
Towards Establishment of a Rice Stress Response Interactome
Rice (Oryza sativa) is a staple food for more than half the world and a model for studies of monocotyledonous species, which include cereal crops and candidate bioenergy grasses. A major limitation of crop production is imposed by a suite of abiotic and biotic stresses resulting in 30%–60% yield losses globally each year. To elucidate stress response signaling networks, we constructed an interactome of 100 proteins by yeast two-hybrid (Y2H) assays around key regulators of the rice biotic and abiotic stress responses. We validated the interactome using protein–protein interaction (PPI) assays, co-expression of transcripts, and phenotypic analyses. Using this interactome-guided prediction and phenotype validation, we identified ten novel regulators of stress tolerance, including two from protein classes not previously known to function in stress responses. Several lines of evidence support cross-talk between biotic and abiotic stress responses. The combination of focused interactome and systems analyses described here represents significant progress toward elucidating the molecular basis of traits of agronomic importance.
Published in the journal:
. PLoS Genet 7(4): e32767. doi:10.1371/journal.pgen.1002020
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002020
Summary
Rice (Oryza sativa) is a staple food for more than half the world and a model for studies of monocotyledonous species, which include cereal crops and candidate bioenergy grasses. A major limitation of crop production is imposed by a suite of abiotic and biotic stresses resulting in 30%–60% yield losses globally each year. To elucidate stress response signaling networks, we constructed an interactome of 100 proteins by yeast two-hybrid (Y2H) assays around key regulators of the rice biotic and abiotic stress responses. We validated the interactome using protein–protein interaction (PPI) assays, co-expression of transcripts, and phenotypic analyses. Using this interactome-guided prediction and phenotype validation, we identified ten novel regulators of stress tolerance, including two from protein classes not previously known to function in stress responses. Several lines of evidence support cross-talk between biotic and abiotic stress responses. The combination of focused interactome and systems analyses described here represents significant progress toward elucidating the molecular basis of traits of agronomic importance.
Introduction
A major limitation of crop production is imposed by a suite of abiotic and biotic stresses resulting in 30%–60% yield losses globally each year [1]. The burgeoning field of systems biology provides new methodologies to make sense of plant stress responses, which are often controlled by highly complex signal transduction pathways that may involve tens or even thousands of proteins [2]. Complementary to large-scale approaches to delineate organisms' entire interactomes [3], we have developed a focused, high-quality Y2H-based interactome around the following key proteins that control the rice responses to disease and flooding: XA21 [4], NH1 (NPR1 homolog1/OsNPR1) [5], [6], SUB1A and SUB1C (submergence tolerance 1A, 1C) [7] (Figure 1A, Table S1). XA21 is a host sensor (also called a pattern recognition receptor (PRR)) of conserved microbial signatures that confers resistance to the Gram-negative bacterium Xanthomonas oryzae pv. oryzae (Xoo) [4], [8], [9]. Overexpression of Nh1 in rice also enhances resistance to Xoo [5]; whereas reduced expression of Nh1 impairs benzothiadiazole-induced resistance to Pyricularia oryzae [10]. SUB1A and SUB1C are ethylene response transcription factors that regulate response to prolonged foliar submergence [7]. Much remains to be learned about the signaling pathways controlled by these pivotal stress response proteins.
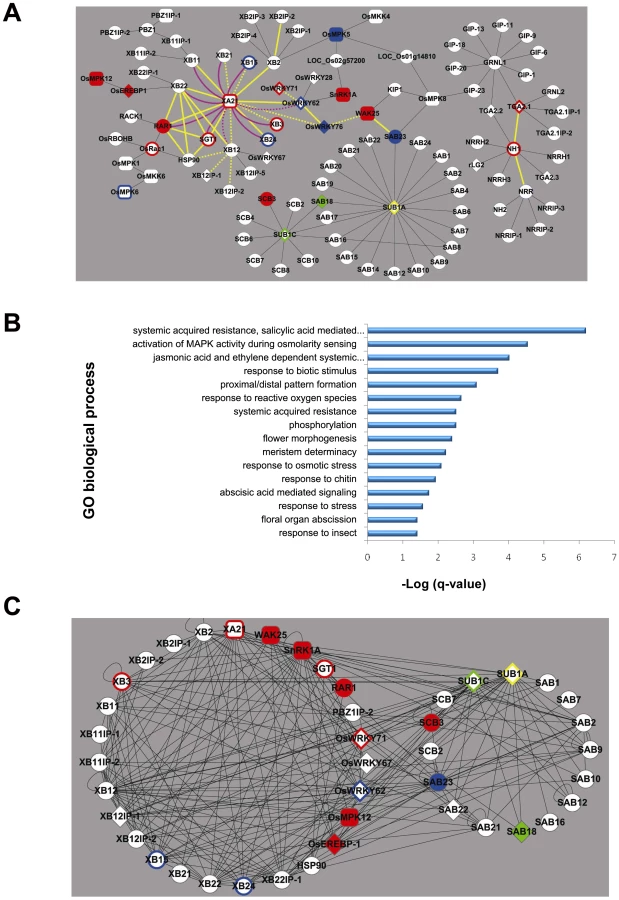
To identify components of these signaling pathways, we carried out yeast two hybrid screening to construct a rice response interactome. We then validated the robustness of the interactome using bimolecular fluorescence complementation [11], yeast mating-based split ubiquitin system assays [12], and phenotypic analysis. Transgenic analysis of genes encoding key proteins coupled with correlation analysis of transcriptomics data and protein-protein interactions revealed ten interactome members that function as positive or negative regulators of biotic or abiotic stress tolerance in rice. Fourteen additional members of the interactome have previously been reported to function in stress tolerance. The high-quality interactome and systems-level analyses described here represent significant progress toward elucidating the molecular basis of traits of agronomic importance.
