
Genetic Rearrangements Can Modify Chromatin Features at Epialleles
Analogous to genetically distinct alleles, epialleles represent heritable states of different gene expression from sequence-identical genes. Alleles and epialleles both contribute to phenotypic heterogeneity. While alleles originate from mutation and recombination, the source of epialleles is less well understood. We analyze active and inactive epialleles that were found at a transgenic insert with a selectable marker gene in Arabidopsis. Both converse expression states are stably transmitted to progeny. The silent epiallele was previously shown to change its state upon loss-of-function of trans-acting regulators and drug treatments. We analyzed the composition of the epialleles, their chromatin features, their nuclear localization, transcripts, and homologous small RNA. After mutagenesis by T-DNA transformation of plants carrying the silent epiallele, we found new active alleles. These switches were associated with different, larger or smaller, and non-overlapping deletions or rearrangements in the 3′ regions of the epiallele. These cis-mutations caused different degrees of gene expression stability depending on the nature of the sequence alteration, the consequences for transcription and transcripts, and the resulting chromatin organization upstream. This illustrates a tight dependence of epigenetic regulation on local structures and indicates that sequence alterations can cause epigenetic changes at some distance in regions not directly affected by the mutation. Similar effects may also be involved in gene expression and chromatin changes in the vicinity of transposon insertions or excisions, recombination events, or DNA repair processes and could contribute to the origin of new epialleles.
Published in the journal:
. PLoS Genet 7(10): e32767. doi:10.1371/journal.pgen.1002331
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002331
Summary
Analogous to genetically distinct alleles, epialleles represent heritable states of different gene expression from sequence-identical genes. Alleles and epialleles both contribute to phenotypic heterogeneity. While alleles originate from mutation and recombination, the source of epialleles is less well understood. We analyze active and inactive epialleles that were found at a transgenic insert with a selectable marker gene in Arabidopsis. Both converse expression states are stably transmitted to progeny. The silent epiallele was previously shown to change its state upon loss-of-function of trans-acting regulators and drug treatments. We analyzed the composition of the epialleles, their chromatin features, their nuclear localization, transcripts, and homologous small RNA. After mutagenesis by T-DNA transformation of plants carrying the silent epiallele, we found new active alleles. These switches were associated with different, larger or smaller, and non-overlapping deletions or rearrangements in the 3′ regions of the epiallele. These cis-mutations caused different degrees of gene expression stability depending on the nature of the sequence alteration, the consequences for transcription and transcripts, and the resulting chromatin organization upstream. This illustrates a tight dependence of epigenetic regulation on local structures and indicates that sequence alterations can cause epigenetic changes at some distance in regions not directly affected by the mutation. Similar effects may also be involved in gene expression and chromatin changes in the vicinity of transposon insertions or excisions, recombination events, or DNA repair processes and could contribute to the origin of new epialleles.
Introduction
Epialleles are heritable states of different gene expression from sequence-identical genes and have been described in several organisms [1]–[3]. Like genetically different alleles, epialleles contribute to phenotypic heterogeneity [4]–[5]. While the mutagenic processes creating DNA sequence allele variations are relatively well understood, little is known about how and when epialleles originate, and it is difficult to investigate this in statu nascendi. In plants, epialleles were described as natural variants [6]–[9], mutation-induced [10]–[12], or associated with tissue-culture [13]–[15]. Once established, epialleles can acquire stability over many generations; however, they have much higher reversion rates than genetic alleles. Therefore, analyzing the switch from one epigenetic state to the other at well-characterized epialleles can provide insight into their natural origin.
Pairs of epialleles are characterized by antithetic histone modifications at the associated nucleosomes, transcriptional activity at the expressed form, and transcriptional gene silencing (TGS) at the other. In some fungi, mammals, and higher plants, the latter is connected with cytosine methylation at the epiallele [e.g. 6], [16]–[17]. Several pairs of epialleles in plants define easily scored phenotypes like morphology [6], [10], development [9], pigmentation [7], [18], or reporter gene expression [19]–[20]. Some epialleles, as well as many other epigenetically controlled genes, have been used for mutant screens and have helped identify many different proteins and RNAs whose presence or absence can cause transient or stable changes of epiallele expression, or influence epigenetic regulation in general. There is also a wealth of data on the influence of drug treatments, sequence determinants, and the role of genomic neighborhood, on epigenetic regulation.
Arabidopsis thaliana has been the plant model of choice for genetic analysis of switching between epiallelic states, based on the rich genetic and genomic resources available. The experimental system in our study is based on a pair of epialleles in Arabidopsis thaliana containing either an expressed or silent hygromycin phosphotransferase gene (HPT). Active transcription confers resistance to the antibiotic while the inactive epiallele renders the plant sensitive. Gene expression can be selected for on antibiotic-containing medium but does not affect the plants during non-selective growth. The epialleles were found in tetraploid plants obtained by regeneration from protoplasts [20]. While some lines had resistant progeny and expressed the HPT gene, other lines had silenced the HPT and produced only sensitive progeny. The R and S epialleles (determining resistance and sensitivity on hygromycin, respectively) were maintained in their particular expression state after diploidization and for all generations of self-pollination analyzed so far (Figure S1). Beside their differences in transcription, they also differ in DNA methylation [21]. We screened for a switch between the epialleles, by scoring for restored hygromycin resistance after T-DNA mutagenesis of the diploid S line. We identified two trans-acting factors whose nature indicated an epigenetic ‘double lock’ at the silent epiallele [22]. In contrast to many other silent genes, silencing could only be released by simultaneous interference with methylation of DNA and histones. Six mutations from the same screen were mapped to the resistance gene itself. These cis-mutations provided the opportunity to study the nature and effect of DNA sequence changes on gene expression, chromatin organization, and genetic stability. We describe these new alleles in detail and compare them with the R and S epialleles. We show that different, and non-overlapping, sequence changes downstream of the HPT gene can restore the expression of the upstream promoter, to a similar extent as the mutations interfering with the chromatin factors in trans. Such small sequence alterations that cause epigenetic changes at some distance may also be involved in gene expression and chromatin changes in the vicinity of transposon insertions/excisions, recombination events, or DNA repair processes and may thereby contribute to the origin of new epialleles.
