
Critical Functions of Rpa3/Ssb3 in S-Phase DNA Damage Responses in Fission Yeast
Replication Protein A (RPA) is a heterotrimeric, single-stranded DNA (ssDNA)–binding complex required for DNA replication and repair, homologous recombination, DNA damage checkpoint signaling, and telomere maintenance. Whilst the larger RPA subunits, Rpa1 and Rpa2, have essential interactions with ssDNA, the molecular functions of the smallest subunit Rpa3 are unknown. Here, we investigate the Rpa3 ortholog Ssb3 in Schizosaccharomyces pombe and find that it is dispensable for cell viability, checkpoint signaling, RPA foci formation, and meiosis. However, increased spontaneous Rad11Rpa1 and Rad22Rad52 nuclear foci in ssb3Δ cells indicate genome maintenance defects. Moreover, Ssb3 is required for resistance to genotoxins that disrupt DNA replication. Genetic interaction studies indicate that Ssb3 has a close functional relationship with the Mms1-Mms22 protein complex, which is required for survival after DNA damage in S-phase, and with the mitotic functions of Mus81-Eme1 Holliday junction resolvase that is required for recovery from replication fork collapse. From these studies we propose that Ssb3 plays a critical role in mediating RPA functions that are required for repair or tolerance of DNA lesions in S-phase. Rpa3 orthologs in humans and other species may have a similar function.
Published in the journal:
. PLoS Genet 6(9): e32767. doi:10.1371/journal.pgen.1001138
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001138
Summary
Replication Protein A (RPA) is a heterotrimeric, single-stranded DNA (ssDNA)–binding complex required for DNA replication and repair, homologous recombination, DNA damage checkpoint signaling, and telomere maintenance. Whilst the larger RPA subunits, Rpa1 and Rpa2, have essential interactions with ssDNA, the molecular functions of the smallest subunit Rpa3 are unknown. Here, we investigate the Rpa3 ortholog Ssb3 in Schizosaccharomyces pombe and find that it is dispensable for cell viability, checkpoint signaling, RPA foci formation, and meiosis. However, increased spontaneous Rad11Rpa1 and Rad22Rad52 nuclear foci in ssb3Δ cells indicate genome maintenance defects. Moreover, Ssb3 is required for resistance to genotoxins that disrupt DNA replication. Genetic interaction studies indicate that Ssb3 has a close functional relationship with the Mms1-Mms22 protein complex, which is required for survival after DNA damage in S-phase, and with the mitotic functions of Mus81-Eme1 Holliday junction resolvase that is required for recovery from replication fork collapse. From these studies we propose that Ssb3 plays a critical role in mediating RPA functions that are required for repair or tolerance of DNA lesions in S-phase. Rpa3 orthologs in humans and other species may have a similar function.
Introduction
Preserving genome integrity in eukaryotic organisms depends on integrated mechanisms of DNA replication, DNA repair and telomere maintenance, which are all overseen by checkpoint control systems. Most genome maintenance proteins target specific types of DNA lesions, but a few have much more generalized functions. Of the latter class, perhaps the best-known example is replication protein A (RPA). Also known as single-stranded DNA-binding protein (SSB) or replication factor A (RFA), RPA consists of Rpa1 (∼70 kDa), Rpa2 (∼36 kDa) and Rpa3 (∼14 kDa), that together comprise the major single-stranded DNA (ssDNA) binding activity in eukaryotic cells [1]–[4]. RPA was originally described as a factor that is essential for replication initiation and elongation of SV40 virus DNA in cell extracts [5]–[7], and has since been shown to be required for nucleotide excision repair (NER) and mismatch repair (MMR) in vitro [8], [9]. It also stimulates the activity of homologous recombination (HR) repair proteins in vitro. Indeed, RPA is thought to be a critical factor in every DNA replication or repair process that involves ssDNA [1], [3].
All the known cellular functions of RPA depend on its ability to bind ssDNA [1], [3], [10], [11]. Although a complete 3-dimensional structure of RPA is lacking, structural and biochemical analyses have provided a detailed picture of its domain organization. In essence, RPA is made of 6 oligosaccharide/oligonucleotide binding (OB)-folds: 4 in Rpa1, and 1 each in Rpa2 and Rpa3. Direct binding to ssDNA is mediated by the OB-folds in Rpa1 and Rpa2. The OB-fold in Rpa3 is thought to mediate protein interactions that are required to stabilize the RPA heterotrimer [12], [13].
Teasing apart the in vivo functions of RPA subunits has been a challenging task because RPA is essential for cell viability. Almost all of this work has been carried out with the budding yeast Saccharomyces cerevisiae, where gene disruption studies established that all 3 subunits are required for cell viability [14]. Most genetic studies have been carried out with Rfa1, the ∼70kDA subunit, in which analyses of temperature sensitive or hypomorphic mutants have uncovered defects in DNA replication, recombination, repair, telomere maintenance, and DNA damage checkpoint signaling [3], [4]. Participation in checkpoint signaling has been traced to an interaction with Mec1-Ddc2 checkpoint kinase, which is orthologous to ATR/ATRIP in mammals and Rad3/Rad26 in the fission yeast Schizosaccharomyces pombe [15], [16]. Studies of Rfa2 mutants in budding yeast demonstrate its importance for DNA replication, recombination, repair, and telomere maintenance, although a checkpoint-signaling defect has yet to be established [1], [3]. In contrast to Rfa1 and Rfa2, very little is known about the function of Rfa3 in vivo, with the analyses limited to an N-terminal truncation mutant and a temperature sensitive allele [17].