Results/Discussion
Construction of the rice stress-response interactome
We initially reconstructed four separate sub-interactomes for NH1, the intracellular kinase domain of XA21 (termed XA21K668 [13]), SUB1A, and SUB1C by screening a rice cDNA library pool. Subsequent rounds of screening with identified interactors, targeted assays with additional proteins identified based on sequence homology, and inclusion of connections from the rice kinase interactome [14] revealed that the NH1-, XA21-, and SUB1-anchored interactomes form a single rice stress interactome (Figure 1A, Table S1).
The four sub-interactomes were constructed by using a high throughput yeast two hybrid (Y2H) approach to identify components of the XA21-, NH1-, and SUB1 - signaling pathways. We identified a total of 8 unique XA21 binding proteins (XBs, Table S1). Five of these XBs, XB2, XB10 (hence forth called OsWRKY62), XB11, XB12 and XB22, were chosen for further screening as baits in the Y2H to identify XB interacting proteins (XBIPs). Using Arabidopsis NPR1 as bait, six interacting proteins (NRR, NRRH1, rTGA2.1, rTGA2.2, rTGA2.3, and rLG2) were isolated by the same approach as described above. With NRR as bait, we isolated an additional six proteins (NH1, NH2, NRRIP-1, NRRIP-2, and NRRIP-3). With rTGA2.1 as bait, 4 interacting proteins were identified (TGA2.1IP-1, TGA2.1IP-2, GRNL1 and GRNL2). GRNL1 was used as bait to isolate nine interacting proteins (rTGA2.1, rTGA2.2, GIP-1, GIP-6, GIP-9, GIP-11, GIP-13, GIP-18, GIP-20, and GIP-23). Using SUB1A and SUB1C as baits, we identified 20 SUB1A binding proteins (SABs) and 9 SUB1C binding proteins (SCBs) (Table S1). Two proteins, SAB8 (SCB5) and SAB18 (SCB9), were identified using both SUB1A and SUB1C as baits. All identified proteins were repeatedly confirmed through secondary screenings were further characterized.
Additional proteins were incorporated into the XA21 and NH1/NRR interaction based on literature curation and subsequent experimentation. For example, ten interactors identified through our previous rice kinase Y2H screen [14], were incorporated into the the rice stress response interactome (Figure 1A, Table S1). We also demonstrated, through Y2H and co-immunoprecipitation assays, that OsRac1 (rice small GTPase, previously shown to play an important role in the rice defense response) interacts with RAR1 (required for Mla12 resistance), HSP90 (heat shock protein 90), OsRBOHB (rice respiratory burst oxidase homologB), and OsMPK1 [15], [16], [17]. We also showed that OsMPK12 (blast - and wound-induced MAP kinase (BWMK1)), which was previously demonstrated to be induced upon infection by Magnaporthe grisea), interacts with XB22IP-2 (hereafter, called OsEREBP1 (rice ethylene-responsive element-binding protein 1, AP2)) [18]. We tested additional interactions based on of the presence of predicted protein motifs. For example, a tetratricopeptide repeat domain found in XB22 is also found in SGT1 (Suppressor of G-two allele of Skp1). XB12 shows sequence similarity with p23, a protein that modulates Hsp90-mediated folding of key molecules involved in diverse signal transduction pathways [19]. We therefore tested the protein interactions of these two XBs with components of the HSP90/SGT1/RAR1 chaperone complex [20]. Positive interactions were incorporated into the rice stress response interactome. Similarly, because NH1 interacts with NRR, we tested two predicted paralogs (NRRH1 and NRRH2) with NH1.
While a genetic interaction between the NH1 and XA21 signaling pathways has previously been demonstrated [21], signaling components shared between submergence tolerance and Xoo-resistance have not yet been described. The current network is composed of 100 proteins and shows significant enrichment (by q<0.05, Fisher exact test with multiple hypothesis adjustment [22]) for several gene ontology (GO) terms related to both abiotic and biotic stress responses (Figure 1B, Table S2). Among molecular functions, the rice stress response interactome is particularly rich in transcription factors (diamond nodes in Figure 1A, p-value = 7.1×10−5, Fisher exact test), including 5 WRKY proteins, 4 TGA proteins, and 4 AP2 factors.