Results
Epialleles Differ in Chromatin Features and Small RNA Abundance
The HPT gene is inserted in an AT-rich intergenic region on Arabidopsis thaliana chromosome 3 [20]. Previous investigations, and published data from genome-wide screens for chromatin features [20], [23]–[24], indicated that the genomic localization itself is unlikely to influence the epigenetic state of the HPT gene, as no prominent epigenetic modifications are present in the neighborhood of the insertion. Resistant and sensitive Arabidopsis lines with the different epialleles had been generated from the same progenitor line homozygous for the HPT gene, thereby being supposedly isogenic. Nevertheless, the lack of transcription initiation in the hygromycin-sensitive lines could have been due to a DNA sequence mutation in a regulatory region, for example, a transcription factor binding site. Also, the structure of the insert had not been analyzed in detail. Therefore, active and inactive versions were amplified from genomic DNA of the respective lines. Both epialleles are potentially fully functional and have identical sequences. The 35S promoter (P1) is flanked upstream by a 661 bp fragment derived from the plasmid vector (V1). A rearrangement between two vector molecules prior to, or during, the integration of the transgene into the plant genome caused a duplication of the adjacent vector sequence (V2) and the 35S promoter (P2), resulting in two tandem repeats (Figure 1A). The polyadenylation signal from the CaMV 35S terminator following the HPT ORF lacks 151 bp compared to the transformation construct and has therefore lost its termination function (ΔT), causing read through of the P1 transcript into the flanking plant genome sequence (Figure 1A). P2 is followed by a 505 bp non-protein coding fragment (NC) harboring sequences of bovine carrier DNA used to assist PEG-mediated direct gene transfer to mesophyll protoplasts [25], interspersed with 54 nucleotides without homology to known sequences. This heterologous DNA is transcribed by P2, giving rise to a smaller non-coding transcript (P2 transcript) (Figure 1A). Resistant plants produce the longer P1 and the shorter P2 transcripts, while both promoters are inactive in sensitive plants (Figure 1B and Figure S6). Therefore, the isogenic inserts differ only by gene expression, and R and S represent true epialleles.

The different expression states were suspected to originate from distinct chromatin configuration, and previous studies had provided evidence for opposing DNA methylation at the epialleles, especially pronounced at the transcription factor binding sites ([20]–[21], Figure 1C). As DNA methylation and silencing are usually correlated with specific changes of the DNA-associated proteins, we investigated histone modifications and nucleosome occupancy at the epialleles by chromatin immunoprecipitation. This revealed significant differences between the epialleles along the whole transgenic insert. While expressing lines (R) were primarily marked by trimethylation of histone H3 at lysine residue 4 (H3K4me3), typically enriched in euchromatic regions, epialleles in silenced lines (S) have nucleosomes with a modification characteristic of heterochromatin, namely dimethylated lysines at position 9 (H3K9me2) (Figure 1D). These marks, also including low levels of H3 dimethylated at position 27 (H3K27me2), only extend a short distance from the transgene into the flanking plant DNA (Figure S2), indicating limited spreading in transcriptional direction. Beside the specific modifications, we also observed an overall reduced association with H3 in line R compared to S (Figure 1E), probably rendering the promoters more accessible for the transcription machinery. While the epialleles clearly differed in their local chromatin configuration, this did not have any effect on their nuclear localization (Figure S3).
Both epialleles were stably inherited over a minimum of eight generations of self-pollination, without any evidence for spontaneous switches in the germ line. To also study the stability of epialleles in undifferentiated cells, we initiated callus cultures, starting with cotyledons of resistant, sensitive, and non-transgenic plants, and propagated the calli for up to six months under non-selective conditions. We screened callus tissue at several time points for its ability to grow under hygromycin selection for up to 5 weeks. Calli derived from R lines were resistant whereas calli obtained from S or non-transgenic lines died on selection plates. We also determined chromatin modifications and DNA methylation in callus tissue grown on non-selective medium, with results comparable to those of leaf tissue (Figure S4). This demonstrates similar states and stable maintenance of epialleles even upon dedifferentiation.
We screened for the involvement of antisense and/or small RNAs in silencing maintenance. Significant promoter activity of the NC region was excluded (Figure S5A), and specific antisense RNA in line S could also not be detected, neither by northern blotting (Figure S5B) nor by RT-PCR (data not shown). Nevertheless, we generated libraries from size-fractionated 19 nt to 26 nt RNAs prepared from flower buds of plants containing either the sensitive or resistant epiallele. Both libraries were sequenced (Table S1) and the reads screened for alignment with the transgenic insert. The library from the R plants had only 59 reads (3 per 1 million reads) with only one sequence with a match in the epiallele (Figure 2A, Table S3). In line S, we found 2661 (129 per 1 million reads) matching the epiallele, with a predominant length of 24 nucleotides (Figure 2A, Table S2 and Table S3), the size class known to be primarily involved in RNA-directed DNA methylation (RdDM). This is significantly more than in R, but still relatively little, compared to an individual miRNA (820 reads per 1 million for miRNA165) or to siRNA from a repetitive sequence (>1000 reads per 1 million for TSI [26]). The reads in S were distributed along the epiallele but mostly outside the HPT coding region. Importantly, among all reads specific for the silent epiallele we found an sRNA peak (671 reads, 476 antisense and 195 sense) covering 61 bp in the middle of the 505 bp non-coding sequence of the P2 transcript (Figure 2B). The most abundant sRNAs overlap with the 54 nucleotides of unknown origin. However, this sequence encompasses 28 nucleotides that are homologous to the most 5′ end of the 35S promoter (Figure 2B).
