
VEZF1 Elements Mediate Protection from DNA Methylation
There is growing consensus that genome organization and long-range gene regulation involves partitioning of the genome into domains of distinct epigenetic chromatin states. Chromatin insulator or barrier elements are key components of these processes as they can establish boundaries between chromatin states. The ability of elements such as the paradigm β-globin HS4 insulator to block the range of enhancers or the spread of repressive histone modifications is well established. Here we have addressed the hypothesis that a barrier element in vertebrates should be capable of defending a gene from silencing by DNA methylation. Using an established stable reporter gene system, we find that HS4 acts specifically to protect a gene promoter from de novo DNA methylation. Notably, protection from methylation can occur in the absence of histone acetylation or transcription. There is a division of labor at HS4; the sequences that mediate protection from methylation are separable from those that mediate CTCF-dependent enhancer blocking and USF-dependent histone modification recruitment. The zinc finger protein VEZF1 was purified as the factor that specifically interacts with the methylation protection elements. VEZF1 is a candidate CpG island protection factor as the G-rich sequences bound by VEZF1 are frequently found at CpG island promoters. Indeed, we show that VEZF1 elements are sufficient to mediate demethylation and protection of the APRT CpG island promoter from DNA methylation. We propose that many barrier elements in vertebrates will prevent DNA methylation in addition to blocking the propagation of repressive histone modifications, as either process is sufficient to direct the establishment of an epigenetically stable silent chromatin state.
Published in the journal:
. PLoS Genet 6(1): e32767. doi:10.1371/journal.pgen.1000804
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000804
Summary
There is growing consensus that genome organization and long-range gene regulation involves partitioning of the genome into domains of distinct epigenetic chromatin states. Chromatin insulator or barrier elements are key components of these processes as they can establish boundaries between chromatin states. The ability of elements such as the paradigm β-globin HS4 insulator to block the range of enhancers or the spread of repressive histone modifications is well established. Here we have addressed the hypothesis that a barrier element in vertebrates should be capable of defending a gene from silencing by DNA methylation. Using an established stable reporter gene system, we find that HS4 acts specifically to protect a gene promoter from de novo DNA methylation. Notably, protection from methylation can occur in the absence of histone acetylation or transcription. There is a division of labor at HS4; the sequences that mediate protection from methylation are separable from those that mediate CTCF-dependent enhancer blocking and USF-dependent histone modification recruitment. The zinc finger protein VEZF1 was purified as the factor that specifically interacts with the methylation protection elements. VEZF1 is a candidate CpG island protection factor as the G-rich sequences bound by VEZF1 are frequently found at CpG island promoters. Indeed, we show that VEZF1 elements are sufficient to mediate demethylation and protection of the APRT CpG island promoter from DNA methylation. We propose that many barrier elements in vertebrates will prevent DNA methylation in addition to blocking the propagation of repressive histone modifications, as either process is sufficient to direct the establishment of an epigenetically stable silent chromatin state.
Introduction
It has been proposed that genes and gene clusters are organized into chromatin domains that are maintained independent of their surroundings through the establishment of boundaries [1],[2]. These boundaries may be variable in position, resulting from a balance between countervailing chromatin opening and condensing processes. Alternatively, chromatin boundaries of fixed position could be established by specific DNA sequence elements and their associated binding proteins. Such elements, collectively called insulators, possess a common ability to protect genes from inappropriate signals emanating from their surrounding environment [3]–[7].
The chicken β-globin genes are clustered within a thirty kilobase domain of nuclease accessible chromatin, the 5′ boundary of which is marked by a constitutive DNaseI hypersensitive site called HS4 (Figure 1). The HS4 element has two activities that functionally define insulators. First, it can block the action of an enhancer element on a linked promoter, but only when positioned between the two [8]. The protein CTCF mediates the enhancer blocking activity of the HS4 element [9]. Second, the HS4 insulator acts as a barrier to chromosomal position effect silencing [10]. The activities of HS4 have been mapped to a 275 bp “core” element that contains five protein binding sites revealed by DNase I footprinting [9]–[11] (Figure S1). The enhancer blocking and barrier activities of HS4 appear to have different underlying mechanisms as they are separable in assay systems. The CTCF binding site footprint II (FII) is necessary and sufficient for enhancer blocking, but can be deleted from HS4 without affecting barrier activity [9],[12],[13]. The four remaining protein binding sites are all essential for barrier activity (FI, FIII, FIV and FV) but dispensable for enhancer blocking activity [9].

We previously found that the binding of ubiquitous USF proteins to a single site in HS4, footprint FIV, is a necessary component of its barrier activity [14]. USF serves to constitutively recruit several histone modifying enzymes, leading to the enrichment of a panel of histone modifications typically associated with transcriptionally active open chromatin, including H3ac, H4ac, H3K4me2 and H4R3me2as. Knock down of USF expression abolishes the recruitment of active histone modifications and leads to the encroachment of transcriptionally repressive chromatin marked by H3K9me2 and H3K27me3 into the β-globin locus [14],[15]. While USF function is necessary, it is not sufficient for HS4's barrier activity. Deletion of any one of the three remaining HS4 binding sites FI, FIII or FV disrupts barrier activity without affecting USF-mediated recruitment of histone modifications to HS4 [12],[14].
We hypothesized that the FI, FIII and FV sites may contribute to barrier activity by preventing a transcriptional silencing process other than that mediated by repressive histone modifications. We previously observed that transgenes lack promoter DNA methylation when shielded from chromosomal silencing by HS4 elements [16]. Similar results were seen with retroviral transgenes shielded by HS4 [17]. It was not clear from these studies whether the lack of DNA methylation was an indirect consequence of transcriptional activity of insulated transgenes, nor did these studies address whether particular DNA elements or proteins bound at HS4 are specifically responsible for this protection. We now use insulator mutations to demonstrate that HS4 does specifically protect a gene promoter from DNA methylation. We determine which HS4 sequence elements are responsible for protection from methylation and have purified the protein that recognizes these elements in vivo. We also demonstrate that these elements are able to mediate the demethylation of a CpG island promoter.
Results
Three elements within the HS4 insulator protect a promoter from DNA methylation
To investigate whether HS4 acts specifically to counter DNA methylation, we have studied transgenic cell lines that were previously established for the HS4 barrier assay. The assay construct consists of an IL-2R reporter gene driven by an erythroid enhancer and promoter randomly integrated into erythroid 6C2 cells. These transgenes are susceptible to chromosomal silencing over a period of 20–40 days in culture following the removal of selection, with transgenic promoters being subject to DNA methylation and subsequent recruitment of the Mi-2/NuRD co-repressor complex [12],[16]. In contrast, transgenes flanked by wild-type HS4 insulators are protected from silencing and lack promoter DNA methylation [16],[17]. It was unclear from these earlier results whether the lack of methylation was a consequence of transcriptional activity or whether particular HS4 activities mediate protection from methylation.
