
Dementia Revealed: Novel Chromosome 6 Locus for Late-Onset Alzheimer Disease Provides Genetic Evidence for Folate-Pathway Abnormalities
Genome-wide association studies (GWAS) of late-onset Alzheimer disease (LOAD) have consistently observed strong evidence of association with polymorphisms in APOE. However, until recently, variants at few other loci with statistically significant associations have replicated across studies. The present study combines data on 483,399 single nucleotide polymorphisms (SNPs) from a previously reported GWAS of 492 LOAD cases and 496 controls and from an independent set of 439 LOAD cases and 608 controls to strengthen power to identify novel genetic association signals. Associations exceeding the experiment-wide significance threshold () were replicated in an additional 1,338 cases and 2,003 controls. As expected, these analyses unequivocally confirmed APOE's risk effect (rs2075650, ). Additionally, the SNP rs11754661 at 151.2 Mb of chromosome 6q25.1 in the gene MTHFD1L (which encodes the methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 1-like protein) was significantly associated with LOAD (; Bonferroni-corrected P = 0.022). Subsequent genotyping of SNPs in high linkage disequilibrium () with rs11754661 identified statistically significant associations in multiple SNPs (rs803424, P = 0.016; rs2073067, P = 0.03; rs2072064, P = 0.035), reducing the likelihood of association due to genotyping error. In the replication case-control set, we observed an association of rs11754661 in the same direction as the previous association at P = 0.002 ( in combined analysis of discovery and replication sets), with associations of similar statistical significance at several adjacent SNPs (rs17349743, P = 0.005; rs803422, P = 0.004). In summary, we observed and replicated a novel statistically significant association in MTHFD1L, a gene involved in the tetrahydrofolate synthesis pathway. This finding is noteworthy, as MTHFD1L may play a role in the generation of methionine from homocysteine and influence homocysteine-related pathways and as levels of homocysteine are a significant risk factor for LOAD development.
Published in the journal:
. PLoS Genet 6(9): e32767. doi:10.1371/journal.pgen.1001130
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001130
Summary
Genome-wide association studies (GWAS) of late-onset Alzheimer disease (LOAD) have consistently observed strong evidence of association with polymorphisms in APOE. However, until recently, variants at few other loci with statistically significant associations have replicated across studies. The present study combines data on 483,399 single nucleotide polymorphisms (SNPs) from a previously reported GWAS of 492 LOAD cases and 496 controls and from an independent set of 439 LOAD cases and 608 controls to strengthen power to identify novel genetic association signals. Associations exceeding the experiment-wide significance threshold () were replicated in an additional 1,338 cases and 2,003 controls. As expected, these analyses unequivocally confirmed APOE's risk effect (rs2075650, ). Additionally, the SNP rs11754661 at 151.2 Mb of chromosome 6q25.1 in the gene MTHFD1L (which encodes the methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 1-like protein) was significantly associated with LOAD (; Bonferroni-corrected P = 0.022). Subsequent genotyping of SNPs in high linkage disequilibrium () with rs11754661 identified statistically significant associations in multiple SNPs (rs803424, P = 0.016; rs2073067, P = 0.03; rs2072064, P = 0.035), reducing the likelihood of association due to genotyping error. In the replication case-control set, we observed an association of rs11754661 in the same direction as the previous association at P = 0.002 ( in combined analysis of discovery and replication sets), with associations of similar statistical significance at several adjacent SNPs (rs17349743, P = 0.005; rs803422, P = 0.004). In summary, we observed and replicated a novel statistically significant association in MTHFD1L, a gene involved in the tetrahydrofolate synthesis pathway. This finding is noteworthy, as MTHFD1L may play a role in the generation of methionine from homocysteine and influence homocysteine-related pathways and as levels of homocysteine are a significant risk factor for LOAD development.
Introduction
Alzheimer disease (AD) [MIM 104300] is a neurodegenerative disorder characterized by memory and cognitive impairment affecting more than 13% of individuals aged 65 years and older [1], [2] and constitutes the most common form of dementia among older adults. While several major genes contributing to risk of Alzheimer Disease have been identified (APP [3], PS1 [4], PS2 [5]–[7]), all but one (APOE [8]–[10]) contributed predominantly to early-onset forms of AD that cluster within families; other than APOE, few consistent association signals have been observed for late-onset AD (LOAD). Recent estimates of the heritability of LOAD fall between 60% and 80% [11]. However, while APOE ε4-alleles elevate AD risk, only 50% of AD cases carry an APOE ε4 allele, suggesting genetic factors elsewhere in the genome contribute to AD risk [12].