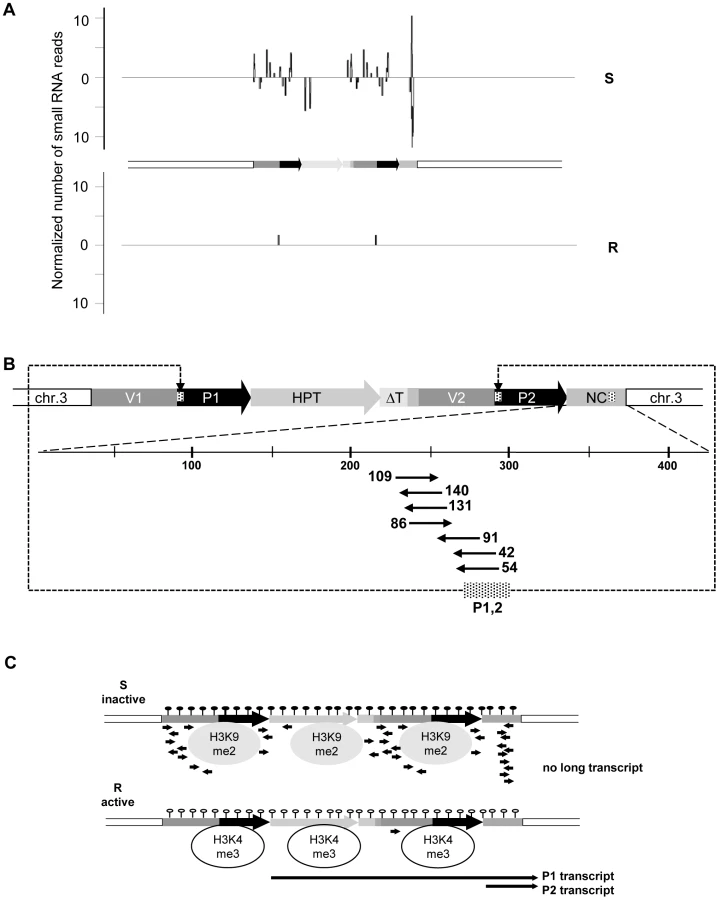
In short, these results indicate very stable and completely isogenic epialleles that differ only in their transcriptional activity. DNA methylation, suppressing chromatin marks, and sRNAs, are specifically enriched at the transcriptionally inactive epiallele; while the counterpart produces high transcript levels, lacks DNA methylation and sRNAs, and carries modifications characteristic of open chromatin (Figure 2C).
Release of Silencing upon Sequence Rearrangement
In addition to the trans-acting mutants identified in a screen for restored HPT expression after mutagenesis of line S [22], we identified six hygromycin-resistant plants in which the mutant phenotype was genetically linked to the resistance gene itself (‘cis-mutations’, RΔ1-6). All these mutants produced progeny that could grow on hygromycin selection plates (Figure 3A), connected with restoration of variable amounts of P1 and P2 transcripts (Figure 3B). Northern blot analysis of cis-mutant RNA revealed P1 transcripts of smaller size in all cis-mutants compared to those from the active R line (Figure 3C). The length is reduced to different extents, indicating several independent mutational changes of the sequence. An extended northern blot analysis, with either total RNA or poly(A)-enriched RNA, showed that the P1 transcript in all lines besides RΔ6 is polyadenylated (Figure S6), likely due to a flanking sequence with some similarity to a polyA signal. While no P2 transcript from the second promoter is detectable in RΔ1, RΔ2, RΔ4, and RΔ6, there is a signal in RΔ3 and RΔ5, including in the poly(A) fraction (Figure S6C, S6D).

To characterize the P1 transcripts, and to identify the transcriptional termination sites in the cis-mutants, we performed 3′-RACE. We also analyzed the genomic DNA of all cis-mutants after amplification of the transgenic insert from genomic DNA and aligned DNA and RNA sequences (Figure 3D). This verified six different sequence rearrangements within the 3′ region: mainly deletions, but also one case of an inserted plant DNA fragment (RΔ3). The mutants RΔ1 and RΔ2 have both lost the duplicated promoter P2 and the NC sequence. The vector duplication was partially (RΔ1) or completely (RΔ2) deleted, as was part of the flanking plant sequence. The deletions in RΔ4, RΔ5, and RΔ6 did not or only partially affect the P2 promoter, and two of them maintain also the NC sequence. The rearrangement in RΔ3 is most complex: here, a 1243 bp plant DNA sequence derived from a position 1.2 kb upstream of the transgene location was inserted between the P1 transcript and the downstream vector fragment. In the mutants RΔ1, RΔ2, RΔ3, and RΔ4, the P1 transcripts are terminated at the (first) site of rearrangement, while the transcripts go beyond the breakpoints in RΔ5 and RΔ6. Only RΔ3 and RΔ5 are able to produce the P2 transcript, as in these cases, the P2 promoter is complete and the heterologous sequence downstream was only slightly affected by mutagenesis (Figure 3D). Nevertheless, the P2 transcript levels are much lower than in the R line (Figure 3B). Interestingly, there is no overlap between the deletions in all individual cis-mutants, but the rearrangements had either affected the second promoter copy (RΔ1, RΔ2, RΔ6), or the DNA template for the P2 transcript (RΔ1, RΔ2 and RΔ4), or the connection between both sequences (RΔ3, RΔ5).
All cis-mutants were tested for effects outside of the epiallele by analyzing the degree of genome-wide methylation at endogenous repeats and by introgressing a transcriptionally silent marker gene coding for β-glucuronidase from line L5, shown to be affected by other epigenetic mutations [27]–[28]. None of the cis-mutants changed the modification or expression of these markers (Figure S7). Therefore, it is unlikely that they have an effect outside of the epiallele.