Validation of the interactome using in vivo assays
Validation of subsets of protein-protein interactions (PPIs) with two additional in vivo assays provides evidence that the interactome is of high quality. Using a mating-based split ubiquitin system that measures interactions with transmembrane proteins [12], we confirmed that 80% (8 out of 10 tested) of the XA21-binding (XB) proteins are able to interact with the full-length, membrane-spanning XA21 (the initial screen was conducted with the truncated XA21K668 protein) (Figure 1A, Figure S1). To assess whether the observed Y2H protein-protein interactions occur in plant cells, we examined 30 candidate proteins pairs using bimolecular fluorescence complementation (BiFC) in rice protoplasts. To rule out false-positive interactions, we tested the interaction of each protein with negative control vectors consisting of half of the yellow fluorescent protein. We found that 14 of the 30 tested showed interactions as detected by fluorescence only in the presence of the interacting rice protein but not in the presence of the negative control. Four proteins fluoresced in the presence of the negative control but displayed greatly enhanced fluorescence intensity in the presence of the interacting rice protein indicating that the interaction could be reproduced in vivo. Together these results indicate that 60% (18/30) of the tested pairs of interactome members interact in rice protoplasts as revealed by BiFC assays (Figure 1A, Figure S2, Table S3).
Interactions among interactome components
Components showing a large number of interactions with other interactome members (high degree) have been hypothesized to be essential for survival of the organism [23] although this finding has been disputed [24]. To identify such key hub proteins, we identified components in the rice stress interactome that displayed high degrees of interactions and then subjected them to pair-wise PPI assays. We tested a 24×20 matrix of 27 biotic stress (XA21) interactome components, a 14×14 matrix of 16 abiotic stress (SUB1) interactome components, and a 24×16 matrix of biotic-abiotic interactome components (Text S1, Table S4). An interaction was considered significant and reproducible if we observed it was replicated in two to three independent assays (Table S4).
Pair-wise PPI assays among interactome members revealed large numbers of possible interactions within and between the biotic and abiotic sub-interactomes (average degree 11±8, Figure 1C, Table S4). These interactomes have a high percentage (21.8%) of interactions beween their components (232 interactions out of 1060 tested) (Table S4). The biotic stress response interactome exhibits the highest level of interactions at 27.5% (132/480). The abiotic stress response interactome and the union between the biotic-abiotic stress response interactomes are even more highly connected [18.9% (37/196) and 16.4% (63/384), respectively]. The high number of interactions observed in the stress response interactome suggests that a large fraction of the components are capable of interacting with each other. These results also suggest that these components serve as members of large and/or changing complexes in vivo [25].
While the high number of interactions we observed is an order of magnitude greater than observed for studies of large-scale interactomes [3], it is comparable to smaller scale, more focused studies, such as that carried out for Arabidopsis MADS box transcription factors. In the MADS-box factor study, an average of only 5.4% of the components showed interactions (272/4998). However, when transcription factors predicted to function in the same biological process were examined, they displayed an increased number of interactions. For example, MADS-box factors predicted to be involved in floral development showed >15% interactions [26].
Consistent with their demonstrated key roles in response to stress, XA21, SUB1A, and SUB1C exhibit a high degree of interactions. In the matrix-based PPI tests, each of these interacted with over 10 additional proteins not initially identified as interactors in the original screen (Table S4). Other proteins with published roles in biotic stress signaling, including XB15 [13], XB3 [27], OsWRKY62 [28], and XB24 [29] are also among those with an above average degree of interaction. Such hubs may have a higher chance of engaging in essential functions because they participate in more interactions [30].
Expression analysis of interactome components
Coexpression network analysis and stress-specific transcriptomics of the interactome components support the validity of the interactome as an integrated module and highlights specific nodes that may function in cross-talk between the abiotic and biotic stress responses (Figure 2). The interactome is highly enriched for genes with correlated or anticorrelated expression compared with the whole genome (Figure 2A and 2C). For this analysis, we built rice biotic and abiotic stress gene transcript coexpression networks for the interactome members based on Pearson's correlation coefficients (PCC) calculated from publically available Affymetrix microarray data (Table S5). We define a correlated or anticorrelated interaction by PCC > |0.5|, a criterion under which 15% of interactome gene pairs interact, compared with ∼5.5% of pairs in the whole rice genome, and no pairs when the expression profiles are randomized (Figure 2A and 2C, Table S5). In both the coexpression networks derived from the abiotic and biotic microarray datasets, many components of the SUB1A (abiotic stress) and the XA21/NH1 (biotic stress) sub-interactomes display highly correlated or anticorrelated expression (Figure 2B and 2D, Table S5). This result further supports cross talk between the abiotic and biotic response networks. Contrasting the networks built from the different array sets, reveals that only a fraction of edges are conserved between the biotic and abiotic gene expression networks. This suggests that the expression of interactome members, and thus their availability to form PPIs with each other, varies depending on the stress regime, consistent with a model of dynamic complex formation [31] (Figure 2B and 2D).