We have performed clonal bisulfite sequencing on single/low-copy transgenes flanked by HS4 insulators that are either wild type or carry deletions in one of five protein binding site ‘footprints’ (Figure 1). We have studied the same stocks of the same cloned transgenic cells that have been characterized for long term expression and histone modification status [12],[14]. The transgene promoter remains free of DNA methylation when insulated from chromosomal silencing by wild type HS4 elements, even following prolonged culture (Figure 2B, WT). Strikingly, we find that transgenic promoters are subject to almost complete DNA methylation if any one of three footprinted sites, FI, FIII or FV, is deleted from the flanking HS4 insulators (Figure 2, Δ1, Δ3 and Δ5). These profiles of DNA hypermethylation are indistinguishable from those observed at non-insulated transgenes [16]. In contrast, deletion of the CTCF (FII) binding site from HS4 results in little de novo DNA methylation of the promoter (Figure 2B, ΔII). The deletion of the CTCF site has no effect on barrier activity and the lack of methylation observed is consistent with the transcriptionally active state of this transgene (Figure 2C, [12]). De novo DNA methylation of the transgene promoter has previously been observed to be a secondary consequence of chromosomal silencing in the absence of insulation [18]. This suggested that deletion of the USF binding site from HS4, which abolishes HS4's barrier activity and results in the loss of active histone modifications [14] and transcriptional silencing [12], should also result in promoter hypermethylation. To our surprise, deletion of the USF (FIV) binding site from HS4 results in little de novo DNA methylation of the promoter (Figure 2B, ΔIV). These results show that the DNA methylation status of a promoter does not necessarily follow its histone modification and transcription status. They also strongly indicate that three protein binding sites at HS4 (FI, FIII and FV) have a specific role to mediate protection from silencing associated with de novo DNA methylation.

Mutant insulators are partially methylated
We next sought to determine how HS4 footprint deletions affect the timing and level of methylation of the mutant insulators in comparison with their linked transgene promoters. The dogma established from previous studies posits that a barrier element like HS4 acts as a passive barrier to the propagation, or spreading, of chromosomal silencing. We therefore expect that when HS4 mutations compromise barrier activity, the mutant insualtors would become methylated either prior to, or coincident with, the transgene promoters they are shielding. We performed bisulfite sequencing of the HS4 elements that are located 5′ of the IL-2R transgenes (Figure 3A). Analyses were made following 30 days of culture, typically the period at which epigenetic silencing of the transgene is being established in non-insulated lines, and after 90 days, at which point any silencing will be complete. We find that wild type HS4 elements remain unmethylated during long term culture, concordant with the lack of transgene methylation (Figure 3C, WT). Deletion of the FI and FV sites results in partial methylation of HS4 (Figure 3C, ΔI and ΔV). This is in line with the effects of these mutations on promoter methylation. The timing of methylation does not fit the spreading models however, as the transgene promoter becomes methylated prior to the flanking insulator (compare ΔI at day 30 in Figure 2B with that in Figure 3C). Furthermore, deletion of the FIII site does not result in methylation of HS4 despite complete promoter methylation resulting from this mutation (compare ΔIII at day 90 in Figure 3C with that in Figure 2B). We also found that deletion of either the CTCF or USF sites leads to partial methylation of HS4, but with no methylation at the promoter (compare ΔII and ΔIV at day 90 in Figure 3C with that in Figure 2B).

We note that the patterns of partial methylation of mutant HS4 elements are heterogeneous, with none of the individually sequenced clones becoming densely methylated (Table S2). The partial methylation of mutant HS4 elements (20%–50%) contrasts with the near total DNA methylation observed at the silenced promoters flanked by FI, FIII or FV site mutant insulators (90%–100%). These findings reveal a disconnect between the level and timing of de novo DNA methylation at a transgene and flanking insulators.
Identification of HS4-binding activities
The footprinted sequences FI, FIII and FV are specifically required for HS4's ability to protect against DNA-methylation-mediated silencing of a transgene. We wished to identify the factors that interact with each of the FI, FIII and FV sites to better understand this activity. We established gel mobility shift assays for insulator-binding activities using nuclear protein extracts of the chicken early erythroid cell line 6C2 (the cell line in which the barrier assay is performed) and adult chicken red blood cells (an abundant source of nuclear protein for purification purposes). Complexes of similar mobility and intensity are observed between the two nuclear extracts and each of the FI, FIII and FV sites (Figure 4, 6C2 data not shown). Competition assays show that these complexes are all specific for homopolymeric dG-dC strings found in each site. Unlabelled wild type FI duplexes compete efficiently with the formation of the major complex with FI, whereas FI duplexes harboring mutations in the (dG-dC)9 string are much less effective as competitors (Figure 4A, arrow, compare lanes 2, 4 and 5). The major complex with FIII specifically interacts with the (dG-dC)6 string at its center (Figure 4B, compare lanes 1, 5, 6 and 7) and the major FV complex also specifically interacts with bases in both of its (dG-dC) strings (Figure 4C, compare lanes 1, 4, 5 and 7).

The same proteins interact with the FI, FIII and FV sites. This is supported by the observation that the three sites can efficiently compete with each other. Unlabelled FIII duplexes compete for nuclear protein interactions with labeled FI (Figure 4A, compare lanes 1 and 8) and FI duplexes efficiently compete for interaction with FIII (Figure 4B, compare lanes 1 and 9), for example. Mutational analysis shows that this cross-competition is dependent upon the dG-dC string bases within each footprint site (data not shown). Competition assays also reveal that the relative affinity of nuclear proteins for the three sites differs somewhat. FI complexes form with approximately 2 - and 5-fold greater affinity than FIII and FV complexes, respectively (data not shown). Together, these observations of similar sequence specificity and comparable complex mobilities indicate that common nuclear proteins interact with all of these sites.
VEZF1 specifically interacts with G-rich sites in the HS4 insulator and the βA-globin promoter
We purified proteins that specifically interact with FI and FIII from chicken red blood cells by conventional chromatography. FI - and FIII-binding activities exactly co-fractionated following ion exchange chromatography with SP - and Heparin sepharose (Figure 5). The active elution peak from Heparin sepharose chromatography was split in two and fractionated in parallel by either FI or FIII DNA affinity chromatography. The resulting purified polypeptides were sequenced by tandem mass spectrometry (Figure S2). The proteins Hsp70, VEZF1, ZF5 and TEF1α were present in both FI and FIII DNA affinity eluates. The proteins SP1 and SP3 were additionally present in the FI DNA affinity eluate.
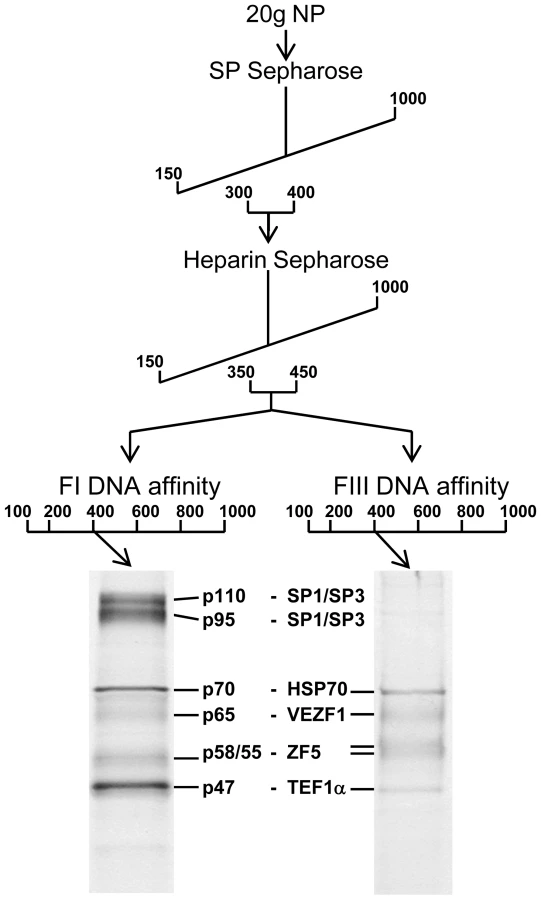
We firstly cloned chicken VEZF1 (Refseq NM_001037827.1) and determined whether it interacts with the G-rich footprinted sites of the HS4 insulator, as it was previously reported that human VEZF1 (also known as DB1) interacts with similar G-rich sites [19],[20]. We find that in vitro translation of chicken VEZF1 cDNA yields a 65 kDa protein that efficiently interacts with the FI, FIII and FV sites (Figure 6A). The complexes formed with recombinant VEZF1 migrate slightly faster than those formed with nuclear extract. This may be a reflection of differing post translational modifications. Nonetheless, recombinant VEZF1 binds with an identical specificity to that observed for nuclear extract proteins. For example, competition of VEZF1 complexes with unlabelled FI and FIII duplexes is disrupted by mutations within their dG-dC strings (Figure 6A, lanes 1–8).