We have been using fission yeast to investigate the cellular responses to DNA damage in S-phase. Many of these studies have focused on the effects of camptothecin (CPT), which is the prototype of a class of anticancer drugs that stabilize covalent DNA-topoisomerase I complexes by preventing the religation step of topoisomerase I [18]. When a replication fork encounters the CPT-Topoisomerase I complex, it can break, either through direct collision with the CPT-Topoisomerase I complex, or through formation of positive supercoils that stall the fork and can lead to its collapse [19]. In either case, the resulting DNA damage is a one-ended DSB that is subsequently repaired by a homologous recombination repair pathway that creates a Holliday junction in the process of reestablishing the replication fork [20]. Notably, the Mus81-Eme1 Holliday junction resolvase is essential for CPT resistance but plays no role in survival of DSBs created by ionizing radiation (IR), which are repaired primarily by a synthesis-dependent strand annealing (SDSA) mechanism that does not require resolution of Holliday junctions [21]–[23]. We also recently described the Mms1-Mms22/Mus7 protein complex in fission yeast, which like its counterpart in budding yeast, appears to play a very important but as yet poorly understood role in the survival of genotoxins that interfere with DNA replication [24]–[28].
In an effort to more fully characterize the response of fission yeast to replication-associated DNA damage, we carried out a genome-wide screen to identify genes required for CPT resistance. Using a haploid deletion library, we identified a group of CPT sensitive mutants, amongst the most sensitive were mutants for mms22 or ssb3, the latter of which encodes Rpa3. In this report we characterize Ssb3 and explore its role in recovery from DNA damage in S-phase.
Results
Rpa3/Ssb3 is dispensable for cell viability in fission yeast
We screened an S. pombe haploid deletion library to identify genes required for CPT resistance. In agreement with another recent study [29], we found that mms22Δ and ssb3Δ mutants were amongst the most CPT-sensitive strains in the library (see below). Identification of mms22Δ was anticipated from other studies [24]–[27]. However, as the three subunits of RPA are essential for cell viability in S. cerevisiae, and at least the large subunit Rad11Ssb1/Rpa1 is essential in S. pombe [30], it was unexpected that an ssb3Δ mutant should be viable in S. pombe.
As some alleles in the Bioneer S. pombe deletion library are incomplete deletions, and errors can arise when screening arrayed mutant libraries, it was important to characterize the structure of the presumptive ssb3::KanMX4 mutant in the library. This analysis revealed that the ssb3::KanMX4 mutant was correctly arrayed in the library but the deletion was incomplete. The Bioneer ssb3::KanMX4 allele can potentially encode a protein having the first 21 amino acids of Ssb3 (S. Cavero and P. Russell, unpublished data). Mindful that the C-terminal 52 amino acids of S. cerevisiae Rfa3 are sufficient for function in vivo [17], we designed a new ssb3::KanMX6 construct that completely eliminates the ssb3+ open reading frame (see Materials and Methods). Haploid cells harboring this allele were viable and sensitive to CPT, confirming that Ssb3 is not required for cell viability in fission yeast but is required for CPT resistance (Figure 1A).
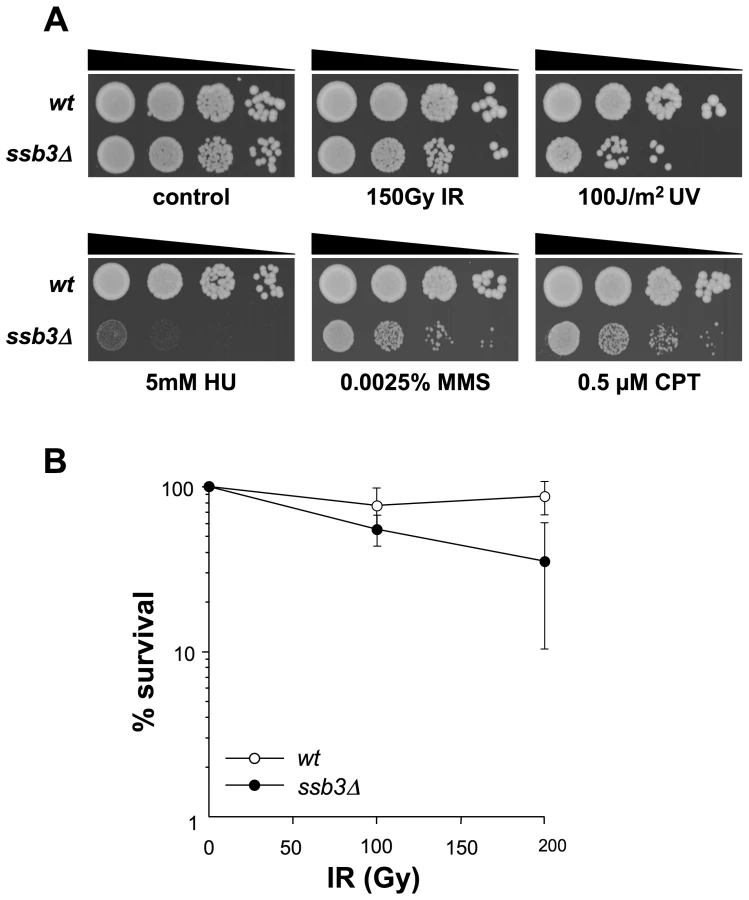
Ssb3 is required for survival of DNA damage in S-phase
We next used serial dilution assays to assess the sensitivity of ssb3Δ cells to a range of genotoxins (Figure 1A). These studies confirmed that ssb3Δ cells are very sensitive to CPT. These cells were also sensitive to a low dose (0.0025%) of methyl methanesulfonate (MMS), which interferes with replication fork progression by alkylating DNA, and to hydroxyrurea (HU), which inhibits DNA replication by poisoning ribonucleotide reductase (Figure 1A). The ssb3Δ cells were also sensitive to UV, which creates cyclobutane dimers and other lesions that impede replication forks. Interestingly, ssb3Δ cells were only modestly sensitive to ionizing radiation (IR), the primary toxic effects of which are DSBs that are repaired in G2 phase (Figure 1A,B). These data show that Ssb3 is required for resistance to a range of genotoxins, particularly those that interfere with DNA replication.
