
SNPs Associated with Cerebrospinal Fluid Phospho-Tau Levels Influence Rate of Decline in Alzheimer's Disease
Alzheimer's Disease (AD) is a complex and multifactorial disease. While large genome-wide association studies have had some success in identifying novel genetic risk factors for AD, case-control studies are less likely to uncover genetic factors that influence progression of disease. An alternative approach to identifying genetic risk for AD is the use of quantitative traits or endophenotypes. The use of endophenotypes has proven to be an effective strategy, implicating genetic risk factors in several diseases, including anemia, osteoporosis and heart disease. In this study we identify a genetic factor associated with the rate of decline in AD patients and present a methodology for identification of other such factors. We have used an established biomarker for AD, cerebrospinal fluid (CSF) tau phosphorylated at threonine 181 (ptau181) levels as an endophenotype for AD, identifying a SNP, rs1868402, in the gene encoding the regulatory sub-unit of protein phosphatase B, associated with CSF ptau181 levels in two independent CSF series . We show no association of rs1868402 with risk for AD or age at onset, but detected a very significant association with rate of progression of disease that is consistent in two independent series . Our analyses suggest that genetic variants associated with CSF ptau181 levels may have a greater impact on rate of progression, while genetic variants such as APOE4, that are associated with CSF Aβ42 levels influence risk and onset but not the rate of progression. Our results also suggest that drugs that inhibit or decrease tau phosphorylation may slow cognitive decline in individuals with very mild dementia or delay the appearance of memory problems in elderly individuals with low CSF Aβ42 levels. Finally, we believe genome-wide association studies of CSF tau/ptau181 levels should identify novel genetic variants which will likely influence rate of progression of AD.
Published in the journal:
. PLoS Genet 6(9): e32767. doi:10.1371/journal.pgen.1001101
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001101
Summary
Alzheimer's Disease (AD) is a complex and multifactorial disease. While large genome-wide association studies have had some success in identifying novel genetic risk factors for AD, case-control studies are less likely to uncover genetic factors that influence progression of disease. An alternative approach to identifying genetic risk for AD is the use of quantitative traits or endophenotypes. The use of endophenotypes has proven to be an effective strategy, implicating genetic risk factors in several diseases, including anemia, osteoporosis and heart disease. In this study we identify a genetic factor associated with the rate of decline in AD patients and present a methodology for identification of other such factors. We have used an established biomarker for AD, cerebrospinal fluid (CSF) tau phosphorylated at threonine 181 (ptau181) levels as an endophenotype for AD, identifying a SNP, rs1868402, in the gene encoding the regulatory sub-unit of protein phosphatase B, associated with CSF ptau181 levels in two independent CSF series . We show no association of rs1868402 with risk for AD or age at onset, but detected a very significant association with rate of progression of disease that is consistent in two independent series . Our analyses suggest that genetic variants associated with CSF ptau181 levels may have a greater impact on rate of progression, while genetic variants such as APOE4, that are associated with CSF Aβ42 levels influence risk and onset but not the rate of progression. Our results also suggest that drugs that inhibit or decrease tau phosphorylation may slow cognitive decline in individuals with very mild dementia or delay the appearance of memory problems in elderly individuals with low CSF Aβ42 levels. Finally, we believe genome-wide association studies of CSF tau/ptau181 levels should identify novel genetic variants which will likely influence rate of progression of AD.
Introduction
Genetic studies have helped to further our understanding of the pathogenic mechanism of several diseases, including AD. To date only the ε4 allele of apolipoprotein E (APOE4), present in 50% of late onset AD (LOAD) cases, has been convincingly demonstrated to influence risk for LOAD. The traditional method for searching for genetic risk factors involves the comparison of genes in AD cases and non-demented elderly controls. AD is a complex and multifactorial disease, and as a result very large datasets have been necessary to identify these genetic risk factors [1]–[2]. An alternative to the standard case-control study design is to use quantitative traits or endophenotypes. Quantitative traits have been used to successfully identify new genetic factors implicated in anemia [3]–[6], osteoporosis [7] and heart disease [8]–[11]. The advantages of quantitative traits are that they provide higher power than regular case-control analyses, a biological model of disease and the possible effects of the associated genetic variation and may decrease the clinical heterogeneity of the samples. This is likely to be true for Alzheimer's Disease (AD) because up to 30% of individuals in screened elderly non-demented control samples show evidence of AD pathology at autopsy [12], and a similar number have biomarker profiles consistent with preclinical AD [13]–[15], thus reducing the power of a case-control design.
Both Aβ and tau protein play an important role in AD, are detectable in cerebrospinal fluid (CSF) in all individuals, and have been used as biomarkers for diagnosis [16]–[18]. Patients with AD show lower CSF Aβ42 levels [19] that inversely correlate with the presence of fibrillar Aβ in the brain (as measured by Pittsburgh Compound B (PET-PIB) retention) in demented individuals [14] and plaque counts in brain samples [20]. Several studies suggest that PET-PIB retention and CSF Aβ42 levels could help to identify individuals with AD pathology before the onset of clinically detectable disease (preclinical AD) [14], [21]. The CSF levels of total tau and tau phosphorylated at threonine 181 (ptau181) are increased in AD [12], [14]. Elevated CSF tau levels are associated with neuronal damage and are also observed in stroke [22] and traumatic brain injury immediately after injury [23], however increases in CSF ptau181 levels appear to be specific to AD [24]–[26]. In several previous studies we have successfully applied this endophenotype-based approach, leveraging the information from both CSF Aβ and tau to identify genetic polymorphisms implicated in AD risk [27]–[29].