The (dG-dC) strings present in the VEZF1 sites at HS4 are reminiscent of a site in the chicken βA-globin promoter that contains a (dG-dC)16 string. A nuclear factor called Beta Globin Protein 1 (BGP1) was previously characterized as interacting with this site [21]. This BGP1 binding site has no effect on transcription in transient assays or on chromatinized templates in vitro, but was considered to indirectly assist in activation by directing nucleosome placement [22]–[24]. BGP1 protein of 66 kDa can be purified using poly(dG)-poly(dC) affinity chromatography [25]. We have now sequenced a purified BGP1 sample (a gift from J. Allan, University of Edinburgh) and find it to be VEZF1. We find that recombinant chicken VEZF1 interacts with the βA promoter site with an identical specificity to that of erythrocyte nuclear protein(s) (Figure 6A, compare lanes 9–15 with 16–22). At least seven contiguous homopolymeric dG-dC base pairs are required for the efficient formation of complexes between recombinant VEZF1 or nuclear proteins and the βA promoter site [25]. Consistent with this, interaction between VEZF1 and the βA site is competed by unlabelled FI which contains a (dG-dC)9 string (Figure 6A, compare lanes 16 and 20). This competition is disrupted by mutation at the centre of the FI dG-dC string (Figure 6A, compare lanes 20 and 21). However, VEZF1 interaction with the βA site is also competed by FIII which contains only a (dG-dC)6 string (Figure 6A, compare lanes 16 and 22). FIII contains a second short (dG-dC)4 string, as does FV (see figure 4D), which may compensate to form a bipartite recognition motif. Recombinant VEZF1 interacts with the contiguous dG-dC strings of the FI and βA sites with approximately 2 - and 5-fold greater affinities than the bipartite dG-dC strings of the FIII and FV sites, respectively (data not shown).
Polyclonal antibodies were raised against a conserved C-terminal fragment of VEZF1, which specifically recognize the 65 kDa VEZF1 polypeptide from chicken nuclear extracts (Figure S7B). VEZF1 antibodies readily supershift/abrogate complexes between recombinant VEZF1 and the FI, FIII, FV and βA sites (Figure 6B). Supershift analysis also reveals that VEZF1 is present in complexes between nuclear extracts and the FI, FIII, FV and βA sites (Figure 6B). VEZF1 appears to be the only factor that interacts with the FIII and FV sites, whereas other factors also appear to bind to the FI and βA sites in vitro.
Chromatin immunoprecipitation (ChIP) analyses were performed to analyze the binding of VEZF1 at the chicken β-globin locus in vivo. Chromatin was prepared from the early erythroid line 6C2, which does not express β-globin and nucleated erythrocytes isolated from 10 day chicken embryos, a stage at which approximately 80% of erythrocytes are definitive and express the βA-globin gene. VEZF1 was found to strongly interact with HS4 in both 6C2 cells and erythrocytes, consistent with our in vitro analyses (Figure 7A and 7B). VEZF1 binding to HS4 therefore does not coincide with transcription of the β-globin genes. VEZF1 does not interact with the 3′HS enhancer blocking element, which does not contain any dG-dC string like motifs and lacks barrier activity [12]. This is in contrast to CTCF, which interacts strongly with both the HS4 and 3′HS insulators in 6C2 cells and erythrocytes (Figure 7A and 7B). We also find that VEZF1 strongly interacts with the βA promoter, consistent with gel mobility shift assays. In contrast to HS4, VEZF1 binding to the βA promoter appears to be restricted to erythrocytes in which the βA gene is expressed (Figure 7A and 7B). None of the other candidate HS4-binding proteins isolated by DNA affinity purification were found to bind in vivo (described in Text S1) (see also Figures S3, S4, S5).

We tested whether VEZF1 requires all three of its sites for binding to HS4 in vivo, as all three VEZF1 binding sites are required for protection from DNA methylation. We found that this was not the case, as VEZF1 remains tightly bound at HS4 when any one of its binding sites is deleted (Figure S6). VEZF1 also remains bound to mutant HS4 elements that have become partially methylated. Consistent with this, we found that VEZF1 binding to its three sites at HS4 is not affected by CpG methylation in vitro (data not shown). We have attempted to disrupt VEZF1 function at HS4 following knockdown by RNAi. We strived to achieve substantial knockdown of VEZF1 to disrupt its binding to the high affinity sites at HS4. Prolonged knockdown was also required as we have previously found that the de novo DNA methylation of these transgenes is a gradual process that takes many days to establish [18]. We were able to knockdown VEZF1 protein to 3% of wild type levels following 2 weeks of stable miRNA expression. However, ChIP analysis revealed that VEZF1 binding to HS4 was not significantly affected following this prolonged and substantial knockdown (Figure S7). Consequently, we observed no de novo DNA methylation of the HS4 element and no change in HS4's ability to protect a transgene from silencing during this period (data not shown). The inadequacy of RNAi to strip constitutive transcription factor binding from high affinity sites has also been observed for CTCF [26]. Unfortunately, we are unable to study the role of murine Vezf1's role in protection from DNA methylation in Vezf1 null ES cells, as we recently found that they are defective for de novo DNA methylation due to the requirement of Vezf1 for full transcriptional activity of the Dnmt3b gene in these cells [27].
VEZF1 elements protect a CpG island from de novo DNA methylation
To address whether VEZF1 elements also protect CpG island (CGI) promoters from DNA methylation, we investigated VEZF1 binding to the APRT gene promoter and its effects on methylation. SP1-like binding elements have been shown to be required to prevent methylation of the mouse and hamster APRT CpG island elements: two earlier papers have shown that deletion of the SP1-like elements is sufficient to induce methylation in these islands [28],[29]. Furthermore, pre-methylated fragments of the hamster APRT CGI that contain three SP1-like elements are subject to demethylation upon integration into mouse ES cells [28]. The SP1 transcription factor itself is not required for the unmethylated state of CGIs however, with the APRT gene remaining unmethylated and expressed normally in Sp1 null ES cells and embryos [30]. Given that VEZF1 recognizes G-rich sequences that are similar to SP1 motifs, we hypothesized that VEZF1 may interact with the APRT CGI elements (Figure 8A). We performed ChIP analyses for the binding of SP1 and VEZF1 to the 720 bp hamster APRT CGI stably integrated into mouse ES cells (Figure 8B). We found SP1 binding at both sites 1/2 and site 3, but site 3 was also occupied by VEZF1. Supershift analysis also shows VEZF1 interaction with site 3 in vitro (Figure S8). Site 3 contains a motif (CCCCCCTTTCCCC) that is reminiscent of the VEZF1-specific bipartite footprint III site found at the HS4 insulator element (CCCCCCGCATCCCC).

