
The Elongator Complex Regulates Neuronal α-tubulin Acetylation
Although acetylated α-tubulin is known to be a marker of stable microtubules in neurons, precise factors that regulate α-tubulin acetylation are, to date, largely unknown. Therefore, a genetic screen was employed in the nematode Caenorhabditis elegans that identified the Elongator complex as a possible regulator of α-tubulin acetylation. Detailed characterization of mutant animals revealed that the acetyltransferase activity of the Elongator is indeed required for correct acetylation of microtubules and for neuronal development. Moreover, the velocity of vesicles on microtubules was affected by mutations in Elongator. Elongator mutants also displayed defects in neurotransmitter levels. Furthermore, acetylation of α-tubulin was shown to act as a novel signal for the fine-tuning of microtubules dynamics by modulating α-tubulin turnover, which in turn affected neuronal shape. Given that mutations in the acetyltransferase subunit of the Elongator (Elp3) and in a scaffold subunit (Elp1) have previously been linked to human neurodegenerative diseases, namely Amyotrophic Lateral Sclerosis and Familial Dysautonomia respectively highlights the importance of this work and offers new insights to understand their etiology.
Published in the journal:
. PLoS Genet 6(1): e32767. doi:10.1371/journal.pgen.1000820
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000820
Summary
Although acetylated α-tubulin is known to be a marker of stable microtubules in neurons, precise factors that regulate α-tubulin acetylation are, to date, largely unknown. Therefore, a genetic screen was employed in the nematode Caenorhabditis elegans that identified the Elongator complex as a possible regulator of α-tubulin acetylation. Detailed characterization of mutant animals revealed that the acetyltransferase activity of the Elongator is indeed required for correct acetylation of microtubules and for neuronal development. Moreover, the velocity of vesicles on microtubules was affected by mutations in Elongator. Elongator mutants also displayed defects in neurotransmitter levels. Furthermore, acetylation of α-tubulin was shown to act as a novel signal for the fine-tuning of microtubules dynamics by modulating α-tubulin turnover, which in turn affected neuronal shape. Given that mutations in the acetyltransferase subunit of the Elongator (Elp3) and in a scaffold subunit (Elp1) have previously been linked to human neurodegenerative diseases, namely Amyotrophic Lateral Sclerosis and Familial Dysautonomia respectively highlights the importance of this work and offers new insights to understand their etiology.
Introduction
Microtubules (MTs) are polymers of α/β-tubulin heterodimers that associate head-to-tail and laterally to form hollow tubes. MTs are usually highly dynamic and undergo rapid turnover by exchange of subunits where the ends of MTs undergo transitions between growth and shrinkage. It has been postulated that this “dynamic instability” provides a space-probing mechanism that creates contact between MTs and target organelles [1]. However, the cytoplasmic network also contains a subpopulation of stable MTs [2],[3],[4] that have an undefined cellular function, possibly required for cellular morphogenesis [5]. A distinguishing and evolutionarily-conserved feature of stable MTs is that they acquire posttranslational modifications (PTMs) in a time-dependent manner [6]. An example of a PTM is the acetylation of α-tubulin on a lysine residue at position 40. HDAC6 and SIRT2 have been shown to act as microtubule de-acetylases [7],[8],[9].
Elimination of acetylation has no obvious phenotypic consequences in Chlamydomonas or Tetrahymena, thus leading to the notion that α-tubulin acetylation is per se not required for cell survival. In Caenorhabditis elegans the expression of a non-acetylatable α-tubulin rescues the touch insensitivity phenotypes of neurons lacking MEC-12, the only C. elegans α-tubulin that contains lysine at position 40 [10],[11],[12]. However, by using other systems, acetylation of α-tubulin has been proposed to control transport mechanisms such as e.g. the dynein/dynactin transport of aggresomes [13],[14] or the selective transport of the Kinesin-1 cargo JIP1 [15]. Furthermore, MT acetylation may also be important for cell motility since overexpression of HDAC6 leads to decreased acetylation and increased cell motility, the opposite is true when HDAC6 is chemically inhibited [7],[16]. In vertebrates, a family of GTPases (Rac, Rho and Cdc42) triggers localized changes in actin and MT dynamics resulting in changes in cell motility [17]. Moreover, downstream effectors of these GTPases include the MT plus-end tracking proteins (+TIPs) that “capture” and “stabilize” the ends of MTs stimulating the detyrosination and acetylation of MTs in fibroblasts [18],[19],[20]. These data suggest that GTPases are involved in MT dynamics and PTMs. In C. elegans the Rac GTPase ced-10 and the Rac-like GTPase mig-2 have overlapping functions in axon guidance and cell migration [21], but their role in MT function has not yet been studied.
The Elongator, a component of the hyperphosphorylated holoenzyme RNA polymerase II (RNAPII), was originally identified in yeast [22]. Significantly, one subunit of Elongator, Elp3, harbors motifs found in the GNAT family of histone acetyltransferases (HATs) [23]. Both yeast and human Elongator have HAT activity in vitro, primarily directed toward histone H3 [24],[25],[26], and yeast elp3 mutation results in decreased histone H3 acetylation levels in chromatin in vivo [24],[27]. The Elongator is associated with nascent RNA that emanates from elongating RNAPII in yeast [28] and human cells [29],[30], and thus is classified as a key component of transcript elongation. However a substantial portion of Elongator is cytoplasmic [25],[26],[31] and may function as a scaffold involved in exocytosis [32], tRNA modification [33], activation of JNK [31] or actin dynamics [34]. The precise cytoplasmic function of the Elongator remains however controversial [35]. Recently, it was reported that the Elongator can acetylate α-tubulin in vitro [36].
This work uncovers that (i) elongator is a regulator of α-tubulin acetylation in vivo; (ii) elongator is important for MT function in correct loading and velocity of vesicles in vivo and (iii) acetylation has a novel function in fine-tuning intrinsic dynamics of MTs by modulating α-tubulin turnover. Altogether, this concept adds an additional layer of understanding explaining how acetylation by the Elongator can affect MT and neuronal function.
Results
Hyperacetylation of MEC-12/α-tubulin in “gain of function” alleles of the rac-like GTPase mig-2 causes neuronal phenotypes and uncoordinated movement
Using various “loss of function (lf)” and “gain of function (gf)” alleles of the C. elegans Rac GTPases rac-2, mig-2 and ced-10 confirmed that lf and gf alleles of mig-2 hamper movement as revealed by reduction in the frequency of body bends in liquid (Figure 1A, [21],[37]). VD/DD motoneurons are located ventrally and send dorsal projections called “commissures”. Using unc-25::gfp as a marker for these motoneurons [21], it was possible to quantify fully developed commissures in L1 larvae. Whilst wt and mig-2(lf) each displayed an invariable number of 6 commissures (with no differences in neuron morphology), commissures were significantly reduced in mig-2(gf) (Figure 1B and 1C). This supports previous reports [21],[37] that mig-2 is required for correct body movement and that mig-2(gf) exerts uncoordinated body movement (unc) and reduces commissures in VD/DD neurons.

Rac GTPases may affect α-tubulin acetylation [19], a notion that was assessed by western blot analyses and whole mount immunofluorescence in neurons with the monoclonal antibody 6-11B-1 that specifically recognizes MEC-12, a neuronal α-tubulin that is acetylated on a single lysine residue at position 40 and that is expressed also in all motoneurons (Figure S1A, S1B, S1C, S1D, S1E, S1F, S1G). Western blot analysis uncovered that acetylation of MEC-12/α-tubulin is absent in eggs but can be reliably detected in all other stages (Figure 2B). As a control, we observed that acetylation of MEC-12 was absent in all stages within a mec-12(e1607) background, an allele previously described to be a putative null mutant (data not shown, [10]). Whilst wt and mig-2(lf) were indistinguishable from each other (data not shown), there was a marked increase of MEC-12/α-tubulin acetylation in mig-2(gf), which was most pronounced in eggs and L1 stages (Figure 2B asterisks, and Figure 2C; Figure S5A for details and quantification). Although the signals were strongest in touch neurons (Figure 2C), acetylation was also shown to be present in other neurons (e.g. motoneurons, [10]). In mec-3(e1338), where the touch neurons are not specified [38], the developmental acetylation pattern was analogous to wt, although slightly reduced due to the missing contribution of the touch neurons (Figure S1H and S1J). This finding confirms that acetylation of MEC-12/α-tubulin is not limited to touch neurons.

If the increase in acetylation of MEC-12/α-tubulin in mig-2(gf) is central to the observed body movement defects and the neuronal phenotypes, a reduction of acetylation of MEC-12/α-tubulin in a mig-2(gf) background should suppress the phenotypes. This was investigated by overexpressing HDAC-6, an α-tubulin deacetylase [7]. Overall acetylation of MEC-12/α-tubulin was reduced in eggs and L1 stages of wt and mig-2(gf) (Figure 2B). Whilst the overexpression of HDAC-6 did not cause detectable phenotypic changes in a wt background, a suppression of the unc phenotype (as scored by a body bends per minute assay, which simply sums the number of “left to right” and “right to left” movements of worms put into water [see also Methods], Figure 1D) and an amelioration of the neuronal morphology (Figure 1E) was apparent in the mig-2(gf) background (notably, the suppression of the morphological defects was not complete thus suggesting that mig-2 may also be involved in other pathways). This finding was shown to be specific to the regulation of acetylation of MEC-12/α-tubulin, as overexpression of HDAC-6 in a mec-12/α-tubulin mutant background abolished the suppression of the movement defects in mig-2(gf). In addition, a synthetic movement phenotype was observed in the mec-12/α-tubulin mutant strain overexpressing HDAC-6 (Figure 1D). Hyperacetylation of MEC-12/α-tubulin in mig-2(gf) can therefore be linked to the observed movement and neuronal phenotypes.