Due to the hygromycin selection in the screen, all cis-mutants were expected to have a functional resistance marker gene. Indeed, the upstream promoter P1 and the HPT coding region were intact and identical in RΔ1-6 and hence potential new epialleles of the resistance gene. Therefore, we compared the chromatin state in this region. We found reduced DNA methylation levels in cis-mutants compared to S (Figure 4A), and a detailed bisulfite methylation analysis confirmed an overall reduction of DNA methylation in the promoter region of cis-mutants (Figure 4B, 4C). However, the degree of hypomethylation, and the distribution of the remaining methylated cytosine residues, do not support a direct and linear correlation with expression levels. Although RΔ2, RΔ3, and RΔ4 show the strongest reduction of CG methylation, especially at the transcription factor binding sites (Figure 4B, asterisk), and have expression levels comparable to R (Figure 3B), methylation in RΔ5 is similar to RΔ3 and RΔ4, although P1 transcript expression is much lower. Also, RΔ3 and RΔ4 have even gained CHH methylation in the 5′ region. Concomitant with the loss of DNA methylation, the modification specific for the silent state (H3K9me2) was changed in favor of the active mark (H3K4me3) in P1 and P1-transcribed regions, as demonstrated by ChIP (Figure 4D). One mutant (RΔ1) maintained a high level of H3K9me2 similar to that of the silent epiallele. Nonetheless, it also acquired a remarkable amount of H3K4me3, although less than other cis-mutants. Independent of the modifications, and similar to the resistant line, cis-mutants showed a decreased level of H3 association, indicating that the sequence rearrangements had also affected the nucleosome density (Figure 4E).

On the whole, the cis-mutants demonstrate that structural rearrangements can cause significant changes in transcriptional activation and chromatin configuration at the previously silent epiallele. These changes are surprisingly divergent and reflect specific effects of similar but not overlapping deletions.
Stability of Silencing after Sequence Rearrangement
The extreme stability of R and S epialleles through many generations and in callus cultures raised the question of expression stability in the cis-mutants. Most structurally rearranged derivatives displayed similar stability and provided comparable hygromycin resistance over several generations of homozygous cis-mutants (S4 to S6 tested). RΔ2, RΔ3, and RΔ4 produced resistant progeny in consecutive generations. Resistance in RΔ5 and RΔ6 was lower in S4 (56% and 61%, respectively), but maintained this level up to S6. In contrast, RΔ1 plants that were clearly hygromycin-resistant in S4 (84%) generated partially sensitive S5 and fully sensitive S6 progeny (Figure 5A). This correlates well with the loss of unmethylated sites at the transgenic insert (Figure 5B), similar to gradual loss of resistance over 5 generations described for another marker gene [29]. The instability in RΔ1 does not correspond with additional sequence changes, as the same rearranged structure (Figure 3D) is maintained in subsequent generations. Rather, it correlates with the epigenetic state, since RΔ1 was characterized by the bivalent histone modifications (Figure 4D).

The re-silencing in generation S6 of RΔ1 allowed us to compare silencing maintenance at promoter 1 between this line and the S epiallele. We tested plants of both lines after growth in the presence of zebularine [reducing DNA methylation, 30] or DZNep [reducing histone methylation and also DNA methylation via SAHH]-[inhibition, 22,31]. Zebularine alone did not reactivate promoter P1 in line S, but in RΔ1S6, and DZNep-induced activation was twice as high in RΔ1S6 compared to S (Figure 5C). This indicates that S and RΔ1S6 differ in the stringency of silencing, either due to presence or absence of the P2 promoter and transcript, or to the lineage history of RΔ1S6 from a recently active state. The presence of the P2 promoter in RΔ3 - 6 and the expression of the P2 transcript in RΔ3 and 5, which do not cause re-silencing in later generations, make the latter explanation more likely.
Discussion
The thorough analysis of the HPT transgene in its two opposite expression states has revealed sequence identity over the full length of the insertion, significant differences in chromatin modifications and few, but silencing-specific, small RNA molecules. Chromatin differences are restricted to the affected sequence, with no hint of genome-wide changes or modified localization of the genomic region within the nucleus. Together with heritability of the expression states over many generations, and their maintenance even upon de-differentiation, the data prove the transcriptionally active and the silenced version to be authentic epialleles. Their occurrence in Arabidopsis, the best studied model for epigenetic research in plants, and the easy assay for the selectable hygromycin resistance conferred by the active state, made this pair of epialleles convenient tools for studying maintenance and switching of epigenetic states.
After mutagenesis, we identified several hygromycin-resistant plants in which mutations in the epiallele sequence downstream of the HPT coding region had reactivated the previously silenced epiallele. Combining DNA and RNA sequence analysis and characterization of chromatin modifications, we found that these structural changes of the DNA sequence caused substantial upstream changes in chromatin and transcriptional activity. Beyond the complex and mutually dependent interplay of chemical modifications of the DNA and the associated histones, and longer and small, coding and non-coding RNAs described in numerous cases, the results presented here have shown that even small and non-overlapping modifications of the genomic template, outside of the promoter and open reading frame, can modify transcription and chromatin states in the vicinity. These changes are not minor: the bacterial gene HPT coding for hygromycin phosphotransferase is a selectable marker gene applied in numerous plant transformation experiments [32], but plants need a significant amount of HPT transcript to produce enough protein to detoxify the antibiotic. Minor reactivation in the background of some epigenetic mutants tested in a reverse genetic approach (data not shown) was not sufficient. Therefore, the stringent assay for restored hygromycin resistance required a substantial change, as in the case of the trans-acting mutants from the same screen that revealed a double lock of two simultaneous chromatin modifications [22]. HPT expression levels are indeed similar between cis-and trans-acting mutants.