Ssb3 is not required for meiosis
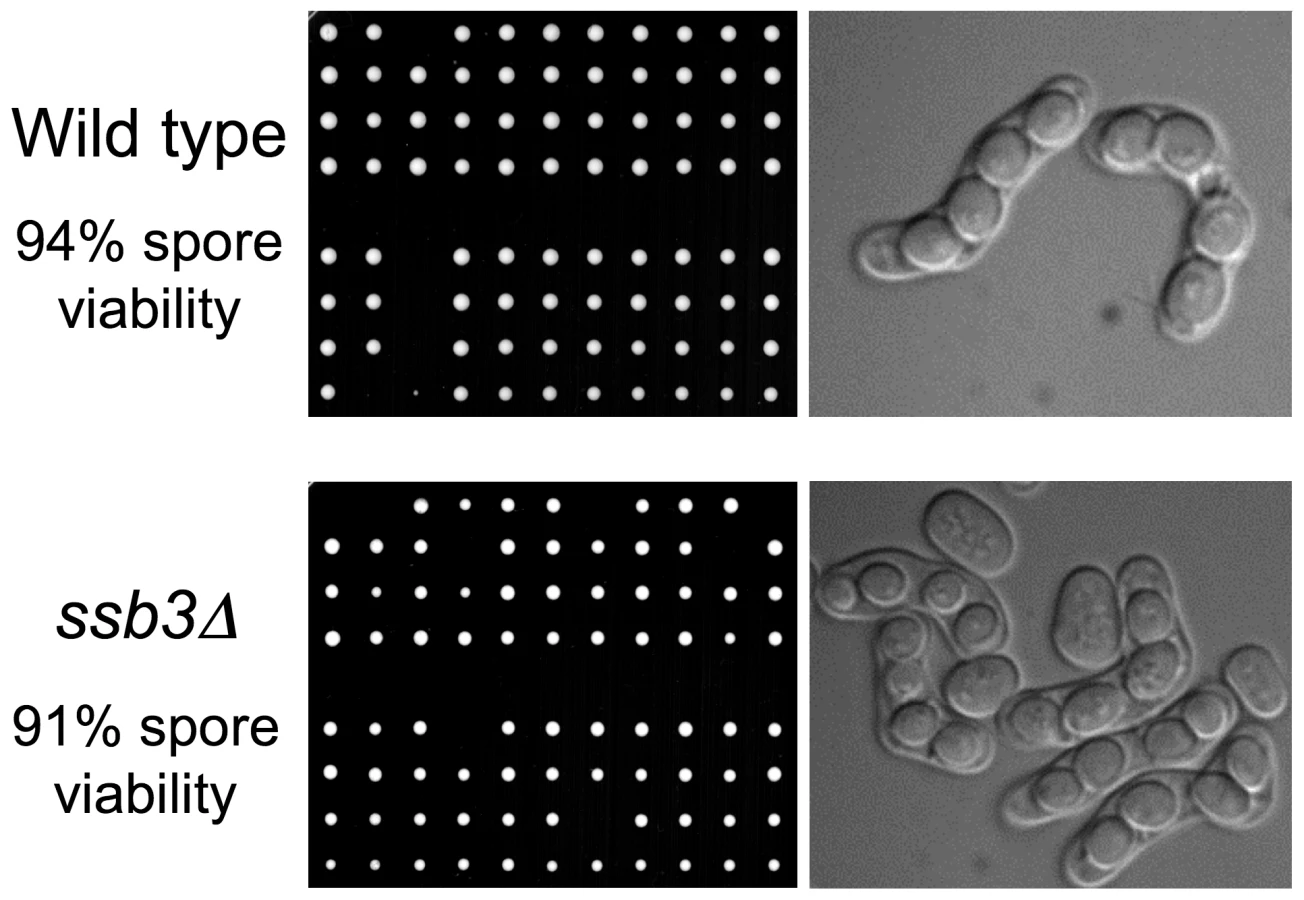
The weak IR sensitivity of ssb3Δ cells suggested that Ssb3 is largely dispensable for homologous recombination-mediated repair of DSBs in mitotic cells. To confirm and extend these findings we analyzed meiosis, in which homologous recombination repair and crossover resolution of programmed meiotic DSBs is required for proper chromosome segregation and spore viability [31]. Tetrad analysis of an ssb3Δ×ssb3Δ cross yielded 91% spore viability, which was only a slight decrease from the 94% spore viability of a ssb3+×ssb3+ cross (Figure 2). Consistent with this high spore viability, microscopic observation showed that the asci from the ssb3Δ×ssb3Δ cross were indistinguishable from wild type. From these results we conclude that Ssb3 is not required for meiotic DSB repair.
DNA damage checkpoint signaling is intact in ssb3Δ cells
Whilst ssb3Δ cells are viable and have only a slightly reduced growth rate, they are elongated relative to wild type (Figure 3A). As this phenotype typically results from activation of a cell cycle checkpoint, we crossed ssb3Δ into strains lacking the Cds1Rad53/Chk2 DNA replication checkpoint kinase or the Chk1 DNA damage checkpoint kinase [32]. Microscopic analysis revealed that the ssb3Δ elongated cell morphology phenotype required Chk1 but not Cds1 (Figure 3A). Various types of DNA damage activate Chk1, whereas Cds1 is primarily activated in response to stalled replication forks. Therefore, these findings indicate that the elongated morphology of ssb3Δ cells is caused by spontaneous DNA damage activating a DNA damage checkpoint that delays the onset of mitosis.

The suppression of the ssb3Δ elongated cell morphology phenotype by chk1Δ suggested that the DNA damage checkpoint is intact in the absence of Ssb3. To further test this hypothesis, we observed the response of ssb3Δ cells to CPT or HU treatment. Both wild type and ssb3Δ cells underwent cell cycle arrest in response to these treatments, whereas cells lacking Rad3Mec1/ATR failed to arrest division (Figure 3B). Consistent with these findings, we observed that Chk1 became hyper-phosphorylated in response to IR or CPT treatment (Figure 3C), which is indicative of an intact DNA damage checkpoint response [33].
