
Genetic Deletion of the Desmosomal Component Promotes Tumor Microinvasion in a Mouse Model of Pancreatic Neuroendocrine Carcinogenesis
We used the RIP1-Tag2 (RT2) mouse model of islet cell carcinogenesis to profile the transcriptome of pancreatic neuroendocrine tumors (PNET) that were either non-invasive or highly invasive, seeking to identify pro - and anti-invasive molecules. Expression of multiple components of desmosomes, structures that help maintain cellular adhesion, was significantly reduced in invasive carcinomas. Genetic deletion of one of these desmosomal components, desmoplakin, resulted in increased local tumor invasion without affecting tumor growth parameters in RT2 PNETs. Expression of cadherin 1, a component of the adherens junction adhesion complex, was maintained in these tumors despite the genetic deletion of desmoplakin. Our results demonstrate that loss of desmoplakin expression and resultant disruption of desmosomal adhesion can promote increased local tumor invasion independent of adherens junction status.
Published in the journal:
. PLoS Genet 6(9): e32767. doi:10.1371/journal.pgen.1001120
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001120
Summary
We used the RIP1-Tag2 (RT2) mouse model of islet cell carcinogenesis to profile the transcriptome of pancreatic neuroendocrine tumors (PNET) that were either non-invasive or highly invasive, seeking to identify pro - and anti-invasive molecules. Expression of multiple components of desmosomes, structures that help maintain cellular adhesion, was significantly reduced in invasive carcinomas. Genetic deletion of one of these desmosomal components, desmoplakin, resulted in increased local tumor invasion without affecting tumor growth parameters in RT2 PNETs. Expression of cadherin 1, a component of the adherens junction adhesion complex, was maintained in these tumors despite the genetic deletion of desmoplakin. Our results demonstrate that loss of desmoplakin expression and resultant disruption of desmosomal adhesion can promote increased local tumor invasion independent of adherens junction status.
Introduction
The ability of a tumor to invade into the surrounding normal tissue marks a critical step in the transition from benign to malignant tumor growth. The acquisition of this hallmark of cancer is associated with poor prognosis for many human cancers and is often considered a precursor to the development of metastases [1]. As such, considerable effort has been directed towards identifying invasion promoting and suppressing molecules and the mechanisms by which they modulate a tumor's invasive phenotype [2].
Amongst the discernible barriers to the acquisition of an invasive growth phenotype is cell-cell adhesion, and cellular alterations that result in disrupted, reduced, or otherwise functionally altered cellular adhesion are strongly associated with the progression to a malignant tumor phenotype [3]–[5]. The importance of sustaining cellular adhesion for homeostasis, particularly in epithelial tissues, is evident in the number of distinct structures whose primary function is to maintain cell-cell interconnections, which include the adherens junctions (AJs), desmosomes, and tight junctions [6], [7]. These complexes share many structural similarities, including the presence of transmembrane proteins – typified by the cadherins – that mediate adhesive connections with neighboring cells as well as intracellular molecules – exemplified by the catenin and the plakin families – that connect these transmembrane components to the cytoskeleton [6], [7]. In particular, changes in the expression and/or function of AJ components have been associated with malignant cancers, and numerous studies have focused on the role of AJs in restricting invasive growth [3], [8], [9].
In this study, we utilized the RIP1-Tag2 (RT2) mouse model of cancer to identify the mechanisms by which tumors acquire invasive growth capabilities. RT2 mice develop multiple pancreatic neuroendocrine tumors (PNET) by 12–14 weeks of age due to the expression of the SV40 T antigen oncoprotein (Tag) in the pancreatic β cells [10]. This model has proven useful in characterizing many aspects of tumorigenesis due to its relatively synchronous and predictable progression through distinctive lesional stages that culminate in invasive carcinomas [11]–[13]. We used this model to identify pro - and anti-invasive molecules in an unbiased fashion by comparing the non-invasive islet tumors to highly invasive carcinomas using microarray profiling of the mRNA transcriptome. We identified several components of desmosomes whose expression was significantly decreased in invasive tumors, implicating attenuation of desmosomal function in malignant progression. To assess this hypothesis, we engineered into the oncogene-expressing cancer cells in RT2 mice a genetic deletion of desmoplakin (Dsp; MGI: 109611), an intracellular protein critical for desmosomal stability [14]. Loss of Dsp led to an increased incidence of invasive carcinomas providing strong evidence that desmosomal adhesion acts as a distinct barrier to invasive tumor growth.
Results
Expression of desmosomal components is lost in invasive RT2 tumor lesions
We chose to use the RT2 mouse model of cancer to characterize mechanisms governing the switch from benign to invasive tumor growth since a broad spectrum of invasive tumor lesions develop in end-stage RT2 animals. These include the non-invasive islet tumor (IT), the focally invasive carcinoma type-1 (IC1), and the broadly invasive carcinoma type-2 (IC2) [15].