Tau activity depends on its state of phosphorylation [30], which is regulated by several kinases, phosphatases and other tau-related proteins [31]. Hyperphosphorylation of tau destabilizes the microtubule network, leading to impaired axonal transport and ultimately to neurofibrillary tangle formation and neuronal death (For review see [32]). In the present study we have evaluated 355 single nucleotide polymorphisms (SNPs) in 34 genes involved in tau modification or metabolism for association with CSF levels of ptau181, then determined the effects of those variants on AD risk, onset and rate of progression.
Results
Association with CSF ptau181 levels: Initial screening
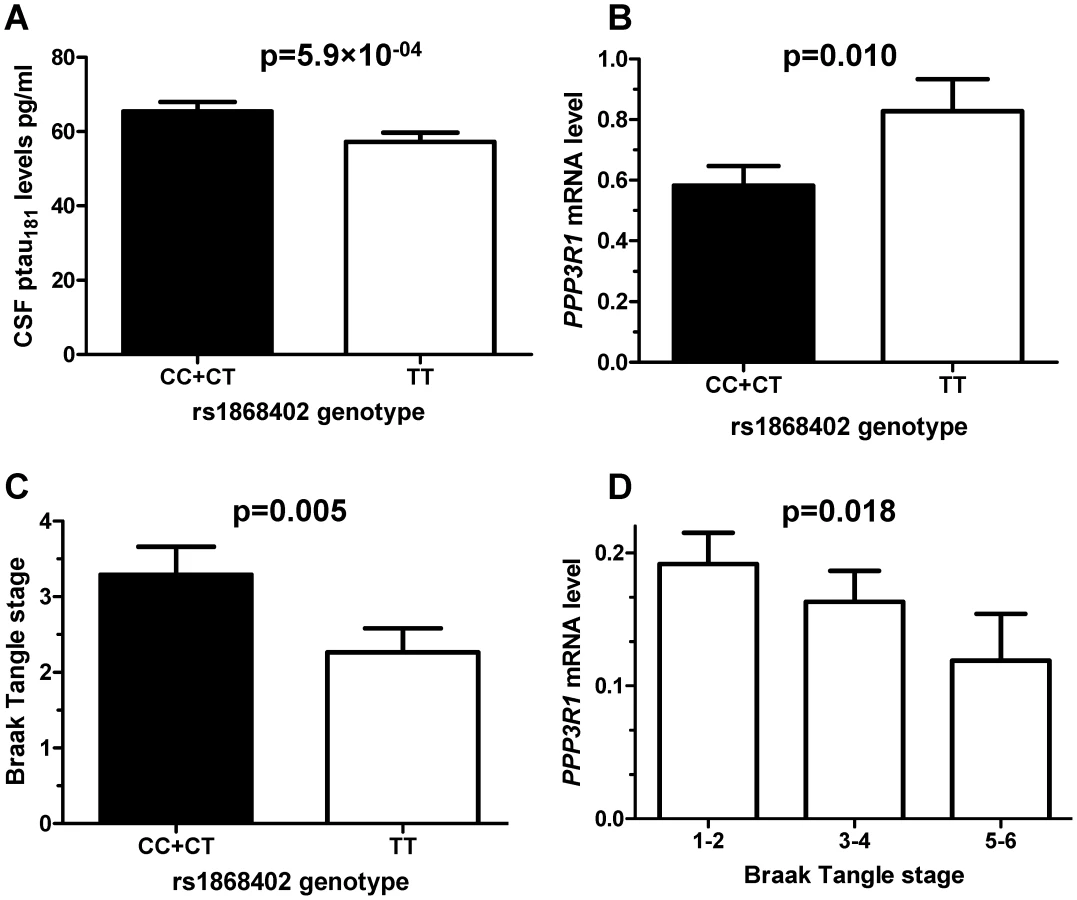
Based on bibliographic data we selected 384 SNPs localized in 34 genes related to tau metabolism (tau kinases, phosphatases, tau O-glcNAcylation or tau degradation (Table S1). 355 SNPs passed quality control (Hardy-Weinberg equilibrium and call rate >95%). Association of SNPs with CSF ptau181 levels was evaluated by ANCOVA in 353 CSF samples from the Washington University Alzheimer Disease Research Center (WU-ADRC-CSF) (Tables 1 and 2). Clinical Dementia Rating (CDR), age and APOE ε4 genotype were included as covariates in the analyses. Eighteen SNPs, located in 7 different genes showed significant association with CSF ptau181 levels in the WU-ADRC-CSF series after multiple test correction (Table 3). The SNP with the most significant p-value, rs1868402, is located in intron 5 of the regulatory subunit of the protein phosphatase B gene, also known as calcineurin B (PPP3R1; MIM#: 601302). The association of rs1868402 with CSF ptau181 levels showed the best fit in the dominant model, with minor alleles carriers showing significantly higher CSF ptau181 levels (P = 5.90×10−04, Figure 1A and Figure S1). All subsequent analyses for rs1868402 used the dominant model. Six other SNPs in PPP3R1, which are in high linkage disequilibrium (LD) with rs1868402 (Figure S2, and Table S2), also showed association with CSF ptau181. Based on the linkage disequilibrium (LD) in PPP3R1, we selected rs1868402 and rs6546366 for replication. The remaining eleven SNPs that were significant after multiple test correction were also selected for replication (Table 3).



