 : 1000; Sigma), mouse mAb anti-HA-tag (1 : 5000; Life Technologies), mouse mAb anti-ORF57 (1 : 1000; Santa Cruz), rabbit mAb anti-Cullin 2 (1 : 1000; Life Technology), mouse mAb anti-PARP-1 (1 : 1000; Cell Signaling Technology), mouse mAb anti-GAPDH (1 : 5000; Sigma), mouse mAb anti-Lamin B (1 : 1000; Santa Cruz), rabbit mAb anti-Phospho-IκBα (ser32) (1 : 1000; Cell Signaling Technology), mouse mAb anti-p65/RelA (1 : 1000; Santa Cruz), rabbit mAb anti-Caspase 3 (8G10) (1 : 1000; Cell Signaling Technology), rabbit mAb anti-active Caspase 3 (Abcam; 1 : 250), mouse mAb anti-Caspase 9 (Cell Signaling Technology), mouse mAb anti-Cdt-1 (1 : 1000; Santa Cruz), mouse mAb anti-γH2Ax (ser139) (1 : 1000, Santa Cruz) rabbit mAb anti-Phospho-p53 (ser15) (1 : 1000; Cell Signaling Technology). Signals were detected using chemiluminescence and densitometry (ImageJ) was used to semi-quantify expression levels.
Quantitative RT-PCR (qRT-PCR)
As previously reported [56], total cellular RNA was extracted from cells using Trizol (Life Technologies) according to the manufacturer’s instructions and contaminating DNA was removed using the DNA-free kit (Ambion). Complimentary DNA (cDNA) was generated from 1 μg RNA in 20 μl reaction volumes using M-MuLV reverse transcriptase (RT; NEB) according to the manufacturer’s recommendations with 5 ng oligo(dT). In parallel, negative control reactions were performed for each RNA by omitting RT in order to confirm that quantification represented cDNA and not contaminating DNA. Quantitative PCR reaction mixes (20 μl) included 1x SensiMix SYBR green master mix (Bioline), 0.5 μM each primer and 1 μl cDNA reaction mix. Cycling was performed in a RotorGene Q machine (Eppendorf) and included an initial 10 min denaturation step at 94°C, followed by 40 cycles of 30 s at 94°C, 30 s at 60°C and 30 s at 72°C. Melting curve analysis was performed between 65 and 95°C (with 0.2°C increments) to verify amplicon specificity. Quantification of GAPDH mRNA was used to normalize between samples, and the average cycle threshold (CT) was determined from three independent RNA samples from independent cultures. Relative expression compared to non-treated or DMSO-treated control cells was calculated using the ΔΔCT method.
Indirect immunofluorescence microscopy
Cells were cultured on poly-L-lysine coated coverslips in 12-well plates for 24 h at 37°C, gently washed with PBS, fixed using 4% formaldehyde (in PBS) for 10 min, permeabilized with PBS, 1% Triton X-100 for 10 min and washed three times with PBS as previously reported [57,58]. Where stated, some experiments involved the removal of soluble nuclear proteins (i.e. those not tightly bound to chromatin) using an “extraction first” method: here coverslips were treated (as previously reported [24]) with CSK buffer (10 mM PIPES, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2) containing 0.5% Triton X-100 for 2 min followed by 2 washes with CSK buffer alone. These cells were then fixed and processed as normal. Primary antibodies were diluted in PBS, 2% BSA, added to cells and incubated in humidity chambers for 2 h at 37°C or overnight at 4°C followed by 5 washed with PBS. The appropriate secondary antibodies (Alexa Fluor 488 or 594; Life Technologies) were diluted 1 : 500 in PBS, 2% BSA and incubated with cells for 1 h at 37°C followed by 5 washed with PBS. Coverslips were mounted in VECTORSHEILD with DAPI (Vectorlabs). Where experimentally possible, double staining was performed by incubating the coverslips with both primary antibodies, and following washing, both secondary antibodies at the same time. Images were captured using an LSM700 laser scanning microscope (Carl Zeiss) and processed using ZEN imaging software (Carl Zeiss). Antibodies included: RTA rabbit antisera (1 : 100), NF-κB RelA mouse mAb (1 : 250; Cell Signaling), anti-ubiquitinylated proteins clone FK2 mouse mAb (1 : 250; Millipore), mouse mAb anti RNA Pol II (Millipore).