To evaluate potential mechanisms regulating invasive tumor growth in this model, we isolated tissue from IT and IC2 lesions in end-stage RT2 animals by laser capture microdissection and then profiled the mRNA transcriptome. The IC2 class showed widespread transcriptional changes as compared to the IT class (Dataset S1). We chose to focus our attention on differentially expressed genes encoding components of two cell-cell adhesion structures, namely adherens junctions and desmosomes (Table 1), since elements of each were prominently downregulated. The expression of cadherin 1 (Cdh1, also known as E-cadherin; MGI: 88354), a molecule previously demonstrated to restrict invasive growth in this and other models [8], [16], was decreased in IC2 lesions as expected. Interestingly, Cdh1 was the only member of AJs that was significantly altered in IC2 lesions (Table 1). In contrast, multiple genes encoding components of desmosomes were significantly reduced in IC2 lesions (Table 1). Moreover, the expression of several desmosomal genes in addition to Cdh1 was progressively reduced in the distinctive stages of PNET tumorigenesis in RT2 mice as well as in human PNETs as compared to normal human pancreatic islets, when total lesional stages, in particular ungraded tumors, were analyzed (Figure S1) [13]. Although the expression of these genes was reduced in ungraded whole tumors in comparison to normal islets, their levels were further reduced in the microdissected invasive IC2 lesions (Table 1). Based on these results, we sought to determine what role desmosomal adhesion might play in regulating invasive tumor growth in this mouse model of cancer.

To confirm the microarray results, we performed immunohistochemistry for multiple desmosomal components. Staining for Dsp and for one of the desmosomal cadherins, desmoglein 2 (Dsg2; MGI: 1196466), as well as for Cdh1 demonstrated that these molecules are expressed in the pancreatic islets as well as in the pancreatic ducts and the exocrine pancreas of wild-type animals (Figure 1 and Figure S2). In tumors of end-stage RT2 animals, the expression of all three molecules was maintained in IT lesions and was largely extinguished in IC2 lesions (Figure 1 and Figure S2). In contrast to Cdh1, expression of catenin beta 1 (Ctnnb1; MGI: 88276), another component of AJs, was maintained in both IT and IC2 lesions, comparable to wild-type islets (Figure S3). This result is consistent both with the microarray result demonstrating that Cdh1 was the only AJ component to show any change in expression and with a previous study suggesting that Ctnnb1 does not contribute to RT2 tumorigenesis [17]. Collectively, these data confirm the microarray results and suggest the hypothesis that loss of desmosomal adhesion might contribute to the development of an invasive phenotype.

β cell specific deletion of Dsp in RT2 animals
To address the hypothesis raised by the microarray and immunohistochemistry results, we asked whether functionally disrupting desmosomal activity in vivo would promote invasive tumor growth in RT2 mice. To accomplish this, we chose to genetically delete Dsp since there is a single Dsp gene as compared to other components of desmosomes for which there are multiple non-allelic genes [6]. Furthermore, ablation of Dsp in vivo has previously been shown to impair desmosome function [14]. Since the Dsp whole body knockout is embryonic lethal [14], we employed the Cre/loxP system to ablate the Dsp gene specifically in the pancreatic β cells, the same cells that express the Tag oncogene in RT2 mice. In combination with a DspFlox allele [18], we used a mouse line in which a tamoxifen-regulatable Cre recombinase is controlled by the pancreatic duodenal homeobox gene 1 promoter (Pdx1-CreER) [19]. Pdx1 is expressed in all pancreatic lineages during development and is variably expressed in the adult pancreas, in particular being widely expressed in β cells [20], [21].
We intercrossed RT2+; DspFlox/WT with Pdx1-CreER+; DspFlox/WT mice to generate the appropriate genotypes, and all expected genotypes and genders were observed in approximate Mendellian ratios (Table S1 and Table S2). To induce Cre activity, all Pdx1-CreER positive mice were given tamoxifen for five consecutive days beginning at 10 weeks of age when incipient tumors are first observed in RT2 mice [22]. In the absence of the RT2 transgene, genetic ablation of Dsp resulted in uniform loss of Dsp expression in the pancreatic islets, as determined by immunohistochemistry (Figure S4). Deletion of Dsp did not cause any change in Cdh1 expression or in the gross morphological appearance of the non-oncogene-expressing islets (Figure S4). Loss of Dsp was accompanied by significantly reduced Dsg2 expression in the pancreatic islets whereas the expression of insulin (Ins), the hormone produced by β cells, did not appear to be affected (Figure S5). These results are consistent with compromised desmosomal adhesion, although we cannot strictly rule out the possibility that some residual desmosomal function persists in the absence of Dsp. Ablation of Dsp in normal pancreatic islets did not affect multiple physiological parameters, such as body mass and fasting glucose levels, and its expression in this tissue compartment is apparently dispensable in adult mice (Figure S6), setting the stage to assess the impact of its loss on PNETs arising from such islets. Lastly, the tamoxifen induction regimen by itself had no obvious effect on any aspect of RT2 tumorigenesis examined, including tumor invasion, when tamoxifen was applied to RT2 mice that lacked the Pdx1-CreER and DspFlox alleles (Figure S7).