Association with CSF ptau181 levels: Replication in an independent CSF series and combined analyses
In a replication series of 493 independent CSF samples from ADNI (ADNI-CSF) and University of Washington (UW) (Tables 1 and 2) only the SNP located in calcineurin B, rs1868402, replicated and passed the FDR filter (P = 0.005 ptau181, Table 3, Figure S1). In this series rs1868402 also showed the best fit in the dominant model and minor allele carriers have higher CSF ptau181 levels. In the replication CSF series, rs6546366 (PPP3R1), showed no association with CSF ptau181 levels (Table 3). The lack of association of rs6546366 with CSF ptau181 levels is probably due to a lower level of LD with rs1868402 in the replication series (r2 = 0.65) compared with the LD between these two SNPs in the WU-ADRC-CSF series (r2 = 0.80). This result indicates that rs1868402, or another unknown variant in LD with rs1868402, is the variant that drives the association with CSF ptau181 levels.
We also performed a combined analysis by combining the residuals for CSF ptau181 after correcting for the covariates (see Materials and Methods). In this analysis we did not included site or platform as a covariate because there were no significant differences between datasets and/or platform for the ptau181 residuals. Inclusion of site/platform as a covariate did not significantly change the p-value. In the combined data, rs1868402 showed the most significant association with CSF ptau181 levels , with a p-value that was significant after Bonferroni correction for the entire study . Minor allele carriers have a 2.4 fold increased risk of being in the highest quartile of the CSF ptau181 distribution compared to the lowest quartile (odds ratio = 2.37, 95% confidence interval 1.59–3.54). None of the other SNPs were significant after Bonferroni correction (Table 3).
Association with CSF ptau181 levels: Context-dependent effects
It has been demonstrated in our longitudinal data and that of others that the increase in CSF tau and ptau181 levels seen in mild AD is preceded by decreases in CSF Aβ42 levels [14], [21]. This likely reflects deposition of Aβ in the brain [14]. Individuals with CSF Aβ42 levels less than 500 pg/ml in the WU-ADRC-CSF, and less than 192 pg/ml in the ADNI-CSF series, have evidence of Aβ deposition in the brain, as detected by PET-PIB [14], [21]. We used these CSF Aβ42 thresholds to stratify the WU-ADRC-CSF and ADNI-CSF samples into individuals with low or high CSF Aβ42 levels (with and without likely Aβ deposition in the brain). The difference in CSF Aβ42 threshold levels between the WU-ADRC-CSF and ADNI-CSF series is due to different antibodies and procedures used to measure the CSF levels (see Materials and Methods). The data necessary to examine the correlation between the CSF Aβ42 levels and PET-PIB signal is not available in the UW CSF series and therefore this dataset could not be included in this analysis. In these analyses we calculated the p-value and the Odds ratios for rs1868402 with CSF ptau181 levels by comparing the frequency of this SNP in the lowest versus the highest quartile of the CSF ptau181 levels after correcting for the covariates. When we stratified the WU-ADRC-CSF and ADNI-CSF series by CSF Aβ42 levels, we observed very significant association for rs1868402 with CSF ptau181 in the low Aβ42 stratum (, Odds Ratio 3.48; 95% confidence interval 1.8–6.7) and a nominally significant association in the high Aβ42 stratum (combined analysis P = 0.023; Odds Ratio 2.54; 95% confidence interval 1.0–6.75, Table 4).. Both the high and low Aβ42 level strata have sufficient power to detect the association between rs1868402 with CSF ptau181 levels (high Aβ42 0.985; low Aβ42 = 0.975; α = 0.05). The nominally significant p-value in the high Aβ stratum may indicate a moderate effect on CSF ptau181 levels in healthy individuals, but it is clear that when AD pathology is present the effect of this SNP is more marked.

Rs1868402 explains 4.62% of the variability in CSF ptau181 levels in individuals with low CSF Aβ42 levels in the WU-ADRC-CSF+ADNI-CSF samples, which is similar to the variability explained by other SNPs and endophenotypes [3]–[11]. It is important to note that in the low CSF Aβ42 group there are individuals diagnosed with DAT (CDR>0, n = 183, 58%) and non-demented individuals (CDR = 0, n = 134, 42%) with possible Aβ deposition in the brain and brain atrophy (presymptomatic AD) [12]. In the high Aβ42 stratum 80% of the samples (n = 242) had a CDR = 0.