We also generated microarray data to monitor transcriptional responses of Xa21-expressing and Nh1 - and Nrr-overexpressing rice (NRR binds NH1 and is a negative regulator of resistance [21]) before and after Xoo infection. Analysis of this dataset as well as a previously reported Sub1a-specific response dataset [32], reveals that interactome members are significantly enriched among differentially expressed genes (p<0.05, Fisher exact test, Figure 2E, Figure 3, Table S6, Figure S3).

Phenotypic assays of key interactome components
The interactome includes fourteen components that have previously been shown to regulate resistance to Xoo, further supporting the high quality of the interactome (Figure 1A, Table S7). We measured the Xoo and/or submergence response phenotypes of mutant rice lines for twenty additional interactome members, focusing primarily on genes encoding proteins with a high degree of PPIs (Table S7). Note that because of this bias in our experimental design, we are unable to test for correlation between a high degree of PPIs and a functional role in rice stress tolerance. Our phenotypic results show that nine out of seventeen genes (53%) that we assayed for a role in resistance to Xoo showed altered defense response phenotypes. Only one out of nine genotypes assayed showed altered tolerance to submergence, possibly due to the absence of SUB1A in the genotypes we examined (Table 1, Figure 3A–3H, Figures S4, S5, S6, S7, S8, S9, S10, S11, S12, S13).

Importantly, our phenotypic analysis revealed roles for two protein classes that, to our knowledge, were previously unknown to function in the plant stress response. on sequence similarities, SAB18 is a SANT-domain transcription factor, and, SCB3, is an enzyme involved in lysine biosynthesis (Table 1). SAB18 is a negative regulator of submergence tolerance suggesting that it may modulate the antagonistic activities of its two binding partners, SUB1A and SUB1C (Figure 3G and 3H, Figure S13). SCB3 serves as a positive regulator of resistance to Xoo (Figure S8). This result together with an earlier report showing that lysine levels increase in the Xoo-challenged Xa21 rice compared to mock treated controls [33], suggests that lysine plays an important, although undefined, role in the rice innate immune response.
The remaining eight proteins that we demonstrate to be involved in rice innate immunity have similarity to known stress-response factors (Table 1, Table S7, Text S1). Though many of these proteins were identified due to association with XA21 or an XB, modification of the expression of four of these genes gives altered resistance phenotypes in the absence of XA21 (Table 1), suggesting that they function in multiple biotic stress-response signaling pathways. Of particular significance, knockdown or knockout experiments show a role for three proteins, (RAR1, WAK 25 (wall associated kinase 25), and SnRK1 (sucrose non-fermenting-related protein kinase 1)), in XA21-mediated immunity.
The chaperone complex, HSP90/RAR1/SGT1 has been long known to play a positive role in intracellular NBS-LRR-mediated immunity [34]. RAR1 and HSP90 have also been shown to play a role in Arabidopsis FLS2-mediated signaling [35] and maturation of the rice chitin extracellular receptor OsCERK1 [36], respectively. Our observation that RAR1 serves as a positive regulator of XA21-mediated immunity (Figure 3A and 3B, Figure S6) further affirms that this complex contributes to host sensor-mediated immunity.
Wak25 (LOC_Os03g12470), compromises XA21-mediated immunity (Figure S10), indicating that WAK25 is a positive regulator of this process. WAKs have previously been shown to function as positive regulators of plant defense responses [37]. Although we do not yet know how WAK25 serves to regulate XA21-mediated immunity, there is precedence for interaction of PRRs with other receptor kinases. For example, the Arabidopsis FLS2 PRR interacts with the BRI1-associated kinase (BAK1) to transduce the immune response [38].
We also found that OsMPK5, previously demonstrated to serve as a negative regulator of resistance to the fungus, Magnaporthe grisea, and the bacteria, Burkholderia glumae [39], also negatively regulates resistance to Xoo (Figure S4). In contrast, the Arabidopsis protein with highest similarity to OsMPK5, AtMPK3, acts downstream of the Arabidopsis host sensor FLS2 and is a positive regulator of camalexin-mediated resistance to Botrytis cinera [40], [41]. The opposite regulatory roles for these Arabidopsis and rice predicted MPK orthologs underlines the limitations of extrapolating function between plant species.