Virus reactivation assays
Virus reactivation was determined by two complimentary assays—identification of viral replication compartments by imaging the incorporation of EdU into replicating viral DNA and by quantitative PCR analysis of viral genome amplification. Incorporation of EdU was performed using a Click-iT EdU Imaging Kit (Life Technologies). Briefly, 1 x 106 TREx-BCBL-1-RTA cells were drug-treated as appropriate, virus reactivation was induced by the addition of doxycycline and the cells were plated onto poly-L-lysine-coated coverslips. After 16 h, cells were pulsed for 45 min with 10 μM EdU, washed, fixed and permeablized according to the manufacturer’s recommendations. After detection of EdU (according to the manufacturer’s protocol), the cells were washed and further incubated with RTA antisera (1 : 100 in PBS, 2% BSA) for 1 h at 37°C, washed again and incubated with Alexa Fluor 488 goat anti-rabbit antibody (Life Technologies; 1 : 500 in PBS, 2% BSA) for 1 h at 37°C. Finally, DNA was stained using Hoechst 33342 (1 : 2000 in PBS) and mounted in VECTORSHIELD (Vectorlabs). Images were captured using an LSM700 laser scanning microscope (Carl Zeiss) using 639 nm, 488 nm and 405 nm lasers for the detection of EdU, RTA and DNA respectively and processed using ZEN imaging software (Carl Zeiss).
For quantitative PCR analysis 1 x 106 TREx-BCBL-1-RTA cells were drug-treated as appropriate and virus reactivation was induced by the addition of doxycycline. At 24 h post-reactivation, total DNA was extracted using a DNA Minikit (Qiagen) and quantified by UV spectrophotometry. Viral DNA was quantified using 3.4 ng DNA, in 20 μl reaction volumes as described above for qRT-PCR, using primers specific for the ORF57 gene. Quantification of GAPDH was used to normalize between samples, and the average cycle threshold (CT) was determined from three independent samples from independent cultures. Relative levels of viral DNA was calculated using the ΔΔCT method.
Immunoprecipitation (NEDDylation assay)
HEK293T cells were plated into 6-well dishes and co-transfected with the indicated plasmids with pcDNA-Myc-NEDD8 or pFLAG-CMV-4-NEDD8 [54] for 24 h. Cells were washed in PBS and proteins extracted in 1 ml lysis buffer (see above) and incubated with anti-c-Myc agarose (Sigma) or goat anti-DDDDK conjugated agarose (for detection of FLAG-tagged proteins; Abcam) following the manufacturer’s recommendations. Immunoprecipitated (NEDDylated) proteins were eluted in Laemmli buffer and subject to immunoblot analysis (see above).
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation (ChIP) assays were carried out as previously stated [59] using the EZ-ChIP chromatin immunoprecipitation kit (Millipore). Per condition, 107 cells were treated with 1% formaldehyde for 10 min prior to three washes with ice-cold PBS. Cells were then resuspended in 3 ml SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris, [pH 8.1]) containing 1x protease inhibitor cocktail II and sonicated 15 time with 20 s pulses at 4°C. Insoluble material was removed by centrifugation for 10 min at 12,000 xg, 4°C. For each immunoprecipitation (IP), 100 μl of prepared chromatin was added to 900 μl of dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, [pH 8.1], 167 mM NaCl) containing 1x protease inhibitor cocktail II. The lysates were then pre-cleared with Protein G agarose before being incubated with appropriate antibodies (Myc-tag and RNA Pol II) overnight at 4°C. The following day 60 μl of Protein G agarose was added to each IP and rotated for 1 h at 4°C. The Protein G agarose-antibody/chromatin complexes were then washed sequentially in 1 ml of the provided buffers; low salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, [pH 8.1], 150 mM NaCl), high salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, [pH 8.1], 500 mM NaCl), LiCl immune complex wash buffer (0.25 M LiCl, 1% IGEPAL CA630, 1% deoxycholic acid sodium salt, 1 mM EDTA, 10 mM Tris, [pH 8.1]) and TE buffer (10 mM Tris-HCl, [pH 8.0], 1 mM EDTA). The DNA/protein complexes were eluted in 200 μl elution buffer (1% SDS, 0.1 M NaHCO3) before reversal of crosslinks with 5 M NaCl for 4 h at 65°C. Samples were then treated with RNase A at 37°C for 30 min followed by incubation with Proteinase K for 2 h at 45°C. The resulting DNA was purified over the provided spin filter columns, with elution in 50 μl of elution buffer C. The purified DNA was then subject to qPCR amplification using primers directed to the OriLyt (Forward primer: 5’ - ACG GGC CTG GAA TCT CGC CTC TGG-3’ and Reverse primer: 5’ - ATG GGC GTA ACC GTA GGA CAA GCT G-3’). Each qPCR reaction performed in triplicate containing 5 μl purified DNA and was quantified as described above.