The elongator is required for correct acetylation of MEC-12/α-tubulin
Based on the observation that a reduction of MEC-12/α-tubulin acetylation in mig-2(gf) partially (but nevertheless clearly) suppresses the movement defects, a whole genome RNAi suppression screen was performed to possibly identify regulators of α-tubulin acetylation (data not shown). We found a series of suppressors, including elpc-1, which upon RNAi distinctly improved the movement and neuronal phenotype of mig-2(gf) (Figure 2D and 2E) in an allele unspecific manner (since it suppressed the gf alleles: gm38 and gm103, data not shown).
ELPC-1, the homologue of human IKAP/hELP1, is predicted to be a subunit of the elongator complex, of which (based on sequence homology) 4 members can be identified in C. elegans: namely elpc-1 – 4 (Figure S2A). The core complex is made up of ELPC-1 – 3 and ELPC-4 is a member of the accessory complex (Figure 2A).
Western blot analyses of the elongator mutants confirmed that the overall acetylation was reduced especially in L1 stage (elpc-1) and L1–L4 stage (elpc-3) when tested in a wt background (Figure 2B blue asterisks, and Figure 2C) and in eggs and L1 in the mig-2(gf) background (Figure 2B blue asterisks, and Figure 2C) [for quantification of immunohistochemical data and Western blots see Figure S5]. Residual acetylation by a yet-to-be-defined pathway can be seen primarily in later stages of development.
RNAi knock-downs of individual members of the elongator complex demonstrated that all four were able to suppress the movement phenotypes of mig-2(gf) (Figure 2D). Worms harboring a chromosomal deletion of elpc-1 and elpc-3 or a point mutation in elpc-3 produced by tilling (Figure S2 and Figure S3) showed no gross neuronal or movement phenotype by themselves however, all three were able to suppress the mig-2(gf) phenotypes (Figure 2D and 2E). Interestingly, the double mutant elpc-1; elpc-3 did not enhance the suppression (Figure 2D and 2E) providing supporting evidence that elpc-1and elpc-3 are not involved in parallel pathways.
A palette of translational rescue constructs (Figure S3) revealed that elpc-1 and elpc-3 are expressed ubiquitously. The subcellular localization of ELPC-1 is mainly in the cytoplasm, whereas ELPC-3 is predominantly nuclear but also cytoplasmatic (data not shown). This finding is analogous to the observations in human fibroblasts [25]. To test whether the core complex subunits also associate in C. elegans, co-immunoprecipitation assays (CoIPs) were performed with a specific polyclonal antibody directed against endogenous ELPC-1. In lysates derived from elpc-3 deletion mutants expressing ELPC-3::GFP (Figure S4) or from wt animals expressing GFP alone, ELPC-3::GFP associated specifically with endogenous ELPC-1, while GFP alone did not (Figure 2F). These results were further confirmed by co-precipitation of endogenous ELPC-1 or ELPC-3::GFP (the reciprocal CoIPs) using antibodies against GFP (Figure 2G). We therefore conclude that the core elongator is conserved in worms. Besides the previously identified function in histone acetylation, this work also suggests that the elongator may be involved in the regulation of α-tubulin acetylation.
The elongator genetically interacts with mec-12/α-tubulin in neurons
Suppression of the mig-2(gf) phenotype by the elongator was analyzed in detail by re-introducing rescue constructs as transgenes into the elongator; mig-2(gf) double mutant background (constructs described in Figure S3). The translational gfp fusion construct of elpc-1 was able to rescue the elpc-1 effect in the mig-2(gf) background (Figure 3A). The overexpression of elpc-1 in a wt background had no effect (Figure 3A). A similar observation was obtained with two minigene constructs, where the elpc-1 cDNA was placed under the control of two different neuronal promoters, namely unc-119 and rab-3. This denotes that the suppression of mig-2(gf) by elpc-1 is due to its function in neurons. Interestingly, a truncated version of elpc-1 (which resembles the ELP1 mutation present in patients suffering from Familial Dysautonomia (FD) [39], was not able to rescue (Figure 3A) even though its localization in the cytoplasm was unaffected by the truncation (data not shown). Finally, the Prab-3::elpc-3 minigene in the respective elpc-3(lf); mig-2(gf) background resulted in a complete rescue, demonstrating that elpc-3 is required in neurons.

elongator mutants should behave like strains overexpressing HDAC-6, at least if acetylation is critical and therefore elongator; mec-12/α-tubulin double mutants should be equivalent to HDAC-6-overexpressing mec-12/α-tubulin strains. Indeed, two of the four mec-12/α-tubulin alleles tested (u63 and u76, Table S1) showed synthetic movement defects with elpc-1(lf) (Figure 3B and 3C). Only the alleles with altered structure of mec-12/α-tubulin showed a synthetic phenotype with elpc-1(lf). [This is due to the fact that mec-12/α-tubulin is accumulated in elpc-1(lf) therefore compromising neuronal function (the detailed analysis and the reasons of this synthetic phenotype are presented later in Figure 7 and Figure 8)].
Moreover, all four mec-12/α-tubulin alleles completely abolished the suppression in the elpc-1(lf); mig-2(gf) double mutant (Figure 3B and 3C) thus highlighting that neurons require MEC-12/α-tubulin for elpc-1 to correctly exert its function. The suppression of mig-2(gf) seems to require a precise equilibrium between MEC-12/α-tubulin and its acetylation. Interfering with this equilibrium by introducing point mutations into MEC-12/α-tubulin in fact abolishes the suppression.
Moreover, a genetic interaction was observed in the elpc-3(lf); mec-12/α-tubulin double mutant. Furthermore, the suppression of mig-2(gf) by elpc-3 required wt mec-12/α-tubulin (Figure 3D). Taken together, these findings reveal that the elongator is likely a regulator of neuronal function mediated by MEC-12/α-tubulin.
The movement phenotypes analyzed here are independent from touch neuron function where mec-12/α-tubulin is required [10]. This is confirmed by the fact that elpc-1 can suppress mig-2(gf) phenotypes in a mec-3(e1338) background, where the touch neurons are not specified (Figure S1I). Moreover, elpc-1(lf) and elpc-3(lf) are normal in touch sensitivity (Figure S4A) and it has been shown that acetylation of mec-12/α-tubulin is not critical for touch sensitivity [10].
Besides neuronal function, mig-2 is also involved in vulva development and mig-2(gf) alleles are egg laying defective (egl) [37]. In isolation, elongator alleles did not display notable egg laying defects and the elongator; mig-2(gf) strains could not suppress the egl phenotype (data not shown). Furthermore, it is known that mig-2 is required for correct distal tip migration and phagocytosis of apoptotic cell corpses in the gonad [21],[40]. Various combinations of elongator, mig-2 and mec-12/α-tubulin mutants were tested, but no phenotypes were observed (Table S2 and Table S3). This suggests that elongator and mec-12/α-tubulin are not required for vulval development, distal tip migration or cell corpse phagocytosis.
Molecular association of the elongator to α-tubulin
Endogenous ELPC-1 and α-tubulin co-immunoprecipitated in a wt and elpc-3 deleted background (Figure 3E). This suggests that the ELPC-1/α-tubulin association is independent of ELPC-3. The association between ELPC-1 and α-tubulin was further analyzed by expressing full length and FD truncated GFP tagged versions of ELPC-1 in elpc-1(lf) mutants followed by the CoIP of α-tubulin. A similar result with α-tubulin was observed with both variants of ELPC-1::GFP (Figure 3F). This is not surprising since FD-ELPC-1::GFP localized like the full length protein. ELPC-3::GFP was also shown to co-immunoprecipitated with α-tubulin (Figure 3F), while GFP alone did not (data not shown). Immunoprecipitation with 6-11B-1, the antibody directed against acetylated α-tubulin, revealed that ELPC-1::GFP did not associate with the acetylated form of α-tubulin (data not shown). This suggests that the affinity of ELPC-1 to α-tubulin is lost after acetylation.
The observed genetic interaction between elongator and mec-12/α-tubulin, the physical association of the core Elongator and α-tubulin and the fact that MEC-12/α-tubulin is less acetylated in elongator mutants suggest that elongator plays a role in α-tubulin acetylation.
Reduction of acetylation of Lysine 40 in neuronal mec-12/α-tubulin is critical for the suppression of mig-2(gf)
To further analyze the role of acetylation of mec-12/α-tubulin in neurons of mig-2(gf) animals we expressed gfp::mec-12/α-tubulin mutated at lysine 40 (K40Q). Overexpressing a non acetylable gfp::mec-12/α-tubulin in mig-2(gf) should out-compete the overacetylated endogenous mec-12/α-tubulin and therefore reduce the overall acetylation. This should suppress the mig-2(gf) phenotypes. The results shown in Figure 4A show how expression of the gfp::mec-12/α-tubulin K40Q transgene suppresses the mig-2(gf) phenotypes in a dose dependent manner. High doses are deleterious, as was also seen with an integrated version of mec-12/α-tubulin K40Q used in Figure 4B and Figure 7A and 7C (data not shown). elongator mutants should behave like strains overexpressing HDAC-6, at least if acetylation is critical and therefore elongator; mec-12/α-tubulin double mutants should be equivalent to HDAC-6-overexpressing mec-12/α-tubulin strains. Indeed, two of the four mec-12/α-tubulin alleles tested (u63 and u76, Table S1) showed synthetic movement defects with elpc-1(lf) (Figure 3B and 3C). Only the alleles with altered structure of mec-12/α-tubulin showed a synthetic phenotype with elpc-1(lf). [This is due to the fact that mec-12/α-tubulin is accumulated in elpc-1(lf) therefore compromising neuronal function (the detailed analysis and the reasons of this synthetic phenotype are presented later in Figure 7 and Figure 8)].