Loss of Dsp does not affect tumor growth parameters in RT2 mice
Induced loss of Dsp at 10 weeks of age did not affect any of the tumor growth parameters in RT2 mice that were sacrificed 4 weeks later. No significant changes were observed in the number of tumors that developed nor in the collective tumor burden when comparing RT2+; Pdx1-CreER+; DspFlox/Flox mice and littermate controls (Figure 2A–B). Furthermore, the rates of tumor proliferation and tumor apoptosis, as judged by the levels of the proliferation marker Ki67 and the TUNEL assay respectively, were indistinguishable between groups (Figure 2C–J). Thus, we conclude that the loss of Dsp does not affect tumor growth in this model.

Loss of Dsp leads to increased local tumor invasion in RT2 mice
While conditional genetic ablation of Dsp in the angiogenic islet dysplasias and incipient solid tumors of RT2 mice had no discernible effects on tumor formation and subsequent tumor growth parameters, it did lead to an increase in tumor invasion. RT2 mice develop a spectrum of tumor lesions, including non-invasive (IT), focally invasive (IC1), and broadly invasive (IC2) lesions (Figure 3A–F) [15]. Loss of Dsp resulted in a greater frequency of invasive tumors and a concomitant reduction in the percentage of non-invasive IT tumors in mice analyzed four weeks after genetic ablation of Dsp in incipient solid tumors (Figure 3G–H). Whereas ∼40% of total tumors could be classified as invasive carcinomas in control mice, greater than 60% of all tumors fell into this category in RT2+; Pdx1-CreER+; DspFlox/Flox mice (Figure 3G). Interestingly, this shift appears to result from selective progression to the focally invasive IC1 but not to the widely invasive IC2 tumors. Indeed, while there is no significant change in the development of IC2 lesions (approximately 10% of all tumors fall into this class regardless of Dsp status), more than 50% of tumors can be classified as IC1 lesions in RT2+; Pdx1-CreER+; DspFlox/Flox mice versus ∼30% in control mice (Figure 3H).

We confirmed that Dsp was in fact lost in these tumors by examining the recombination status of the Dsp allele by PCR. Tumors that were genotypically DspFlox/Flox showed near universal recombination of the Dsp allele, confirming that Dsp was lost in these tumors (Figure 3I). Tumors isolated from control DspWT/WT or DspFlox/WT mice showed no recombination or were heterozygous for the recombined and wild-type Dsp alleles respectively. Thus, we conclude that the conditional genetic ablation of Dsp in incipient tumors of RT2 mice leads to increased local tumor invasion.
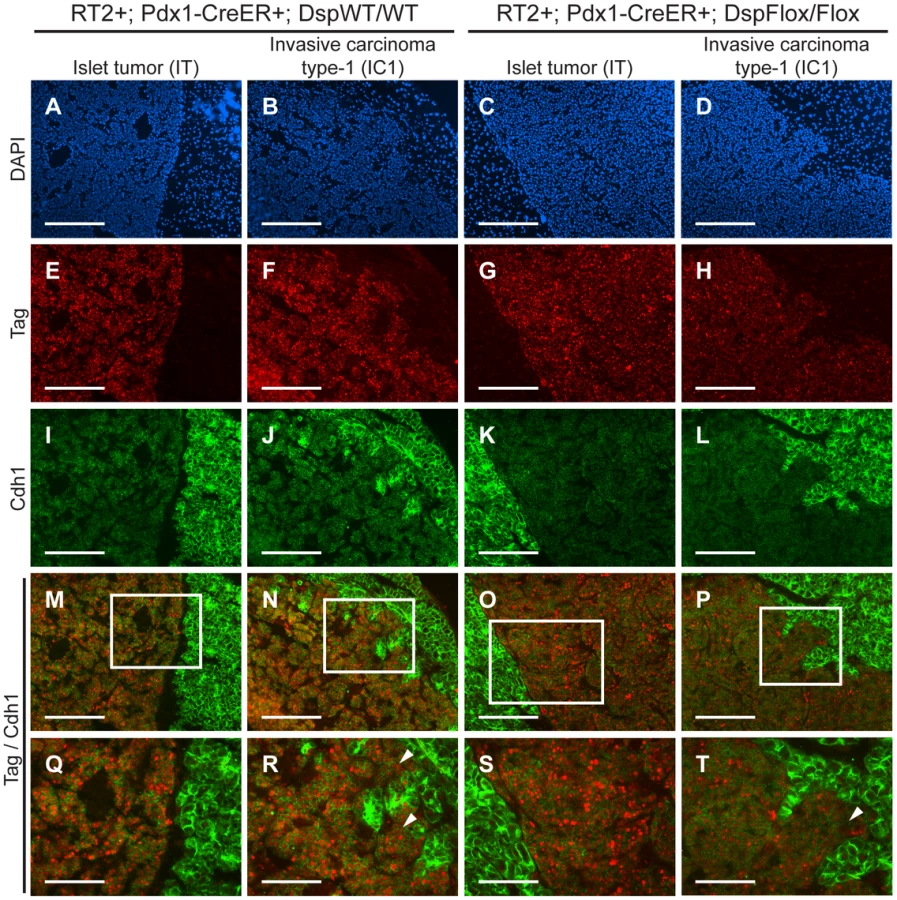
Cdh1 expression is maintained in IC1 tumor lesions regardless of Dsp status
We were intrigued that loss of Dsp led to an increase in the IC1 class but not in the IC2 class of invasive tumors. Since Cdh1 also acts as a dominant invasion suppressor in this model, we examined its status in the tumors from RT2+; Pdx1-CreER+; DspFlox/Flox mice and littermate controls by immunohistochemistry. We found that Cdh1 expression was maintained in the IT and IC1 tumors that developed regardless of Dsp status (Figure 4I–L). Tumor margins and regions of invasion were identified by staining for the Tag oncoprotein (Figure 4E–H). Indeed, Cdh1 appeared to be expressed at comparable levels in IT and IC1 tumor lesions regardless of Dsp status (Figure 4M–T). Expression in IT and IC1 lesions of a second component of AJs, junction plakoglobin (Jup, also known as gamma catenin; MGI: 96650), was also unaffected by Dsp status (Figure S8), consistent with AJ function being maintained in these lesions despite the absence of Dsp and impaired/ablated desmosomal function. Lastly, cadherin 2 (Cdh2, also known as N-cadherin; MGI: 88355), a marker of epithelial-mesenchymal transition (EMT), was expressed at readily detectable and comparable levels in IT and IC1 tumors regardless of Dsp status, as well as in the IC2 tumors that did not express Cdh1 (Figure S9), consistent with the results of a previous study investigating determinants of progression to invasive carcinoma [8]; notably, there is no indication that activation of the invasive growth capability in this pathway involves an EMT, as reflected in differential expression of Cdh2 or other markers of EMT. Given that the expression of both Dsp and Cdh1 was lost in IC2 lesions, the most invasive class of RT2 tumors, both in unmodified RT2 mice and in tamoxifen-treated RT2+; Pdx1-CreER+; DspFlox/Flox mice (Figure 1 and data not shown), we infer that loss of Dsp by itself is sufficient to promote the development of focally invasive tumors while the additional loss of Cdh1 is required to develop a more aggressive invasive tumor phenotype.