These data showed that Ssb3 is not required for the Rad3-Chk1 branch of the DNA damage checkpoint pathway. To confirm this proposition, we carried out genetic epistasis studies with rad3Δ, chk1Δ and cds1Δ mutations. By crossing rad3Δ and ssb3Δ mutants, we discovered a strong genetic interaction between the two mutations, with the double mutant growing much slower than either single mutant (untreated panel in Figure 3D). Although the chk1Δ ssb3Δ double mutant did not have a strong growth defect in the absence of genotoxins, there were obvious synergistic interactions in the presence of IR, UV, HU, MMS and CPT (Figure 3D). The interactions between cds1Δ and ssb3Δ were more complicated: we observed no interactions in response to IR or UV, an additive effect in HU, and suppression in the presence of MMS or CPT (Figure 3D).
Increased spontaneous DNA damage in ssb3Δ cells
The Chk1-dependent cell elongation in ssb3Δ cells suggested that they suffer spontaneous DNA damage. To explore this idea further, we monitored Rad22-YFP foci in ssb3Δ cells. Rad22 is the fission yeast ortholog of Rad52, which is essential for homologous recombination (HR) repair, and many mutants that have genome maintenance defects have increased numbers of Rad22 foci [34]–[36]. We observed a large increase in cells with Rad22-YFP foci in the ssb3Δ strain (∼34%) compared to wild type (∼6%) (Figure 4). We consistently observed that a large fraction of the Rad22-YFP foci in ssb3Δ cells were brighter than in wild type, indicating more extensive recruitment of Rad22. Approximately 33% of the cells with Rad22-YFP foci were septated or attached, which correlates with S-phase in fission yeast, whilst most of the other cells with Rad22-YFP foci appeared to be in G2 phase (Figure 4). These findings suggest that ssb3Δ cells suffer increased rates of DNA damage in S-phase.

Ssb3 colocalizes with Rad22
To address whether Ssb3 relocalizes to sites of DNA damage, we created a strain in which genomic ssb3+ was modified to encode Ssb3-GFP. This strain was not noticeably sensitive to CPT, indicating that Ssb3 function is undisturbed by the C-terminal GFP fusion (S. Cavero and P. Russell, unpublished data). By live cell analysis we observed that Ssb3-GFP was exclusively nuclear, with ∼7.5% of the cells in an asynchronous population having a bright nuclear focus. This pattern is typical of HR repair proteins such as Rad22 [36]. Upon treatment with CPT, there was a large increase in the number of cells with one or more Ssb3-GFP foci (Figure 5A). Again, this is typical of HR proteins, indicating that Ssb3 is a subunit of an RPA complex involved in homologous repair of DSBs.

To confirm whether Ssb3-GFP localizes at sites of ongoing DSB repair, we created a strain that co-expresses Ssb3-GFP and Rad22-RFP from their genomic loci. In the absence of genotoxic stress, ∼50% of the Ssb3-GFP foci co-localize with Rad22-RFP foci (Figure 5B–D). However, following CPT treatment, there was nearly complete overlap of the Ssb3-GFP and Rad22-RFP foci. This result indicates that Ssb3-GFP foci represent sites of actual DSBs and support a direct role for this protein in the repair of broken replication forks. It should be noted that Ssb3-GFP has a higher nuclear fluorescence, with brighter foci than Rad22-RFP, especially in the absence of DNA damaging agents. This fluorescence difference may cause an underestimation of Rad22-RFP foci, especially in the absence of genotoxins, and hence an underestimation of colocalization of Ssb3-GFP and Rad22-RFP foci.
Genetic interactions involving Ssb3 and Rad11
To further investigate the role of Ssb3 in RPA function, we performed a genetic cross to express GFP-tagged Rad11RPA1 in ssb3Δ cells. Although the rad11-GFP and ssb3Δ parents grew well at the standard growth temperature of 30°C, tetrad analysis uncovered a strong synthetic lethal interaction between the two alleles at 30°C (Figure 6A). Microscopic analyses performed revealed that the rad11-GFP ssb3Δ spores formed microcolonies of very elongated cells at 30°C (Figure 6A). A similar effect was observed with HA-tagged rad11 (S. Cavero and P. Russell, unpublished data). These data indicated that the tags slightly impair Rad11 function or destabilize the RPA complex, making it highly dependent on Ssb3. In support of this idea, we found that spore germination at 25°C yielded healthier rad11-GFP ssb3Δ cells that were suitable for localization studies. As expected, the growth of these cells was temperature sensitive (S. Cavero and P. Russell, unpublished data).

We carried out live-cell analysis of Rad11-GFP localization, in the absence or presence of CPT, in wild type (wt) or ssb3Δ cells grown at 25°C. About 5% of wild type cells had Rad11-GFP nuclear foci in the absence of DNA damage (Figure 6B). This number increased ∼3.5-fold when cells were incubated with 30µM CPT for 4 hours, indicating that Rad11-containing RPA complex localizes to DSBs, as expected. Interestingly, the percentage of nuclei with Rad11-GFP foci in the absence of DNA damage was greater in ssb3Δ cells, probably resulting from increased spontaneous DNA damage. Similarly, after CPT treatment, the increase in Rad11-GFP foci formation was also greater in ssb3Δ cells compared to wild type (Fig. 6B). Therefore, the relocalization of RPA complex to DSBs does not depend on Ssb3, and instead it appears to be enhanced.
It should be noted that the frequency of Rad11-GFP foci observed in wild type cells in this experiment was less than seen for Ssb3-GFP in Figure 5. This difference most likely arises from the effect of temperature; indeed, the frequency of Ssb3-GFP foci is reduced at 25°C versus 30°C (S. Cavero and P. Russell, unpublished data). Moreover, co-expression of Rad11-GFP and Ssb3-RFP revealed almost 100% overlap of nuclear foci (Figure 6C).