![Effects of <i>mec-12</i>/<i>α-tubulin</i>[K40Q] and cytoplasmic requirement of ELPC-3.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/f968f6bc07e64f3b253ccb40e90a48a1.png)
The dose of gfp::mec-12/α-tubulin K40Q required for suppression of mig-2(gf) in a mig-2(gf);mec-12(e1607) double background appears to be higher than in a single mig-2(gf) mutant. We hypothesize that the difference observed in the two backgrounds is due to negative effects of MEC-12/α-tubulin overexpression. In the mig-2(gm38) background, the total levels of functional MEC-12/α-tubulin protein are higher, because of the concomitant presence of the endogenous mec-12 gene. For this reason the mig-2(gm38) worms reach a threshold of toxicity at lower mec-12/a-tubulin array expression levels. The general reduction of movement seen in the left panel in mig-2(gm38) is probably due to negative effects of the array in this background. Moreover, the expression of gfp::mec-12/α-tubulin K40Q also suppressed the defects in commissure formation (Figure 4B). This indicates a possible role of mec-12/α-tubulin acetylation during neuronal development. We conclude that acetylation on MEC-12 K40 is an important (but probably not the sole) aspect of the elongator phenotype.
Cytoplasmic requirement of the ELPC-3
To study the cytoplasmic function of the Elongator, neuronally expressed elpc-3 were tagged with the well characterized nuclear localization signal [NLS] (to localize elpc-3 into the nucleus) or with a nuclear export signal [NES] (in order to direct the expression mainly in the cytoplasm). No well-established NES has been characterized in worms yet. NESs are leucine-rich sequences with an evolutionary conserved consensus LX1–3 LX2–3 LXL [41].
For the purpose of this study the well known NES of HIV Rev protein was chosen. If elpc-3 is required in the cytoplasm to exert its function with mec-12/α-tubulin then only the NES version, but not the NLS version, should be able to rescue. The correct nuclear and/or cytoplasmic localization was assayed by microscopy since both construct are translational gfp fusions and expressed in predicted subcellular compartment (data not shown). Figure 4C shows the body bends assays confirming the hypothesis that NES::ELP-3 was able to rescue the suppression in the mig-2(gf); elp-3(lf) background, whereas NLS::ELP-3 was not.
To further confirm the cytoplasmic requirement of the Elongator in our system we carried out body bends assays of worms treated with Leptomycin A and B. Leptomycin B has previously been shown to be an important tool in the study of nuclear-cytoplamic transport in C. elegans [42]. The molecular composition of Leptomycin A is very similar to Leptomycin B and both were used to substantiate the results. We hypothesized that treating mig-2(gf) animals with Leptomycin would sequester the Elongator to the nucleus and therefore reduce the acetylation of mec-12/α-tubulin, which in turn should suppress the body bends defects of mig-2(gf). Worms were raised in the presence of the drugs, so ensuring that possible developmental effects were not excluded when the locomotion tests were performed (at the young adult stage). The correct nuclear localization of elpc-3 upon drug treatment was assayed by microscopy using a transgenic strain bearing a translational rescuing elpc-3::gfp fusion which correctly localized to the nucleus in worms treated with Leptomycin (data not shown). The movement data obtained not only confirmed the hypothesis (Figure 4D), but also show that the suppression of mig-2(gf) is dependent on mec-12/α-tubulin, since the suppression was abolished in a mig-2(gf); mec-12/α-tubulin double mutant.
Rescue experiments were performed using neuronally expressed elpc-3::gfp in elpc-3(lf); mig-2(gf) double mutants. As seen in Figure 4D, the elpc-3::gfp construct is functional. Furthermore, this experiment confirmed the cytoplasmic requirement of the Elongator, since the rescue was progressively lost by increasing the concentration of Leptomycin A (Figure 4D). The effect of Leptomycin A was persistently higher than Leptomycin B, which was toxic at high concentrations (data not shown).
The elongator is required in CAN neurons for proper cell migration and posterior process outgrowth
It has been shown that mig-2 is important for cell migration and axon pathfinding in the CAN neuron (Lundquist et al and references therein [21]). Since the body sizes of elongator mutants and wt worms are identical, the position of the CAN cell body was measured to assay cell migration and posterior process outgrowth in L1 larvae (Figure 3G). In elpc-1 and elpc-3 mutants the CAN cell migrated less and the posterior process was shorter than in wt. In the elpc-1; elpc-3 double mutant the phenotypes were not enhanced, indicating that elpc-1 and elpc-3 are not involved in parallel pathways (Figure 3G). No misguidance or branching phenotypes were observed. Notably, elongator mutants were able to ameliorate the CAN cell migration defect, but not the posterior process outgrowth of mig-2(gf) (Figure 3G). These results suggest that the elongator is required for correct neuronal migration and axonal extension.
The velocity of Dense Core Vesicles is regulated by elongator
Acetylation of MTs has been proposed to be important for vesicle transport [15]. In C. elegans a unique tool to measure vesicle velocities exists: namely IDA-1::GFP which marks the dense core vesicles in some neurons [43]. The velocity of dense core vesicles (DCVs) along MT tracks was measured in the ALA lateral process of living worms using a fast time-lapse method [43] (Figure 5A). If the acetylation of MT positively regulates DCV velocity, then a reduced function of the elongator should slow down the vesicles. The mean velocity of vesicles measured in wt animals reflected the published data for wt animals (2,25 µm/sec. vs 2,10 µm/sec. [43]). Indeed, reducing the function of the elongator diminished the average velocity of DCVs by about 35% of wt young adults (Figure 5B). In contrast, the mean velocity of mig-2(gf) mutant was increased by about 13%. In addition, the phenotype of mig-2(gf) was suppressed in the mig-2(gf); elpc-1(RNAi) double mutant. Analogous to this, the increased velocity of mig-2(gf) was suppressed by mec-12(u76), even though mec-12(u76) on its own had no velocity phenotype (Figure 5B). Moreover mec-12(e1607) in which mec-12 is not acetylated showed a marked reduction of DCV velocity (Figure 5B). These experiments confirm that a pathway with mig-2, elongator and mec-12 affects vesicle transport in vivo. It is known that transport of DCV relies, in part, on the same mechanisms as the transport of clear core vesicles [43]. Clear core vesicles transport neurotransmitters are important for movement (see below), however, tools to measure the transport of clear core vesicles directly are not available yet. Since MT are also acetylated in motoneurons, acetylation may play a role in regulating transport of clear core vesicles and therefore also movement.

The acetylcholine concentration is decreased at synaptic clefts of neuromuscular junctions in elongator mutants
In C. elegans movement is controlled by the major neurotransmitter acetylcholine (ACh). Aldicarb, a potent ACh esterase inhibitor that potentiates ACh response, can be used to test for changes in ACh concentration at neuromuscular junctions (NMJs). In a standard test, which utilizes different concentrations of aldicarb, the sensitivity of adult elongator mutants and wt were identical ([44]; and data not shown). However, a differential sensitivity towards aldicarb was observed when exposure time was reduced from 4 hours to 1 hour. Compared to wt elpc-1, elpc-3 and the elongator double mutant less sensitive to aldicarb (Figure 6A–6C). The double mutant was indistinguishable from single mutants (Figure 6C). All elongator mutants suppressed the aldicarb phenotype of mig-2(gf) mutants (Figure 6D–6F). The sensitivity of mig-2(gf) was not as pronounced as in goa-1(n363) [a classical hypersensitive mutant [45] that displays a strong phenotype (Figure 6G)], this might at least in part be due to developmental defects. The resistance to aldicarb of elpc-1 mutants was comparable to the syntaxin mutant unc-64(e264) (Figure 6G). elpc-1(lf) could be rescued by re-expressing elpc-1 in neurons (Figure 6G). This proves that pre-synaptic elpc-1 is required to ensure correct ACh concentrations at NMJs. The reason for the reduction of ACh at NMJs might in part be due to reduction of velocity of vesicles. On the other hand the hypersensitive phenotype of mig-2(gm38) might be due to the overacetylation. Although this is a possible explanation it has to be considered that developmental defects in mig-2(gm38) might further influence the phenotype (e.g. by wrong NMJ wiring). Since no method exists to date to directly measure the ACh flow towards the NMJs other functions than acetylation may be additionally responsible for the mig-2(gf) phenotype in these tests.