OsMPK12 -and OsEREBP1 - are also positive regulators of resistance to Xoo (Figure S5, Figure S12). OsMPK12 was previously shown to phosphorylate OsEREBP1 [18]. OsEREBP1, as phosphorylated by OsMPK12, exhibits enhanced binding to the GCC box element of pathogenicity-related (PR) gene promoters. Overexpression of OsMPK12 in tobacco enhances expression of PR genes and increases resistance to Pseudomonas syringae and Phytophthora parasitica infection [18]. Thus, our results together with previously published studies indicate that OsMPK12 and OsEREBP1 are positive regulators of resistance to many pathogens.
We have also demonstrated a negative regulatory function for OsWRKY76 (Figure 3E and 3F, Figure S11), as has previously been shown for OsWRKY62 [28]. These two OsWRKYs are in the same WRKY subgroup (IIA) and are orthologs of barley HvWRKY1 and HvWRKY2, which serve as negative regulators of resistance to Blumeria graminis [42]. Along with our observation that the OsWRKY IIA proteins interact with members of the XA21 and SUB1 sub-interactomes [28], [43], these data are consistent with the WRKYIIA proteins playing a key role in fine-tuning grass defense responses.
SAB23 is a plant homeobox domain - (PHD) containing protein, which is known to function in development [44] and has been linked to response to pathogen stress [45] (Table 1). SAB23 serves as a negative regulator of resistance to Xoo (Figure 3C and 3D, Figure S7). This result supports previous observations that components regulating XA21-mediated resistance are also involved in developmental regulation [21], [46], [47]
SnRK1A, a well-known regulator of sugar sensing [48], was identified as a positive regulator in XA21-mediated immunity (Figure S9). Arabidopsis SnRK1 has been identified as a key regulator in sugar sensing and abscisic acid (ABA) signaling [49]. Though ABA has typically been found to act as a positive regulator of abiotic stress responses and a negative regulator of biotic stress responses [50], several positive regulators of the rice biotic stress response including SnRK1A and OsMPK12 participate in ABA signaling. Genes with ABA-related GO annotations are also up-regulated in Nh1-overexpressing and Sub1a-expressing transgenic rice (q = 1.3×10−2 and q = 5.3×10−10, respectively, Fisher exact test, multiple hypothesis adjustment) (Table S9). Together these observations support the hypothesis that ABA also has important functions in resistance to Xoo and tolerance to submergence in rice.
Comparable to analyses that show a correlation between essentiality and network degree centrality for essential genes [51] and negative regulators of growth (i.e., tumor suppressors) [52], we found that the rice interactome proteins with a validated role in the stress response have a significantly higher degree centrality in the abiotic co-expression network compared with those for which we were unable to measure a phenotype (Figure 3i, p = 3.7×10−2, Wilcoxon signed rank test, Table S8). Thus, interactome members that serve as central hubs as measured by co-expression analysis are more likely to function in the stress response than those members that do not serve as central hubs. This observation indicates the power of using the “guilt-by-association principle” to guide experiments based on co-expression maps [53], [54].
Conclusions
Here, we constructed a rice stress response interactome composed of 100 proteins governing the rice response to biotic and abiotic stress. Integration of protein-protein interaction assays, co-expression studies, and phenotypic analyses allowed us to efficiently identify ten novel proteins regulating the rice stress response.
Materials and Methods
Yeast two-hybrid screening
The XA21 kinase fragment K668 was cloned into the Y2H bait vector pMC86. SUB1A and SUB1C were also cloned into pMC86. Sequence information is provided in Table S1. The Y2H screening experiments for SUB1A and SUB1C were conducted in the same manner as those for XA21. Bait constructs were transformed into yeast strains HF7c MATa, plated on selective medium, and screened as described (Clontech's Matchmaker Pretransformed Libraries User Manual). Colonies from the HF7c baits were grown to approximately 2×108 cfu/mL in 50 mL synthetic dextrose (SD: 6.7 g Difco yeast nitrogen base w/o amino acids, 2% glucose, 1X drop out solution [supplemented with appropriate amino acids], pH 5.8) lacking Tryptophan (Trp) media for use in the primary screens. Cells of HF7c baits were pelleted, washed once with sterile H2O and resuspended in 50 mL rich yeast media, YPAD (20 g Difco peptone, 10 g yeast extract, 40 mg Adenine hemisulfate, 2% glucose, pH 5.8). Target yeast (Y187) were transformed with cDNAs from a Hybrizap (Stratagene) Y2H library derived from seven-week-old IRBB21 (Indica cultivar containing Xa21) leaf mRNA. One aliquot of the Y187 target yeast was mixed with the Hf7c bait yeast in 50 mL YPAD and poured into a tissue culture flask. Yeast strains were allowed to mate for 20 to 24 hrs at 28°C with slight shaking. Yeast were then isolated and washed twice with sterile water and plated on SD medium lacking Histidine (His), Tryptophan (Trp), Leucine (Leu) and supplemented with 2 mM 3-amino-1, 2, 4-triazole (3-AT). Putative positive diploids from the primary screens were isolated and plasmids extracted. Confirmation of interacting proteins through plasmid re-transformation eliminates many false positives; a step often dispensed of in high throughput Y2H studies due to the encumbrance of bacterial transformation and plasmid propagation [14]. Yeast plasmids were transformed into E. coli DH5α to amplify plasmids. Amplified plasmids were then re-transformed into the yeast strain AH109 (Clonetech) to confirm interactions. Transformed yeast for the secondary screens were first plated on selective medium lacking Leu and Trp. Once yeast colonies appeared, they were then streaked on selective medium lacking His, Leu, and Trp, plus 2 mM 3-AT and medium lacking Ade, Leu, and Trp. Prey plasmids were isolated and sequenced only after confirmation in secondary screens. The PPI datasets were submitted directly to DIP and assigned the International Molecular Exchange identifier IM-15311[55].