Oligonucleotides
Oligonucleotide primer sequences are available upon request.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol. 2012;19 : 99–115. doi: 10.1016/j.chembiol.2012.01.003 22284358
2. Boulanger E, Meignin V, Oksenhendler E. Bortezomib (PS-341) in patients with human herpesvirus 8-associated primary effusion lymphoma. Br J Haematol. 2008;141 : 559–561. doi: 10.1111/j.1365-2141.2008.07057.x 18341641
3. Brown HJ, McBride WH, Zack JA, Sun R. Prostratin and bortezomib are novel inducers of latent Kaposi's sarcoma-associated herpesvirus. Antivir Ther. 2005;10 : 745–751. 16218174
4. Matta H, Chaudhary PM. The proteasome inhibitor bortezomib (PS-341) inhibits growth and induces apoptosis in primary effusion lymphoma cells. Cancer Biol Ther. 2005;4 : 77–82. 15662128
5. Sarosiek KA, Cavallin LE, Bhatt S, Toomey NL, Natkunam Y, et al. Efficacy of bortezomib in a direct xenograft model of primary effusion lymphoma. Proc Natl Acad Sci U S A. 2010;107 : 13069–13074. doi: 10.1073/pnas.1002985107 20615981
6. Dittmer DP, Damania B. Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)—an update. Curr Opin Virol. 2013;3 : 238–244. doi: 10.1016/j.coviro.2013.05.012 23769237
7. Mesri EA, Cesarman E, Boshoff C. Kaposi's sarcoma and its associated herpesvirus. Nat Rev Cancer. 2010; 10 : 707–719. doi: 10.1038/nrc2888 20865011
8. La Ferla L, Pinzone MR, Nunnari G, Martellotta F, Lleshi A, et al. Kaposi' s sarcoma in HIV-positive patients: the state of art in the HAART-era. Eur Rev Med Pharmacol Sci. 2013;17 : 2354–2365. 24065230
9. Simonelli C, Spina M, Cinelli R, Talamini R, Tedeschi R, et al. Clinical features and outcome of primary effusion lymphoma in HIV-infected patients: a single-institution study. J Clin Oncol. 2003;21 : 3948–3954. 14581418
10. Gantt S, Casper C. Human herpesvirus 8-associated neoplasms: the roles of viral replication and antiviral treatment. Curr Opin Infect Dis. 2011;24 : 295–301. doi: 10.1097/QCO.0b013e3283486d04 21666458
11. Bhatt S, Ashlock BM, Toomey NL, Diaz LA, Mesri EA, et al. Efficacious proteasome/HDAC inhibitor combination therapy for primary effusion lymphoma. J Clin Invest. 2013;123 : 2616–2628. doi: 10.1172/JCI64503 23635777
12. Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458 : 732–736. doi: 10.1038/nature07884 19360080
13. Sarikas A, Hartmann T, Pan ZQ. The cullin protein family. Genome Biol. 2011;12 : 220. doi: 10.1186/gb-2011-12-4-220 21554755
14. Bennett EJ, Rush J, Gygi SP, Harper JW. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell. 2010;143 : 951–965. doi: 10.1016/j.cell.2010.11.017 21145461
15. Rabut G, Peter M. Function and regulation of protein neddylation. 'Protein modifications: beyond the usual suspects' review series. EMBO Rep. 2008;9 : 969–976. doi: 10.1038/embor.2008.183 18802447
16. Wimmer P, Schreiner S, Dobner T. Human pathogens and the host cell SUMOylation system. J Virol. 2012;86 : 642–654. doi: 10.1128/JVI.06227-11 22072786
17. Everett RD, Boutell C, Hale BG. Interplay between viruses and host sumoylation pathways. Nat Rev Microbiol. 2013;11 : 400–411. doi: 10.1038/nrmicro3015 23624814
18. Gastaldello S, Hildebrand S, Faridani O, Callegari S, Palmkvist M, et al. A deneddylase encoded by Epstein-Barr virus promotes viral DNA replication by regulating the activity of cullin-RING ligases. Nat Cell Biol. 2010;12 : 351–361. doi: 10.1038/ncb2035 20190741
19. Stanley DJ, Bartholomeeusen K, Crosby DC, Kim DY, Kwon E, et al. Inhibition of a NEDD8 Cascade Restores Restriction of HIV by APOBEC3G. PLoS Pathog. 2012;8: e1003085. doi: 10.1371/journal.ppat.1003085 23300442
20. Romani B, Engelbrecht S, Glashoff RH. Antiviral roles of APOBEC proteins against HIV-1 and suppression by Vif. Arch Virol. 2009;154 : 1579–1588. doi: 10.1007/s00705-009-0481-y 19669862
21. Yu X, Yu Y, Liu B, Luo K, Kong W, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302 : 1056–1060. 14564014