At present, eleven studies have tested association with LOAD on genome-wide panels of single nucleotide polymorphisms (SNPs). Most [13]–[22], but not all [23], of these studies indirectly observed associations with APOE on chromosome 19q with strong experiment-wide statistical significance. However, only a few of the studies observed associations at other loci exceeded experiment-wide statistical significance thresholds. A follow-up study [15] to Coon et al. [14] stratifying cases and controls by APOE genotype detected strong associations with GAB2 (MIM:606203) SNPs, and in follow-up work observed altered GAB2 transcript levels in vulnerable neurons, and an effect of GAB2 levels on tau phosphorylation; replication studies observed mixed results. In a family-based study of LOAD, Bertram et al. [17] observed four SNP associations exceeding adjusted experiment-wide thresholds for statistical significance, including one for the chromosome. Our group reported a SNP association with experiment-wide statistical significance on chromosome 12q13 [18]. A GWAS originating from the Mayo Clinic [19] identified a novel signal on the X chromosome in the gene PCDH11X (MIM: 300246), encoding a protocadherin, a cell-cell adhesion molecule expressed in the brain. Generally, these earlier reports have not been consistently replicated in other studies, possibly due to sample sizes that are substantially smaller than those of GWAS studies that have successfully identified genes for other complex disorders [24], [25]. Two large collaborative GWAS of LOAD examined many thousands of cases and controls [20], [21] and both identified novel association signals in the gene CLU (aka APOJ, MIM: 185430; Apolipoprotein J or Clusterin), as well as signals in CR1 (MIM: 120620, Complement Component Receptor 1) and in PICALM (MIM: 603025, Phosphatidylinositol-Binding Clathrin Assembly Protein), reporting some of the most consistent results for LOAD to date.
Even with the increased sample sizes and improved statistical power to detect loci with moderate effect sizes, it remains unlikely that these studies, incorporating cases and controls from multiple samples with varying case/control inclusion criteria, have identified all loci with modest effect sizes in LOAD. We analyzed genome-wide association in a discovery dataset of 931 cases and 1,104 controls and performed replication analysis on the strongest associations (P<10−5) using genotype data from four existing studies totaling 1,338 cases and 2,003 controls.
Results
Dataset Characteristics
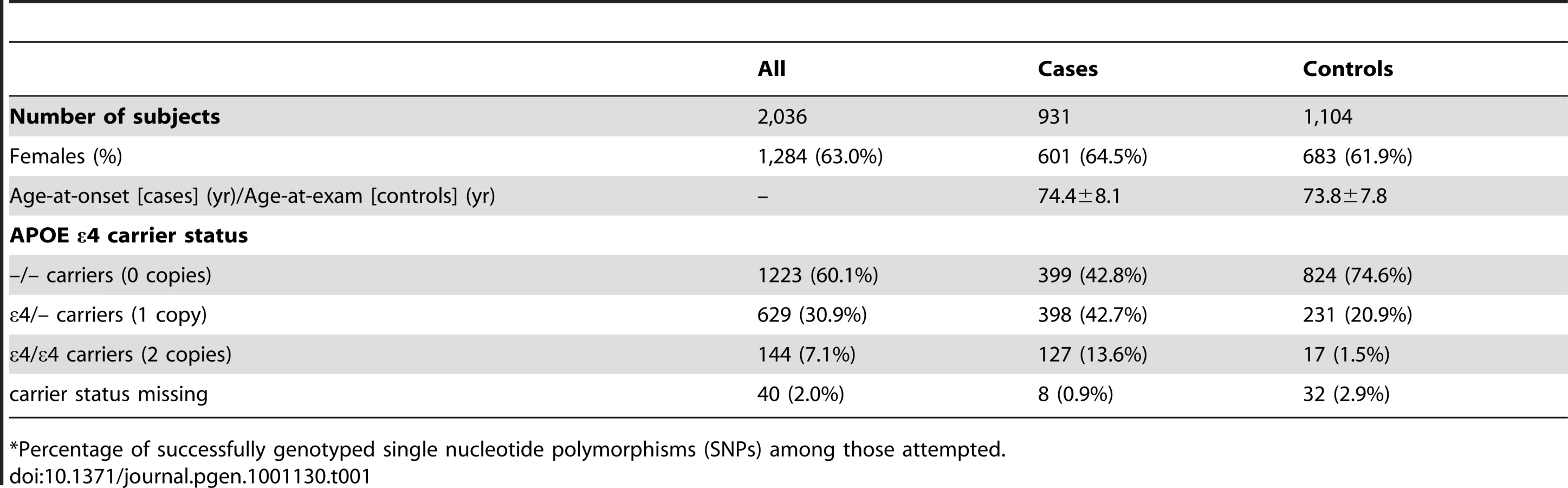
Table 1 depicts the demographic characteristics of the case and control samples examined in initial association analyses. We examined 931 LOAD cases, average age 74.4 years at onset (standard deviation: ±8.1 years), and 1,104 cognitive controls, average age 73.8 years at exam (±7.8 years) (Table 1). Cases were 64.5% female, while controls were 61.9% female.