To address whether VEZF1 elements could protect the APRT CGI from methylation, we replaced each of the three SP1-like elements with the VEZF1-specific FIII element from the HS4 insulator (Figure 8A). ChIP analysis shows that VEZF1 binding replaces that of SP1 at the mutant APRT CGI integrated into ES cells (Figure 8B). Supershift analysis also shows that VEZF1 interacts with the mutant sites 1&2 and site 3, while SP1/SP3 binding is lost (Figure S8). We then tested the ability of wild type and mutant APRT CGIs to resist DNA methylation. Firstly, we confirmed that the de novo DNA methylation machinery functioned normally in the ES cells as non-island sequences from the APRT gene body succumb to de novo methylation (Figure 8C). Consistent with previous results [28],[29], we find that the wild type APRT CGI is protected from DNA methylation when stably integrated into ES cells (Figure 8D). Furthermore, a pre-methylated wild type APRT CGI is demethylated upon integration (Figure 8E). It has previously been shown that mutation of the SP1-like elements results in the de novo methylation of the APRT CGI [28]. Our results show that substitution of the SP1-like elements with VEZF1-specific FIII elements from HS4 restores the ability of the mutant APRT CGI to be both protected from de novo methylation and to remove pre-methylation (Figure 8E and 8F). VEZF1 elements from HS4 are therefore sufficient to mediate the demethylation and protection of a CGI from DNA methylation.
Discussion
Here we have studied the paradigm HS4 element to address our hypothesis that a barrier element in vertebrates must be capable of defending a gene from silencing by DNA methylation and have identified a novel CpG island factor. We have presented six findings: 1) a vertebrate barrier element protects a gene promoter from DNA methylation-mediated silencing, 2) the essential transcription factor VEZF1 is a barrier/anti-methylation factor, 3) there is a modular division of labor at the compound HS4 insulator as VEZF1-mediated protection from methylation is separable from CTCF-mediated enhancer blocking and USF-mediated recruitment of active histone modifications, 4) the de novo DNA methylation activity prevented by the HS4 barrier does not appear to spread from its chromosomal neighborhood, 5) a promoter can be protected from DNA methylation even when it lacks active histone modifications and transcriptional activity, and 6) short DNA elements bound by VEZF1 mediate the demethylation and protection of a CpG island from DNA methylation.
The barrier activity of HS4 consists of separable activities which prevent silencing mediated by either histone or DNA methylation
We have previously demonstrated that the HS4 insulator acts as a barrier to the spread of histone methylation marks associated with repressive chromatin [14],[15]. While we found that active histone modifications recruited by USF proteins are an essential component of HS4's barrier activity, they are not sufficient [14]. Three addition protein binding sites are essential for barrier activity but are not required for the recruitment of active histone modifications [12],[14]. These findings, summarized in Figure 9, indicated that there was an additional and separable component to HS4's barrier activity. Here, we show that all three sites are bound by VEZF1 and are required for HS4's ability to protect a linked promoter from de novo DNA methylation.

It was previously shown that the transgenes used in this study become marked by dense promoter DNA methylation upon chromosomal position effect silencing [16]. Promoter DNA methylation occurred subsequent to histone deacetylation and transcriptional inactivation of the promoter [18]. While flanking HS4 elements perfectly shield the transgene from silencing and DNA methylation, it was unclear from these experiments whether the lack of promoter methylation was simply a readout of the promoter's transcription status. We show that VEZF1-mediated protection from DNA methylation of a transgene promoter is retained even when USF site mutations at HS4 lead to histone deacetylation and transcriptional silencing (Figure 9, ΔUSF). The separation of DNA methylation protection from a promoter's histone modification and transcriptional status is a strong indication that the VEZF1 sites at HS4 possess a bona fide activity that is protective against DNA methylation.
De novo DNA methylation of a transgene does not appear to propagate via a continuous DNA methylation-dependent spreading mechanism
Determining the source of de novo DNA methylation is key to our understanding of how VEZF1 binding at HS4 could protect a promoter from epigenetic silencing. Previous studies using the same transgene system studied here found that non-insulated transgenes, regardless of integration site, are consistently subject to promoter methylation upon chromosomal silencing, and that flanking with HS4 elements can shield transgenes from this methylation [16],[18]. The simplest explanation of these results is that HS4 is acting as a barrier to the encroachment, or spreading, of a silencing mechanisms that results in DNA methylation. The ability of repressive histone modifications and associated chromatin factors to mediate the spreading of gene silencing is well documented for many systems [31]. In the case of the chicken β-globin locus, the spreading of repressive histone modifications is observed upon perturbation of active histone modification recruitment at the HS4 barrier [14],[15]. Analyses of progressive CpG island methylation during tumor progression are consistent with models that describe the spreading of DNA methylation [32],[33].
Should de novo DNA methylation arise via spreading from the chromosomal integration site in our transgene system, we would expect to see high levels of methylation at compromised mutant insulators either coincident with, or prior to promoter methylation. However, we observe that promoters become methylated prior to the insulators, which remain unmethylated or become partially methylated. The observed independence of methylation states between insulator and promoter argue against spreading and clearly show that there can be no single mechanism that controls the methylation state of both the insulator and promoter. It remains possible that VEZF1 elements at HS4 are acting as a barrier to the spreading of a DNA methylation mechanism, but that additional processes prevent the accumulation of methylation at HS4 itself. An alternative possibility is that DNA methylation does not result from spreading, and that the insulator directly interacts with the promoter to deliver VEZF1 co-factors that prevent promoter methylation. In this model, the promoter itself would have its own program to recruit de novo DNA methylation, and VEZF1 would act as a factor that mediates inhibition of methylation. This would distinguish the activity of VEZF1 from those of USF1/USF2, which bind elsewhere in the insulator element and recruit a number of enzymes that deliver active histone modifications to the reporter gene [14],[15],[34]. It will be of interest in future to determine whether VEZF1 elements potentiate the expression of nearby genes through the control of DNA methylation.
Intact insulator protein complexes maintain HS4 as a CpG island
The 275 bp “core” HS4 element comprises a CpG island (CGI) that is free of DNA methylation regardless of neighboring gene expression [11],[35],[36], as well as when it is inserted into the mouse Igf2/H19 domain [37]. Consistent with this, we found that wild type transgenic HS4 elements remain unmethylated during long term culture. The processes that maintain the unmethylated state of the insulator appear to be complex, as we find that the mutation of all insulator protein binding sites results in some degree of de novo DNA methylation of HS4. It has previously been shown that DNA binding proteins can prevent the methylation of their binding sites simply by steric hindrance of de novo DNA methyltransferases (DNMTs) [38]. It is possible that the HS4 deletions studied here disrupt cooperative interactions between insulator proteins, thus permitting DNMT access. We performed ChIP experiments on HS4 mutants and found that deletion of any one insulator binding site does not lead to the loss of binding of another (Figure S6). This is in agreement with functional assays which also found that deletion of any one insulator protein binding site does not lead to the loss of function associated with another site [9],[12],[14]. These findings argue against a simple steric protection of HS4 DNA from DNMTs by transcription factor binding. The degree of methylation at mutant HS4 elements was typically moderate (20–50%) and did not increase to the near total methylation seen at the transgene promoter (90–100%) following long term culture. These observations are consistent with a balance between activities that add and remove DNA methylation at HS4.
VEZF1 elements at CpG island promoters
All constitutively expressed genes and ∼40% of genes with tissue-restricted expression have CGI promoters [39]. CGIs are typically unmethylated, especially in the germ line, which ensures that these CpGs are not subject to mutation by spontaneous deamination. It remains to be determined how CGIs resist global de novo methylation during early development, and how they remain hypomethylated irrespective of transcriptional status. Recent epigenomic profiling studies have begun to reveal a significant portion of CGIs that are subject to varying degrees of tissue-specific methylation in human somatic tissues [40]–[42]. These findings point to the existence of processes that protect CGIs from de novo methylation, which can be selectively inactivated during development and may become defective during cancer progression [43]. Definition of the cis-regulatory elements and trans-acting factors that control CGI methylation status is key to unraveling these processes.