Although the transgenic marker allowed this convenient selection for drastic epigenetic switches, without affecting plants under non-selective conditions, it could have been considered not representative for other, plant-endogenous or general cases. However, a recent publication [33] describes an interesting mutation that affects expression of the gene for nodulation factor SUNN in Medicago truncatula. The mutation is closely linked to the SUNN gene, acts only in cis but does not change the DNA sequence of the SUNN gene itself. Although the nature of this mutation is not yet identified, it could exert its effect in a similar way to the cis-mutants described here, especially since the ‘like sunn supernodulator’ mutant phenotype is occasionally unstable, like the hygromycin resistance in RΔ1, 5, and 6. Other examples may be found upon further inspection of natural transcript level variation between regions with very similar gene sequences in plants [e.g. 8] or in the connection between chromatin structure and trinucleotide repeat expansion in mammals [for review 34].
Transcriptional gene silencing is often associated with the presence of homologous sequences in the genome [e.g. 35]–[37], and intentional rearrangements from complex inserts to single copies by site-specific recombinase eliminate silencing [e.g. 38]. Therefore, when we started the analysis of the sequence changes in the cis-mutants, we were expecting a clear dependence of reactivation on loss of the duplicated region. This is not the case, since all cis-mutants, with the exception of RΔ2, still retain some duplicated regions. Also against expectation, a loss of the non-coding sequence homologous to the most abundant small RNAs is not a prerequisite for reactivation (RΔ3, RΔ5, and RΔ6). Furthermore, a loss of the small transcript starting from the P2 promoter is not necessary (RΔ3 and RΔ5), although its level in these mutants is not as high as in R plants. It should be kept in mind that neither the tandem sequence duplications, nor either of the two transcripts, are sufficient to initiate silencing, since R plants (with the complete insert and substantial transcription from P1 and P2) are fully resistant and stable. This is distinct from the FWA gene where tandem repeats are necessary and sufficient for silencing and DNA methylation [39]. Considering the lack of DNA methylation and small RNAs at the HPT insert in R plants, it is possible that the initial steps of silencing do not occur, are not efficient enough to start the reinforcing mechanism [39], or are inhibited by efficient transcription [40]. However, such conditions must have been overruled on the rare occasions that produced the silent epiallele in the first place.
The deletions in the different cis-mutants do not overlap in a specific region, and the smallest change is the loss of just 65 bp (RΔ5). Apparently, rather than affecting a specific sequence, the rearrangements change the overall organization at this locus. These changes can have variable consequences for the upstream promoter, causing either decisive, stable epigenetic switches (RΔ2, RΔ3, RΔ4) or leading to ambivalent states that can later fall back into silencing (RΔ1). How such small genetic heterogeneity, that does not affect coding or regulatory regions, can cause extreme epigenetic diversity at a promoter elsewhere remains an open question. The sequence changes could exert their effect by modifying the distance to flanking regulatory regions, the nucleosome arrangement or density, the association with DNA-binding molecules, or any higher order structure within the DNA. It is clearly different from the ‘spreading’ effect of silencing often associated with RdDM [41]–[42]: it causes activation (not silencing), goes against (not along with) the direction of transcription, and the most abundant of the relatively few small RNAs does not match the affected sequence of the upstream promoter. The results emphasize the mutual dependence between genetic and epigenetic factors, while indicating that these do not necessarily act at overlapping genomic sites. Similar effects might explain some of the associated changes in gene expression in the vicinity of small or large sequence modifications by transposon or recombination events. One example at a similar distance might be the transposon-dependent loss and gain of DNA methylation and inverse gene expression regulating sex determination in melon, at a site just 1.5 kb away from the insertion/excision site [43].
The relatively high number of cis-mutants in the screen was plausible in retrospective: mutations outside of the epiallele released silencing only if they reduce two epigenetic marks simultaneously. This is achieved by a few special mutations [22] or theoretically by rare double mutations and explains the low number of trans-acting mutants. In the study here, the genetic changes were found after mutagenesis by Agrobacterium-mediated T-DNA transformation [22], although none of the cis-mutations was connected with an integrated fragment of the incoming T-DNA. T-DNA transformation is also known to create mutations unlinked, or independent, from the site of integration [44] and can cause complex chromosome rearrangements [45]–[46]. Successful, and possibly also attempted, integrations occur at sites of microhomologies between T-DNA and plant DNA [47]–[48]. The incoming T-DNA [49] has some homology with the terminator sequences in the epiallele (ΔT), and in fact, the deletion sites in two cis-mutants (RΔ2, RΔ3) are near, or in, this sequence. The other deletions are close to promoter copy P2 that has no homology with the T-DNA, but potentially reflect a recombination hotspot in the 35S promoter sequence [50]. Alternatively, the double strand breaks connected with completed or aborted integration might stimulate repair via homologous recombination between the duplicated sequences of the epiallele (RΔ3). This would indeed have selected for 3′ rearrangements since those affecting the upstream copy are likely to lose the functional HPT cassette.
All together, the R and S epialleles described here provide an example of identical DNA sequences with converse expression states and specific epigenetic configuration that are faithfully transmitted to progeny. However, sequence changes in the vicinity of the silent epiallele can induce an epigenetic switch to the opposite state. These can have different degrees of stability, depending on the complex interplay between the nature of the sequence alteration, the consequences for transcription and transcripts, and the chromatin organization (Figure 6). This also illustrates a tight dependence of epigenetic regulation on local structures and makes it likely that DNA rearrangements can potentially change or induce new epialleles outside the affected region.