GWAS Results
11 SNPs had association p-values (P)<10−5 after adjustment for population substructure (Table 2; Q-Q plot for all association results in Figure 1; P<10−4 in Tables S1, S2; all association results in Figure S1). Although the SNPs defining the APOE ε2, ε3, and ε4 alleles, rs429358 and rs7412, were not included on our genotyping platforms, we independently genotyped these SNPs and tested the association of APOE ε4 with LOAD risk (OR (95% CI): 4.18 (3.51, 4.97); ). SNPs adjacent to the APOE haplotype on chromosome 19 otherwise demonstrated the highest associations observed, with the peak association being rs2075650 with , confirming the expected effect of APOE on LOAD risk in this sample. The most significant non-APOE SNP in our previous GWAS [18] (Table S3) was rs11610206 on 12q13 (45.92 Mb) with ; in this study, this SNP was still strongly assoFciated with LOAD (OR (95% CI): 0.67 (0.54, 0.85); ), but not with experiment-wide statistical significance.


The SNP rs11754661, located at 151.2Mb of chromosome 6q25.1 in the gene MTHFD1L, was significantly associated with LOAD (; Bonferroni-corrected P = 0.022). To ensure that this association was not spurious due to differences between subsets of genotyped samples, we performed several post-hoc quality control analyses. We examined clustering plots from genotype calling by platform to determine if misclassification could have affected the associations observed for the top 11 SNPs with P<10−5, and observed discrete clustering by genotype for all 11 SNPs. We found no evidence of a difference in genotype frequencies among controls across subsets by genotyping platform (Fisher's exact test P = 0.71) or by study center (Fisher's exact test P = 0.95, Table S4). We also examined differences in dataset characteristics including variation in age, sex, and APOE ε4 genotype distributions, and found limited differences between subsets by study center, autopsy - or clinical-confirmation of case or control status, and by genotyping platform (Table S5). Subsequently, we examined the first hundred principal components generated from EIGENSTRAT to determine if any of the principal components were associated with both differences in genotyping platform subset and disease status at P<0.05 as markers of potential systematic bias. While two principal components other than those used to adjust for population substructure showed association with both genotyping platform subset and LOAD, additional adjustment for these principal components did not change the strength of association between rs11754661 and LOAD (data not shown). Models further adjusting for age, sex, and APOE ε4 carrier status (+/−) (Table S6) only marginally diminished the effect size and statistical significance of the association of rs11754661 with LOAD (adjustment for age and sex, OR (95% CI): 2.03 (1.56, 2.64), ; adjustment for age, sex and APOE ε4 (+/−), OR (95% CI): 2.01 (1.51, 2.67), ).
Furthermore, we examined the associations in 4 SNPs in linkage disequilibrium (LD) (Figures S2) of D'>0.8 with rs11754661, which demonstrated variable patterns of association with LOAD (Figure 2; rs2839947, P = 0.0479; rs11757561, P = 0.000684; rs2073066, P = 0.768; rs13201018, P = 0.185). It should be noted that due to the low minor allele frequency (MAF) of rs11754661 (MAF = 0.07), only one of these SNPs, rs11757561 (MAF = 0.20), had an r2>0.10 (r2 = 0.23). This SNP had a similar direction of association as rs11754461 (OR (95% CI): 1.31 (1.12, 1.53)).

Based on the pattern of LD in the vicinity of rs11754661, we examined several haplotypes of MTHFD1L which included this SNP (described in Text S1) to identify potential markers for untyped variants associated with LOAD. Two haplotypes (the first comprising rs2073066-rs11754661-rs13201018, the second comprising rs2839947-rs11757561-rs2073066-rs11754661-rs13201018) both containing the risk-increasing A allele of rs11754661 had highly statistically significant associations similar to the genotypic association of rs11754661 ( and , respectively) (Table S7). Both haplotypes had similar frequencies (MHF) to the A allele of rs11754661 (MHF = 0.0696 and MHF = 0.0629, respectively).
In order to ensure that the association we observed at rs11754661 was not merely due to genotyping error, we genotyped four additional SNPs in MTHFD1L proximal to and in high LD (r2>0.8) with rs11754661. All SNPs but one (rs7765521, P = 0.055) demonstrated associations with nominal statistical significance (rs803424, P = 0.016; rs2073067, P = 0.030; and rs2072064, P = 0.035). Figure 2 shows the −log10-transformed P-values for single SNP tests of association in the MTHFD1L and 50kb flanking region (151.2Mb–151.3Mb) surrounding the chromosome 6 association signal at rs11754661, among both SNPs genotyped in the initial GWAS and those genotyped subsequently.
Association analyses of pooled datasets combining data on 1,242 cases and 1,737 controls confirmed experiment-wide statistically significant associations for SNPs in/near APOE (replication from to P = 0.00187) for all but one SNP (rs8106922; discovery , replication P = 0.108) (Table 2), however the direction of association in the replication was consistent across each of these SNPs. The association of the MTHFD1L SNP rs11754661 in the replication was both statistically significant (P = 0.00187) and showed similar strength and direction (discovery OR (95%CI): 2.03 (1.58, 2.62); replication OR (95%CI): 2.34 (1.37, 3.98)).