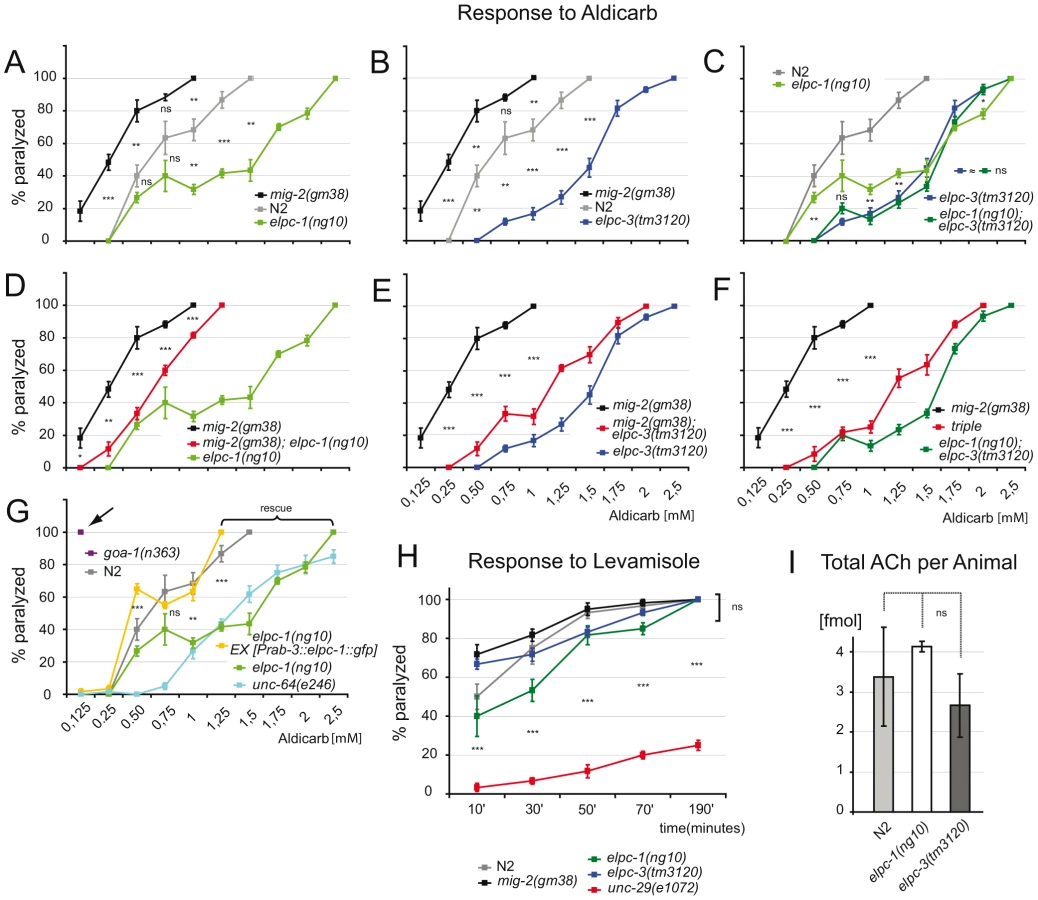
Using levamisole, an agonist of the nicotinic ACh receptor [44], it was possible to assess neurotransmission and function of the postsynaptic apparatus in elongator mutants. The response of adult elongator mutants and mig-2(gf) animals was statistically equal to wt but clearly different to unc-29(e1072), a mutant with a defective nicotinic ACh receptor (Figure 6H). These data underline the notion that the elongator and the mig-2(gf) mutants regulate ACh at NMJs due to presynaptic defects. Interestingly the degree of sensitivity to aldicarb correlated with the level of acetylation of the MTs. Whether changes of ACh levels at NMJs is a result of the direct or indirect regulation of acetylation of microtubules and changes of vesicle behavior remains to be analyzed in other systems. To verify that the change of ACh at NMJs is not due to a defect in ACh synthesis, a chromatographic method was devised capable of measuring ACh concentrations directly in worm extracts. The amount of ACh per adult worm was determined to approximate 3 fmol, a concentration statistically invariable in the elongator mutants (Figure 6I). Therefore the phenotypes are unlikely a result of differences in presynaptic ACh production.
Acetylation of Lysine 40 fine-tunes the levels of MEC-12/α-tubulin and resistance of microtubules to nocodazole
Even though elongator and mec-12/a-tubulin were shown to interact genetically (Figure 3), DNA microarray and RT-PCR of elpc-1(ng10) and wt revealed no differential regulation of mec-12/α-tubulin transcripts (data not shown). The levels of MEC-12/α-tubulin were analysed via a functional GFP-tagged version (to date a specific antibody to endogenous MEC-12 has not been raised). Surprisingly, the amount of GFP::MEC-12 protein was found to be inversely proportional to the degree of acetylation (the cell body of the touch neuron ALM was chosen to measure the GFP signal in L1 of all genetic backgrounds). In detail, compared to wt, the GFP signal of elongator and mig-2(gf) mutants was about 20% higher and 20% lower, respectively (Figure 7A). The reduced level of GFP::MEC-12 signal observed in mig-2(gf) mutants could be suppressed by crossing in elongator mutants (Figure 7A). The double mutant elpc-1; elpc-3 did not enhance the effect (Figure 6A), suggesting that elpc-1 and elpc-3 act together in this function. We wanted to investigate whether this effect is transcriptional or posttranslational. The first evidence that this regulation is posttranslational was provided by the fact that GFP::MEC-12 cDNA driven by a heterologous promoter with a heterologous 3′ UTR did not change the relative levels of GFP::MEC-12. This analysis was performed using the CAN-neuron specific promoter ceh-10 (to rule out possible effects of differences in mec-12 expression in mechanosensory and non-mechanosensory neurons). As before, the signal from this GFP::MEC-12 minigene was increased in the elpc-1 and mig-2(gf); elpc-1 background, but diminished in the mig-2(gf) background (Figure 7B). In a previous experiment, a point mutation that changed lysine 40 to glutamine (Q) in MEC-12/α-tubulin was still able to rescue a touch sensitivity phenotype, therefore MEC-12(K40Q) was considered to be structurally functional [10]. Follow up experiments presented here were used to investigate whether lysine 40 has an impact on the regulation of GFP::MEC-12 levels. Given that the regulation was abolished in GFP::MEC-12(K40Q) transgenic animals (Figure 7C), provides strong evidence that the acetylation of lysine 40 is important for the posttranslational fine-tuning of MEC-12/α-tubulin levels presented here.

These observations also explain the unexpectedly high levels of acetylation in L4 and young adult worms. This might be due to the elevated amount of MEC-12/α-tubulin levels leading to a misinterpretation of the Western data. In fact we observe an accumulation of MEC-12/α-tubulin in adults (see Figure 8).

To decipher whether the increase of MEC-12/α-tubulin alters dynamics of MTs in vivo, the movement phenotype was scored in worms grown in the presence of sublethal concentrations of the MT depolymerizing drug nocodazole or the MT stabilizing drug taxol [46]. This assay was designed to uncover possible effects of the drug during development, e.g. commissure formation defects that persist from L1 to adulthood (since the worms developed through all stages in the presence of the drug). In wt, movement was impaired by nocodazole in a linear dose dependent manner. elpc-1 and to a lesser extent elpc-3 mutants were both more resistant to nocodazole treatment with no additive effect seen in the double mutant elpc-1; elpc-3 (Figure 7D). Compared to mig-2(gf), the suppression of mig-2(gf) by single or double elongator mutants resulted in an increased resistance to nocodazole (Figure 7D). To gain an insight into whether the observed nocodazole resistance in elongator mutants is neuron specific or due to a broader effect, elpc-1 was re-introduced under the control of a neuron specific promoter into elpc-1 mutant worms. The resistance to nocodazole was abolished and essentially wt, suggesting that this phenotype of elpc-1 is neuron specific (Figure 7D).
If stabilization of MTs is fundamental for the suppression of mig-2(gf) by elongator mutants (as suggested by the nocodazole experiment) then MT stabilization by taxol should have a similar effect. In turn, elongator mutants may be expected to have an increased sensitivity towards taxol. Indeed, exposure to taxol suppressed the movement phenotype in mig-2(gf), an effect that was dependent on mec-12/α-tubulin. In contrast, elongator mutants by themselves displayed an increased sensitivity to high concentrations of taxol (Figure 7E). In conclusion, these experiments show that elongator fine-tunes the amount of MEC-12/α-tubulin protein, which in turn may regulate the sensitivity of MTs to depolymerizing and stabilizating drugs revealing a change in their dynamic properties.
Regulation of microtubule structure in elongator mutants
Resistance to nocodazole in elpc-1(lf) may arise due to MTs being stabilized by MT associated proteins. However this is unlikely, as the comparative expression analysis of wt and elpc-1 mutants by DNA microarray technology yielded no MT associated proteins (data not shown). Alternatively, MTs number or length may be increased, a notion that was addressed by transmission electron microscopy (TEM). An ideal target for investigation proved to be the ALM neuron because firstly, it has an unusually high number of special MTs (Figure 8A) and secondly, MEC-12/α-tubulin is highly expressed [10],[47]). In randomly selected TEM sections, the overall number of MTs was indistinguishable in wt and mig-2(gf) but significantly increased in elpc-1(lf) (Figure 8A and 8B). This was confirmed in serial sections (in 3.2 µm increments) of single wt and elpc-1(lf) mutant worms (Figure 8C). The identified variation in MT number along the axon was fed into a mathematical model (see Experimental Procedures for details) which suggested that, the difference identified in elpc-1(lf) may be also the result of MTs being longer rather than solely based on an increase of MT number (Figure S5B). This is, as said above, further supported by the fact that the DNA microarray data could not pinpoint MT nucleation factors (e.g. γ-tubulin) in elpc-1(lf) that are instrumental for the increase in MT numbers. In summary, this provides at least circumstantial evidence that an increase in MTs number is unlikely (Figure 8A–8C).
If increasing the amount of MEC-12/α-tubulin causes longer MTs and subsequently suppresses mig-2(gf) movement defects, any alternative means that increases MEC-12/α-tubulin levels should be able to replicate these observations. Treatment with lactacystin, a potent and specific inhibitor of the proteasome used in C. elegans [48], imposed no movement effect on wt animals, but was able to partially suppress the movement phenotype of mig-2(gf) at 15 µM (Figure 8D). As mentioned before, the treatment was continuous during development of the worms, thereby ensuring that defects during early larval stages impose effects on locomotion at young adult stage. Moreover, the suppression was dependent on MEC-12/α-tubulin (Figure 8D), thus (re)confirming the taxol data (Figure 7E). Interestingly, compared to wt, mec-12/α-tubulin and elongator mutants were all sensitive to high concentrations of lactacystin (Figure 8D). In both cases high levels of lactacystin are predicted to impair MT function, where accumulation of a mutated MEC-12/α-tubulin and “overstabilized” MTs in elongator mutants alter MT function; findings that are analogous to the taxol experiments (Figure 7E).