Mating based-split ubiqutin system (mb-SUS) assays
For mating based-split ubitquitin assays, we followed protocols and used vectors and yeast strains as described previously [12]. In brief, using Gateway LR Clonase (Invitrogen) we constructed the bait by transferring XA21cDNA from pENT/D into pMetYC_Gate and the preys through transfer of the corresponding cDNA from pENT/D into pNX_Gate32-3HA. Primers for these constructs are described in Table S10. For identification of positive interaction via yeast mating, the bait and prey constructs were transformed to yeast strain THY.AP5 and THY.AP5, respectively by using the yeast transformation kit, Frozen-EZ yeast transformation II (Zymo Research). Positive interactions were selected by colony growth in minimal SD/Ade-/Leu-/Trp-/His - media (Figure S1).
Bimolecular fluorescence complementation (BiFC) assays
We conducted BiFC assays as described in Ding et al. [14]. As negative controls, we included the both empty vectors (735 (YC)-EV and 736 (YN)-EV) for each pair-wise test. The BiFC assays are summarized in Table S3 and Figure S2.
Construction of the co-expression network
We calculated Pearson correlation coefficient (PCC) scores to measure tendency of coexpression between genes based on two sets of publicly available Affymetrix microarray data—219 rice abiotic and 179 rice biotic category data—for 37,993 genes which have Affymetrix probe set matched, of which 34,016 have unique Affymetrix probe set available and only these genes were included in this database (Table S5). The raw Affymetrix data was downloaded from NCBI Gene Expression Omnibus [56] and EBI ArrayExpress [57]. We processed raw Affymetrix data using the MAS 5.0 R-package. The trimmed mean target intensity of each array was arbitrarily set to 500, and the data were then log2 transformed. The Rice Multiple-platform Microarray Element Search was used to map the Affymetrix probesets to rice genes [58]. Distributions of PCC scores of 578,527,120 pairs of rice genes with processed microarrays or with randomized microarrays (by random shuffling of arrays) are summarized in Figure 2A and 2C and Table S5.
Transcriptional profiling of Xa21-, Nh1-, and Nrr-overexpressing rice
We grew TaiPei309 (TP309), Xa21::Xa21 106-17-3-37, LiaoGeng (LG), Ubi::Nh1 LG 11, and Ubi::Nrr 64 LG plants for six weeks in the greenhouse. We then transferred the plants to a growth chamber set for a 14-h daytime period, a 28/26°C temperature cycle and 90% humidity. We employed the scissors dip method with multiple cuts to inoculate the plants using a suspension (OD600 of 0.5) of PXO99 Xoo. One and two days after inoculation, mock-inoculated and inoculated leaves were harvested for gene expression profiling using the NSF45K array. The replicate mRNAs for the comparisons of Ubi::Xa21 TP309 vs TP309, Ubi::Nh1 LG vs. LG, and Ubi::Nrr LG vs. LG were labeled with either Cy3 or Cy5 dyes, resulting in one technical replicate and three biological replicates per genotype pair. Gene expression data were processed as previously described [58]. The microarray data have been deposited to NCBI GEO and have the accession number GSE22112.