Implication of SNPs associated with CSF ptau181 levels in AD: Association with rate of progression of AD but not risk for AD or age at onset
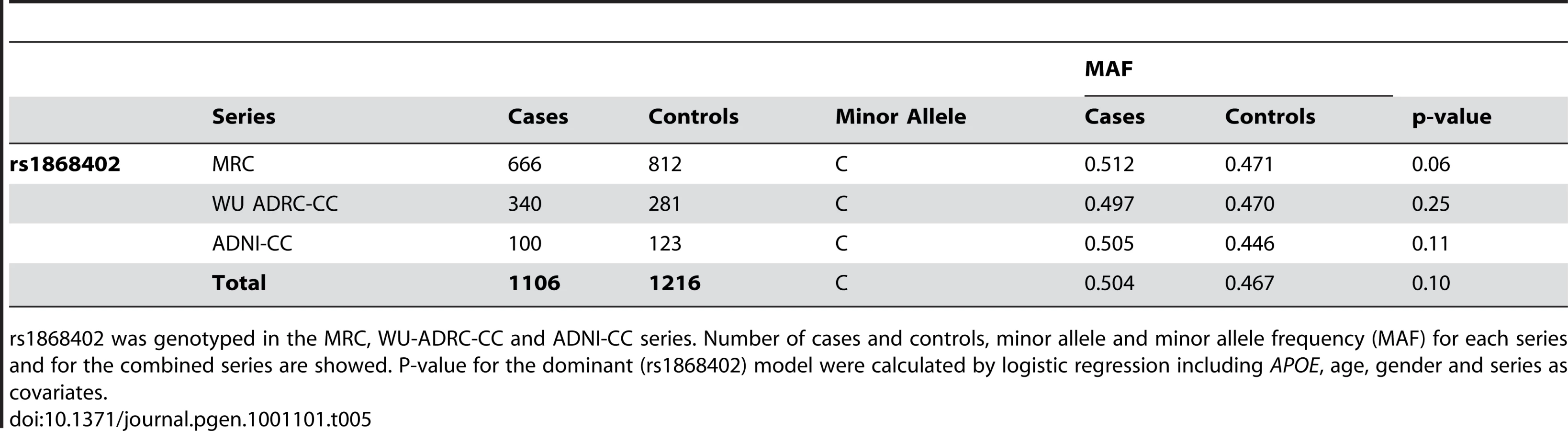
The premise of this endophenotype-based approach is that a SNP, such as rs1868402 that shows strong, replicable association with an important AD biomarker should also modulate risk, onset and/or progression of AD. We tested whether rs1868402 influences risk for AD, age at onset and disease progression. We found no association between rs1868402 and risk for AD (P = 0.10, Table 5) or age at onset (P = 0.19 Figure 2) in 1106 cases and 1216 controls of European descent.

To examine disease progression we used two longitudinal datasets: 109 subjects from WU-ADRC-CSF (399 observations) and 150 subjects from ADNI-CSF (620 observations). Association with rate of progression was evaluated by comparing the change in sum of boxes of the CDR (SB-CDR) per year (slope) by genotype including age at the first visit, gender, APOE genotype and initial CDR as covariates. CSF ptau181 and Aβ42 levels were also included in the model to correct for the potential association between these phenotypes with progression [33]. Because the association of rs1868402 with CSF ptau181 was mainly in individuals with low CSF Aβ42 levels, association with progression was analyzed in individuals with low CSF Aβ42 levels (less than 500 pg/ml in the WU-ADRC-CSF, and less than 192 pg/ml in the ADNI-CSF series), and an initial CDR of 0 or 0.5. Individuals with CDR 0 and 0.5 were selected to maximize the amount of information on progression per individual and to avoid possible ceiling effects from individuals who began the study with advanced levels of dementia.
Carriers of the rs1868402 allele associated with higher CSF ptau181 levels showed an increase of 0.58 SB-CDR per year, which is six-fold faster than the rate seen in individuals homozygous for the allele associated with low CSF ptau181 levels (P = 0.0026; Table 6, Figure 3A and Figure S3), and almost two times faster than the average change for the entire series (SB-CDR per year for the entire series 0.31). The association of rs1868402 with progression replicated in the ADNI-CSF series (P = 0.014) with a Pcombined = 1.96×10−05. In addition, we also used the ADNI samples with no CSF data (ADNI-CC, Table 6) to replicate the association with rate of progression in an independent sample. In this dataset rs1868402 also showed a significant association with rate of progression (P = 0.018, Table 6) and in the same direction as in the previous analyses.


We tested whether rs1868402 interacts with rs3785883, a SNP located in MAPT, which is also associated with CSF ptau181 levels [27]. Rs3785883 also showed a p-value for association with CSF ptau181 levels of 0.008 in the combined series of this study.We found significant epistasis between these SNPs. Individuals carrying alleles associated with higher CSF ptau181 for rs1868402 and rs3785883 showed an increase of 1.02 SB-CDR per year on average, whereas those carrying the alleles associated with lower CSF ptau181 at each SNP showed essentially no change in the SB-CDR per year (; Figure 3B; Table 6). The interaction between rs1868402 and rs3785883 also replicated in the ADNI-CSF and ADNI-CC series (Table 6).
Gene expression
Since rs1868402 is located in a region that encodes a tau phosphatase, we tested next whether the SNP is associated with PPP3R1 mRNA levels and tau pathology in brain. We extracted total RNA from the parietal lobe of 82 AD cases and 39 non-demented elderly individuals. The allele of rs1868402, associated with higher CSF ptau181 levels, showed significantly lower PPP3R1 mRNA levels (P = 0.010; Figure 1B), and higher tangle pathology as measured by Braak stage (P = 0.005; Figure 1C) in brain samples with Aβ pathology, but not in neuropathologically normal samples. We also found that there was a correlation between PPP3R1 mRNA levels and Braak stage in these samples (P = 0.018; Figure 1D). Rs12713636, in LD with rs1868402 (D′ = 1, R2 = 0.75) also shows association with PPP3R1 mRNA levels and in the same direction in the publicly available GEO GSE8919 [34] dataset (P = 0.015).