22. Cai QL, Knight JS, Verma SC, Zald P, Robertson ES. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. 2006;2: e116. 17069461
23. Hannah J, Zhou P. Regulation of DNA damage response pathways by the cullin-RING ubiquitin ligases. DNA Repair (Amst). 2009;8 : 536–543. doi: 10.1016/j.dnarep.2009.01.011 19231300
24. O'Dowd JM, Zavala AG, Brown CJ, Mori T, Fortunato EA. HCMV-infected cells maintain efficient nucleotide excision repair of the viral genome while abrogating repair of the host genome. PLoS Pathog. 2012;8: e1003038. doi: 10.1371/journal.ppat.1003038 23209410
25. Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer. 2010;1 : 708–716. doi: 10.1177/1947601910382898 21779466
26. Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, et al. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011;71 : 3042–3051. doi: 10.1158/0008-5472.CAN-10-2122 21487042
27. Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116 : 1515–1523. doi: 10.1182/blood-2010-03-272567 20525923
28. de Oliveira DE, Ballon G, Cesarman E. NF-kappaB signaling modulation by EBV and KSHV. Trends Microbiol. 2010;18 : 248–257. doi: 10.1016/j.tim.2010.04.001 20452220
29. Majerciak V, Kruhlak M, Dagur PK, McCoy JP, Zheng ZM Jr.. Caspase-7 cleavage of Kaposi sarcoma-associated herpesvirus ORF57 confers a cellular function against viral lytic gene expression. J Biol Chem. 2010;285 : 11297–11307. doi: 10.1074/jbc.M109.068221 20159985
30. Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, et al. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J Virol. 1999;73 : 2232–2242. 9971806
31. Pearce M, Matsumura S, Wilson AC. Transcripts Encoding K12, v-FLIP, v-Cyclin, and the MicroRNA Cluster of Kaposi's Sarcoma-Associated Herpesvirus Originate from a Common Promoter. J Virol. 2005;79 : 14457–14464. 16254382
32. Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18 : 621–663. 10837071
33. Jha HC, Upadhyay SK, M AJP, Lu J, Cai Q, et al. H2AX phosphorylation is important for LANA-mediated Kaposi's sarcoma-associated herpesvirus episome persistence. J Virol. 2013;87 : 5255–5269. doi: 10.1128/JVI.03575-12 23449797
34. Lei X, Bai Z, Ye F, Xie J, Kim CG, et al. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol. 2010;12 : 193–199. doi: 10.1038/ncb2019 20081837
35. Jin J, Ang XL, Shirogane T, Wade Harper J. Identification of substrates for F-box proteins. Methods Enzymol. 2005;399 : 287–309. 16338364
36. Lee DF, Kuo HP, Liu M, Chou CK, Xia W, et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol Cell. 2009;36 : 131–140. doi: 10.1016/j.molcel.2009.07.025 19818716
37. Yaron A, Hatzubai A, Davis M, Lavon I, Amit S, et al. Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature. 1998;396 : 590–594. 9859996
38. Nakagawa T, Xiong Y. X-linked mental retardation gene CUL4B targets ubiquitylation of H3K4 methyltransferase component WDR5 and regulates neuronal gene expression. Mol Cell. 2011;43 : 381–391. doi: 10.1016/j.molcel.2011.05.033 21816345
39. Ma T, Chen Y, Zhang F, Yang CY, Wang S, et al. RNF111-dependent neddylation activates DNA damage-induced ubiquitination. Mol Cell. 2013;49 : 897–907. doi: 10.1016/j.molcel.2013.01.006 23394999
40. Rossetto CC, Susilarini NK, Pari GS. nteraction of Kaposi's sarcoma-associated herpesvirus ORF59 with oriLyt is dependent on binding with K-Rta. J Virol. 2011;85 : 3833–3841. doi: 10.1128/JVI.02361-10 21289111
41. Wang Y, Li H, Chan MY, Zhu FX, Lukac DM, et al. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J Virol. 2004;78 : 8615–8629. 15280471
42. Wang Y, Li H, Tang Q, Maul GG, Yuan Y. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: involvement of host cellular factors. J Virol. 2008;82 : 2867–2882. doi: 10.1128/JVI.01319-07 18199640
43. Chen W, Hilton IB, Staudt MR, Burd CE, Dittmer DP. Distinct p53, p53:LANA, and LANA complexes in Kaposi's Sarcoma—associated Herpesvirus Lymphomas. J Virol. 2010;84 : 3898–3908. doi: 10.1128/JVI.01321-09 20130056