Supporting Information
Zdroje
1. DhlaminiZSpillaneCMossJPRuaneJUrquiaN 2005 Status of Research and Application of Crop Biotechnologies in Developing Countries. Rome, Italy Food and Agriculture Oganization of the United Nations Natural Resources Management and Environment Department
2. FujitaMFujitaYNoutoshiYTakahashiFNarusakaY 2006 Crosstalk between abiotic and biotic stress responses: a current view from the points of convergence in the stress signaling networks. Curr Opin Plant Biol 9 436 442
3. RualJFVenkatesanKHaoTHirozane-KishikawaTDricotA 2005 Towards a proteome-scale map of the human protein-protein interaction network. Nature 437 1173 1178
4. SongWYWangGLChenLLKimHSPiLY 1995 A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science 270 1804 1806
5. ChernMFitzgeraldHACanlasPENavarreDARonaldPC 2005 Overexpression of a rice NPR1 homolog leads to constitutive activation of defense response and hypersensitivity to light. Mol Plant Microbe Interact 18 511 520
6. DurrantWEDongX 2004 Systemic acquired resistance. Annu Rev Phytopathol 42 185 209
7. XuKXuXFukaoTCanlasPMaghirang-RodriguezR 2006 Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 442 705 708
8. LeeSWHanSWSririyanumMParkCJSeoYS 2009 A type I-secreted, sulfated peptide triggers XA21-mediated innate immunity. Science 326 850 853
9. RonaldPCBeutlerB 2010 Plant and animal host sensors of conserved microbial signatures. Science In Press
10. ShimonoMSuganoSNakayamaAJiangCJOnoK 2007 Rice WRKY45 plays a crucial role in benzothiadiazole-inducible blast resistance. Plant Cell 19 2064 2076
11. Bracha-DroriKShichrurKKatzAOlivaMAngeloviciR 2004 Detection of protein-protein interactions in plants using bimolecular fluorescence complementation. Plant J 40 419 427
12. GrefenCLalondeSObrdlikP 2007 Split-ubiquitin system for identifying protein-protein interactions in membrane and full-length proteins. Curr Protoc Neurosci Chapter 5 Unit 5 27
13. ParkCJPengYChenXDardickCRuanD 2008 Rice XB15, a protein phosphatase 2C, negatively regulates cell death and XA21-mediated innate immunity. PLoS Biol 6 e231 doi:10.1371/journal.pbio.0060231
14. DingXRichterTChenMFujiiHSeoYS 2009 A rice kinase-protein interaction map. Plant Physiol 149 1478 1492
15. NakashimaAChenLThaoNPFujiwaraMWongHL 2008 RACK1 functions in rice innate immunity by interacting with the Rac1 immune complex. Plant Cell 20 2265 2279
16. ThaoNPChenLNakashimaAHaraSUmemuraK 2007 RAR1 and HSP90 form a complex with Rac/Rop GTPase and function in innate-immune responses in rice. Plant Cell 19 4035 4045
17. WongHLPinontoanRHayashiKTabataRYaenoT 2007 Regulation of rice NADPH oxidase by binding of Rac GTPase to its N-terminal extension. Plant Cell 19 4022 4034
18. CheongYHMoonBCKimJKKimCYKimMC 2003 BWMK1, a rice mitogen-activated protein kinase, locates in the nucleus and mediates pathogenesis-related gene expression by activation of a transcription factor. Plant Physiol 132 1961 1972
19. ZhuSTytgatJ 2004 Evolutionary epitopes of Hsp90 and p23: implications for their interaction. FASEB J 18 940 947
20. ShirasuK 2009 The HSP90-SGT1 chaperone complex for NLR immune sensors. Annu Rev Plant Biol 60 139 164
21. ChernMCanlasPEFitzgeraldHARonaldPC 2005 Rice NRR, a negative regulator of disease resistance, interacts with Arabidopsis NPR1 and rice NH1. Plant J 43 623 635
22. StoreyJDTibshiraniR 2003 Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100 9440 9445
23. JeongHMasonSPBarabasiALOltvaiZN 2001 Lethality and centrality in protein networks. Nature 411 41 42
24. GandhiTKZhongJMathivananSKarthickLChandrikaKN 2006 Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 38 285 293
25. ParkCJHanSWChenXRonaldPC 2010 Elucidation of XA21-mediated innate immunity. Cell Microbiol 12 1017 1025
26. de FolterSImminkRGKiefferMParenicovaLHenzSR 2005 Comprehensive interaction map of the Arabidopsis MADS Box transcription factors. Plant Cell 17 1424 1433
27. WangYSPiLYChenXChakrabartyPKJiangJ 2006 Rice XA21 binding protein 3 is a ubiquitin ligase required for full Xa21-mediated disease resistance. Plant Cell 18 3635 3646
28. PengYBartleyLEChenXDardickCChernM 2008 OsWRKY62 is a negative regulator of basal and Xa21-mediated defense against Xanthomonas oryzae pv. oryzae in rice. Mol Plant 1 446 458
29. ChenXChernMCanlasPERuanDJiangC 2010 An ATPase promotes autophosphorylation of the pattern recognition receptor XA21 and inhibits XA21-mediated immunity. Proc Natl Acad Sci U S A 107 8029 8034
30. HeXZhangJ 2006 Why do hubs tend to be essential in protein networks? PLoS Genet 2 e88 doi:10.1371/journal.pgen.0020088