Materials and Methods
Plant Material, Growth, and Chemical Treatments
Arabidopsis thaliana lines with R and S epialleles in accession Zürich and mutagenesis of line S were described previously [20], [22]. Stratified seeds were surface-sterilized with 5% sodium hypochlorite and 0.05% Tween-80 for 6 min, washed and air-dried overnight. Sterilized seeds were germinated and grown in Petri dishes containing agar-solidified germination medium (GM) in growth chambers under 16 h light/8 h dark cycles at 21°C. For drug treatments, seeds were sown and plants grown on GM plates with hygromycin (10 µg/ml, Calbiochem), zebularine (40 µM, Sigma) or 3-deazaneplanocin (DZNep, 2 µM, donated by Dr. Victor Marquez) under the conditions described above.
Nucleic Acid Isolation and Gel Blot Analysis
Genomic DNA was isolated from 3 week-old seedlings using either DNeasy Plant Mini Kit (Qiagen) or Phytopure (Amersham), following the manufacturers' protocols, except that genomic DNA was eluted in sterile water. Total RNA extraction from 3 week-old seedlings was performed with RNeasy Plant Mini Kit (Qiagen) including an on-column DNase I digest (Qiagen). For Southern blot analysis, 10 µg of genomic DNA were digested overnight with 20 U restriction enzymes. For methylation-specific Southern blot analysis, the methylation-sensitive restriction enzymes (HpaII, blocked by mCG and mCHG, and MspI, blocked only by mCHG) were used. Digested samples were electrophoretically separated on 1.2% TAE agarose gels, depurinated for 10 min in 250 mM HCl, denaturated for 30 min in denaturation solution containing 0.5 M NaOH and 1.5 M NaCl and neutralized twice in 0.5 M Tris, 1.5 M NaCl and 1 mM EDTA at pH7.2 for 15 min. For northern blot analysis of total and poly(A) RNA, 5 µg of RNA were denatured with 15% glyoxal and 50% DMSO for 1 h at 50°C and separated using 1.5% agarose gels in 10 mM sodium phosphate buffer pH7 in a Sea2000 circular flow electrophoresis chamber (Elchrom Scientific). DNA and RNA gels were blotted onto Hybond N+ (Amersham) membranes overnight with 20× SSC, washed and UV-crosslinked using a Stratalinker (Stratagene). Hybridization was performed as described [51]. Radioactively labeled sequence-specific probes were synthesized from 25 ng of DNA using the Rediprime labeling kit (Amersham) and 50 µCi dCTP-α-32P (Amersham or Hartmann Analytic) and purified on G50 Probequant (Amersham) columns. Signals were detected with phosphoimager screens (Bio-Rad) and scanned with a Molecular Imager FX (Bio-Rad).
Rapid Amplification of cDNA 3′ Ends
3′-RACE was performed with the SMART RACE cDNA Amplification Kit (Clontech) according to the instructions. Total RNA (700 ng) was treated with DNaseI (Fermentas), then reverse-transcribed with RevertAidRT (Fermentas) with 3-RACE A primer (5–AAGCAGTGGTATCAACGCAGAGTAC(T)30V N–3) in a 20 µl reaction. Two µl of cDNA reaction were used as template in 3′-RACE PCR. For this, Advantage 2 PCR Kit (Clontech) was used according to instructions. A control primer (Actin, Act2F primer: 5-GCCATCCAAGCTGTTCTCTC-3) and gene-specific primers were used in combination with UniA_45 (5–CTAATACGACTCACTATAGGGCAAGCAGTGGTATCAACGCAGAGT–3).
Reverse Transcription PCR and Quantitative Real-Time PCR
RNA samples were treated with DNase I (MBI Fermentas) for 30 min at 37°C to remove residual DNA contamination. The reaction was inactivated by addition of EDTA and incubation at 65°C for 10 min. Reverse transcription was performed on 1 µg of RNA with 0.2 µg of random hexamer primers (MBI Fermentas) using 1 U RevertAid H Minus M-MuLV-RTase (MBI Fermentas) in the presence of 20 U RiboLock Ribonuclease inhibitor at 42°C for 1.5 h. Real time PCR analysis was performed with the 2× SensiMix Plus SYBR & Fluorescein Kit (Quantace) protocol using an iQ5 Real-Time-PCR System (BioRad Laboratories). The obtained Ct values were analyzed with the iQ5 Optical System Software Version 2.0 (Bio-Rad), applying the mathematical model for relative quantification in Excel (Microsoft) as described [52]. All primer sequences are listed in Table S4.
Bisulphite Conversion, Sequencing, and Evaluation
After treatment with RNase A and proteinase K, 1–2 µg of genomic DNA were digested overnight with BamHI (MBI Fermentas). Subsequent bisulphite conversion was carried out using the Epitect Conversion Kit (Qiagen) and controlled for completion as described [21], [53]. Converted DNA was used for PCR amplification. PCR-amplified DNA was cloned using pGEM-Teasy (Promega) and ligation mixes transformed into DH5α cells (Invitrogen) and sequenced by terminal-labeling using BigDye Terminator v3.1 (Applied Biosystems). The sequence information obtained was analyzed with CyMATE, www.gmi.oeaw.ac.at/cymate [54], and Excel (Microsoft).