Ubiquitin mediated proteolysis is the key player in the control of cytoplasmic protein turnover. To investigate whether impaired proteasomal proteolysis of α-tubulin might be linked to the phenotypes observed in elongator mutants, immunoprecipitated α-tubulin from different genetic backgrounds was probed with an anti ubiquitin antibody (Figure 8E). The ubiquitination level was higher in elpc-3 mutants compared to worms with an elpc-1 background. Interestingly, this observation parallels the strong effects on acetylation levels in elpc-3 mutants (Figure 2B). These differences in post-translational modification are possibly due to the fact that ELPC-3 is the catalytic subunit of the elongator complex. This experiment demonstrated that α-tubulin is strongly modified by endogenous ubiquitin in elongator mutants. We conclude that elongator is a critical factor for α-tubulin turnover.
Discussion
elongator regulates α-tubulin acetylation
The results presented here demonstrate how a genetic animal model can be used to study acetylation of MTs. In detail, it was possible to corroborate that mig-2 regulates α-tubulin acetylation and ascertain that the elongator is a new regulator of α-tubulin acetylation. Recently, Creppe and colleagues showed that elongator is involved in this process in mouse cortical neurons [36]. Their data confirm the hypothesis, presented here, namely that elongator activity regulates α-tubulin function. In our work, regulation of acetylation by the elongator was shown to be strong in early stages of development and acetylation of α-tubulin lower, but nevertheless still present, in late stages of elongator mutants. The fact that elongator also regulates other processes and that some acetylation remains in the absence of elongator suggests the presence of some degree of redundancy. It should be noted that during embryonic development and early larval stages numerous neurons undergo dramatic morphological change and in consequence, a tight control of acetylation by elongator has a deep impact on neuronal structure during development. This notion is discussed below.
Regulation of MT acetylation and neuronal shape
Gain of function alleles of the Rac-like GTPase mig-2 display defects in neuronal shape due to the hyperacetylation of α-tubulin. The defects are partially suppressed (although not fully) when acetylation is reduced either by overexpression of HDAC-6, the α-tubulin deacetylase, or by knocking out the elongator. This implies that mig-2 modulates other pathways as well. For example mig-2 is also involved in actin dynamics to regulate neuronal shape and axon pathfinding [49]. But if acetylation of MTs is regarded as a distinct fact, how can acetylation of MTs affect cell shape? It has been shown in fibroblasts and neurons that Rac GTPases regulate MT dynamics by different mechanisms. One of those is the recruitment of +TIPs such as APC and EB1 to promote stabilization via Rac effector mDia [17]. The same +TIPs are proposed to act in the linkers between actin cortex and the protruding MTs in the growth cone of neurons [50]. Although acetylation of MTs seems to be secondary to stabilization [6], it has not been excluded that RacGTPases may actively regulate acetylation.
If α-tubulin is hyperacetylated, as in the Rac gain of function mutants, the cellular consequences are twofold: firstly, neurons fail to project their axons correctly and secondly neuronal migration is, at least in some cases, downregulated. The discovery that acetylation affects α-tubulin levels by regulating its turnover is important, since the dynamics of MT may also be altered in a system where α-tubulin is “hyperacetylated”. This may be the result of MTs being too short or prone to depolymerization. Whilst a regulation in number of MT cross-sections was not seen in the mig-2(gf) mutant, it was possible to suppress its phenotypes by either raising the stability of the MTs with taxol or by mimicking an α-tubulin accumulation by blocking the proteasome. This supports the hypothesis that the regulation of α-tubulin levels affects the dynamics of MTs. In contrast, if acetylation is reduced, as seen in the elpc-1 mutants, then the length of MTs was probably increased. This evidence was supported by the observed resistance to the depolymerizing drug nocodazole and the increased sensitivity to taxol and proteasome inhibitor. In addition, a significant change in neuron shape was seen in elongator mutants. Altogether, the results advocate that the formation of stable MTs requires the fine-tuning of α-tubulin levels. Moreover, since Rac GTPases are upstream of these effects, it is possible that they contribute to the coordination of multiple events that lead to changes in cell shape, namely: a) actin cytoskeleton dynamics, b) stabilization of MT ends and c) fine-tuning of MT dynamics via regulation of α-tubulin turnover.
A further aspect is that axons of mig-2(gf) mutants do not only elongate less, but also show misguidance phenotypes [21]. Since acetylation of α-tubulin affects vesicle loading and transport, it is conceivable that extracellular axon guidance cues might be deregulated by transport problems due to incorrect acetylation of MTs [6]. Recently it has been shown that mig-2 and vab-8 (kinesin-like motor protein) are required to regulate the correct subcellular localization of UNC-40 (a homolog of the netrin receptor) which specifies cell polarity of neurons [51]. In addition, the authors showed that UNC-40 was mislocalized in a mig-2 gain of function mutant. Of course MTs were hyperacetylated in that background, as shown by the results presented here. This leads to the assumption that UNC-40 was in fact mislocalized due to the improper loading and/or velocity of VAB-8 dependent vesicles. A more direct involvement of elongator complex in the regulation of intracellular trafficking that is critical for cell shape remodeling during migration and terminal branching of mouse cortical neurons (corticogenesis) has recently been shown [36]. In summary, this adds further evidence that acetylation may affect neuronal shapes by regulating a) the dynamics of MTs and their propensity to be stabilized via the turnover of α-tubulin and b) the loading and transport of vesicles (and their cargo).
Stability and function of microtubules
The elongator mutants were shown to be resistant to the MT depolymerizing drug nocodazole. Based on this alone, one may conclude that MTs are more stable in elongator mutants, however further experiments showed that the resulting increase of α-tubulin levels also altered the dynamics of MTs. This can be confused with the notion that the “stability” of MTs requires an involvement of active stabilizing factors (such as +TIPs) which in the end change the half life of MTs [6],[50]. Although not the subject of this study, the possible link between elongator and +TIPs is an interesting point that should be investigated in the future.
Others have shown that changes in acetylation levels (mostly achieved by blocking the α-tubulin deacetylase HDAC6) modify the stability of MTs [8],[52]. Indeed, the experiments presented here confirm this point of view, and in addition allow a re-interpretation, namely that their findings are likely a result of the α-tubulin pool being regulated and not due to the direct regulation of MT stability.
Protein turnover and acetylation
Deregulation of acetylation not only affects vesicle dynamics but also α-tubulin turnover (summarized in model Figure 8F). Indeed, this is important for cellular function as revealed by the movement defect of the elongator; α-tubulin double mutant. It is noteworthy that the defects can only be seen when crossing structural mutants of α-tubulin and not regulative or null mutants (Figure 3B–3D, Table S1). These defects may thus be the consequence of the accumulation of damaged α-tubulin, which in turn alters MT and neuronal function. In contrast, precise regulation of the levels of functional α-tubulin is required in the suppression experiments, since all mutations in α-tubulin abolish suppression. This provides further evidence that acetylation is important for the regulation of α-tubulin levels. Besides this work, a significant and growing amount of studies identifies acetylation of various proteins as a key regulator for their turnover. A very interesting finding is that molecules that participate in protein deacetylation, for example HDAC6, can directly interact with CDC48, a factor required for quality control and polyubiquitination of substrates. This reveals a link between control of acetylation states, active deacetylation and degradation [53],[54].
Amyotrophic Lateral Sclerosis and Familial Dysautonomia
Recently it has been shown that elp3 was linked to neurodegenerative diseases and more specifically to Amyotrophic Lateral Sclerosis (ALS) in three families. ALS is the most common adult onset human motor neuron disease typically resulting in death from respiratory muscle weakness within three years [55]. Furthermore, mutations that cause tissue-specific exon skipping thereby truncating human Elp1 result in the autosomal recessive Familial Dysautonomia (FD), one of the most frequent hereditary neuropathies [39],[56]. Affected individuals are born with a reduced number of neurons within the autonomic and sensory nervous system. However, penetrance of the FD mutation is typically incomplete and a low level of full-length IKAP protein can prevail in brain tissues from FD patients [57],[58]. DNA microarray analysis on human cells revealed that the depletion of Elongator (via RNAi of ELP1) in fibroblasts modulates the expression of genes, several notably linked to motility and migration. This led to the conclusion that impaired cell movement within the nervous system might be involved in the neuropathology of FD patients [59]. Recently, a mouse knock out system showed that complete ablation of ELP1 leads to embryonic lethality due to defects in neurolation and blood vessel development [60]. As the worm knock-out of elpc-1 is viable, it is arguably at present, the most suitable system to study and model the ELP1 function in neurons. Indeed, it may at first seem surprising that the defects in the worm nervous system are more similar to the human disease, while the mouse model has such a severe phenotype. However, several differences within the systems may offer plausible explanations. In humans, ELP1 is only downregulated, rather than completely deleted as in the mouse ELP1 experiment. Moreover, whilst only one α-tubulin is acetylable in the nematode, namely MEC-12/α-tubulin (expressed exclusively in neurons, which are not critical for survival as in mouse), all vertebrate α-tubulins possess the acetylatable lysine 40. Therefore regulation of vertebrate turnover of α-tubulin may have a greater impact at the cellular and organismal level and contribute to the severe phenotype observed in the knock out mouse. The mouse phenotypes may all be a result of defective cytoskeletal dynamics, such as reduced polarization and/or vesicular transport due to the downregulation of α-tubulin acetylation. The results presented here offer exquisite support for this line of argumentation, but clearly need further investigations into the transcriptional function of the elongator in vertebrates.