Association in Combined Discovery and Replication Datasets
In combined analyses, associations in and around the APOE locus were unequivocally strengthened, with the p-values observed ranging from to . Variation at rs11754461 was strongly associated () with an elevated risk of LOAD with OR = 2.10 (95% CI: 1.67, 2.64). Several adjacent SNPs also demonstrated nominal associations with similar direction of effect, including rs11757561 (P = 0.000846) with OR = 1.31 (95% CI: 1.12, 1.53) and rs12195069 (P = 0.0432) with OR = 1.25 (95% CI: 1.01, 1.55).
Two SNPs with only modest statistical significance of association in the discovery GWAS demonstrated highly statistically significant association in analyses combining both discovery and replication datasets (Tables S1 and S2). SNPs rs4676049 and rs17034806, located at 109Mb on chromosome 2q13, had associations of OR = 1.62 () and OR = 1.61 () respectively in the discovery dataset. However, combining discovery and replication datasets, the SNP associations gained modest strength in effect size (OR = 1.76 for rs4676049 and OR = 1.75 for rs17034806), but the associations now exceeded the threshold for experiment-wide statistical significance, with for rs4676049 and for rs17034806.
Discussion
Although associations with experiment-wide statistical significance have not been observed for MTHFD1L in previous GWAS of LOAD, biological evidence suggests a role for this gene in dementia and AD pathology. MTHFD1L, which encodes the methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 1-like protein, is involved in tetrahydrofolate (THF) synthesis, catalyzing the reversible synthesis of 10-formyl-THF to formate and THF, an important step in homocysteine conversion to methionine [26]. Elevated plasma homocysteine levels have been implicated in AD [27], [28] and other neurodegenerative disease including Parkinson's [29], and have been recognized as a risk factor for pre-eclampsia [30], diabetic complications [31], and heart disease [32]. Interestingly, a recent GWAS of coronary artery disease (CAD) identified MTHFD1L as a CAD risk factor in both British and German populations studied [33]. Several potential mechanisms may explain this connection: hyperhomocysteinaemia may influence AD dementia by causing vascular alterations [34]; it may cause cholinergic deficit due to toxicity to cortical neurons [35]; several lines of evidence suggest that elevated homocysteine contributes to AD risk through increased oxidative stress [36]–[38]. On-going biological investigations are continuing to elucidate the pathways connecting elevated homocysteine with AD.
Mthfd1l protein has been reported to be decreased in the hippocampus in a mouse model of AD using a proteomic approach [39]. Homocysteic acid, derived from homocysteine and methionine, is elevated in these mice and treatment with antibodies to homocysteic acid reduced amyloid burden and inhibited cognitive decline in these animals [40]. B6-deficient diets lead to further increases in homocysteic acid in these mice.
That we observed an experiment-wide statistically significant association in MTHFD1L in addition to the associations of a number of APOE SNPs with LOAD risk is consistent with results from previous work. MTHFD1L is located on chromosome 6q25.1, near linkage signals observed in two prior genome-wide linkage studies of LOAD [41], [42]. The previous GWAS performed by our group [18], from which nearly 1,000 individuals in the current study were drawn, observed a strong, but not experiment-wide, statistically significant association between the same MTHFD1L SNP and LOAD at P = 2.01×10−5. Experiment-wide statistical significance for this association was observed with the addition of another 1,047 individuals in this study.
We did not observe associations with LOAD with experiment-wide statistical significance in any of the peak non-APOE signals identified in previous GWAS studies, including the APOJ/CLU SNP rs11136000 that was identified in both Harold et al. and Lambert et al. studies (analysis of this dataset reported elsewhere (Jun et al., in preparation)). Given the observed OR = 0.86 of rs11136000 for LOAD, in our sample of 931 cases and 1,104 controls, we had <1% power to detect the observed effect at the Bonferroni-corrected threshold for experiment-wide statistical significance, α = 1.03×10−7, suggesting that most significant associations of small or modest effect size would be missed in this study. The association of variation in PCDH11X and GAB2 was not observed in this dataset; the findings of these analyses are reported elsewhere [43]. In addition, we observed a strong association of the chromosome 12 SNP rs11610206 with LOAD, but not with genomewide statistical significance as observed in our previous GWAS [18], suggesting that the findings of the Beecham et al. study, as with previous LOAD GWAS, may be subject to the “winner's curse” [44].
Despite a wealth of evidence for the role of chromosome 2 loci in Alzheimer's Disease, the chromosome 2q13 SNPs identified with experiment-wide statistically significant associations in the combined analyses, rs4676049 and rs17034806, do not fall in the vicinity of chromosome 2 regions of interest [22], .
Based on the patterns of studies emerging in other complex diseases, GWAS studies with sample sizes greater than the combined Lambert et al. and Harold et al. datasets may be necessary to validate associations observed in smaller GWAS studies and to identify susceptibility variants with more modest effects. This approach has been taken in type 2 diabetes, where a meta-analysis of 54,000 subjects confirmed multiple susceptibility loci [43]. Other approaches to identify new susceptibility variants are exploring the Common Disease-Rare Variants (CDRV) hypothesis, which aim to identify novel susceptibility loci for disease by assessing the aggregate effects of multiple rare variants in single genes on disease risk [46].