Discussion
To date, much of the work on desmosomes in human disease has focused on their role in maintaining heart and skin integrity, where desmosomal defects are associated with cardiomyopathy and skin blistering conditions respectively [23]. More recently, a potential role for desmosomes in cancer progression has been suggested based on a variety of experimental clues [24]. For example, in vitro cell culture assays demonstrated that inhibiting desmosomal adhesion via blocking peptides caused morphological disorganization [25] while introduction of desmosomal components into a nonadhesive cell line resulted in increased cell aggregation and reduced cellular invasion in vitro [26]. These studies suggested that loss of desmosomal function might contribute to tumor invasion and malignancy, consistent with their role in maintaining cellular adhesion. (Our attempts to perform similar in vitro experiments using cell lines derived from RT2 tumors [βTCs] were hindered by the fact that βTC cell lines express desmosomal components at low levels, presumably due to adaptations to culture, and generally perform poorly in migration/invasion assays – data not shown). In further support of the proposed role of desmosomes as a barrier to malignant progression, several pathology studies characterizing human cancers have shown that decreased or altered expression of desmosomal components, including Dsp, correlates with increased tumor invasion, advanced tumor grade, and poor patient prognosis, particularly in oral cancers where expression of desmosomal components are highly expressed in the normal oral mucosa [4], [5], [27]. Additionally, our bioinformatic analysis of human cancer databases confirmed that the expression of desmosomal genes is often decreased in a variety of human epithelial cancers as compared to normal tissues and is occasionally further decreased in more advanced grades of tumors (Table S3). The present study substantively extends this current state of knowledge by demonstrating that desmosomal adhesion can indeed act as a distinct barrier to the development of an invasive tumor phenotype in the in vivo setting of a genetically engineered mouse model of cancer.
We identified several components of desmosomes – Dsp, Dsg2, desmocollin 2 (Dsc2; MGI: 103221), and plakophilin 2 (Pkp2; MGI: 1914701) – whose expression was significantly downregulated in the highly invasive tumor lesions that develop in the RT2 mouse model of PNET. These changes were reflected at the protein level as determined by immunostaining of non-invasive IT lesions and broadly invasive IC2 lesions. The simultaneous decrease in expression for multiple desmosomal genes suggests that there may be coordinated transcriptional regulation of desmosomal components. Prime candidates for such regulation include the transcription factors that regulate EMT, such as the Snail and Twist families of transcription factors [28]. Notably, however, we did not detect significant differential expression of such transcription factors in our microarray analysis comparing non-invasive IT and highly invasive IC2 PNETs (Dataset S1), and the expression of one prominent marker of EMT, Cdh2, was not obviously different between IT and IC2 lesions, consistent with the results of a previous study investigating determinants of the invasive phenotype using this same model of PNET [8]. Thus, the current evidence suggests that the acquisition of an invasive phenotype in this tumor type does not involve a classical EMT. Our results clearly demonstrate that the conditional genetic deletion of a single core desmosomal component, Dsp, promotes increased local tumor invasion in RT2 mice, producing a phenocopy of such inferred transcriptional regulation in the normal circumstances of tumor progression.