A further aspect is the degeneration of neurons present in FD patients as well as in ALS patients. In the worm model α-tubulin turnover is regulated through acetylation. Degenerative aspects may arise from deregulation of transcription as both ELP1 (involved in FD) and Elp3 (involved in ALS) are part of the same transcriptional elongator complex. Nevertheless, accumulation of “hypoacetylated” targets may be an additional explanation, since acetylation is important in the regulation of protein turnover. In addition, the reduced transport along MTs might lead to an accumulation of proteins destined for degradation. Indeed, the results show that transport is hampered in elongator mutants (Figure 4), that the elongator regulates α-tubulin acetylation and that downregulation of acetylation leads to accumulation of α-tubulin which in turn alters MT dynamics. Accumulation of α-tubulin, as well as the resulting changes in MT dynamics, may constitute an additional stress leading to degeneration and cell death.
Crosstalk between microtubule and histone acetylation
The HAT activity of the Elongator, which is directed toward histone H3, has been proven in various systems to be crucial for transcription and affects genes important for cell movement in human fibroblasts [59]. Furthermore, it has been speculated that defects in this cellular function may underlie FD. In normal cells (fibroblasts and neurons) cell migration is always the concerted interaction between transcription and cytoskeletal dynamics. In this study α-tubulin was identified as an additional genetic target of the elongator. By acetylating histone H3 and regulating α-tubulin acetylation, it is conceivable that the elongator links and “synchronizes” the two functions to modulate the migration event. Rac GTPases may be involved in the response to outer signals and by doing so control the function of the elongator. To date, nothing is known about the regulation of elongator activation. This study highlights that acetylation in mig-2(gf) mutants is increased. However whether this is due to the activation of pathways leading to acetylation regulated by the elongator, to the downregulation of HDAC-6 function or an indirect consequence of upregulation of stabilizing +TIPs still awaits to be unraveled.
Conclusion
This study uncovers that the elongator is linked to the regulation of α-tubulin acetylation. In addition the results presented here indicate that elongator is not only important for vesicle transport but also for α-tubulin turnover. This in turn seems to affect MT dynamics. These observations are instrumental for the introduction of a novel point of view, ultimately explaining how neuronal function is perturbed in neuropathies of Amyotrophic Lateral Sclerosis and Familial Dysautonomia. These new insights offer intriguing cues that targeting microtubules may lead to changes in the design of future therapies.
Material and Methods
C. elegans strains
Worms were handled according to standard procedures [61]. Strains: N2 (wt), mig-2(mu28)X, mig-2(gm38)X, mig-2(gm103)X, rac-2(ok326)IV, ced-10(n1993)IV, mec-12(u76)III, mec-12(u63)III, mec-12(e1605)III, mec-12(e1607)III, unc-29(e1072)I, goa-1(n363)I, unc-64(e246)III, unc-104(e1265)II, mec-3(e1338)IV. elpc-1(ng10)I and elpc-3(ng15)V were generated by ourselves: elpc-1(ng10)I by TMP/UV mutagenesis and elpc-3(ng15)V by tilling. elpc-3(tm3120)V was kindly provided by S. Mitani.
Expression constructs and generation of transgenic strains
GABA-ergic motoneurons were visualized by crossing the strain juIs76[Punc-25::gfp], analogously the CAN neuron was marked using lqIs4[Pceh-10::gfp] (gifts from E. A. Lundquist). Dense core vesicles were visualized using the strain BL5752 bearing the double insertion: inIs181[ida-1::gfp] and inIs182[ida-1::gfp]. Transgenics made by ourselves: (plasmid GU327): ngEx2[elpc-1::gfp] (50ng co-injected with 75ng of ttx-3::gfp), (plasmid GU329): ngEx8[Ex[Punc-119::elpc-1::gfp] (50ng co-injected with 50ng of ttx-3::gfp), (plasmid GU330): ngEx43[Prab-3::elpc-1::gfp] (20ng co-injected with 80ng of ttx-3::rfp and 20ng of lin-15), (plasmid GU446): ngEx94[Prab-3::elpc-1-FD::gfp] (20ng co-injected with 80ng of ttx-3::rfp and 20ng of lin-15), (plasmid GU320): ngEx19[elpc-3::gfp] (20ng co-injected with 80ng of ttx-3::gfp) and (plasmid GU448): ngEx33[Prab-3::elpc-3] (20ng co-injected with 80ng of ttx-3::rfp) (see Figure S3); (construct GU326): ngIs14[hdac-6::gfp]I (20ng co-injected with 80ng of ttx-3::gfp) (wormbase.org: “HDAC-6” = F41H10.6c), (construct GU312): ngIs9[Pmec-12::gfp::mec-12]III (5ng co-injected with 80ng of ttx-3::rfp and 20ng of lin-15), (construct GU331): ngIs10[Pceh-10::gfp::mec-12]X (20ng co-injected with 50ng of ttx-3::rpf and 50ng of lin-15) and (construct GU314): ngIs11[Pmec-12::gfp::mec-12(K40Q)]IV (5ng co-injected with 80ng of ttx-3::rfp and 20ng lin-15) (see Figure 6A–6C). The construct GU314 was also used for the differential expression of mec-12 in Figure 4A: ngEx98 showed no detectable GFP signal (control strain for no expression of mec-12::gfp), ngEx99 and ngEx100 showed weak, ngEx101 medium and ngEx102 strong expression of mec-12::gfp (3ng co-injected with 80ng of ttx-3::rfp and 20ng lin-15). Constructs for nuclear or cytoplasmic expression of elpc-3: (construct GU449): ngEx103 [Prab-3::NLS::elpc-3::gfp] (containing the NLS sequence from the Fire Lab vector kit) (20 ng co-injected with 80 ng of ttx-3::rfp and 20 ng of lin-15), (construct GU450): ngEx104 [Prab-3::NES::elpc-3::gfp] (20 ng co-injected with 80 ng of ttx-3::rfp and 20 ng of lin-15). Constructs for ectopic expression of mec-12, elpc-1 and elpc-3 were designed using the promoter regions of ceh-10 for CAN neuron expression [21], unc-119 and rab-3 for panneuronal expression (Figure S4). Plasmids GU327, GU329 and GU330 contain PCR amplified elpc-1 cDNA cloned into pPD95.75 using the PstI and XmaI sites. Prab-3::elpc-1-FD::gfp (GU446) was obtained deleting the 3′ end of Prab-3::elpc-1::gfp (GU330) using the convenient HpaI site (in the cDNA) and the MscI site (in the vector) and blunt ligated, in frame, with gfp (Figure S4). GU320 contains the whole elpc-3 operon amplified by PCR and cloned into the XmaI site of pPD95.75. GU448 contains the elpc-3 cDNA under the control of the rab-3 promoter cloned into the NotI XmaI site of pPD95.75 vector. The NLS and NES versions of elpc-3 were obtained by PCR amplification of the respective sequences and cloned into vector GU448. The coding sequence for the NES was: CTT CCA CCA CTC GAG AGG CTT ACG CTT. GU326 was obtained by PCR amplification of F41H10.6c (long isoform of HDAC-6) and cloning into pPD95.75 using the SphI and XbaI sites. GU312 contains mec-12 genomic locus (including the putative promoter) cloned into PstI and KpnI sites of pPD117.01, followed by insertion of gfp into BglII site in-frame with mec-12. Pmec-12::gfp::mec-12(K40Q) (GU314) was obtained by standard point mutation method using Pmec-12::gfp::mec-12 as template and subsequently sequenced. GU331 contains ceh-10 promoter inserted via SalI and BamHI sites into pPD117.01 and subsequently mec-12 cDNA was added with XbaI site into the compatible NheI site. Primer sequences used for PCR amplifications are available upon request. All expression constructs were cloned into the vector pPD95.75 except for mec-12 and mec-12(K40Q) cloned in pPD117.01 (kind gift from A. Fire).
Worm mutagenesis and integration of constructs
The elpc-1(ng10) deletion allele was performed as described [62]. elpc-3(ng15) was isolated by tilling as described [63]. Integration of constructs GU326, GU312, GU331 and GU314 was obtained by UV-irradiation (30,000J) of L4 stage worms. Fertile young adults were singled the next day and subsequently starved for 2 weeks to lose the non-integrated extrachromosomal array. Starved worms were transferred onto fresh growth plates and scored for stable inheritance of the respective ttx-3::rfp marker. Integrants were outcrossed twice then mapped by crossing with strain DA438 containing markers for each chromosome (kindly provided by CGC and constructed by L. Avery).