Genetic epistasis studies with ssb3Δ
Having found that Ssb3 is required for survival of S-phase DNA damage and that it colocalizes with Rad22 and RPA at CPT-induced DNA damage sites, we lastly performed a genetic epistasis analysis of ssb3Δ with genes involved in different pathways of DNA replication or repair. Double mutant strains were created by mating and assessed for growth in dilution series on rich growth medium in the absence or presence of various genotoxins (Figures 7 and 8). The data are summarized in Table 1. In the absence of genotoxins, we detected negative genetic interactions with the Swi1Tof1-Swi3Csm3 replication fork protection complex (FPC), which is required for stable fork pausing and efficient activation of Cds1 [34], [37]; Ctf18, which is a subunit of an alternative Replication Factor C (RFC) complex that is involved in fork stabilization [38]; Rfc3, a subunit of RFC [39], [40]; Rad2, the 5′ flap endonuclease required for Okazaki fragment processing during DNA replication; Rhp55Rad55, a Rhp51Rad51 paralog that forms a heterodimer with Rhp57Rad57 that mediates the formation and/or stabilization of the Rad51-DNA filament required for HR repair [40]; and Brc1Rtt107, a 6-BRCT domain protein that is involved in survival after DNA damage in S-phase and binds phospho-histone H2A (γH2A) at sites of DNA damage [41], [42]. Under the same conditions we detected only weak or no negative genetic interactions with Rad13 and Swi10, which are required for nucleotide excision repair (NER); Rhp51, which is the Rad51 ortholog required for most types of HR repair; or with Mms22, which is required for recovery after DNA damage in S-phase [24], [25]. In the presence of genotoxins, additional negative genetic interactions with ssb3Δ were revealed for Rad13, Swi10 and Rhp51. Mms22 did not show a genetic interaction with Ssb3 in the presence of genotoxins, with the exception of a weak interaction in cells treated with UV (Figures 7 and 8).



Mms22 forms a protein complex with Mms1, and we have found that mms1Δ mms22Δ double mutants behave identically to either single mutant [28]. From these genetic relationships we predicted that ssb3Δ and mms1Δ should have an epistatic genetic interaction in all conditions except for UV, as was seen for ssb3Δ and mms22Δ (Figure 8). Genetic studies confirmed this prediction (Figure 9A).

Mutations that inactivate Mms1 and Mms22 have negative genetic interactions with deletions of many genome maintenance genes. One exception is Mus81, which together with Eme1 forms a Holliday junction resolvase that is required for recovery from collapsed replication forks and resolution of crossovers in meiosis [21]–[23], [43]. Having found that ssb3Δ has an epistatic relationship with mms1Δ and mms22Δ, we explored the genetic interactions involving ssb3Δ and mus81Δ. In the absence of genotoxins, mus81Δ cells grew slowly compared to wild type, but this defect was not enhanced by eliminating Ssb3. Similarly, in the presence of genotoxins, deleting Ssb3 appeared to only slightly enhance the growth defect of mus81Δ cells, indicating a close functional relationship between Ssb3 and Mus81 (Figure 9B).
Our studies identified a negative genetic interaction between ssb3Δ and rhp55Δ, with the double mutant being similar to the rhp51Δ strain (Figure 8A). Rhp55-Rhp57 protein complex is one of two Rhp51 mediators, with the other consisting of Sfr1 and Swi5, which form a protein complex that serves as an alternative mediator for Rhp51 [43], [44]. Inactivating both mediators severely impairs Rhp51 function. In view of these relationships, we tested the genetic interactions between ssb3Δ and sfr1Δ or swi5Δ. In contrast to the interaction between ssb3Δ and rhp55Δ in the absence of genotoxins (Figure 8A), our data indicated there was little or no interaction between ssb3Δ and sfr1Δ or swi5Δ in the absence of genotoxins (Figure 9C). However, there were obvious negative interactions between ssb3Δ and sfr1Δ or swi5Δ in the presence of MMS or CPT, and weaker interaction in double mutant cells treated with UV or HU (Figure 9C). The possible interpretations of these data are discussed below.
Discussion
As a central component of all DNA transactions involving ssDNA, RPA has been the subject of many genetic, biochemical and structural studies. The large majority of the in vivo functional studies have been carried out with S. cerevisiae, in which it is clearly established that all 3 subunits of RPA are essential for cell viability. It was reasonable to assume the same was true in all organisms. We were therefore surprised to find that the small subunit of RPA was apparently not essential in fission yeast. To eliminate doubts, we reengineered an ssb3Δ mutation to completely eliminate the ssb3+ open reading frame, and found that this mutant was also viable. From these studies we conclude that a heterodimeric complex consisting of Rad11Rpa1and Ssb2Rpa2 can carry out the essential DNA replication functions of RPA in fission yeast. As functional studies unfold in other model organisms, it will be interesting to determine whether the essentiality of Rpa3 is the rule or the exception.
Whilst our studies demonstrate that Ssb3 is not required for the essential functions of RPA, they nevertheless show that Ssb3 has important effects on RPA function. One observation supporting this conclusion is the temperature sensitive genetic interaction between Rad11-GFP and ssb3Δ. It is likely that both alleles modestly destabilize or impair the function of RPA complex, to the degree that combining the alleles causes an acute temperature sensitive phenotype. This hypothesis is consistent with data indicating that Rpa3 mediates protein interactions that help to stabilize the RPA heterotrimer [12], [13].