While desmosomes play an integral role in maintaining epithelial integrity, they are by no means the only structure involved in cellular adhesion. In addition to desmosomes, several related structures, including AJs, contribute to maintaining cell-cell adhesion [7]. However, while desmosomes and AJs play related biological roles in terms of maintaining cellular adhesion and have similar structural compositions, it is worth noting that there are clear differences in the consequences of impaired desmosome adhesion versus impaired AJ adhesion on tumor phenotypes. An elegant functional genetic study demonstrated that Cdh1, a core member of AJs, acts as an invasion suppressor in vivo; targeting a transgene encoding a dominant-negative Cdh1 molecule to the oncogene-expressing pancreatic β cells markedly accelerated tumor progression and led to significantly increased frequencies of invasive carcinomas and to the development of lymph node metastasis in this same mouse model of PNET [8]. In comparison, deletion of Dsp led to an increase in the frequency of the focally invasive IC1 grade of islet carcinomas but not the more widely aggressive IC2 carcinomas, and distant metastases were not observed (data not shown). One possible explanation for the differences in these phenotypic outcomes is the different roles that Dsp and Cdh1 play within their respective adhesion complex. While Cdh1 is a transmembrane protein that directly links cells together by forming homotypic interactions with other Cdh1 molecules on neighboring cells [29], Dsp is an intracellular molecule that contributes to the overall stability of the desmosomal plaque and links this structure to the intermediate filaments [14]. Therefore, deletion of Dsp may attenuate but not totally abolish desmosomal function; if so, then the specific deletion of one of the desmosomal cadherins, Dsc2 or Dsg2, might have a more pronounced effect on invasiveness. An additional explanation for the increase in the focally invasive IC1 fraction but not the broadly invasive IC2 fraction of invasive tumors following ablation of Dsp involves the observed maintenance of Cdh1 and AJs. Expression of Cdh1 as well as a second component of AJs, Jup, was retained in both the non-invasive IT tumors and in the now more prevalent focally invasive IC1 tumors following genetic deletion of Dsp. It would seem likely, in light of the aforementioned functional study in this same mouse model of cancer [8], that the preservation of Cdh1 expression and of AJ function serves to maintain an additional, stronger brake on tumor invasion. Thus, while loss of Dsp and impairment of desmosomal adhesion leads to the focal invasion observed in IC1 lesions, the development of the broadly invasive phenotype found in IC2 lesions evidently requires the concomitant loss of Cdh1. Indeed, the IC2 tumor lesions that normally develop in RT2 mice show a coordinated reduction in the expression of Cdh1 and multiple desmosomal components (Table 1, Figure 1, and Figure S2). The apparently independent regulation of desmosomal and AJ adhesion is notable since AJ stability has been proposed to affect desmosomal stability and vice versa in other contexts [18], [30], [31], whereas Cdh1 and Jup are evidently not affected by the deletion of Dsp during PNET tumorigenesis in RT2 mice.
Interestingly, the genetic deletion of Dsp had no consequential effects on the other parameters of RT2 tumorigenesis beyond invasion. Although it has been suggested that Dsp and other desmosomal components can affect cellular proliferation and apoptosis [32], [33], we did not observe any changes in tumor growth parameters following the genetic deletion of Dsp (Figure 2). Our results are consistent with one of the earliest studies to examine the role of Dsp in vivo, wherein a skin-specific deletion of catenin alpha 1 (Ctnna1; MGI: 88274), the AJ homologue of Dsp, led to increased skin proliferation and hyperplasia whereas ablation of Dsp did not [34]. Thus, with regards to the RT2 model of PNET and possibly other forms of cancer, it appears that desmosomes primarily serve to maintain cell-cell adhesion and hence suppress the acquisition of an invasive growth capability such that the observed downregulation of desmosomal genes results in the impairment of desmosomal function and a concomitant weakening in cellular adhesion without affecting other parameters of tumorigenesis.
Finally, it is important to set these results into the broader context of knowledge about malignant progression to an invasive growth state in this stereotypical pathway of multistep tumorigenesis. While disrupted cell-cell adhesion caused by the reduced expression of Cdh1 [8] and/or desmosomal genes (this report) clearly promotes invasive tumor growth, other factors are involved as well. Thus for example, increased expression of the type-1 insulin-like growth factor receptor (Igf1r; MGI: 96433) can drive these PNETs to acquire a highly invasive phenotype [15]. Additionally, the recruitment of immune cells to the margins of these PNETs has been shown to promote invasiveness, in part by supplying cathepsin proteases and heparanase (Hpse; MGI: 1343124) [35]–[37]. As such, multiple factors can impact the progression to invasiveness by varying degrees (Figure 5), and future research may well identify additional components. Irrespective, our results demonstrate that loss of desmosomal adhesion, as exemplified by the genetic deletion of Dsp, can enable a tumor to acquire an invasive phenotype. The functional study presented herein establishes desmosomal adhesion as a distinct and ostensibly independent suppressor of invasive tumor growth. This knowledge will likely contribute to a better understanding of the mechanisms governing tumor progression to an invasive growth state and may prove useful in evaluating invasive states of human cancers.