Discussion
In the present study we have used a novel and powerful endophenotype-based approach to identify a novel genetic factor implicated in AD. Most genetic studies in AD have focused on identifying genetic factors that modulate risk or age at onset of disease [1]–[2]. The genetic factors influence other important facets of AD, such as rate of progression or disease duration remain poorly understood. Here we report that the minor allele of rs1868402 shows significant, replicable association with higher CSF ptau181 levels and faster rate of progression of AD. We failed to detect evidence for association with risk for disease or age at onset. This is consistent with the known pathobiology of AD, in which Aβ aggregation and deposition is an early preclinical event, followed by increased CSF tau and ptau181 levels and tau pathology during the clinical phase of the disease. Under this model, factors that affect tau phosphorylation and aggregation may be expected to modify disease progression but not risk for disease, while genes that influence Aβ aggregation, such as APOE4 [35], would be expected to influence CSF Aβ42 levels and risk/age at onset of disease but not CSF tau/ptau181 levels or rate of progression of disease. Table 7 illustrates that this is the case in our data. Genetic analyses of CSF Aβ42 and tau variation therefore allow identification of genetic factors that influence different components of the disease process.

Our results suggest that rs1868402, or another variant in LD with it, may reduce calcineurin expression/activity leading to an increase in tau phosphorylation increasing tau pathology and neurodegeneration in individuals with Aβ deposition. The resulting increase in tau-related pathology would then increase the rate of progression of AD. Several studies provide support for a role of calcineurin in AD pathogenesis. Inhibition of calcineurin in mouse brains by cyclosporin A or FK506 or rat brain by antisense oligonucleotides led to enhanced tau phosphorylation [36]–[38]. In mice the increase in tau phosphorylation was accompanied by impaired spatial memory, a characteristic feature of AD [36]. Finally, in AD patients, calcineurin activity is decreased and correlates with neuropathologic changes [39].
Our analyses suggest that genetic variants associated with CSF Aβ42 levels also influence risk and age at onset (e.g. APOE) but variants associated with CSF ptau181 levels have a greater impact on rate of progression (Table 7). Genome-wide association studies of CSF tau/ptau181 levels should identify novel genetic variants which will likely influence rate of progression of AD. Variants that influence disease progression may have significant clinical benefit. For example, these variants have the potential to predict more accurately the time from diagnosis to functional impairment that may require nursing home placement. Stratification of samples by such SNPs will enable cheaper and more efficient clinical trials by selecting individuals expected to have faster rates of progression. By targeting different facets of AD biology this approach can identify a broader range of potential therapeutic targets than a conventional case-control design. Drugs that inhibit or decrease tau phosphorylation would be expected to decrease cognitive decline in individuals with very mild dementia or delay the appearance of memory problems in elderly individuals with low CSF Aβ42 levels. Finally, we believe that this approach is applicable to other common neurological and psychiatric disorders, where biomarkers of disease have been identified, and underlines the value and importance of finding such markers in other diseases.
Materials and Methods
Subjects and endophenotypes
The cerebrospinal fluid discovery series includes 353 individuals enrolled in longitudinal studies at the WU-ADRC. CSF collection and Aβ42, tau and ptau181 measurements were performed as described previously [14]. Table 1 shows the demographic data for the CSF series and Table 2 shows a description of the CSF biomarker in each dataset. The CSF replication series consists of 236 individuals from the ADNI dataset and 257 individuals from the University of Washington (UW, Seattle). All CSF samples were from individuals of European descent. Written consent was obtained from all participants. While there are differences in the absolute levels of the biomarker measurements between the two studies that likely reflect differences in the methods used for quantification (regular ELISA vs Luminex), ascertainment, and/or in handling of the CSF after collection, CSF ptau181 levels in the WU-ADRC-CSF, ADNI-CSF, and UW samples show similar characteristics. CSF ptau181 levels show a 10–17 fold difference between individuals in each dataset, are normally distributed after log-log transformation, and have similar covariates in each dataset (see statistical analyses).
Risk for disease and age at onset analyses were analyzed in a total of 1106 late-onset AD (LOAD) cases and 1216 age-gender-ethnicity matched non-demented controls (Table 5). These samples were ascertained at the WU-ADRC, MRC genetic resource for late-onset AD (UK, MRC Sample [40]), and ADNI. Cases received a diagnosis of dementia of the Alzheimer's type (DAT), using criteria equivalent to the National Institute of Neurological and Communication Disorders and Stroke-Alzheimer's Disease and Related Disorders Association for probable AD [41]–[42]. All individuals were of European descent and written consent was obtained from all participants.
Association of rate of progression of dementia with genetic variants was tested in two longitudinal series from the WU-ADRC and ADNI. The WU-ADRC-CSF series includes 109 individuals with clinical data from at least two time points starting with a CDR of 0 or 0.5 in the first interview, and a diagnosis of DAT (dementia Alzheimer Type) at the last visit. There are an average of 3.8 observations per individual, which varies from 2 to 14 with an average follow up time of 3.2 years. The second series with longitudinal data is the ADNI series: 459 individuals (236 with CSF data and another 223 with no CSF data) have had a clinical examination at least two time points with an average of 4.1 observations per individual, which varies between two and six observations, however the average follow up time is only 1.9 years. To study the association with progression rate in the CSF samples we analyze only the 150 samples with low (<192pg/ml) CSF Aβ42, as explained in the Statistical Analyses section.