In this genome-wide association study of LOAD, we identified a novel association with experiment-wide statistical significance in a gene with a potential biological role, MTHFD1L. We replicated this association in additional publicly-available genomewide association datasets, and observed statistically significant association with a similar effect size and direction at this SNP. In summary, MTHFD1L is an excellent candidate for LOAD on account of its involvement in folate-pathway abnormalities linked with homocysteine, a significant biological risk factor for AD.
Methods
Ethics Statement
After complete description of the study to the subjects, written informed consent was obtained from all participants, in agreement with protocols approved by the institutional review board at each contributing center.
Ascertainment
Discovery dataset cases and controls were clinically ascertained through the Collaborative Alzheimer's Project (CAP) comprising the University of Miami John P. Hussman Institute for Human Genomics (HIHG) and the Vanderbilt University Center for Human Genetics Research (CHGR), and autopsy-verified cases and controls were collected through the Mount Sinai Brain Bank (MSBB) at the Mount Sinai School of Medicine (see [47]). Additional controls were also identified in the National Cell Repository for Alzheimer's Disease (NCRAD). 266 cases and 643 controls genotyped in the discovery dataset from the NCRAD, HIHG, and CHGR [48] and NCRAD were independent from previously published data sets including those from our group's previously published GWAS [18]. All CAP-ascertained cases and controls were recruited and evaluated using standardized criteria and protocols, and case adjudication in the CAP was performed jointly by a Clinical Advisory Board (CAB) composed of both HIHG and CHGR members, with controls evaluated jointly as well.
All cases and controls from the HIHG, CHGR, and NCRAD met selection criteria described in the Beecham et al. study [18]. Briefly, the study was described and written informed consists were obtained from all participants, in accordance with institutional review board protocols at each study center. Each individual classified as a LOAD case met the NINCDS-ADRDA criteria for probable or definite AD and had an age at onset greater than 60 years of age [49], as determined from specific questions within the clinical history answered by a reliable family informant or from documented significant cognitive impairment in the medical record. Vascular dementia was diagnosed according to contemporary standards [50] by the CAB, and individuals with confirmed vascular dementia or phenotypic uncertainty were excluded from analyses. Cognitive controls were individuals who showed signs of dementia in clinical history or upon interview, and were drawn from spouses, friends, and other biologically unrelated individuals of cases, were frequency-matched by age and gender to the cases, and were located in the same clinical catchment areas. All cognitive controls were examined, and none showed signs of dementia in clinical history or upon interview. Also, each cognitive control had a documented Mini-Mental State Exam (MMSE) score ≥27 or a Modified Mini-Mental State (3MS) Exam score ≥87. Clinical history and interview data for NCRAD controls, including MMSE scores, were made available and collected along with whole blood for DNA extraction for inclusion in our study.
306 cases and 81 controls identified in the MSBB were recently deceased patients at the Mount Sinai Medical Center in New York, NY, and had affection status verified through clinical review and brain autopsy. Neither the cases nor controls examined have been used in previously published studies. Covariates including age at death and sex were abstracted from reviews of medical charts performed by members of the MSBB.
In total, 572 new cases and 724 new controls were genotyped in this study, and after quality controls measures, combined with data on 492 cases and 496 controls from the previous GWAS [18] for analysis. We also had available for replication from the HIHG, a dataset of 246 cases and 69 cognitively normal controls from a previously described dataset [51].
Genotyping
We extracted DNA for individuals ascertained by the HIHG, CHGR, MSBB, and NCRAD from whole blood by using Puregene chemistry (QIAGEN, Germantown, MD, USA). We performed genotyping using the Illumina Beadstation and the Illumina Infinium Human 1M beadchip on 530 cases and 393 controls following the recommended protocol, only with a more stringent GenCall score threshold of 0.25. Genotyping on 248 controls from the PD GWAS dataset [48] was performed using the Illumina Infinium Human 610-Quad beadchip. Genotyping efficiency was greater than 99%, and quality assurance was achieved by the inclusion of one CEPH control per 96-well plate that was genotyped multiple times. Technicians were blinded to affection status and quality-control samples. We used Taqman Genotyping Assays for SNPs +3937/rs429358 and +4075/rs7412 and performed allelic discrimination/genotype calling on the ABI 7900 Taqman system, the results of which were used to determine APOE ε2/ε3/ε4 genotypes.
After excluding samples which failed quality control (described in the next section) with low genotyping call rates, genotype data was available on 870,954 SNPs (after quality control) using the Illumina 1M BeadChip on 440 cases and 437 controls, while genotype data on 490,960 SNPs (after quality control) from the Illumina 610Quad BeadChip was available on 172 controls. Combining these data with the 522,366 SNPs on 492 cases and 496 controls in our previous GWAS [18], a set of 483,399 SNPs common to all platforms was generated that passed quality control for each subset individually and in a pooled dataset. The Bonferroni-corrected threshold for experiment-wide statistical significance was thus set at Bonferroni-corrected .