Materials and Methods
Ethics statement
All mice used in this study were housed and maintained in accordance with the University of California, San Francisco (UCSF) institutional guidelines governing the care of laboratory mice.
Genetically engineered mice
The generation and characterization of the RIP1-Tag2 (RT2) [10], DspFlox [18], and Pdx1-CreER [19] mouse lines have been previously reported. All mice were backcrossed a minimum of six generations into the C57Bl/6 (B6) background (Charles River, Wilmington, MA) and then intercrossed to generate the specified genotypes. To induce CreER activity, mice were injected intraperitoneally with 100 µl of 10 mg/ml tamoxifen (Sigma, St. Louis, MO) suspended in peanut oil for five consecutive days beginning at 10 weeks of age. To relieve the effects of hypoglycemia induced by the insulin-secreting tumors, all RT2 mice received 50% sugar food (Harlan Teklad, Madison, WI) beginning at 10 weeks of age.
Tissue preparation, tumor analysis, and histology
Pancreata were isolated from 14-week-old mice and embedded in OCT (Sakura Finetek, Torrance, CA) on dry ice. Tumor number and tumor volume were quantified as previously described [12]. For histological analysis, frozen tissues were sectioned at 10 µm thickness, and every tenth section was stained with hematoxylin and eosin (Surgipath Medical Industries, Richmond, IL) using standard methods. Tumors were classified as a non-invasive islet tumor (IT), a focally invasive carcinoma type-1 (IC1), or a broadly invasive carcinoma type-2 (IC2) using a previously defined grading scheme [15].
Laser capture microdissection and RNA purification and amplification
Fresh-frozen pancreatic sections (10 µm) from 14-week-old RT2 B6 mice were fixed in cold 70% ethanol for 16 hours prior to laser capture microdissection (LCM). Sections were stained using a modified hematoxylin and eosin stain that preserves RNA integrity while allowing for the microscopic visualization of pancreatic structures [38]. LCM was performed using an Arcturus PixCell II laser capture microscope system (Molecular Devices, Sunnyvale, CA). Total RNA was isolated using the Arcturus PicoPure RNA Isolation kit (Molecular Devices, Sunnyvale, CA) and DNase I treated (Qiagen, Valencia, CA). Equal amounts of RNA (8 ng/lesion) from three independent IT or IC2 tumor lesions were pooled, and then cDNA was generated, amplified, and biotinylated using the Ovation Biotin System (NuGen, San Carlos, CA). Three independent pools per tumor class were generated for subsequent microarray analysis.
Microarray analysis
Labeled cDNA was hybridized to Affymetrix Mouse Genome 430 2.0 arrays (Affymetrix, Santa Clara, CA) according to the manufacturer's specifications. Data were analyzed by the UCSF Helen Diller Family Comprehensive Cancer Center Biostatistics and Computational Biology Core. The data were normalized using a robust multi-chip averaging method utilizing the freely available R language. Linear models were fit for each pair of groups to be compared with log2 expression as the response and the tumor phenotype indicator as the independent variable using the limma package in Bioconductor. Moderated t-statistics were used, and p-values were adjusted by controlling the false discovery rate. A change in gene expression was identified as significant if the false discovery rate was less than 0.05, meaning that fewer than 5% of false findings would be expected among the genes declared to be differentially expressed.
Immunohistochemical staining and analysis
Frozen tissues were sectioned at 10 µm thickness. For immunofluorescence staining, sections were fixed in cold acetone. For colorometric staining, sections were fixed in 10% Zn-buffered formalin (Medical Chemical Corporation, Torrance, CA), subjected to antigen retrieval using the Antigen Unmasking Solution (Vector Laboratories, Burlingame, CA), and blocked for endogenous peroxidase activity. Antibodies used in this study were as follows: rat anti-cadherin 1 (Invitrogen, Carlsbad, CA); mouse anti-desmoplakin I/II, mouse anti-desmoglein 1/2 (Fitzgerald, Concord, MA); mouse anti-catenin beta 1, mouse anti-cadherin 2, mouse anti-junction plakoglobin (BD Biosciences, San Jose, CA); guinea pig anti-insulin (Millipore, Billerica, MA); rabbit anti-T-antigen (Hanahan laboratory preparation); rabbit anti-Ki67 (Novus Biologicals, Littleton, CO); rhodamine red-X-conjugated donkey anti-mouse IgG, rhodamine red-X-conjugated donkey anti-rabbit IgG, FITC-conjugated donkey anti-rat IgG, FITC-conjugated donkey anti-guinea pig IgG, biotin-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA). For mouse antibodies, non-specific binding was blocked using the Mouse on Mouse Blocking Reagent (Vector Laboratories, Burlingame, CA). Fluorescently labeled tissues were mounted with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA) to visualize cell nuclei. The TdT-mediated dUTP-digoxigenin nick-end labeling (TUNEL) assay was used to assess tumor apoptosis as previously described [15]. For colorometric staining, signal was amplified using the Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA), visualized using Nova Red substrate (Vector Laboratories, Burlingame, CA), and counterstained with hematoxylin. For Ki67 and TUNEL quantification, two to three random fields were obtained using a 40× objective lens from at least two tumors per mouse and at least five mice per group. The proliferation or apoptosis index was calculated as the percentage of total cells per field that were Ki67 - or TUNEL-positive respectively using the MetaMorph software package (Molecular Devices, Sunnyvale, CA). For all other immunohistochemical analysis, two to three tumors per mouse from a minimum of five mice per indicated group were analyzed per staining condition. All images were captured using an Axio Imager bright field microscope or an Axio Scope fluorescence microscope and the AxioVision LE software package (Carl Zeiss, Thornwood, NY).