SNP selection and genotyping
Based on bibliographic data, we selected 384 SNPs in the most relevant tau kinases, phosphatases, and in other genes implicated in other posttranslational modifications of tau, or tau degradation [45]–[47] (Table S1). Tagging SNPs (r2>0.8), based on CEU-HapMap data, were selected for each of these genes. We used Pupasuite software [48] to select potentially functional variants in the selected genes and flanking regions. SNPs were genotyped using the Illumina Golden Gate, Sequenom and/or Taqman genotyping technologies. Only SNPs with a genotyping call rate higher than 95% and in Hardy-Weinberg equilibrium were used in the analyses.
Gene expression
Expression studies were carried out using cDNA obtained from the parietal lobe of 82 AD cases and 39 non-demented individuals (CDR = 0) obtained through the WU-ADRC Neuropathology Core (Brain samples; Table 1). AD changes were measured using Braak and Braak staging [43]. All AD cases had a Braak and Braak score of 5 or 6. Among the non-demented individuals 24 brains had a Braak and Braak score ranging from 1–4 indicating the presence of some tangle pathology.
Total RNA was extracted from the parietal lobe of 82 AD cases and 39 non-demented individuals, using the RNeasy mini kit (Qiagen) following the manufacturer's protocol. cDNAs were prepared from the total RNA, using the High-Capacity cDNA Archive kit (ABI). Gene expression was analyzed by real-time PCR, using an ABI-7500 real-time PCR system. Real-time PCR assays were used to quantify PPP3R1 cDNA levels. Taqman assays for GAPDH (sequences available on request) PPP3R1 (ABI: C_12044272_10) and cyclophilin A (ABI: 4326316E) were used to quantify the gene expression levels. Each real-time PCR run included within-plate duplicates and each experiment was performed, at least twice for each sample. Real-time data were analyzed using the comparative Ct method. The Ct values of each sample were normalized with the Ct value for the housekeeping genes, GADPH and cyclophilin, and were corrected for the PCR efficiency of each assay [44], although the efficiency of all reactions was close to 100%. Only samples with a standard error of <0.15% were analyzed.
Statistical analyses
CSF ptau181 values were log-log transformed to approximate a normal distribution. Analysis of the covariance (ANCOVA) was used to test for association between genotypes and CSF ptau181 levels. In order to identify the covariates that affect CSF ptau181 levels, we performed a stepwise discriminant analysis including CDR, age, gender and APOE genotype. CDR, age, and APOE genotype were identified as significant covariates in the WU-ADRC-CSF series and, CDR and APOE genotype in the replication series (this series has a narrower age range than the WU-ADRC-CSF series). These covariates were included in the respective ANCOVA. Each SNP was tested using an additive model with minor allele homozygotes coded as 0, heterozygotes coded as 1, and major allele homozygotes coded as 2. When the additive model was significant after multiple test correction, dominant and recessive models were tested to determine whether the y were a better fit. Because the CSF ptau181 levels in the WU-ADRC-CSF, ADNI-CSF and UW samples were measured using different platforms (Innotest plate ELISA vs AlzBia3 bead-based ELISA, respectively) we were not able to combine the raw data, rather we combined the residual values of the CSF ptau181 obtained after correcting for the covariates. No significant differences in the residuals from the different series were found, indicating that the differences in the CSF levels due to the different platforms were corrected by using the residuals.
Multiple test correction: Initial tests for association of SNPs with CSF ptau181 levels were evaluated using a False Discovery Rate (FDR) filter of 0.1 to correct for multiple testing [49]. In the initial screening only p-values more significant than passed FDR filter of 0.1. In the replication series p-values more significant than passed FDR filter. To reduce the probability of false positives we also used the more stringent Bonferroni correction to adjust the alpha level in the analysis of association with CSF ptau181 levels in the combined samples. In this case, the threshold for Bonferroni correction for the combined sample is . No multiple test correction was applied for association with risk for disease, age at onset or progression because only one or two SNPs with specific hypotheses were tested for association. To calculate the impact of rs1868402 on the CSF ptau181 levels, we calculated the Odds Ratio (OR) of this SNP by comparing its frequency in the highest vs lowest quartile of the residuals for CSF ptau181 levels.
Allelic association with risk for AD was tested using logistic regression including APOE, gender, age and series as covariates. Association with AAO was carried out using the Kaplan-Meier method and tested for significant differences, using a log-rank test. Association with rate of disease progression was evaluated as described previously [33]. Briefly, progression of disease was measured by the change in sum of boxes on the CDR (clinical dementia rate; SB-CDR) per year. CDR is a global measurement of the severity of symptoms of dementia. CDR evaluates cognitive and functional performance in six areas (memory, orientation, judgment and problem solving, community affairs, home and hobbies and, personal care), each of these areas has a possible score of 0, 0.5, 1, 2 or 3. The sum of boxes can vary between 0 and 18. Higher scores indicate more significant memory problems and correlate with neurodegeneration [50]. The change in SB-CDR per year fitted a linear model in both series and therefore we used a mixed linear model (PROC MIXED; SAS Institute Inc) to determine whether there is a relationship between the slope of the SB-CDR score and time as a function of genotype after controlling for initial age, gender, APOE, initial CDR, CSF ptau181 and Aβ42 levels. Because the association between rs1868402 and CSF ptau181 was driven by individuals with low CSF Aβ42 levels, association with progression in the CSF datasets was analyzed only in individuals with low CSF Aβ42 levels (less than 500 pg/ml in the WU-ADRC-CSF series [14] or 192 pg/ml in the ADNI-CSF series [21]). We analyzed whether the combination of genotypes of rs1868402 and rs3785883 predicts the rate of progression better than either of these SNPs alone. To do that we included an interaction term rs1868402*rs3785883 in the “proc mixed” SAS program, that combine the genotypes for the two SNPs. We also combined the data from the WU-ADRC-CSF and ADNI-CSF to increase the statistical power.