In the absence of exogenous DNA damaging agents, ssb3Δ mutants have a modest growth defect, moderate cell elongation dependent on Chk1, and an elevated number of Rad11Rpa1 and Rad22Rad52 foci. These phenotypes most likely result from defects in DNA replication, leading to DNA structures recognized as DNA lesions by the DNA damage checkpoint and HR repair machinery. However, ssb3Δ and chk1Δ mutations do not have an obvious synergistic growth defect. These data suggest that the DNA lesions leading to a Chk1-dependent mitotic delay in ssb3Δ cells are unlikely to be DSBs. In support of this conclusion, we did not detect a obvious genetic interaction between ssb3Δ and rhp51Δ when mutants were tested in the absence of genotoxins. Thus, it is likely that ssb3Δ cells accumulate gapped ssDNA structures that activate the DNA damage checkpoint and are substrates for Rad22.
We found that cds1Δ partially suppresses the MMS and CPT sensitivity of ssb3Δ cells (Figure 3D). This genetic interaction was unexpected. The toxicity of MMS and CPT is thought to derive primarily from replication fork collapse; therefore, these data suggest that activation of the replication checkpoint is detrimental to restoration of collapsed replication forks in ssb3Δ cells. It will be interesting to discover which substrates of Cds1 kinase mediate this effect.
Whilst ssb3Δ cells are sensitive to CPT, MMS and UV, they display only weak sensitivity to IR. The toxic effects of CPT, MMS and UV are thought to arise primarily from creating DNA lesions that interfere with DNA replication, whereas IR directly causes DSBs in all phases of the cell cycle. These data suggest that Ssb3 is not critical for typical HR-mediated DSB repair in mitotic cells, which occurs through a synthesis-dependent strand-annealing (SDSA) pathway requiring Rad22Rad52, Rhp51Rad51, and other core HR proteins. Furthermore, ssb3Δ cells exhibit only a very minor defect in spore viability. Since formation of viable spores depends on carrying out HR repair of programmed meiotic DSBs through a double-strand break repair (DSBR) pathway requiring resolution of Holliday junctions by Mus81-Eme1 resolvase [21], our findings show that both pathways of HR repair are largely functional in the absence of Ssb3. However, these data do not exclude the possibility that deletion of ssb3+ might have modest effects in meiotic recombination.
The HR-mediated repair of CPT-induced broken replication forks also depends on Rad22, Rhp51 and Mus81, and our data indicate that Ssb3 plays a relatively important role in this pathway. It is unclear why Ssb3 should appear to be more important for repair of CPT-induced damage than IR-induced DSBs. Ssb3 might have an important role in stabilizing stalled replication forks, such as those that are proposed to form as the result of positive supercoils forming ahead of the fork in cells treated with CPT [19]. It is noteworthy that previous genetic studies have identified other genome maintenance factors that are not required for repair of IR-induced DSBs but are critical for survival of CPT treatment. The most relevant factors may be Mus81-Eme1 and Mms1-Mms22 protein complexes, which are critical for survival of CPT treatment but are not required for repair of IR-induced DSBs [21]-[23], [25], [28]. In this respect, it is likely to be particularly significant that ssb3Δ displays little or no synergistic genetic interactions with mus81Δ, mms1Δ and mms22Δ mutations in the absence or presence of genotoxins, indicating that there are likely to be close functional connections between the Ssb3-dependent functions of RPA and the activities of Mus81-Eme1 Holliday junction resolvase and Mms1-Mms22 protein complex. This possibility is entirely consistent with studies showing that mms1Δ and mms22Δ have negative genetic interactions with mutations of many genome maintenance genes, but not with mus81Δ [24], [25], [28]. However, it is important to note that unlike Mus81, Ssb3 is not required for resolution of Holliday junctions in meiosis, and thus it is unlikely that Ssb3 has an integral role in the Mus81-dependent resolution of Holliday junctions that form upon restoration of collapsed replication forks [21], [44].
Regardless of the genotoxins assayed in our studies, the phenotypes of ssb3Δ cells do not match that of an rhp51Δ mutant that is severely defective in HR-mediated DSB repair. However, the phenotypes of ssb3Δ and rhp55Δ mutants are similar, and there is a strong genetic interaction when ssb3Δ and rhp55Δ are combined. In fact, the ssb3Δ rhp55Δ double mutant is approximately equivalent to rhp51Δ. These data suggest that there is a nearly complete breakdown of HR in the ssb3Δ rhp55Δ double mutant. Rhp55 is a Rad51 paralog that forms a heterodimer with Rhp57. Studies of the analogous Rad55-Rad57 complex in budding yeast have shown that it can function as a mediator in the strand-exchange reaction necessary for Rad51 to nucleate on ssDNA in the presence of RPA [45]. One possible interpretation of these findings is that both ssb3Δ and rhp55Δ mutations cause defects in Rad51 nucleation on ssDNA, resulting in a synergistic interaction of the mutations. RPA foci are actually enhanced in ssb3Δ cells, suggesting that a key role of Ssb3 may be promoting the disassembly of RPA from ssDNA.
Interestingly, in the absence of exogenous genotoxins, ssb3Δ does not have obvious genetic interactions with mutations deleting genes encoding Sfr1 or Swi5, which form an alternative mediator complex for Rhp51 [46], [47]. These data are consistent with the possibility that Ssb3 might act with Sfr1-Swi5 complex. However, when tested in the presence of genotoxins, there are clear negative genetic interactions involving ssb3Δ and sfr1Δ or swi5Δ. The same is true for ssb3Δ and rhp51Δ mutations. Thus, Ssb3 might act with the Sfr1-Swi5 complex, but it also has functions that do not involve Rhp51 and its mediators.
Materials and Methods
Strains and genetic methods
The strains used in this study are listed in Table S1. Standard procedures and media for S. pombe genetic and biochemical analyses were used as previously described [48]. Cells were cultured in YES (yeast extract, glucose and supplements) or EMM (defined minimal medium with supplements) as described [48]. The complete deletion of the ssb3 open reading frame was made using pFA6a-KanMX6 as template and the primers “Ssb3 KanMX6 forward” (5′-TCG TGT CAA CAA GTA GTT AAC TAC CTG GTC TGA TAC ATA CTT CAC TTC CAC CAC TTT ATA AAC AAC GCG TAT AAA ATA ATC GGA TCC CCG GGT TAA TTA A-3′) and “Ssb3 KanMX6 reverse” (5′-CGT TTA TTC TTC CAT GTT TAT TTG TAC TGT GCA TGA GAA ATG AAA GAG AAA TCT GTG TTG TAT GAT CCA TAA AAT GTT TCG AAT TCG AGC TCG TTT AAA C-3′). The PCR product was transformed into PR110 (h+ leu1-32 ura4-D18) cells by electroporation, G418-resistant colonies were obtained, and PCR and DNA sequencing verified ssb3 disruption.