We have revisited the well established example of the APRT gene CGI promoter. It was previously shown that SP1-like binding elements are required to prevent CGI methylation [28],[29], although surprisingly the SP1 transcription factor itself is not required [30]. These findings suggested that other factors function at the G-rich SP1-like motifs, which are commonly found at CpG islands [44]. We show that VEZF1 interacts with site 3 of the hamster APRT CGI. A promoter-less APRT CGI fragment containing only site 3 remains protected from DNA methylation [28]. We were able to abrogate SP factor binding while retaining VEZF1 binding by introducing VEZF1-specific elements. The VEZF1-specific mutant retained its ability to mediate demethylation and protection from de novo DNA methylation. Thus, VEZF1 binding elements can protect a CGI from DNA methylation. We attempted to definitively address the requirement for VEZF1, but discovered that global de novo DNA methylation mechanisms are defective in Vezf1 null ES cells [27]. We also show that VEZF1 interacts with the CGI promoter of the DHFR gene (Figure S5). Furthermore, ChIP-array analysis in somatic human cells reveals that VEZF1 predominantly interacts with CGI promoters and regulates genes with diverse functions (R.S. and A.W., unpublished observations). It remains to be determined whether VEZF1 plays a widespread role in the control of DNA methylation and what contribution this epigenetic control makes to developmental gene regulation and cancer progression.
DNA methylation and the multiple roles of chromatin boundaries
Experimental evidence has demonstrated that chromatin barrier elements can employ a number of different mechanisms to limit the spread of transcriptionally repressive chromatin; including tethering, nucleosome gaps/masking and histone code manipulation [6]. The constitutive recruitment of histone modifications such as acetylation is considered to be sufficient to establish barrier activity in eukaryotes that do not methylate their genomes, as demonstrated at synthetic barriers in yeast [45]. Our finding that HS4 also mediates protection from de novo DNA methylation adds another tier to understanding the mechanism of barrier elements in vertebrates. It is well established that densely methylated DNA abrogates transcription factor binding and is sufficient to establish all the features of repressive chromatin, including repressive histone modifications [46],[47]. We propose that a barrier element in higher eukaryotes must be capable of preventing de novo DNA methylation in addition to blocking the propagation of silencing histone modifications, as either event, if not inhibited, is sufficient to direct the establishment of an epigenetically stable silent chromatin state. We have shown here that a fully effective vertebrate barrier combines both of these properties in a single multi-component element.
Materials and Methods
Bisulfite sequencing analysis
Chicken 6C2 erythroleukemia cells carrying IL-2R reporter transgenes (8103, wild type HS4; 10401, ΔFI; 10506, ΔFII; 10615, ΔFIII; 10901, ΔFIV; 8d5, ΔFV) were cultured and assayed for IL-2R expression by FACS as described previously [12],[14]. Genomic DNA was extracted from cell lines after 30 and 90 days of culture and bisulfite modified (EZ Methylation, Zymo Research, CA). The upstream double-core HS4 elements and the IL-2R promoter were PCR amplified from each line. We were unable to amplify bisulfite modified double-core HS4 elements from the 10506, ΔFII line to sufficient levels to provide representative sequence data. We therefore opted to amplify the outermost HS4 copy only. The 8d5, ΔFV line also only contains one copy of HS4 in the upstream location [12]. All PCR products were gel purified and cloned, followed by sequencing (GATC Biotech, Konstanz, Germany) of 10 clones for each region of interest.
Gel mobility shift assays were performed as described previously [14]. Recombinant VEZF1, SP1, SP3 and ZF5 were produced by in vitro translation using rabbit reticulocyte lysate (Promega).
Protein purification
FI - and FIII-binding proteins were purified from adult chicken red blood nuclear protein extracts by ion exchange chromatography. Throughout the purification, eluate fractions were analyzed for FI - and FIII-binding activity with gel mobility shift assays. The binding specificity of partially purified proteins was checked by competition analysis after each purification step. FI - and FIII-binding activities co-fractionated following ion exchange chromatography with SP XL and Heparin sepharose (GE Healthcare). Phosphocellulose, Q and Sephacryl S300 columns were used in early purification attempts to resolve FI - and FIII-binding activities but they co-fractionated in each case (data not shown). FI - and FIII-binding activities both eluted in two distinct fractions of approximately 200 and 400 kDa following gel filtration (data not shown). The active fractions eluted from heparin sepharose were pooled then split into two and fractionated in parallel by FI or FIII DNA affinity as described previously [14]. Polypeptides electrophoresed on 7% Tris-acetate gels (Invitrogen) were excised, digested with trypsin and sequenced at the Harvard Microchemistry Facility by microcapillary reverse-phase HPLC nano-electrospray tandem mass spectrometry (μLC/MS/MS) on a Finnigan LCQ DECA quadrupole ion trap mass spectrometer.
cDNA cloning
Chicken VEZF1/BGP1 cDNA was cloned following RT-PCR from 6C2 cell total RNA based on an assumption of conservation of 5′ cDNA sequence with human VEZF1/DB1. The oligonucleotide Adaptor-A 5′CATGCCGCTCGAGCGGTTTTTTTTTTTTTTTTT was used in first strand cDNA synthesis with Superscript II reverse transcriptase (Invitrogen). The primers VEZF1_5′, 5′CCATGACCCATGGGCAGAGCCAAAGT and Adaptor 5′CATGCCGCTCGAGCGG were used to amplify a full length chicken VEZF1 cDNA by PCR which was TA cloned into pCRII (Invitrogen). VEZF1 cDNA was sub-cloned into pCITE4b (Novagen) to generate p4bVEZFfull for the purpose of in vitro transcription. cDNA encoding chicken ZF5 was isolated by RT-PCR from 6C2 cell total RNA using primers designed from the published sequence (U51640). We found that the bases CpG 1306-7 in the published sequence were GpC in our clone, causing codon 436 to translate as alanine instead of arginine. We obtained the vectors pCDNA3-ZF5 and pEVRFO-ZF5 (a kind gift of W. Stumph, San Diego State University) and we also found the CpG to GpC conflict with the published sequence. Full length chicken SP1 and SP3 cDNAs cloned in the pBluescript-based vectors pH-SP1 and pH-SP3-3 were a kind gift from Marc Castellazzi (INSERM, Lyon).
Antibodies
Polyclonal antibodies were raised (Rockland Immunochemicals) against chicken VEZF1 (Ser376-Ala547) and chicken ZF5 peptides (Ser131-Lys248) produced in E.coli (QIAexpress, Qiagen). Peptides were produced in M15 [pREP4] E. coli followed by rapid lysis with B-PER reagent (Pierce). ZF5 peptide was soluble and purified on Ni-NTA agarose (Qiagen). VEZF1 peptide was insoluble and resulting inclusion bodies were prepared using B-PER reagent (Pierce), solubilized with 6M guanidium hydrochloride and immobilized on TALON Sepharose resin (Clontech) at pH 7. VEZF1 peptide was renatured in a stepwise manner with 6, 4, 3, 2, 1 and 0.5 M guanidium hydrochloride prior to elution. VEZF1 and ZF5 polyclonal IgG antibodies (Rockland Immunochemicals) were purified from rabbit serum using PROSEP-A media (Montage, Millipore). Anti-full length chicken VEZF1 antibodies were raised previously [27]. Antibodies raised against CTCF (06-917), SP1 (PEP2X, H-225X), SP3 (D20X) and USF1(B01) were obtained from Millipore, Santa Cruz Biotechnology and Abnova, respectively.