Statistical analysis
Fisher's exact test and the chi-square test were used to compare tumor invasion metrics. The Mann-Whitney test was used to compare tumor burden, tumor number, tumor proliferation rates, tumor apoptosis rates, and body mass metrics. The Mann-Whitney and the Wilcoxon matched pairs test was used to compare fasting glucose metrics. For all statistical tests, a p-value of p≤0.05 was considered significant. All statistics were performed using the Prism software package (GraphPad Software, La Jolla, CA).
Fasting glucose measurements
Animals were fasted overnight for 14–16 hours prior to the first tamoxifen injection and one week following the final tamoxifen injection. Fasting glucose levels were measured using a FreeStyle Freedom glucose meter (Abbott Laboratories, Abbott Park, IL).
Tumor genotype analysis by polymerase chain reaction (PCR)
Tumor tissue was isolated directly from OCT embedded tissues, and genomic DNA was isolated using the QIAmp DNA Micro kit (Qiagen, Valencia, CA). PCR was performed using standard methods. Primers used were as follows: Cre (forward: 5′-CATGTTCAGGGATCGCCAGG-3′ and reverse: 5′-TGCGGTGCTAACCAGCGTTTT-3′); β2 microglobulin (forward: 5′-CACCGGAGAATGGGAAGCCGAA-3′ and reverse: 5′-TCCACACAGATGGAGCGTCCAG-3′); Dsp-WT/Flox (forward: 5′-GGTTGGGCCTCTCGAATCATGAGTGTCTAGCG-3′ and reverse: 5′-TGTCTGTTGCCATGTGATGCC-3′); Dsp-Recombined/Non-Recombined (forward: 5′-ACAGGCCAGATGAGATCACC-3′ and reverse: 5′-TGTCTGTTGCCATGTGATGCC-3′).
Real-time quantitative PCR
Normal islets were isolated from six-week-old wild-type B6 mice, and hyperplastic islets were isolated from six-week-old RT2 B6 mice as previously described [39]. Angiogenic islets were isolated from nine-week-old RT2 B6 mice by selection based on their red, hemorrhagic appearance following collagenase digestion of pancreata [39]. Islet tumors were excised from the surrounding exocrine pancreas from 14-week-old RT2 B6 mice. Total RNA was purified using the RNeasy Mini kit (Qiagen, Valencia, CA) and DNase I treated (Qiagen, Valencia, CA). cDNA was synthesized using the iScript cDNA Synthesis kit (Bio-Rad Laboratories, Hercules, CA). Real-time quantitative PCR was performed using a 7900HT system (Applied Biosystems, Foster City, CA) (see Table S4 for a complete list of primers used in this study) according to the manufacturer's specifications.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HanahanD
WeinbergRA
2000 The hallmarks of cancer. Cell 100 57 70
2. ChristoforiG
2006 New signals from the invasive front. Nature 441 444 450
3. HirohashiS
1998 Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J Pathol 153 333 339
4. HirakiA
ShinoharaM
IkebeT
NakamuraS
KuraharaS
1996 Immunohistochemical staining of desmosomal components in oral squamous cell carcinomas and its association with tumour behaviour. Br J Cancer 73 1491 1497
5. ShinoharaM
HirakiA
IkebeT
NakamuraS
KuraharaS
1998 Immunohistochemical study of desmosomes in oral squamous cell carcinoma: correlation with cytokeratin and E-cadherin staining, and with tumour behaviour. J Pathol 184 369 381
6. GarrodD
ChidgeyM
2008 Desmosome structure, composition and function. Biochim Biophys Acta 1778 572 587
7. HartsockA
NelsonWJ
2008 Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta 1778 660 669
8. PerlAK
WilgenbusP
DahlU
SembH
ChristoforiG
1998 A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 392 190 193
9. VleminckxK
VakaetLJr
MareelM
FiersW
van RoyF
1991 Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 66 107 119
10. HanahanD
1985 Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 315 115 122
11. ChristoforiG
NaikP
HanahanD
1994 A second signal supplied by insulin-like growth factor II in oncogene-induced tumorigenesis. Nature 369 414 418
12. InoueM
HagerJH
FerraraN
GerberHP
HanahanD
2002 VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic beta cell carcinogenesis. Cancer Cell 1 193 202
13. OlsonP
LuJ
ZhangH
ShaiA
ChunMG
2009 MicroRNA dynamics in the stages of tumorigenesis correlate with hallmark capabilities of cancer. Genes Dev 23 2152 2165
14. GallicanoGI
KouklisP
BauerC