44. Alkharsah KR, Singh VV, Bosco R, Santag S, Grundhoff A, et al. Deletion of Kaposi's sarcoma-associated herpesvirus FLICE inhibitory protein, vFLIP, from the viral genome compromises the activation of STAT1-responsive cellular genes and spindle cell formation in endothelial cells. J Virol. 2011;85 : 10375–10388. doi: 10.1128/JVI.00226-11 21795355
45. Grossmann C, Podgrabinska S, Skobe M, Ganem D. Activation of NF-kappaB by the latent vFLIP gene of Kaposi's sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J Virol. 2006;80 : 7179–7185. 16809323
46. Izumiya Y, Izumiya C, Hsia D, Ellison TJ, Luciw PA, et al. NF-kappaB serves as a cellular sensor of Kaposi's sarcoma-associated herpesvirus latency and negatively regulates K-Rta by antagonizing the RBP-Jkappa coactivator. J Virol. 2009;83 : 4435–4446. doi: 10.1128/JVI.01999-08 19244329
47. Jackson BR, Noerenberg M, Whitehouse A. A novel mechanism inducing genome instability in Kaposi's sarcoma-associated herpesvirus infected cells. PLoS Pathog. 2014;10: e1004098. doi: 10.1371/journal.ppat.1004098 24788796
48. Karttunen H, Savas JN, McKinney C, Chen YH, Yates JR 3rd, et al. Co-opting the Fanconi Anemia Genomic Stability Pathway Enables Herpesvirus DNA Synthesis and Productive Growth. Mol Cell. 2014;55 : 111–22. doi: 10.1016/j.molcel.2014.05.020 24954902
49. Turnell AS, Grand RJ. DNA viruses and the cellular DNA-damage response. J Gen Virol. 2012;93 : 2076–2097. doi: 10.1099/vir.0.044412-0 22855786
50. Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell. 2013;49 : 795–807. doi: 10.1016/j.molcel.2013.01.017 23416108
51. Bu W, Carroll KD, Palmeri D, Lukac DM. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta lytic switch protein functions as a tetramer. J Virol. 2007;81 : 5788–5806. 17392367
52. Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, et al. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol. 2003;77 : 4205–4220. 12634378
53. Vieira J O'Hearn PM. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology. 2004;325 : 225–240. 15246263
54. Jubelin G, Taieb F, Duda DM, Hsu Y, Samba-Louaka A, et al. Pathogenic bacteria target NEDD8-conjugated cullins to hijack host-cell signaling pathways. PLoS Pathog. 2010;6: e1001128. doi: 10.1371/journal.ppat.1001128 20941356
55. Hall KT, Giles MS, Calderwood MA, Goodwin DJ, Matthews DA, et al. The Herpesvirus Saimiri open reading frame 73 gene product interacts with the cellular protein p32. J Virol. 2002;76 : 11612–11622. 12388722
56. Jackson BR, Boyne JR, Noerenberg M, Taylor A, Hautbergue GM, et al. An interaction between KSHV ORF57 and UIF provides mRNA-adaptor redundancy in herpesvirus intronless mRNA export. PLoS Pathog. 2011;7: e1002138. doi: 10.1371/journal.ppat.1002138 21814512
57. Boyne JR, Jackson BR, Taylor A, Macnab SA, Whitehouse A. Kaposi's sarcoma-associated herpesvirus ORF57 protein interacts with PYM to enhance translation of viral intronless mRNAs. EMBO J. 2010;29 : 1851–1864. doi: 10.1038/emboj.2010.77 20436455
58. Goodwin DJ, Hall KT, Giles MS, Calderwood MA, Markham AF, et al. The carboxy terminus of the herpesvirus saimiri ORF 57 gene contains domains that are required for transactivation and transrepression. J Gen Virol. 2000;81 : 2253–2265. 10950983
59. Gould F, Harrison SM, Hewitt EW, Whitehouse A. Kaposi's sarcoma-associated herpesvirus RTA promotes degradation of the Hey1 repressor protein through the ubiquitin proteasome pathway. J Virol. 2009;83 : 6727–6738. doi: 10.1128/JVI.00351-09 19369342
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 3
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Bacterial Immune Evasion through Manipulation of Host Inhibitory Immune Signaling
- BILBO1 Is a Scaffold Protein of the Flagellar Pocket Collar in the Pathogen
- Antimicrobial-Induced DNA Damage and Genomic Instability in Microbial Pathogens
- Attenuation of Tick-Borne Encephalitis Virus Using Large-Scale Random Codon Re-encoding