Chromatin immunoprecipitation (ChIP) analysis
ChIP analysis of transcription factor binding in chicken cells was performed using formaldehyde crosslinked chromatin prepared from chicken 10 day embryonic erythrocytes isolated from fertilized White Leghorn eggs (CBT Farms, Chestertown, MD) or cultured 6C2 erythroleukemia cells. 10 day erythrocytes were washed and resuspended in 25 mls of PBS (2×107 cells/ml) and fixed with a final concentration of 0.25% formaldehyde at room temperature for 30 seconds. 6C2 cells were harvested in mid-exponential growth phase, divided into 30 ml aliquots containing 1×108 cells in fresh media and fixed with a final concentration of 0.8% formaldehyde at room temperature for 5 minutes. Reactions were stopped by adding glycine to a final concentration of 0.125 M. The cells were washed in PBS and resuspended in cell lysis buffer (0.25% Triton X-100, 10 mM EDTA, 0.5 mM EGTA, 10 mM Tris pH 8.0) to isolate nuclei. Chromatin was prepared following washing (0.2 M NaCl, 1 mM EDTA, 0.5 mM EGTA, 10 mM Tris pH 8.0) and lysis (NLB: 50 mM Tris-HCl pH 8.0, 10 mM EDTA, 0.5% SDS) of nuclei. Crosslinked chromatin was fragmented by sonicaton (Misonix) for a total time of 10 minutes in regular 10 second pulses at 4°C. Debris was removed by centrifugation at 15000 g for 10 minutes and chromatin was diluted in 10 volumes of X-ChIP buffer (1.1% TX-100, 1.2 mM EDTA, 16.7 mM Tris pH 8.1, 167 mM NaCl). Agarose gel electrophoresis was used to confirm that chromatin fragments were ∼500 bp in length on average.
Chromatin was pre-cleared with 100 µl of protein A agarose (50% slurry in X-ChIP buffer, Millipore) and 50 µg of normal rabbit IgG (Santa Cruz) for 3 hours at 4°C on a rotating wheel. Aliquots of chromatin were taken to generate input DNA and protein for western analysis. Individual ChIPs were performed using chromatin from 1×107 cells in a total volume of 1 ml by diluting the pre-cleared chromatin with modified X-ChIP buffer (1 part NLB to 9 parts X-ChIP). Incubation with antibodies was performed overnight at 4°C on a rotating wheel. Between 10 and 30 µg of specific antibodies or 10 µg of normal rabbit IgG (Santa Cruz) were used per ChIP. Chromatin was precipitated with protein A agarose (50 µl slurry in X-ChIP, Millipore) for 4 hours at 4°C with rotation. Immunoprecipitated chromatin was collected, washed, eluted and crosslinks reversed. DNA was then extracted by phenol-chloroform and ethanol precipitated in the presence of 10 µg glycogen.
Relative DNA enrichments were quantified by TaqMan real-time qPCR using the comparative Ct method relative to input DNA and normalized to the primer set 10.35 within the 16 kb condensed chromatin region upstream of the chicken β-globin locus as described previously [14]. The TaqMan primer sets 10.35 (“16 kb”), 21.54 (HS4 core, “end HS4”), 39.806 (beta-adult promoter), 50.861 (3′HS) and PGI5′ (5′ HS4 elements on IL-2R transgene, “trans HS4”) were used in this study.
10.35_For: GGAACAAGTTGGCAAGGTCCTAT
10.35_Rev: TCTTCTGCCCTGCCCGTAT
10.35_TM: FAM-TGCAGTTCCCTGTTCATGTGCTTTTCG-TAMRA
21.54_For: TCCTGGAAGGTCCTGGAAG
21.54_Rev: CGGGGGAGGGACGTAAT
21.54_TM: 6FAM-CCCAAAGCCCCCAGGGATGT-TAMRA
39.8_For: CTGTGGTCTCCTGCCTCACA
39.8_Rev: AGGCTGGGTGCCCCTC
39.8_TM: FAM-CAATGCAGAGTGCTGTGGTTTGGAACTG-TAMRA
PGI_5′_For CACAGGAAACAGCTATGACATGATT
PGI_5′_Rev TCTGCCTTCTCCCTGATAACG
PGI_5′_TM 6FAM-AATTCCTGCCCACACCCTCCTGC-TAMRA
ChIP analysis of transcription factor binding in murine E14Tg2A.4 ES cell lines was performed using formaldehyde crosslinked chromatin prepared from 1×109 cells treated with 1% formaldehyde for 5 minutes. Chromatin was prepared as described above, where fragments sizes ranged from 500–700 bp. Semi-quantitative PCR was performed using the following primers
APRT1/2_For: AAAGGCGTGCGGGAGCCAGAAAT
APRT1/2_Rev: CCTTGGTAGGTGGGG
APRT3_For: CCCTGTTCCTGGGCTCC
APRT3_Rev: TGACTGGCCAGGAGG
ChIP analysis from human embryonic kidney 293-T cell line SD5 that contains a stably integrated copy of the 275 bp HS4 core chicken insulator was performed as described for cultured chicken cells above. SD5 cells were crosslinked with 1.6% formaldehyde for 5 minutes. The following primers were used in SYBR quantitative PCR analysis:
HS4_21.726_F: CGGGATCGCTTTCCTCTGA
HS4_21.726_R: CCGTATCCCCCAGGTGTCT
P_DHFR_F: TCGCCTGCACAAATAGGGAC
P_DHFR_R: AGAACGCGCGGTCAAGTTT
Control
VEZF1_CDS_F: GACAGCAGCCGAACTTCGTT
VEZF1_CDS_R: TGGTGCCCGAGGAAGATG
APRT elements. The following elements were amplified from Hamster liver genomic DNA:
Wild-type APRT CpG island
Sp1-like motifs in red. AvaII and HpaII restriction sites in bold.
CTAGAGGATCCGGACAACACCCACACCGGCCCCTCCAGGTCCAGAAAGCTGGCCCTGCGAGAAGCGGGACTGAAAAGGCGTGCGGGAGCCAGAAATCCAAAAGGGTGCCAAGGCATGCGTCCTTTTTCCACCCAGAAATAACCCCAGGCTTTCAATTTGAGGTTATTTCAATATCCAGCAAATGCGTTACTTCCTGCCAAAAGCCAGCCTCCCCGCAACCCACTCTCCCAGAGGCCCCGCCCCGTCCCGCCCCCTCCCGGCCTCTCCTCGTGCTGGATCGCTCCCTAAGGACGCCCCGCTCCAGAAGCCCCACCTACCAAGGACGCCCCACCCTTGTTCCCGGACTGGTATGACCCCAGCCTGCTGACATCCCTCCGCCCTTTCTCGTGCACGCGGCTATGGCGGAATCTGAGTTGCAGCTGGTGGCGCAGCGATCCGCAGTTTCCCCGACTTCCCCATCCCCGGCGTGCTGTTTAGGTGAGATCACGAGCCAGCAAGGCGTTGGAGCCCTGTTCCTGGGCTCCCGGCGAGGCGCATGGGCAGTCTCGGGGATCTTGTGGGGTCTCCGCCCCCCTTTCCCCGGCCACCAGCCTCTCCTTGTTCCCAGGGATATCTCGCCCCTCCTGAAGGACCCCGCCTCCTTCCGAGCTTCCATCCGCCTCCTGGCCAGTCACCTTAAGTCCACGCATGGCGGCAAGATCGACTACATCGCAGGTCTA
Mutant APRT CpG island
Mutated bases in blue were introduced by site directed mutagenesis. Overall CpG content increased from 46 to 48 following these mutations. AvaII and HpaII restriction sites in bold.