Sample Quality Control
After genotyping, multiple quality controls were performed including assessment of sample efficiency, which is the proportion of valid genotype calls to attempted calls within a sample. Samples with efficiency less than 0.98 were dropped from the analysis. Reported gender and genetic gender were examined with the use of X-linked SNPs; 32 inconsistent samples were dropped from the analysis. Relatedness between samples was tested via the program Graphical Representation of Relatedness (GRR) [52], and 3 related samples were dropped from the analysis.
To determine if population substructure exists in the case-control sample, a set of 10,000 SNPs with MAF>0.25, selected for minimal between-SNP linkage disequilibrium (r2<0.20), and spread evenly across the autosomal chromosomes were analyzed using the program STRUCTURE [53], [54] (burn in: 5,000, iterations: 25,000) assuming different number of assumed subpopulations (K). The −log likelihood for K was maximized at K = 3, suggesting population substructure. Further analysis was performed in EIGENSTRAT [55], where principal components analysis on the sample of 10,000 SNPs was used to generate principal component loadings for samples and remove outliers by using the top ten principal components over 5 iterations with a threshold of six standard deviations. The top three principal component loadings were used as covariates to account for population structure in the association analysis.
Removing genotyped individuals with low genotype call rates, incorrect reported gender, high relatedness with other samples, and extreme outliers in substructure analyses, 440 cases and 608 controls remained for inclusion in analysis, and were combined with 492 cases and 496 controls from the previous GWAS.
SNP Quality Control
Quality control was performed to remove any low quality SNPs. Genotype clusters were redefined using signal intensities of samples with efficiency greater than 0.98, and genotypes were recalled on the basis of these new clusters per the manufacturer's recommendation. Efficiency of individual SNPs was estimated as the proportion of samples with genotype calls for a given SNP, and SNPs with efficiency less than 0.95 were dropped from analysis. Due to concerns of low statistical power to detect association, SNPs with MAF<0.005 were dropped from analysis. Hardy-Weinberg Disequilibrium (HWD) statistics were calculated among controls with the Fisher's exact test in the PLINK software package [56]; SNPs with P<10−6 for HWD were dropped from analysis. In addition, due to concerns with the spurious association originating from the use of different genotyping platforms on samples in the previous and current GWAS studies, distributions of genotype frequencies at each SNP in each study were examined among controls using a Fisher's exact test, and SNPs with highly-differing genotype distributions across genotyping subsets (P<0.001) were dropped prior to analysis. After these quality control measures, 483,399 SNPs remained for association analysis.
Association Analysis
Association analysis was performed using logistic regression to test association of genotypes with LOAD under an additive model. Logistic regression was used to permit covariate adjustment for loadings taken from the first three principal components identified in EIGENSTRAT to account for population substructure. Here we report results from logistic regression models adjusting only for population substructure with principal components. Further regression modeling was also performed on SNPs with initial associations of P<10−5, extending models to adjust for APOE genotype (designated as the number of ε4 alleles), age-at-onset in cases and age-at-exam in controls, and gender as covariates (Table S6). All analyses were performed using the PLINK software package [56].
Quantile-quantile plots of the associations were made (Figure 1), and suggest the absence of systematic bias in the tests of association.
Imputation and Replication Analysis
To provide independent replication of the associations observed in the discovery dataset, genome-wide genotyping data were combined from four additional datasets (one unpublished and three publicly-available datasets) and missing genotype data imputed using IMPUTE v1.0 [57] (Table S8). SNPs with differing genotypic distributions between datasets were excluded from imputation using the Fisher's exact test approached described earlier [58]. Both primary and replication datasets were imputed to a HapMap reference of over 2.5 million SNPs. Individual genotypes with probability less than 0.90 were not included, and SNPs missing >10% of genotypes within either data set were dropped. In addition to using the combined Hapmap Phase III CEPH Utah pedigree (CEU) and Tuscan (TSI) haplotype reference panels for imputation, for imputation within each study, we used genotype data on controls from other datasets to improve imputation accuracy, and Affymetrix 5.0 genotype data on 105 individuals genotyped in an independent Ashkenazi Jewish genotyping panel [59].
We analyzed existing pooled and imputed datasets of unrelated individuals from several studies: 147 cases and 182 controls from the Alzheimer's Disease Neuroimaging Initiative (ADNI) [60], 86 cases and 1,200 controls (all unrelated) from the Framingham Study SHARe dataset [61], and 859 cases and 552 controls from the Reiman et al. [15] LOAD GWAS dataset, and a set of 246 LOAD cases and 69 cognitively normal controls previously described [51] and genotyped on the Affymetrix 6.0 genotyping platform on which results have not been previously published.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. Alzheimer's Association 2009 2009 Alzheimer's Disease Facts and Figures. Washington, D.C.