Association between cDNA levels, tau pathology (Braak tangle stage) and genotypes were carried out using ANCOVA. Stepwise discriminant analysis was used to determine the significant covariates (age, gender, postmortem interval, APOE genotype, and CDR). One-tailed P-values were calculated, because a priori predictions were made based on the associations with CSF ptau181 levels. We also used the GEO dataset GSE8919 [51] to analyze the association between rs12713636, in LD with rs1868402 (D′ = 1, R2 = 0.75) and PPP3R1 gene expression. In this dataset there are genotype and expression data from 486 Late onset Alzheimer Diseases cases and 279 neuropathologically confirmed controls. We only extracted the normalized PPP3R1 mRNA levels and the genotype data for rs12713636. Genotypes for rs12713636 were used because rs1868402 was not included in this dataset.
ADNI material and methods
Data used in the preparation of this article were obtained from the ADNI database (www.loni.ucla.edu/ADNI). The ADNI was launched in 2003 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the Food and Drug Administration, private pharmaceutical companies and non-profit organizations, as a $60 million, 5-year public-private partnership. The Principal Investigator of this initiative is Michael W. Weiner, M.D. ADNI is the result of efforts of many co-investigators from a broad range of academic institutions and private corporations, and subjects have been recruited from over 50 sites across the U.S. and Canada. The initial goal of ADNI was to recruit 800 adults, ages 55 to 90, to participate in the research -approximately 200 cognitively normal older individuals to be followed for 3 years, 400 people with MCI to be followed for 3 years, and 200 people with early AD to be followed for 2 years.” For up-to-date information see www.adni-info.org.
Supporting Information
Zdroje
1. HaroldD
AbrahamR
HollingworthP
SimsR
GerrishA
2009 Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 41 1088 1093
2. LambertJC
HeathS
EvenG
CampionD
SleegersK
2009 Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41 1094 1099
3. ChambersJC
ZhangW
LiY
SehmiJ
WassMN
2009 Genome-wide association study identifies variants in TMPRSS6 associated with hemoglobin levels. Nat Genet 41 1170 1172
4. BenyaminB
FerreiraMA
WillemsenG
GordonS
MiddelbergRP
2009 Common variants in TMPRSS6 are associated with iron status and erythrocyte volume. Nat Genet 41 1173 1175
5. SoranzoN
SpectorTD
ManginoM
KuhnelB
RendonA
2009 A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet 41 1182 1190
6. GaneshSK
ZakaiNA
van RooijFJ
SoranzoN
SmithAV
2009 Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat Genet 41 1191 1198
7. RivadeneiraF
StyrkarsdottirU
EstradaK
HalldorssonBV
HsuYH
2009 Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat Genet 41 1199 1206
8. CohenJC
KissRS
PertsemlidisA
MarcelYL
McPhersonR
2004 Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science 305 869 872
9. CohenJC
PertsemlidisA
FahmiS
EsmailS
VegaGL
2006 Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc Natl Acad Sci U S A 103 1810 1815
10. RomeoS
PennacchioLA
FuY
BoerwinkleE
Tybjaerg-HansenA
2007 Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet 39 513 516
11. RomeoS
YinW
KozlitinaJ
PennacchioLA
BoerwinkleE
2009 Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest 119 70 79
12. PriceJL
MorrisJC
1999 Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol 45 358 368
13. FaganAM
HeadD
ShahAR
MarcusD
MintunM
2009 Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol 65 176 183
14. FaganAM
MintunMA
MachRH
LeeSY
DenceCS
2006 Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol 59 512 519
15. MorrisJC
RoeCM
XiongC
FaganAM
GoateAM
2010 APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 67 122 131
16. HanssonO
ZetterbergH
BuchhaveP
AndreassonU
LondosE
2007 Prediction of Alzheimer's disease using the CSF Abeta42/Abeta40 ratio in patients with mild cognitive impairment. Dement Geriatr Cogn Disord 23 316 320
17. KapakiEN
ParaskevasGP
TzerakisNG
SfagosC
SeretisA
2007 Cerebrospinal fluid tau, phospho-tau181 and beta-amyloid1-42 in idiopathic normal pressure hydrocephalus: a discrimination from Alzheimer's disease. Eur J Neurol 14 168 173
18. FaganAM
RoeCM
XiongC
MintunMA
MorrisJC
2007 Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 64 343 349
19. NoguchiM
YoshitaM
MatsumotoY
OnoK
IwasaK
2005 Decreased beta-amyloid peptide42 in cerebrospinal fluid of patients with progressive supranuclear palsy and corticobasal degeneration. J Neurol Sci 237 61 65
20. IkonomovicMD
KlunkWE
AbrahamsonEE
MathisCA
PriceJC
2008 Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain 131 1630 1645
21. JagustWJ
LandauSM
ShawLM
TrojanowskiJQ
KoeppeRA
2009 Relationships between biomarkers in aging and dementia. Neurology 73 1193 1199
22. HesseC
RosengrenL
AndreasenN
DavidssonP