CTAGAGGATCCGGACAACACCCACACCGGCCCCTCCAGGTCCAGAAAGCTGGCCCTGCGAGAAGCGGGACTGAAAAGGCGTGCGGGAGCCAGAAATCCAAAAGGGTGCCAAGGCATGCGTCCTTTTTCCACCCAGAAATAACCCCAGGCTTTCAATTTGAGGTTATTTCAATATCCAGCAAATGCGTTACTTCCTGCCAAAAGCCAGCCTCCCCGCAACCCACTCTCCCAGAGACCCCCCGCATCCCCGACGCTACCCCCCGCATCCCCGATCTCCTCGTGCTGGATCGCTCCCTAAGGACGCCCCGCTCCAGAAGCCCCACCTACCAAGGACGCCCCACCCTTGTTCCCGGACTGGTATGACCCCAGCCTGCTGACATCCCTCCGCCCTTTCTCGTGCACGCGGCTATGGCGGAATCTGAGTTGCAGCTGGTGGCGCAGCGATCCGCAGTTTCCCCGACTTCCCCATCCCCGGCGTGCTGTTTAGGTGAGATCACGAGCCAGCAAGGCGTTGGAGCCCTGTTCCTGGGCTCCCGGCGAGGCGCATGGGCAGTCTCGGGGATCTTGTGGGGTCCGCTCCCCCCGCATCCCCGACACCAGCCTCTCCTTGTTCCCAGGGATATCTCGCCCCTCCTGAAGGACCCCGCCTCCTTCCGAGCTTCCATCCGCCTCCTGGCCAGTCACCTTAAGTCCACGCATGGCGGCAAGATCGACTACATCGCAGGTCTA
Non-CpG island from APRT locus
HpaII site in bold.
TCTAGATTGCTAGGAGTAGCACCTAAGATGAACTAGATGCTAAAAAATGCTGTATCTTTGGGGCACACGAGGGCATGCCTGGGCAGGCTTAGAGCCTGGTAGTCTCAGGGGCTGCACCAAAGTGTAATTCTTGTGCTAAATAACTTTCACTTACCAGTGCCAAGCACGGGCTTCAGAAACACCCTAGGGTCGCTGAATGTCCACCAGGGGAGTCAGACATGTCCAGAGGGTGAGAACCCCAGAGAATTCGGTAGCCCTGACATGTGCTACAATTACTGATGCCCACTTCCTACTGGTTCCTCCTGGCCATACCTCAGGAATTAGGGCATGCTTTCTGCCTGCTACAGTAGCTCATCCTCCCTGGAAGTGACCCCAGACATATACCCTGAACTGTAACCGATAAAGTGCGCCTGGGCAGATGTATTTGAGAGGTGGCAAAAGTAAACCATAGGTGTCCCCGAGCTAGATACAGAAGGCAGATAACATCCCCAAGGCTAAGCTGCTGCCCCAATAGCCATCAGCCTTCTAGTTATAGCTAGTAAGACCTAGTATTCCTGGTCAATACTATTCACTCAATCCTTACACCTCAGCCCTAACACGCCCCCTCTCTCATCCTAACAGGCCTAGACTCCAGGGGATTCTTGTTTGGCCCCTCCCTAGCTCAGGAGCTGGGCCTGGGCTGTGTGCTCATCCGGAAGCGAGGGAAGCTGCCAGGCCCCACAGTGTCAGCCTCCTATGCTCTCGAGTATGGCAAGGTAAGCAGGCAGTGGGTAGCTGTCTAGGAGTAAATGTGGGGGCTCAGAGAGGTTAAGTCATCAGGCCAGGTTTATACCACCAGGAAACATGGAGAAGCTAGGGGTGGTGGTTCTAGA
CpG island methylation assays
∼720 bp wild type and mutant APRT CpG islands and an 870 bp non-island region of the APRT gene were cloned into pUC19. Each element was PCR amplified, half of which was subject to in vitro CpG methylation by M.SssI methyltransferase(New England Biolabs). In vitro methylation was validated by HpaII and MspI digestion. 1 µg of each APRT element was transfected into murine E14Tg2A.4 ES cells (BayGenomics) with 1 µg of the XbaI fragment of pREP7 (Invitrogen). Hygromycin resistant clones were grown in LIF without feeder co-culture. Genomic DNA was extracted after two weeks of culture. Probes for Southern blotting were generated using Ready-to-go dCTP beads (GE Healthcare).
Oligonucleotide sequences
Oligonucleotides were generated on an ABI 394 DNA synthesizer.
The top strand sequences for each of the duplexes used for gel mobility shift analyses were:
FI wt 5′ GGAGCTCACGGGGACAGCCCCCCCCCAAAGCCCCCAGGGA,
FIII wt 5′ aggcgcgccCCGGTCCGGCGCTCCCCCCGCATCCCCGAGCCGGggcgcgcct,
FV wt 5′ CCTGCAGACACCTGGGGGGATACGGGGAAAAAGCTTTAGG,
Sp1 5′ ATTCGATCGGGGCGGGGCGAGC,
glo wt 5′ AATTGCAGAGCTGGGAATCGGGGGGGGGGGGGGGGCGGGTGGTGGTGTGG,
glo 7G 5′ AATTGCAGAGCTGGGAATCGGGGGGGCGGGTGGTGGTGTGG,
glo 6G 5′ AATTGCAGAGCTGGGAATCGGGGGGCGGGTGGTGGTGTGG.
Asc I restriction sites used for cloning FIII sites in an earlier study are shown as lower case. All FI and FIII oligonucleotides were identical to the wt sequences above except for mutations indicated in Figure 1E.
DNA affinity columns were prepared using the following oligonucleotides
FI-DA TOP 5′ gatcTCACGGGGACAGCCCCCCCCCAAAGCCCCCA
FI-DA BOTTOM 5′ gatcTGGGGGCTTTGGGGGGGGGCTGTCCCCGTGA
FIII-DA TOP 5′ gatcGGTCCGGCGCTCCCCCCGCATCCCCGAGCCGGCA
FIII-DA BOTTOM 5′ gatcTGCCGGCTCGGGGATGCGGGGGGAGCGCCGGACC
PCR primers used in bisulfite sequencing were
HS4 5′ double forward 5′ GGTATTAGAGTAGATTGTATTGAGAGTGTA
HS4 5′ double reverse 5′ CATAACTATTTCCTATATAAATCCCC
HS4 5′ single forward 5′ AGAGTAGATTGTATTGAGAGTGTATTATA
HS4 5′ single reverse 5′ ACATCCCTAAAAACTTTAAAAAAAA
IL-2R forward 5′ GTTAAGGTTGGGGGTTTTTT
IL-2R reverse 5′ AAAACTCTACCTAACAACCAAACAC
PCR primers used in RT-PCR gene expression analysis were
GgVEZF805anti 5′CAGTGCACGTTTGGCATTTGAAG
GgVEZF716sense 5′GAAAAGGCTTCTCGAGGCCTGATC
GgGAPDH-247T 5′-6FAM-TCCAGGAGCGTGACCCCAGCA-TAMRA
GgGAPDH-226F 5′ ATGGGCACGCCATCACTATC
GgGAPDH-302R 5′ AACATACTCAGCACCTGCATCTG
GgB-ACTIN_F 5′ TGCTGCGCTCGTTGTTGA
GgB-ACTIN_R 5′ CATCGTCCCCGGCGA
GgB-ACTIN_T 5′-6FAM-TGGCTCCGGTATGTGCAAGGCC-TAMRA
The primers used for methylation specific PCR analysis of the APRT non-island element were:
APRTNI_5′ TCTAGATTGCTAGGAGTAGC
APRTNI_3′ TCTAGAACCACCCCTAGC
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. KellumR
SchedlP
1991 A position-effect assay for boundaries of higher order chromosomal domains. Cell 64 941 950
2. StalderJ
LarsenA
EngelJD
DolanM
GroudineM
1980 Tissue-specific DNA cleavages in the globin chromatin domain introduced by DNAase I. Cell 20 451 460
3. GasznerM
FelsenfeldG
2006 Insulators: exploiting transcriptional and epigenetic mechanisms. Nat Rev Genet 7 703 713
4. ValenzuelaL
KamakakaRT
2006 Chromatin insulators. Annu Rev Genet 40 107 138
5. WallaceJA
FelsenfeldG
2007 We gather together: insulators and genome organization. Curr Opin Genet Dev 17 400 407
6. WestAG
FraserP
2005 Remote control of gene transcription. Hum Mol Genet 14 Spec No 1 R101 111
7. WestAG
GasznerM
FelsenfeldG
2002 Insulators: many functions, many mechanisms. Genes Dev 16 271 288
8. ChungJH
WhiteleyM
FelsenfeldG
1993 A 5′ element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell 74 505 514
9. BellAC
WestAG
FelsenfeldG
1999 The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 98 387 396
10. PikaartMJ
Recillas-TargaF
FelsenfeldG
1998 Loss of transcriptional activity of a transgene is accompanied by DNA methylation and histone deacetylation and is prevented by insulators. Genes Dev 12 2852 2862
11. ChungJH
BellAC
FelsenfeldG
1997 Characterization of the chicken beta-globin insulator. Proc Natl Acad Sci U S A 94 575 580
12. Recillas-TargaF
PikaartMJ
Burgess-BeusseB
BellAC
LittMD
2002 Position-effect protection and enhancer blocking by the chicken beta-globin insulator are separable activities. Proc Natl Acad Sci U S A 99 6883 6888