Microscopy
Cells were photographed using a Nikon Eclipse E800 microscope equipped with a Photometrics Quantix CCD camera and IPlab Spectrum software. All fusion proteins were expressed at their own genomic locus. Rad22-YFP and Ssb3-GFP/Rad22-RFP-expressing strains were cultured in EMM, while Ssb3-GFP and Rad11-GFP-expressing strains were grown in YES until mid-log phase for foci quantification assays. Experiments with Rad11-GFP expressing strains were carried out at 25°C because of the thermosensitivity of the rad11-GFP ssb3Δ strain. In the case of CPT treatment, 30µM CPT in DMSO (or DMSO alone as a control, 0.3% final concentration) was added to mid-log phase cultures for 4 hrs at 30°C, washed out and cells were resuspended in YES for foci quantification. In all cases, at least 800 nuclei were scored in three independent experiments. All microscopy was conducted with live cells. Cell length, nuclei number and position, and the presence of a division plate were used to assess cell cycle position.
Survival assays
In chronic exposures to drugs, mid-log phase cultures were resuspended to 0.5 OD600 and serially diluted tenfold. Dilutions were spotted onto YES agar plates containing the indicated amounts of CPT, MMS or HU. Note that the effective concentration of these drugs, particularly MMS, can vary depending on the age and source of the stock solution, which accounts for the different concentrations used in some of the survival assays. For exposure to IR, cells were irradiated using a 137Cs source with the indicated dose and then serially diluted onto triplicate YES plates. In the case of UV treatment, cells were serially diluted onto YES plates and irradiated using a Stratagene Stratalinker UV source. Cell survival was determined after 3–4 days at 30°C unless otherwise indicated.
Immunoblotting
Whole cell extracts were prepared from exponentially growing cells disrupted in lysis buffer (50mM Tris-HCl pH 8, 150mM NaCl, 2.5mM EDTA, 10% Glycerol, 0.2% NP-40, 50mM NaF, 5mM PMSF, and Complete Protease Inhibitor tablets (Roche)) with a glass bead beater and resolved in 8% SDS-PAGE. Proteins were transferred to nitrocellulose membranes, blocked with 5% dry milk, 0.05% Tween-20 in TBS (150mM NaCl, 50mM Tris-HCl pH 7.6) and probed with HA (Roche) antibody.
Supporting Information
Zdroje
1. BinzSK
SheehanAM
WoldMS
2004 Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair 3 1015 1024
2. RichardDJ
BoldersonE
KhannaKK
2009 Multiple human single-stranded DNA binding proteins function in genome maintenance: structural, biochemical and functional analysis. Crit Rev Biochem Mol Biol 1 19
3. WoldMS
1997 Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem 66 61 92
4. IftodeC
DanielyY
BorowiecJA
1999 Replication protein A (RPA): the eukaryotic SSB. Crit Rev Biochem Mol Biol 34 141 180
5. WobbeCR
WeissbachL
BorowiecJA
DeanFB
MurakamiY
1987 Replication of simian virus 40 origin-containing DNA in vitro with purified proteins. Proc Natl Acad Sci U S A 84 1834 1838
6. FairmanMP
StillmanB
1988 Cellular factors required for multiple stages of SV40 DNA replication in vitro. EMBO J 7 1211 1218
7. WoldMS
KellyT
1988 Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc Natl Acad Sci U S A 85 2523 2527
8. HeZ
HenricksenLA
WoldMS
InglesCJ
1995 RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature 374 566 569
9. LinYL
ShivjiMK
ChenC
KolodnerR
WoodRD
1998 The evolutionarily conserved zinc finger motif in the largest subunit of human replication protein A is required for DNA replication and mismatch repair but not for nucleotide excision repair. J Biol Chem 273 1453 1461
10. Bastin-ShanowerSA
BrillSJ
2001 Functional analysis of the four DNA binding domains of replication protein A. The role of RPA2 in ssDNA binding. J Biol Chem 276 36446 36453
11. BochkarevA
BochkarevaE
2004 From RPA to BRCA2: lessons from single-stranded DNA binding by the OB-fold. Curr Opin Struct Biol 14 36 42
12. HenricksenLA
UmbrichtCB
WoldMS
1994 Recombinant replication protein A: expression, complex formation, and functional characterization. J Biol Chem 269 11121 11132
13. StiggerE
DeanFB
HurwitzJ
LeeSH
1994 Reconstitution of functional human single-stranded DNA-binding protein from individual subunits expressed by recombinant baculoviruses. Proc Natl Acad Sci U S A 91 579 583
14. BrillSJ
StillmanB
1991 Replication factor-A from Saccharomyces cerevisiae is encoded by three essential genes coordinately expressed at S phase. Genes Dev 5 1589 1600
15. ZouL
ElledgeSJ
2003 Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300 1542 1548
16. BallHL
EhrhardtMR
MordesDA
GlickGG
ChazinWJ
2007 Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol Cell Biol 27 3367 3377
17. ManiarHS
WilsonR
BrillSJ
1997 Roles of replication protein-A subunits 2 and 3 in DNA replication fork movement in Saccharomyces cerevisiae. Genetics 145 891 902
18. PommierY
2006 Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 6 789 802
19. KosterDA
PalleK
BotES
BjornstiMA
DekkerNH