Protein biochemistry
Worms grown in liquid at 20°C to the appropriate stage were collected in buffer A (20 mM Tris pH 7.4, 200 mM NaCl, 10% (v/v) glycerol, 1% Triton) and stored at −80°C. Protein extracts were prepared adding 1 mM PMSF, 1× complete protease inhibitor (Roche) and 300 nM TSA (Sigma T8552). Acid-washed glass beads (Sigma G8772) and FastPrep-24 sample preparation system (MP Biomedicals) were used to homogenize worms (4.0 m/s for 45 seconds at RT). After centrifugation at 4°C, the protein concentration of the supernatants was determined by Bradford assay (Biorad) and 15 µg total protein were resuspended in Laemmli buffer. Proteins separated on 10% SDS-PAGE gels were detected by immunoblotting using ECL. Worm extracts for immunoprecipitation were prepared in HNNG buffer (15 mM Hepes pH 7.5, 250 mM NaCl, 1% NP - 40, 5% glycerol, 1 mM PMSF, 10 mM sodium butyrate, -Sigma B5887-, 300 mM TSA and 1× protease inhibitors cocktail). Lysates (1–2 mg/ml) were incubated overnight with 1 µg of antibody at 4°C. Immunocomplexes were collected with protein A plus sepharose beads (Amersham Biosc), protein G plus sepharose beads (Zymed), anti-α-tubulin - or anti-ELPC-1-conjugated agarose beads, sequentially washed at 4°C with HNNG buffer and finally resuspended in Laemmli buffer. anti-α-tubulin and anti-ELPC-1 crosslinked resins have been produced by Cogentech (Consortium for Genomic Technologies, Milan, Italy) using standard procedures. Antibodies used for western blots and/or immunoprecipitations: anti-α tubulin (Sigma T5168), anti-acetylated-α-tubulin (Sigma, T6793), anti-actin (MP biomedicals, #69100), anti-GFP (Torrey Pines Biolabs Inc, TP401) and anti-Ubi (Upstate, # 05-944). Rabbit polyclonal anti-ELPC-1 antibody was produced by Cogentech. anti-rabbit or anti-mouse IgG HRP-conjugated antibodies were both from Cell Signaling Tech (# 7074 and # 7076, respectively). Western blots were performed at least twice to confirm the results.
TEM, GFP quantification and immunolocalization, Dense Core Vesicles velocity
TEM was performed using standard procedures. Briefly, worms were washed in M9 and anesthetized in 8% ethanol in M9 for 5 min. They were placed in a fixative (2.5% glutaraldehyde, 1% paraformaldehyde in 0.1M sucrose, and 10 mM PBS, pH 7.4), cut open with a needle at the anterior and posterior ends, and fixed for 2 h. Worms were embedded in 2% agarose, cut into small blocks, and washed three times in PBS. Subsequently, pieces were fixed with a second solution (1% osmium tetroxide, 1.5% potassium ferrocyanide in PBS) for 2 h and washed three times in water. Worms were stained with 1% uranyl acetate for 1 h. Samples were dehydrated in ethanol (10 min in 50% ethanol, 10 min in 70% ethanol, 10 min in 90% ethanol, and 10 min in 100% ethanol) and acetone (10 min). Blocks with worms were embedded in Epon resin (Fluka, Buchs, Switzerland): first in Epon-acetone (1∶1) for 1–2 h and then in pure resin for 2–4 h. Samples polymerized for 24–48 h at 60°C and in 60-nm sections were prepared with Ultracut E. Sections were stained in uranyl acetate for 60 min and then 2 min in Millonig's lead acetate stain. Pictures were taken on Philips Morgagni 80 KV microscope (Eindhoven, The Netherlands) [64].
GFP signal was quantified by taking pictures of ALM or CAN neuronal body of anesthetized L1 at 63× magnification at fixed exposure time (2 sec.) The total signals were measured using ImageJ software. Whole mount staining of acetylated MEC-12/α-tubulin using the specific monoclonal antibody (see below) was performed according to previous protocols [10]. Cy3 conjugated secondary antibodies from Jackson Lab. were used. Primary antibody dilution: 1/500. Secondary antibody dilution: 1/200. Analysis of dense core vesicles was performed as described [43]. Immunolocalizations were repeated 3× per genotype.
Interpretation of TEM data
In Figure 7C we show the two sets of measurements for the number of intersections of microtubules on eight independent sections of ALM in a wild type and an elpc-1(lf) mutant. In wt the average number of intersections and its standard deviation are μ1 = 67 and σ1 = 9; for elpc-1(lf) μ2 = 85 and σ2 = 5. The means are statistically different: μ1 differs from μ2 (one-sided t test, P = 2·10−4; 11–Inf is 95% confidence interval for μ2−μ1). These measurements confirm that the two individuals of different genotype show similar MT intersection levels than the respective ones in Figure 7B. Based on these data, homogeneity of variances cannot be discarded (one-sided F test, P = 0.1). Interpretation model: Assuming that microtubules have a fixed length h, parallel to the axis of the axon and distributed uniformly along an axon of a given length L leads to following calculations: the probability for a microtubule to cross any section is p = h/L and follows the Poisson distribution (h/L equals the ways a “stick” of length h can be placed in a longer “tube” of length L). Performing N trials of a Poisson process with probability p, the expected value and the variance of the number of successes for a random variable X are E(X) = Np and VAR(X) = Np(1−p), respectively. Thus, if we have m microtubules, the expected value for the number of intersections is E(X) = mh/L, while the variance is VAR(X) = mh/L(1−h/L). Since the elpc-1(lf) mutant has, on average, more microtubules intersections than wild type, using E(X) = mh/L we obtain the condition m2h2>m1h1 (1 = wt; 2 = elpc-1(lf)). In other words, elpc-1(lf) have more microtubules, or the microtubules are longer, or both. It seems unlikely that the main change is the number of microtubules, because the assumption that microtubule length is unchanged, i.e. h2 = h1 and m2>m1, requires VAR2(X)>VAR1(X), i.e. σ22>σ21. The later is unsupported by the measurements.
Movement tests and drug treatments
Body bends per minute: Worms grown at 20°C to L4/young adult stage were placed into water. Body bends were counted for 1 minute. Blind scoring was not applied to these tests, because they were highly reproducible and not subjective. The number of body bends per minute for wt worms in our tests reproduced published levels [65]. Drug tests: Standard NGM plates were supplemented with 0.1–0.5 µM nocodazole, 1–5 µM taxol [46], 0–75 µM leptomycin B [42] or 0–40 µM lactacystin [48]. DMSO [for nocodazole and taxol] or water [for leptomycin and lactacystin] alone were used as a control. Worms were grown in the presence of the respective drug and body bend assays were carried out on the F1. The Aldicarb tests were performed as described [44]. Concentration used: 0–40 µM. Modifications to the original protocol: worms were exposed only 1h (instead of 4h) and the test was performed in M9 buffer (this modified procedure increased the sensitivity). Levamisole test was performed as described [44]. Aldicarb and levamisole tests were repeated 5 times using each time 12 animals calculating the average and error. The sensitivity of wt worms when exposed 4h to aldicarb and in the levamisole test reproduced the published data (not shown, [44]). Touch sensitivity: young adult worms were touched 10 times with an eyelash, alternating between the anterior and posterior part of the body. Between touches worms were given 1 min to recover. Positive response by movement away form the stimulation was scored for 10 independent worms per strain.
Apoptotic bodies and distal tip cell migration phenotype analysis
Performed as described [21],[40].
ACh measurement
Young adult worms were flash frozen in water (1000 worms per pellet). Extraction was performed in 0.2 M perchloric acid (PCA) solution with 100 mM EDTA. Ethylhomocholine (0.1 µM) served as internal standard. To homogenize the worms the FastPrep®-24 sample preparation system with beads was used as described above. After centrifugation, the pH of the supernatant was neutralized using 1M KHCO3 and purified using 0.45 mm pore size filters. Final extracts were stored at −80°C and analyzed on an electrochemical detector HTEC-500 (Eicom Co. Kyoto, Japan) as described [66].