13. YaoS
OsborneCS
BharadwajRR
PasceriP
SukonnikT
2003 Retrovirus silencer blocking by the cHS4 insulator is CTCF independent. Nucleic Acids Res 31 5317 5323
14. WestAG
HuangS
GasznerM
LittMD
FelsenfeldG
2004 Recruitment of histone modifications by USF proteins at a vertebrate barrier element. Molecular Cell 16 453 463
15. HuangS
LiX
YusufzaiTM
QiuY
FelsenfeldG
2007 USF1 recruits histone modification complexes and is critical for maintenance of a chromatin barrier. Mol Cell Biol 27 7991 8002
16. MutskovVJ
FarrellCM
WadePA
WolffeAP
FelsenfeldG
2002 The barrier function of an insulator couples high histone acetylation levels with specific protection of promoter DNA from methylation. Genes Dev 16 1540 1554
17. LiCL
EmeryDW
2008 The cHS4 chromatin insulator reduces gammaretroviral vector silencing by epigenetic modifications of integrated provirus. Gene Ther 15 49 53
18. MutskovV
FelsenfeldG
2004 Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. Embo J 23 138 149
19. AitsebaomoJ
Kingsley-KallesenML
WuY
QuertermousT
PattersonC
2001 Vezf1/DB1 is an endothelial cell-specific transcription factor that regulates expression of the endothelin-1 promoter. J Biol Chem 276 39197 39205
20. Koyano-NakagawaN
NishidaJ
BaldwinD
AraiK
YokotaT
1994 Molecular cloning of a novel human cDNA encoding a zinc finger protein that binds to the interleukin-3 promoter. Mol Cell Biol 14 5099 5107
21. LewisCD
ClarkSP
FelsenfeldG
GouldH
1988 An erythrocyte-specific protein that binds to the poly(dG) region of the chicken beta-globin gene promoter. Genes Dev 2 863 873
22. BartonMC
MadaniN
EmersonBM
1993 The erythroid protein cGATA-1 functions with a stage-specific factor to activate transcription of chromatin-assembled beta-globin genes. Genes Dev 7 1796 1809
23. BuckleR
BalmerM
YenidunyaA
AllanJ
1991 The promoter and enhancer of the inactive chicken beta-globin gene contains precisely positioned nucleosomes. Nucleic Acids Res 19 1219 1226
24. JacksonPD
EvansT
NickolJM
FelsenfeldG
1989 Developmental modulation of protein binding to beta-globin gene regulatory sites within chicken erythrocyte nuclei. Genes Dev 3 1860 1873
25. ClarkSP
LewisCD
FelsenfeldG
1990 Properties of BGP1, a poly(dG)-binding protein from chicken erythrocytes. Nucleic Acids Res 18 5119 5126
26. WendtKS
YoshidaK
ItohT
BandoM
KochB
2008 Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451 796 801
27. GowherH
StuhlmannH
FelsenfeldG
2008 Vezf1 regulates genomic DNA methylation through its effects on expression of DNA methyltransferase Dnmt3b. Genes Dev 22 2075 2084
28. BrandeisM
FrankD
KeshetI
SiegfriedZ
MendelsohnM
1994 Sp1 elements protect a CpG island from de novo methylation. Nature 371 435 438
29. MacleodD
CharltonJ
MullinsJ
BirdAP
1994 Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev 8 2282 2292
30. MarinM
KarisA
VisserP
GrosveldF
PhilipsenS
1997 Transcription factor Sp1 is essential for early embryonic development but dispensable for cell growth and differentiation. Cell 89 619 628
31. TalbertPB
HenikoffS
2006 Spreading of silent chromatin: inaction at a distance. Nat Rev Genet 7 793 803
32. IssaJP
2004 CpG island methylator phenotype in cancer. Nat Rev Cancer 4 988 993
33. TurkerMS
2002 Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene 21 5388 5393
34. HuangS
LittM
FelsenfeldG
2005 Methylation of histone H4 by arginine methyltransferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes Dev 19 1885 1893
35. PrioleauMN
NonyP
SimpsonM
FelsenfeldG
1999 An insulator element and condensed chromatin region separate the chicken beta-globin locus from an independently regulated erythroid-specific folate receptor gene. Embo J 18 4035 4048
36. PrioleauMN
GendronMC
HyrienO
2003 Replication of the chicken beta-globin locus: early-firing origins at the 5′ HS4 insulator and the rho - and betaA-globin genes show opposite epigenetic modifications. Mol Cell Biol 23 3536 3549
37. SzaboPE
TangSH
ReedMR
SilvaFJ
TsarkWM
2002 The chicken beta-globin insulator element conveys chromatin boundary activity but not imprinting at the mouse Igf2/H19 domain. Development 129 897 904
38. LinIG
HsiehCL
2001 Chromosomal DNA demethylation specified by protein binding. EMBO Rep 2 108 112
39. AntequeraF
BirdA
1993 Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci U S A 90 11995 11999
40. RakyanVK
DownTA
ThorneNP
FlicekP
KuleshaE
2008 An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Res 18 1518 1529
41. StraussmanR
NejmanD
RobertsD
SteinfeldI
BlumB
2009 Developmental programming of CpG island methylation profiles in the human genome. Nat Struct Mol Biol 16 564 571
42. SuzukiMM
BirdA
2008 DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9 465 476
43. JonesPA
BaylinSB
2007 The epigenomics of cancer. Cell 128 683 692
44. FanS
FangF
ZhangX
ZhangMQ
2007 Putative zinc finger protein binding sites are over-represented in the boundaries of methylation-resistant CpG islands in the human genome. PLoS ONE 2 e1184
45. OkiM
ValenzuelaL
ChibaT
ItoT
KamakakaRT
2004 Barrier proteins remodel and modify chromatin to restrict silenced domains. Mol Cell Biol 24 1956 1967
46. HashimshonyT
ZhangJ
KeshetI
BustinM
CedarH
2003 The role of DNA methylation in setting up chromatin structure during development. Nat Genet 34 187 192
47. JaenischR
BirdA
2003 Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33 Suppl 245 254
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 1
Nejčtenější v tomto čísle
- A Major Role of the RecFOR Pathway in DNA Double-Strand-Break Repair through ESDSA in
- Kidney Development in the Absence of and Requires
- The Werner Syndrome Protein Functions Upstream of ATR and ATM in Response to DNA Replication Inhibition and Double-Strand DNA Breaks
- Alternative Epigenetic Chromatin States of Polycomb Target Genes