2. HebertLE
ScherrPA
BieniasJL
BennettDA
EvansDA
2003 Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 60 1119 1122
3. GoateA
Chartier-HarlinMC
MullanM
BrownJ
CrawfordF
1991 Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349 704 706
4. SherringtonR
RogaevEI
LiangY
RogaevaEA
LevesqueG
1995 Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 375 754 760
5. Levy-LahadE
LahadA
WijsmanEM
BirdTD
SchellenbergGD
1995 Apolipoprotein E genotypes and age of onset in early-onset familial Alzheimer's disease. Ann Neurol 38 678 680
6. Levy-LahadE
WascoW
PoorkajP
RomanoDM
OshimaJ
1995 Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 269 973 977
7. RogaevEI
SherringtonR
RogaevaEA
LevesqueG
IkedaM
1995 Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 376 775 778
8. SaundersAM
StrittmatterWJ
SchmechelD
George-HyslopPH
Pericak-VanceMA
1993 Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology 43 1467 1472
9. CorderEH
SaundersAM
StrittmatterWJ
SchmechelDE
GaskellPC
1993 Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261 921 923
10. StrittmatterWJ
SaundersAM
SchmechelD
Pericak-VanceM
EnghildJ
1993 Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 90 1977 1981
11. GatzM
PedersenNL
BergS
JohanssonB
JohanssonK
1997 Heritability for Alzheimer's disease: the study of dementia in Swedish twins. J Gerontol A Biol Sci Med Sci 52 M117 125
12. HuangW
QiuC
von StraussE
WinbladB
FratiglioniL
2004 APOE genotype, family history of dementia, and Alzheimer disease risk: a 6-year follow-up study. Arch Neurol 61 1930 1934
13. GrupeA
AbrahamR
LiY
RowlandC
HollingworthP
2007 Evidence for novel susceptibility genes for late-onset Alzheimer's disease from a genome-wide association study of putative functional variants. Hum Mol Genet 16 865 873
14. CoonKD
MyersAJ
CraigDW
WebsterJA
PearsonJV
2007 A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer's disease. J Clin Psychiatry 68 613 618
15. ReimanEM
WebsterJA
MyersAJ
HardyJ
DunckleyT
2007 GAB2 alleles modify Alzheimer's risk in APOE epsilon4 carriers. Neuron 54 713 720
16. AbrahamR
MoskvinaV
SimsR
HollingworthP
MorganA
2008 A genome-wide association study for late-onset Alzheimer's disease using DNA pooling. BMC Med Genomics 1 44
17. BertramL
LangeC
MullinK
ParkinsonM
HsiaoM
2008 Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet 83 623 632
18. BeechamGW
MartinER
LiYJ
SliferMA
GilbertJR
2009 Genome-wide association study implicates a chromosome 12 risk locus for late-onset Alzheimer disease. Am J Hum Genet 84 35 43
19. CarrasquilloMM
ZouF
PankratzVS
WilcoxSL
MaL
2009 Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer's disease. Nat Genet 41 192 198
20. LambertJC
HeathS
EvenG
CampionD
SleegersK
2009 Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41 1094 1099
21. HaroldD
AbrahamR
HollingworthP
SimsR
GerrishA
2009 Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 41 1088 1093
22. SeshadriS
FitzpatrickAL
IkramMA
DeStefanoAL
GudnasonV
2010 Genome-wide analysis of genetic loci associated with Alzheimer disease. Jama 303 1832 1840
23. LiH
WettenS
LiL
St JeanPL
UpmanyuR
2008 Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol 65 45 53
24. AltshulerD
DalyMJ
LanderES
2008 Genetic mapping in human disease. Science 322 881 888
25. Welcome Trust Case Control Consortium 2007 Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447 661 678
26. PikeST
RajendraR
ArtztK
ApplingDR
2010 Mitochondrial C1-Tetrahydrofolate Synthase (MTHFD1L) Supports the Flow of Mitochondrial One-carbon Units into the Methyl Cycle in Embryos. J Biol Chem 285 4612 4620
27. Van DamF
Van GoolWA
2009 Hyperhomocysteinemia and Alzheimer's disease: A systematic review. Arch Gerontol Geriatr 48 425 430
28. MorrisMS
2003 Homocysteine and Alzheimer's disease. Lancet Neurol 2 425 428
29. HerrmannW
KnappJP
2002 Hyperhomocysteinemia: a new risk factor for degenerative diseases. Clin Lab 48 471 481
30. RobertsJM
CooperDW
2001 Pathogenesis and genetics of pre-eclampsia. Lancet 357 53 56
31. de LuisDA
FernandezN
ArranzML
AllerR
IzaolaO
2005 Total homocysteine levels relation with chronic complications of diabetes, body composition, and other cardiovascular risk factors in a population of patients with diabetes mellitus type 2. J Diabetes Complications 19 42 46
32. ArnesenE
RefsumH
BonaaKH
UelandPM
FordeOH
1995 Serum total homocysteine and coronary heart disease. Int J Epidemiol 24 704 709
33. SamaniNJ
ErdmannJ
HallAS
HengstenbergC
ManginoM
2007 Genomewide association analysis of coronary artery disease. N Engl J Med 357 443 453