Statistical analysis
If not otherwise stated, statistical significance was performed by two-tailed unpaired Student's t-test. P value>0,05 was scored as “ns” (not significant).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. KirschnerMW
MitchisonT
1986 Microtubule dynamics. Nature 324 621
2. SaxtonWM
StempleDL
LeslieRJ
SalmonED
ZavortinkM
1984 Tubulin dynamics in cultured mammalian cells. J Cell Biol 99 2175 2186
3. SchulzeE
KirschnerM
1986 Microtubule dynamics in interphase cells. J Cell Biol 102 1020 1031
4. SchulzeE
KirschnerM
1987 Dynamic and stable populations of microtubules in cells. J Cell Biol 104 277 288
5. KirschnerM
MitchisonT
1986 Beyond self-assembly: from microtubules to morphogenesis. Cell 45 329 342
6. VerheyKJ
GaertigJ
2007 The tubulin code. Cell Cycle 6 2152 2160
7. HubbertC
GuardiolaA
ShaoR
KawaguchiY
ItoA
2002 HDAC6 is a microtubule-associated deacetylase. Nature 417 455 458
8. MatsuyamaA
ShimazuT
SumidaY
SaitoA
YoshimatsuY
2002 In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. Embo J 21 6820 6831
9. NorthBJ
MarshallBL
BorraMT
DenuJM
VerdinE
2003 The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell 11 437 444
10. FukushigeT
SiddiquiZK
ChouM
CulottiJG
GogoneaCB
1999 MEC-12, an alpha-tubulin required for touch sensitivity in C. elegans. J Cell Sci 112 ( Pt 3) 395 403
11. GaertigJ
CruzMA
BowenJ
GuL
PennockDG
1995 Acetylation of lysine 40 in alpha-tubulin is not essential in Tetrahymena thermophila. J Cell Biol 129 1301 1310
12. KozminskiKG
DienerDR
RosenbaumJL
1993 High level expression of nonacetylatable alpha-tubulin in Chlamydomonas reinhardtii. Cell Motil Cytoskeleton 25 158 170
13. CorcoranLJ
MitchisonTJ
LiuQ
2004 A novel action of histone deacetylase inhibitors in a protein aggresome disease model. Curr Biol 14 488 492
14. KawaguchiY
KovacsJJ
McLaurinA
VanceJM
ItoA
2003 The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115 727 738
15. ReedNA
CaiD
BlasiusTL
JihGT
MeyhoferE
2006 Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol 16 2166 2172
16. HaggartySJ
KoellerKM
WongJC
GrozingerCM
SchreiberSL
2003 Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A 100 4389 4394
17. WatanabeT
NoritakeJ
KaibuchiK
2005 Regulation of microtubules in cell migration. Trends Cell Biol 15 76 83
18. AkhmanovaA
HoogenraadCC
DrabekK
StepanovaT
DortlandB
2001 Clasps are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell 104 923 935
19. PalazzoAF
EngCH
SchlaepferDD
MarcantonioEE
GundersenGG
2004 Localized stabilization of microtubules by integrin - and FAK-facilitated Rho signaling. Science 303 836 839
20. WenY
EngCH
SchmoranzerJ
Cabrera-PochN
MorrisEJ
2004 EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat Cell Biol 6 820 830
21. LundquistEA
ReddienPW
HartwiegE
HorvitzHR
BargmannCI
2001 Three C. elegans Rac proteins and several alternative Rac regulators control axon guidance, cell migration and apoptotic cell phagocytosis. Development 128 4475 4488
22. OteroG
FellowsJ
LiY
de BizemontT
DiracAM
1999 Elongator, a multisubunit component of a novel RNA polymerase II holoenzyme for transcriptional elongation. Mol Cell 3 109 118
23. WittschiebenBO
OteroG
de BizemontT
FellowsJ
Erdjument-BromageH
1999 A novel histone acetyltransferase is an integral subunit of elongating RNA polymerase II holoenzyme. Mol Cell 4 123 128
24. WinklerGS
KristjuhanA
Erdjument-BromageH
TempstP
SvejstrupJQ
2002 Elongator is a histone H3 and H4 acetyltransferase important for normal histone acetylation levels in vivo. Proc Natl Acad Sci U S A 99 3517 3522
25. HawkesNA
OteroG
WinklerGS
MarshallN
DahmusME
2002 Purification and characterization of the human elongator complex. J Biol Chem 277 3047 3052
26. KimJH
LaneWS
ReinbergD
2002 Human Elongator facilitates RNA polymerase II transcription through chromatin. Proc Natl Acad Sci U S A 99 1241 1246
27. KristjuhanA
WalkerJ
SukaN
GrunsteinM
RobertsD
2002 Transcriptional inhibition of genes with severe histone h3 hypoacetylation in the coding region. Mol Cell 10 925 933
28. GilbertC
KristjuhanA
WinklerGS
SvejstrupJQ
2004 Elongator interactions with nascent mRNA revealed by RNA immunoprecipitation. Mol Cell 14 457 464
29. KouskoutiA
TalianidisI
2005 Histone modifications defining active genes persist after transcriptional and mitotic inactivation. Embo J 24 347 357
30. MetivierR
PenotG
HubnerMR
ReidG
BrandH
2003 Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115 751 763
31. HolmbergC
KatzS
LerdrupM
HerdegenT
JaattelaM
2002 A novel specific role for I kappa B kinase complex-associated protein in cytosolic stress signaling. J Biol Chem 277 31918 31928
32. RahlPB
ChenCZ
CollinsRN
2005 Elp1p, the yeast homolog of the FD disease syndrome protein, negatively regulates exocytosis independently of transcriptional elongation. Mol Cell 17 841 853
33. HuangB
JohanssonMJ
BystromAS
2005 An early step in wobble uridine tRNA modification requires the Elongator complex. Rna 11 424 436
34. JohansenLD
NaumanenT
KnudsenA
WesterlundN
GromovaI
2008 IKAP localizes to membrane ruffles with filamin A and regulates actin cytoskeleton organization and cell migration. J Cell Sci 121 854 864
35. SvejstrupJQ
2007 Elongator complex: how many roles does it play? Curr Opin Cell Biol 19 331 336
36. CreppeC
MalinouskayaL
VolvertML
GillardM
CloseP
2009 Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell 136 551 564
37. ZipkinID
KindtRM
KenyonCJ
1997 Role of a new Rho family member in cell migration and axon guidance in C. elegans. Cell 90 883 894
38. ZhangY
MaC
DeloheryT
NasipakB
FoatBC
2002 Identification of genes expressed in C. elegans touch receptor neurons. Nature 418 331 335
39. AxelrodFB
2004 Familial dysautonomia. Muscle Nerve 29 352 363
40. deBakkerCD
HaneyLB
KinchenJM
GrimsleyC
LuM
2004 Phagocytosis of apoptotic cells is regulated by a UNC-73/TRIO-MIG-2/RhoG signaling module and armadillo repeats of CED-12/ELMO. Curr Biol 14 2208 2216
41. KanwalC
LiH
LimCS
2002 Model system to study classical nuclear export signals. AAPS PharmSci 4 E18
42. SegalSP
GravesLE
VerheydenJ
GoodwinEB
2001 RNA-Regulated TRA-1 nuclear export controls sexual fate. Dev Cell 1 539 551
43. ZahnTR
AnglesonJK
MacMorrisMA
DomkeE
HuttonJF
2004 Dense core vesicle dynamics in Caenorhabditis elegans neurons and the role of kinesin UNC-104. Traffic 5 544 559
44. SaifeeO
WeiL
NonetML
1998 The Caenorhabditis elegans unc-64 locus encodes a syntaxin that interacts genetically with synaptobrevin. Mol Biol Cell 9 1235 1252
45. MillerKG
EmersonMD
RandJB
1999 Goalpha and diacylglycerol kinase negatively regulate the Gqalpha pathway in C. elegans. Neuron 24 323 333
46. ZubovychI
DoundoulakisT
HarranPG
RothMG
2006 A missense mutation in Caenorhabditis elegans prohibitin 2 confers an atypical multidrug resistance. Proc Natl Acad Sci U S A 103 15523 15528
47. ChalfieM
ThomsonJN
1979 Organization of neuronal microtubules in the nematode Caenorhabditis elegans. J Cell Biol 82 278 289
48. DingM
ChaoD
WangG
ShenK
2007 Spatial regulation of an E3 ubiquitin ligase directs selective synapse elimination. Science 317 947 951
49. ShakirMA
JiangK
StruckhoffEC
DemarcoRS
PatelFB
2008 The Arp2/3 activators WAVE and WASP have distinct genetic interactions with Rac GTPases in Caenorhabditis elegans axon guidance. Genetics 179 1957 1971
50. WitteH
BradkeF
2008 The role of the cytoskeleton during neuronal polarization. Curr Opin Neurobiol 18 479 487
51. Levy-StrumpfN
CulottiJG
2007 VAB-8, UNC-73 and MIG-2 regulate axon polarity and cell migration functions of UNC-40 in C. elegans. Nat Neurosci 10 161 168
52. TranAD
MarmoTP
SalamAA
CheS
FinkelsteinE
2007 HDAC6 deacetylation of tubulin modulates dynamics of cellular adhesions. J Cell Sci 120 1469 1479
53. KwonHS
OttM
2008 The ups and downs of SIRT1. Trends Biochem Sci 33 517 525
54. SadoulK
BoyaultC
PabionM
KhochbinS
2008 Regulation of protein turnover by acetyltransferases and deacetylases. Biochimie 90 306 312
55. SimpsonCL
LemmensR
MiskiewiczK
BroomWJ
HansenVK
2009 Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum Mol Genet 18 472 481
56. SlaugenhauptSA
GusellaJF
2002 Familial dysautonomia. Curr Opin Genet Dev 12 307 311
57. CuajungcoMP
LeyneM
MullJ
GillSP
LuW
2003 Tissue-specific reduction in splicing efficiency of IKBKAP due to the major mutation associated with familial dysautonomia. Am J Hum Genet 72 749 758
58. SlaugenhauptSA
BlumenfeldA
GillSP
LeyneM
MullJ
2001 Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am J Hum Genet 68 598 605
59. CloseP
HawkesN
CornezI
CreppeC
LambertCA
2006 Transcription impairment and cell migration defects in elongator-depleted cells: implication for familial dysautonomia. Mol Cell 22 521 531
60. ChenYT
HimsMM
ShettyRS
MullJ
LiuL
2009 Loss of mouse Ikbkap, a subunit of elongator, leads to transcriptional deficits and embryonic lethality that can be rescued by human IKBKAP. Mol Cell Biol 29 736 744
61. WoodWB
1988 The Nematode Caenorhabditis elegans Cold Spring Laboratory Press
62. YandellMD
EdgarLG
WoodWB
1994 Trimethylpsoralen induces small deletion mutations in Caenorhabditis elegans. Proc Natl Acad Sci U S A 91 1381 1385
63. CuppenE
GortE
HazendonkE
MuddeJ
van de BeltJ
2007 Efficient target-selected mutagenesis in Caenorhabditis elegans: toward a knockout for every gene. Genome Res 17 649 658
64. TrzebiatowskaA
TopfU
SauderU
DrabikowskiK
Chiquet-EhrismannR
2008 Caenorhabditis elegans teneurin, ten-1, is required for gonadal and pharyngeal basement membrane integrity and acts redundantly with integrin ina-1 and dystroglycan dgn-1. Mol Biol Cell 19 3898 3908
65. JanieschPC
KimJ
MouyssetJ
BarikbinR
LochmullerH
2007 The ubiquitin-selective chaperone CDC-48/p97 links myosin assembly to human myopathy. Nat Cell Biol 9 379 390
66. TakaseK
MitsushimaD
FunabashiT
KimuraF
2007 Sex difference in the 24-h acetylcholine release profile in the premotor/supplementary motor area of behaving rats. Brain Res 1154 105 115
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 1
Nejčtenější v tomto čísle
- A Major Role of the RecFOR Pathway in DNA Double-Strand-Break Repair through ESDSA in
- Kidney Development in the Absence of and Requires
- The Werner Syndrome Protein Functions Upstream of ATR and ATM in Response to DNA Replication Inhibition and Double-Strand DNA Breaks
- Alternative Epigenetic Chromatin States of Polycomb Target Genes