Chromatin Immunoprecipitation
ChIP was performed as described (http://mescaline.igh.cnrs.fr/EpiGeneSys/www/images/protopdf/p13.pdf) using 3 week-old seedlings. The chromatin was immuno-precipitated with antibodies to histone H3 (Abcam, ab1791), H3K4me3 (Upstate, 07-473), H3K9me2 (T. Jenuwein 4677 and Abcam ab1220), and H3K27me2 (Upstate, 07-473). Immunoprecipitated DNA was purified using a Qiagen PCR Purification Kit and eluted in 50 µl of EB buffer. Quantitative real-time PCR was carried out in a total reaction volume of 25 µl and qPCR conditions were according to the 2× SensiMix Plus SYBR & Fluorescein Kit (Quantace) protocol using an iQ5 Real-Time-PCR System (BioRad Laboratories). qPCR data were evaluated as a ratio to input DNA [55].
sRNA Isolation, Library Generation, and Bioinformatic Analysis
Small RNA was isolated from either pooled inflorescences or seedlings (21 days old) using the mirVana miRNA Isolation Kit (Ambion). Small RNA libraries were generated as previously described [56] and sequenced using the Illumina G2 platform. After clipping the adapter sequence by vectorstrip software from EMBOSS package [57], small RNA reads were screened for homology with the epiallele sequence using bowtie [58], allowing only perfect matches (Table S3). Reads homologous to tRNA, rRNA, snRNA, snoRNA, mitochondrial RNAs, and chloroplast RNAs were removed by custom Perl scripts. The total number of reads that mapped to a certain region was computed as sum of 1/N_i (N_i is the number of times the read i was mapped). It was then normalized to indicate the number of each read per million bp (adapted from the RPKM concept in RNA-Seq, [59]. A threshold of 10 reads was chosen for any sequence to be taken into account. For the epiallele region, the normalized number of mapped reads was computed at single bp scale. For a more detailed view on a selected region, the analysis was performed with SiLoMa [60].
Additional methods are described in Text S1.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. FinneganEJ 2002 Epialleles - a source of random variation in times of stress. Current Opinion in Plant Biology 5 101 106
2. KaliszSPuruggananMD 2004 Epialleles via DNA methylation: consequences for plant evolution. Trends in Ecology & Evolution 19 309 314
3. MorganDKWhitelawE 2008 The case for transgenerational epigenetic inheritance in humans. Mammalian Genome 19 394 397
4. FinerSHollandMLNantyLRakyanVK 2011 The Hunt for the Epiallele. Environmental and Molecular Mutagenesis 52 1 11
5. ShibaHTakayaniaS 2007 RNA silencing systems and their relevance to allele-specific DNA methylation in plants. Bioscience Biotechnology and Biochemistry 71 2632 2646
6. CubasPVincentCCoenE 1999 An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401 157 161
7. ManningKTorMPooleMHongYThompsonAJ 2006 A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nature Genetics 38 948 952
8. RangwalaSHElumalaiRVanierCOzkanHGalbraithDW 2006 Meiotically stable natural epialleles of Sadhu, a novel Arabidopsis retroposon. PLoS Genet 2 e36 doi:10.1371/journal.pgen.0020036
9. SoppeWJJJacobsenSEAlonso-BlancoCJacksonJPKakutaniT 2000 The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Molecular Cell 6 791 802
10. JacobsenSEMeyerowitzEM 1997 Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science 277 1100 1103
11. JohannesFPorcherETeixeiraFKSaliba-ColombaniVSimonM 2009 Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet 5 e1000530 doi:10.1371/journal.pgen.1000530
12. ReindersJWulffBBHMirouzeMMari-OrdonezADappM 2009 Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes & Development 23 939 950
13. KrizovaKFojtovaMDepickerAKovarikA 2009 Cell culture-induced gradual and frequent epigenetic reprogramming of invertedly repeated tobacco transgene epialleles. Plant Physiol 149 1493 1504
14. MeinsFJrThomasM 2003 Meiotic transmission of epigenetic changes in the cell-division factor requirement of plant cells. Development 130 6201 6208
15. RheeYSekhonRSChopraSKaepplerS 2010 Tissue Culture-Induced Novel Epialleles of a Myb Transcription Factor Encoded by pericarp color1 in Maize. Genetics 186 843-U151
16. DolinoyDCDasRWeidmanJRJirtleRL 2007 Metastable epialleles, imprinting, and the fetal origins of adult diseases. Pediatric Research 61 30R 37R
17. RhounimLRossignolJLFaugeronG 1992 Epimutation of Repeated Genes in Ascobolus-Immersus. Embo Journal 11 4451 4457
18. PattersonGIThorpeCJChandlerVL 1993 Paramutation, an allelic interaction, is associated with a stable and heritable reduction of transcription of the maize b regulatory gene. Genetics 135 881 894
19. BenderJFinkGR 1995 Epigenetic Control of an Endogenous Gene Family Is Revealed by a Novel Blue Fluorescent Mutant of Arabidopsis. Cell 83 725 734
20. Mittelsten ScheidOAfsarKPaszkowskiJ 2003 Formation of stable epialleles and their paramutation-like interaction in tetraploid Arabidopsis thaliana. Nat Genet 34 450 454
21. HetzlJFoersterAMRaidlGMittelsten ScheidO 2007 CyMATE: a new tool for methylation analysis of plant genornic DNA after bisulphite sequencing. Plant Journal 51 526 536
22. BaubecTDinhHQPecinkaARakicBRozhonW 2010 Cooperation of Multiple Chromatin Modifications Can Generate Unanticipated Stability of Epigenetic States in Arabidopsis. Plant Cell 22 34 47
23. CokusSJFengSZhangXChenZMerrimanB 2008 Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452 215 219
24. ListerRO'MalleyRCTonti-FilippiniJGregoryBDBerryCC 2008 Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133 523 536
25. KareschHBilangRMittelsten ScheidOPotrykusI 1991 Direct gene transfer to protoplasts of Arabidopsis thaliana. Plant Cell Reports 9 571 574