34. SnowdonDA
GreinerLH
MortimerJA
RileyKP
GreinerPA
1997 Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. Jama 277 813 817
35. HoPI
OrtizD
RogersE
SheaTB
2002 Multiple aspects of homocysteine neurotoxicity: glutamate excitotoxicity, kinase hyperactivation and DNA damage. J Neurosci Res 70 694 702
36. McCaddonA
ReglandB
HudsonP
DaviesG
2002 Functional vitamin B(12) deficiency and Alzheimer disease. Neurology 58 1395 1399
37. McCaddonA
HudsonP
HillD
BarberJ
LloydA
2003 Alzheimer's disease and total plasma aminothiols. Biol Psychiatry 53 254 260
38. MattsonMP
SheaTB
2003 Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci 26 137 146
39. MartinB
BrennemanR
BeckerKG
GucekM
ColeRN
2008 iTRAQ analysis of complex proteome alterations in 3xTgAD Alzheimer's mice: understanding the interface between physiology and disease. PLoS One 3 e2750 doi:10.1371/journal.pone.0002750
40. HasegawaT
MikodaN
KitazawaM
LaFerlaFM
2010 Treatment of Alzheimer's disease with anti-homocysteic acid antibody in 3xTg-AD male mice. PLoS One 5 e8593 doi:10.1371/journal.pone.0008593
41. OlsonJM
GoddardKA
DudekDM
2002 A second locus for very-late-onset Alzheimer disease: a genome scan reveals linkage to 20p and epistasis between 20p and the amyloid precursor protein region. Am J Hum Genet 71 154 161
42. BlackerD
BertramL
SaundersAJ
MoscarilloTJ
AlbertMS
2003 Results of a high-resolution genome screen of 437 Alzheimer's disease families. Hum Mol Genet 12 23 32
43. BeechamGW
NajAC
GilbertJR
HainesJL
BuxbaumJD
2010 PCDH11X variation is not associated with late-onset Alzheimer disease susceptibility. Psychiatr Genet In press
44. CapenEC
ClappRV
CampbellWM
1971 Competitive Bidding in High-Risk Situations. Journal of Petroleum Technology 23 641 653
45. LeeJH
ChengR
SantanaV
WilliamsonJ
LantiguaR
2006 Expanded genomewide scan implicates a novel locus at 3q28 among Caribbean hispanics with familial Alzheimer disease. Arch Neurol 63 1591 1598
46. BodmerW
BonillaC
2008 Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet 40 695 701
47. HaroutunianV
PerlDP
PurohitDP
MarinD
KhanK
1998 Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol 55 1185 1191
48. EdwardsTL
ScottWK
AlmonteC
BurtA
PowellEH
2010 Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 74 97 109
49. McKhannG
DrachmanD
FolsteinM
KatzmanR
PriceD
1984 Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 34 939 944
50. RomanGC
TatemichiTK
ErkinjunttiT
CummingsJL
MasdeuJC
1993 Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology 43 250 260
51. SliferMA
MartinER
BronsonPG
Browning-LargeC
DoraiswamyPM
2006 Lack of association between UBQLN1 and Alzheimer disease. Am J Med Genet B Neuropsychiatr Genet 141B 208 213
52. AbecasisGR
ChernySS
CooksonWO
CardonLR
2001 GRR: graphical representation of relationship errors. Bioinformatics 17 742 743
53. PritchardJK
StephensM
DonnellyP
2000 Inference of population structure using multilocus genotype data. Genetics 155 945 959
54. PritchardJK
StephensM
RosenbergNA
DonnellyP
2000 Association mapping in structured populations. Am J Hum Genet 67 170 181
55. PriceAL
PattersonNJ
PlengeRM
WeinblattME
ShadickNA
2006 Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38 904 909
56. PurcellS
NealeB
Todd-BrownK
ThomasL
FerreiraMA
2007 PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81 559 575
57. MarchiniJ
HowieB
MyersS
McVeanG
DonnellyP
2007 A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 39 906 913
58. ZieglerA
2009 Genome-wide association studies: quality control and population-based measures. Genet Epidemiol 33 S45 S50
59. IntraGenDB population genetics database
60. FrankRA
GalaskoD
HampelH
HardyJ
de LeonMJ
2003 Biological markers for therapeutic trials in Alzheimer's disease. Proceedings of the biological markers working group; NIA initiative on neuroimaging in Alzheimer's disease. Neurobiol Aging 24 521 536
61. BachmanDL
WolfPA
LinnR
KnoefelJE
CobbJ
1992 Prevalence of dementia and probable senile dementia of the Alzheimer type in the Framingham Study. Neurology 42 115 119
62. RivaA
KohaneIS
2002 SNPper: retrieval and analysis of human SNPs. Bioinformatics 18 1681 1685
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 9
Nejčtenější v tomto čísle
- Synthesizing and Salvaging NAD: Lessons Learned from
- Optimal Strategy for Competence Differentiation in Bacteria
- Long- and Short-Term Selective Forces on Malaria Parasite Genomes
- Identifying Signatures of Natural Selection in Tibetan and Andean Populations Using Dense Genome Scan Data