YinM
VasioukhinV
1998 Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol 143 2009 2022
15. LopezT
HanahanD
2002 Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 1 339 353
16. JeanesA
GottardiCJ
YapAS
2008 Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene 27 6920 6929
17. HerzigM
SavareseF
NovatchkovaM
SembH
ChristoforiG
2007 Tumor progression induced by the loss of E-cadherin independent of beta-catenin/Tcf-mediated Wnt signaling. Oncogene 26 2290 2298
18. VasioukhinV
BowersE
BauerC
DegensteinL
FuchsE
2001 Desmoplakin is essential in epidermal sheet formation. Nat Cell Biol 3 1076 1085
19. GuG
DubauskaiteJ
MeltonDA
2002 Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129 2447 2457
20. OffieldMF
JettonTL
LaboskyPA
RayM
SteinRW
1996 PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 122 983 995
21. Gidekel FriedlanderSY
ChuGC
SnyderEL
GirniusN
DibeliusG
2009 Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 16 379 389
22. BergersG
JavaherianK
LoKM
FolkmanJ
HanahanD
1999 Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science 284 808 812
23. BazziH
ChristianoAM
2007 Broken hearts, woolly hair, and tattered skin: when desmosomal adhesion goes awry. Curr Opin Cell Biol 19 515 520
24. ChidgeyM
DawsonC
2007 Desmosomes: a role in cancer? Br J Cancer 96 1783 1787
25. RunswickSK
O'HareMJ
JonesL
StreuliCH
GarrodDR
2001 Desmosomal adhesion regulates epithelial morphogenesis and cell positioning. Nat Cell Biol 3 823 830
26. TselepisC
ChidgeyM
NorthA
GarrodD
1998 Desmosomal adhesion inhibits invasive behavior. Proc Natl Acad Sci U S A 95 8064 8069
27. PapagerakisS
ShabanaAH
PollockBH
PapagerakisP
DepondtJ
2009 Altered desmoplakin expression at transcriptional and protein levels provides prognostic information in human oropharyngeal cancer. Hum Pathol 40 1320 1329
28. PeinadoH
OlmedaD
CanoA
2007 Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 7 415 428
29. HalbleibJM
NelsonWJ
2006 Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes Dev 20 3199 3214
30. LewisJE
JensenPJ
WheelockMJ
1994 Cadherin function is required for human keratinocytes to assemble desmosomes and stratify in response to calcium. J Invest Dermatol 102 870 877
31. LewisJE
WahlJKIII
SassKM
JensenPJ
JohnsonKR
1997 Cross-talk between adherens junctions and desmosomes depends on plakoglobin. J Cell Biol 136 919 934
32. NavaP
LaukoetterMG
HopkinsAM
LaurO
Gerner-SmidtK
2007 Desmoglein-2: a novel regulator of apoptosis in the intestinal epithelium. Mol Biol Cell 18 4565 4578
33. WanH
SouthAP
HartIR
2007 Increased keratinocyte proliferation initiated through downregulation of desmoplakin by RNA interference. Exp Cell Res 313 2336 2344
34. VasioukhinV
BauerC
DegensteinL
WiseB
FuchsE
2001 Hyperproliferation and defects in epithelial polarity upon conditional ablation of alpha-catenin in skin. Cell 104 605 617
35. GochevaV
ZengW
KeD
KlimstraD
ReinheckelT
2006 Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev 20 543 556
36. JoyceJA
FreemanC
Meyer-MorseN
ParishCR
HanahanD
2005 A functional heparan sulfate mimetic implicates both heparanase and heparan sulfate in tumor angiogenesis and invasion in a mouse model of multistage cancer. Oncogene 24 4037 4051
37. GochevaV
WangHW
GadeaBB
ShreeT
HunterKE
2010 IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev 24 241 255
38. LawlorER
SoucekL
Brown-SwigartL
ShchorsK
BialuchaCU
2006 Reversible kinetic analysis of Myc targets in vivo provides novel insights into Myc-mediated tumorigenesis. Cancer Res 66 4591 4601
39. ParangiS
DietrichW
ChristoforiG
LanderES
HanahanD
1995 Tumor suppressor loci on mouse chromosomes 9 and 16 are lost at distinct stages of tumorigenesis in a transgenic model of islet cell carcinoma. Cancer Res 55 6071 6076
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 9
Nejčtenější v tomto čísle
- Synthesizing and Salvaging NAD: Lessons Learned from
- Optimal Strategy for Competence Differentiation in Bacteria
- Long- and Short-Term Selective Forces on Malaria Parasite Genomes
- Identifying Signatures of Natural Selection in Tibetan and Andean Populations Using Dense Genome Scan Data