26. SteimerAAmedeoPAfsarKFranszPScheidOM 2000 Endogenous targets of transcriptional gene silencing in arabidopsis. Plant Cell 12 1165 1178
27. MorelJBMourrainPBeclinCVaucheretH 2000 DNA methylation and chromatin structure affect transcriptional and post-transcriptional transgene silencing in Arabidopsis. Curr Biol 10 1591 1594
28. ElmayanTProuxFVaucheretH 2005 Arabidopsis RPA2: a genetic link among transcriptional gene silencing, DNA repair, and DNA replication. Curr Biol 15 1919 1925
29. KilbyNJLeyserHMOFurnerIJ 1992 Promoter Methylation and Progressive Transgene Inactivation in Arabidopsis. Plant Molecular Biology 20 103 112
30. BaubecTPecinkaARozhonWMittelsten ScheidO 2009 Effective, homogeneous and transient interference with cytosine methylation in plant genomic DNA by zebularine. Plant J 57 542 554
31. MirandaTBCortezCCYooCBLiangGNAbeM 2009 DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Molecular Cancer Therapeutics 8 1579 1588
32. MikiBAbdeenAManabeYMacDonaldP 2009 Selectable marker genes and unintended changes to the plant transcriptome. Plant Biotechnology Journal 7 211 218
33. SchnabelEMukherjeeASmithLKassawTLongSR 2010 The lss Supernodulation Mutant of Medicago truncatula Reduces Expression of the SUNN Gene. Plant Physiology 154 1390 1402
34. DionVWilsonJH 2009 Instability and chromatin structure of expanded trinucleotide repeats. Trends in Genetics 25 288 297
35. SekhonRSChopraS 2009 Progressive loss of DNA methylation releases epigenetic gene silencing from a tandemly repeated maize Myb gene. Genetics 181 81 91
36. KinoshitaYSazeHKinoshitaTMiuraASoppeWJ 2007 Control of FWA gene silencing in Arabidopsis thaliana by SINE-related direct repeats. Plant J 49 38 45
37. StamMBeleleCDorweilerJEChandlerVL 2002 Differential chromatin structure within a tandem array 100 kb upstream of the maize b1 locus is associated with paramutation. Genes Dev 16 1906 1918
38. De BuckSPeckIDe WildeCMarjanacGNolfJ 2007 Generation of single-copy T-DNA transformants in Arabidopsis by the CRE/loxP recombination-mediated resolution system. Plant Physiology 145 1171 1182
39. ChanSWZhangXBernatavichuteYVJacobsenSE 2006 Two-step recruitment of RNA-directed DNA methylation to tandem repeats. PLoS Biol 4 e363 doi:10.1371/journal.pbio.0040363
40. BerrettaJMorillonA 2009 Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep 10 973 982
41. DaxingerLKannoTBucherEvan der WindenJNaumannU 2009 A stepwise pathway for biogenesis of 24-nt secondary siRNAs and spreading of DNA methylation. Embo Journal 28 48 57
42. HendersonIRJacobsenSE 2008 Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes & Development 22 1597 1606
43. MartinATroadecCBoualemARajabMFernandezR 2009 A transposon-induced epigenetic change leads to sex determination in melon. Nature 461 1135 1138
44. KonczCNemethKRedeiGPSchellJ 1992 T-DNA Insertional Mutagenesis in Arabidopsis. Plant Molecular Biology 20 963 976
45. NacryPCamilleriCCourtialBCabocheMBouchezD 1998 Major chromosomal rearrangements induced by T-DNA transformation in Arabidopsis. Genetics 149 641 650
46. ParinovSSundaresanV 2000 Functional genomics in Arabidopsis: large-scale insertional mutagenesis complements the genome sequencing project. Curr Opin Biotechnol 11 157 161
47. MatsumotoSItoYHosoiTTakahashiYMachidaY 1990 Integration of Agrobacterium T-DNA into a Tobacco Chromosome - Possible Involvement of DNA Homology between T-DNA and Plant DNA. Molecular & General Genetics 224 309 316
48. MullerAEAtkinsonRGSandovalRBJorgensenRA 2007 Microhomologies between T-DNA ends and target sites often occur in inverted orientation and may be responsible for the high frequency of T-DNA-associated inversions. Plant Cell Rep 26 617 630
49. MengisteTAmedeoPPaszkowskiJ 1997 High-efficiency transformation of Arabidopsis thaliana with a selectable marker gene regulated by the T-DNA 1' promoter. Plant J 12 945 948
50. KohliAGriffithsSPalaciosNTwymanRMVainP 1999 Molecular characterization of transforming plasmid rearrangements in transgenic rice reveals a recombination hotspot in the CaMV 35S promoter and confirms the predominance of microhomology mediated recombination. Plant J 17 591 601
51. ChurchGMGilbertW 1984 Genomic sequencing. Proc Natl Acad Sci U S A 81 1991 1995
52. PfafflMW 2001 A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29 e45
53. FoersterAMMittelsten ScheidO 2010 Analysis of DNA methylation in plants by bisulfite sequencing. Methods in Molecular Biology 631 1 11
54. FoersterAMHetzlJMuellnerCMittelsten ScheidO 2010 Analysis of bisulfite sequencing data from plant DNA using CyMATE. Methods in Molecular Biology 631 13 22
55. HaringMOffermannSDankerTHorstIPeterhanselC 2007 Chromatin immunoprecipitation: optimization, quantitative analysis and data normalization. Plant Methods 3 11
56. BrenneckeJAravinAAStarkADusMKellisM 2007 Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128 1089 1103
57. RicePLongdenIBleasbyA 2000 EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16 276 277
58. LangmeadBTrapnellCPopMSalzbergSL 2009 Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10 R25
59. MortazaviAWilliamsBAMcCueKSchaefferLWoldB 2008 Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5 621 628
60. MoxonSSchwachFDalmayTMacleanDStudholmeDJ 2008 A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics 24 2252 2253
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 10
Nejčtenější v tomto čísle
- The Glycobiome Reveals Mechanisms of Pentose and Hexose Co-Utilization in Bacteria
- Global Mapping of Cell Type–Specific Open Chromatin by FAIRE-seq Reveals the Regulatory Role of the NFI Family in Adipocyte Differentiation
- Genetic Determinants of Serum Testosterone Concentrations in Men
- MicroRNA Expression and Regulation in Human, Chimpanzee, and Macaque Brains