VandersticheleH
2001 Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neurosci Lett 297 187 190
23. OstM
NylenK
CsajbokL
OhrfeltAO
TullbergM
2006 Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology 67 1600 1604
24. BuergerK
EwersM
PirttilaT
ZinkowskiR
AlafuzoffI
2006 CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer's disease. Brain 129 3035 3041
25. UrakamiK
WadaK
AraiH
SasakiH
KanaiM
2001 Diagnostic significance of tau protein in cerebrospinal fluid from patients with corticobasal degeneration or progressive supranuclear palsy. J Neurol Sci 183 95 98
26. AraiH
MorikawaY
HiguchiM
MatsuiT
ClarkCM
1997 Cerebrospinal fluid tau levels in neurodegenerative diseases with distinct tau-related pathology. Biochem Biophys Res Commun 236 262 264
27. KauweJS
CruchagaC
MayoK
FenoglioC
BertelsenS
2008 Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc Natl Acad Sci U S A 105 8050 8054
28. KauweJS
JacquartS
ChakravertyS
WangJ
MayoK
2007 Extreme cerebrospinal fluid amyloid beta levels identify family with late-onset Alzheimer's disease presenilin 1 mutation. Ann Neurol 61 446 453
29. KauweJS
WangJ
MayoK
MorrisJC
FaganAM
2009 Alzheimer's disease risk variants show association with cerebrospinal fluid amyloid beta. Neurogenetics 10 13 17
30. Sato-HaradaR
OkabeS
UmeyamaT
KanaiY
HirokawaN
1996 Microtubule-associated proteins regulate microtubule function as the track for intracellular membrane organelle transports. Cell Struct Funct 21 283 295
31. WangJZ
Grundke-IqbalI
IqbalK
2007 Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci 25 59 68
32. Spires-JonesTL
StoothoffWH
de CalignonA
JonesPB
HymanBT
2009 Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci 32 150 159
33. SniderBJ
FaganAM
RoeC
ShahAR
GrantEA
2009 Cerebrospinal fluid biomarkers and rate of cognitive decline in very mild dementia of the Alzheimer type. Arch Neurol 66 638 645
34. WebsterJA
GibbsJR
ClarkeJ
RayM
ZhangW
2009 Genetic control of human brain transcript expression in Alzheimer disease. Am J Hum Genet 84 445 458
35. BertramL
McQueenMB
MullinK
BlackerD
TanziRE
2007 Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 39 17 23
36. YuDY
LuoJ
BuF
SongGJ
ZhangLQ
2006 Inhibition of calcineurin by infusion of CsA causes hyperphosphorylation of tau and is accompanied by abnormal behavior in mice. Biol Chem 387 977 983
37. LuoJ
MaJ
YuDY
BuF
ZhangW
2008 Infusion of FK506, a specific inhibitor of calcineurin, induces potent tau hyperphosphorylation in mouse brain. Brain Res Bull 76 464 468
38. GarverTD
KincaidRL
ConnRA
BillingsleyML
1999 Reduction of calcineurin activity in brain by antisense oligonucleotides leads to persistent phosphorylation of tau protein at Thr181 and Thr231. Mol Pharmacol 55 632 641
39. LadnerCJ
CzechJ
MauriceJ
LorensSA
LeeJM
1996 Reduction of calcineurin enzymatic activity in Alzheimer's disease: correlation with neuropathologic changes. J Neuropathol Exp Neurol 55 924 931
40. MorganAR
TuricD
JehuL
HamiltonG
HollingworthP
2007 Association studies of 23 positional/functional candidate genes on chromosome 10 in late-onset Alzheimer's disease. Am J Med Genet B Neuropsychiatr Genet 144B 762 770
41. BergL
McKeelDWJr
MillerJP
StorandtM
RubinEH
1998 Clinicopathologic studies in cognitively healthy aging and Alzheimer's disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol 55 326 335
42. McKhannG
DrachmanD
FolsteinM
KatzmanR
PriceD
1984 Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 34 939 944
43. BraakH
BraakE
1991 Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 82 239 259
44. MullerPY
JanovjakH
MiserezAR
DobbieZ
2002 Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques 32 1372 1374, 1376, 1378–1379
45. StoothoffWH
JohnsonGV
2005 Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta 1739 280 297
46. YuzwaSA
VocadloDJ
2009 O-GlcNAc modification and the tauopathies: insights from chemical biology. Curr Alzheimer Res 6 451 454
47. ChungSH
2009 Aberrant phosphorylation in the pathogenesis of Alzheimer's disease. BMB Rep 42 467 474
48. CondeL
VaquerizasJM
DopazoH
ArbizaL
ReumersJ
2006 PupaSuite: finding functional single nucleotide polymorphisms for large-scale genotyping purposes. Nucleic Acids Res 34 W621 625
49. StoreyJD
TibshiraniR
2003 Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100 9440 9445
50. MorrisJC
1993 The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43 2412 2414
51. MyersAJ
GibbsJR
WebsterJA
RohrerK
ZhaoA
2007 A survey of genetic human cortical gene expression. Nat Genet 39 1494 1499
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 9
Nejčtenější v tomto čísle
- Synthesizing and Salvaging NAD: Lessons Learned from
- Optimal Strategy for Competence Differentiation in Bacteria
- Long- and Short-Term Selective Forces on Malaria Parasite Genomes
- Identifying Signatures of Natural Selection in Tibetan and Andean Populations Using Dense Genome Scan Data