2007 Antitumour drugs impede DNA uncoiling by topoisomerase I. Nature 448 213 217
20. RoseaulinL
YamadaY
TsutsuiY
RussellP
IwasakiH
2008 Mus81 is essential for sister chromatid recombination at broken replication forks. Embo J 27 1378 1387
21. BoddyMN
GaillardPH
McDonaldWH
ShanahanP
YatesJR3rd
2001 Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107 537 548
22. BoddyMN
Lopez-GironaA
ShanahanP
InterthalH
HeyerWD
2000 Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol Cell Biol 20 8758 8766
23. DoeCL
AhnJS
DixonJ
WhitbyMC
2002 Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J Biol Chem 277 32753 32759
24. YokoyamaM
InoueH
IshiiC
MurakamiY
2007 The novel gene mus7+ is involved in the repair of replication-associated DNA damage in fission yeast. DNA Repair 6 770 780
25. DoveyCL
RussellP
2007 Mms22 preserves genomic integrity during DNA replication in Schizosaccharomyces pombe. Genetics 177 47 61
26. DuroE
VaisicaJA
BrownGW
RouseJ
2008 Budding yeast Mms22 and Mms1 regulate homologous recombination induced by replisome blockage. DNA Repair 7 811 818
27. ZaidiIW
RabutG
PovedaA
ScheelH
MalmstromJ
2008 Rtt101 and Mms1 in budding yeast form a CUL4(DDB1)-like ubiquitin ligase that promotes replication through damaged DNA. EMBO Rep 9 1034 1040
28. DoveyCL
AslanianA
SofuevaS
YatesJR3rd
RussellP
2009 Mms1-Mms22 complex protects genome integrity in Schizosaccharomyces pombe. DNA Repair 8 1390 1399
29. DeshpandeGP
HaylesJ
HoeKL
KimDU
ParkHO
2009 Screening a genome-wide S. pombe deletion library identifies novel genes and pathways involved in genome stability maintenance. DNA Repair 8 672 679
30. ParkerAE
ClyneRK
CarrAM
KellyTJ
1997 The Schizosaccharomyces pombe rad11+ gene encodes the large subunit of replication protein A. Mol Cell Biol 17 2381 2390
31. SmithGR
BoddyMN
ShanahanP
RussellP
2003 Fission yeast Mus81.Eme1 Holliday junction resolvase is required for meiotic crossing over but not for gene conversion. Genetics 165 2289 2293
32. RhindN
RussellP
2000 Chk1 and Cds1: linchpins of the DNA damage and replication checkpoint pathways. J Cell Sci 113 3889 3896
33. WalworthNC
BernardsR
1996 rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science 271 353 356
34. NoguchiE
NoguchiC
DuLL
RussellP
2003 Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol Cell Biol 23 7861 7874
35. MeisterP
PoidevinM
FrancesconiS
TratnerI
ZarzovP
2003 Nuclear factories for signalling and repairing DNA double strand breaks in living fission yeast. Nucleic Acids Res 31 5064 5073
36. DuLL
NakamuraTM
MoserBA
RussellP
2003 Retention but not recruitment of Crb2 at double-strand breaks requires Rad1 and Rad3 complexes. Mol Cell Biol 23 6150 6158
37. NoguchiE
NoguchiC
McDonaldWH
YatesJR3rd
RussellP
2004 Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol Cell Biol 24 8342 8355
38. AnsbachAB
NoguchiC
KlansekIW
HeidlebaughM
NakamuraTM
2008 RFCCtf18 and the Swi1-Swi3 complex function in separate and redundant pathways required for the stabilization of replication forks to facilitate sister chromatid cohesion in Schizosaccharomyces pombe. Mol Biol Cell 19 595 607
39. ShimadaM
OkuzakiD
TanakaS
TouganT
TamaiKK
1999 Replication factor C3 of Schizosaccharomyces pombe, a small subunit of replication factor C complex, plays a role in both replication and damage checkpoints. Mol Biol Cell 10 3991 4003
40. KhasanovFK
SavchenkoGV
BashkirovaEV
KorolevVG
HeyerWD
1999 A new recombinational DNA repair gene from Schizosaccharomyces pombe with homology to Escherichia coli RecA. Genetics 152 1557 1572
41. SheedyDM
DimitrovaD
RankinJK
BassKL
LeeKM
2005 Brc1-mediated DNA repair and damage tolerance. Genetics 171 457 468
42. WilliamsJS
WilliamsRS
DoveyCL
GuentherG
TainerJA
2010 gammaH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J 29 1136 1148
43. GaillardPH
NoguchiE
ShanahanP
RussellP
2003 The endogenous Mus81-Eme1 complex resolves Holliday junctions by a nick and counternick mechanism. Mol Cell 12 747 759
44. CromieGA
HyppaRW
TaylorAF
ZakharyevichK
HunterN
2006 Single Holliday junctions are intermediates of meiotic recombination. Cell 127 1167 1178
45. SungP
1997 Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev 11 1111 1121
46. AkamatsuY
TsutsuiY
MorishitaT
SiddiqueMS
KurokawaY
2007 Fission yeast Swi5/Sfr1 and Rhp55/Rhp57 differentially regulate Rhp51-dependent recombination outcomes. EMBO J 26 1352 1362
47. KurokawaY
MurayamaY
Haruta-TakahashiN
UrabeI
IwasakiH
2008 Reconstitution of DNA strand exchange mediated by Rhp51 recombinase and two mediators. PLoS Biol 6 e88 doi:10.1371/journal.pbio.0060088
48. ForsburgSL
RhindN
2006 Basic methods for fission yeast. Yeast 23 173 183
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 9
Nejčtenější v tomto čísle
- Synthesizing and Salvaging NAD: Lessons Learned from
- Optimal Strategy for Competence Differentiation in Bacteria
- Long- and Short-Term Selective Forces on Malaria Parasite Genomes
- Identifying Signatures of Natural Selection in Tibetan and Andean Populations Using Dense Genome Scan Data