31. LuscombeNMBabuMMYuHSnyderMTeichmannSA 2004 Genomic analysis of regulatory network dynamics reveals large topological changes. Nature 431 308 312
32. JungKHSeoYSWaliaHCaoPFukaoT 2010 The submergence tolerance regulator Sub1A mediates stress-responsive expression of AP2/ERF transcription factors. Plant Physiol 152 1674 1692
33. SanaTRFischerSWohlgemuthGKatrekarAJungKH 2010 Metabolomic and transcriptomic analysis of the rice response to the bacterial blight pathogen Xanthomonas oryzae pv. oryzae. Metabolomics 6 451 465
34. KadotaYShirasuKGueroisR 2010 NLR sensors meet at the SGT1-HSP90 crossroad. Trends Biochem Sci 35 199 207
35. ShangYLiXCuiHHePThilmonyR 2006 RAR1, a central player in plant immunity, is targeted by Pseudomonas syringae effector AvrB. Proc Natl Acad Sci U S A 103 19200 19205
36. ChenLHamadaSFujiwaraMZhuTThaoNP 2010 The Hop/Sti1-Hsp90 chaperone complex facilitates the maturation and transport of a PAMP receptor in rice innate immunity. Cell Host Microbe 7 185 196
37. BrutusASiciliaFMaconeACervoneFDe LorenzoG 2010 A domain swap approach reveals a role of the plant wall-associated kinase 1 (WAK1) as a receptor of oligogalacturonides. Proc Natl Acad Sci U S A 107 9452 9457
38. ChinchillaDZipfelCRobatzekSKemmerlingBNurnbergerT 2007 A flagellin-induced complex of the receptor FLS2 and BAK1 initiates plant defence. Nature 448 497 500
39. XiongLYangY 2003 Disease resistance and abiotic stress tolerance in rice are inversely modulated by an abscisic acid-inducible mitogen-activated protein kinase. Plant Cell 15 745 759
40. AsaiTTenaGPlotnikovaJWillmannMRChiuWL 2002 MAP kinase signalling cascade in Arabidopsis innate immunity. Nature 415 977 983
41. RenDLiuYYangKYHanLMaoG 2008 A fungal-responsive MAPK cascade regulates phytoalexin biosynthesis in Arabidopsis. Proc Natl Acad Sci U S A 105 5638 5643
42. ShenQHSaijoYMauchSBiskupCBieriS 2007 Nuclear activity of MLA immune receptors links isolate-specific and basal disease-resistance responses. Science 315 1098 1103
43. PengYBartleyLECanlasPERonaldPC 2010 OsWRKY IIa Transcription Factors Modulate Rice Innate Immunity. Rice 3 36 42
44. SaigaSFurumizuCYokoyamaRKurataTSatoS 2008 The Arabidopsis OBERON1 and OBERON2 genes encode plant homeodomain finger proteins and are required for apical meristem maintenance. Development 135 1751 1759
45. KorfhageUTrezziniGFMeierIHahlbrockKSomssichIE 1994 Plant homeodomain protein involved in transcriptional regulation of a pathogen defense-related gene. Plant Cell 6 695 708
46. CenturyKSLagmanRAAdkissonMMorlanJTobiasR 1999 Short communication: developmental control of Xa21-mediated disease resistance in rice. Plant J 20 231 236
47. ParkCJLeeSWChernMSharmaRCanlasPE 2010 Ectopic expression of rice Xa21 overcomes developmentally controlled resistance to Xanthomonas oryzae pv. oryzae. Plant Sci 179 466 471
48. HalfordNGHeySJ 2009 Snf1-related protein kinases (SnRKs) act within an intricate network that links metabolic and stress signalling in plants. Biochem J 419 247 259
49. JossierMBoulyJPMeimounPArjmandALessardP 2009 SnRK1 (SNF1-related kinase 1) has a central role in sugar and ABA signalling in Arabidopsis thaliana. Plant J 59 316 328
50. AsselberghBDe VleesschauwerDHofteM 2008 Global switches and fine-tuning-ABA modulates plant pathogen defense. Mol Plant Microbe Interact 21 709 719
51. LeeILehnerBCrombieCWongWFraserAG 2008 A single gene network accurately predicts phenotypic effects of gene perturbation in Caenorhabditis elegans. Nat Genet 40 181 188
52. CollavinLLunardiADel SalG 2010 p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ 17 901 911
53. HazbunTRFieldsS 2001 Networking proteins in yeast. Proc Natl Acad Sci U S A 98 4277 4278
54. WangPIMarcotteEM 2010 It's the machine that matters: predicting gene function and phenotype from protein networks. J Proteomics 73 2277 2289
55. OrchardSArandaBHermjakobH 2010 The publication and database deposition of molecular interaction data. Curr Protoc Protein Sci Chapter 25
56. BarrettTTroupDBWilhiteSELedouxPRudnevD 2009 NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res 37 D885 890
57. ParkinsonHKapusheskyMKolesnikovNRusticiGShojatalabM 2009 ArrayExpress update—from an archive of functional genomics experiments to the atlas of gene expression. Nucleic Acids Res 37 D868 872
58. JungKHDardickCBartleyLECaoPPhetsomJ 2008 Refinement of light-responsive transcript lists using rice oligonucleotide arrays: evaluation of gene-redundancy. PLoS ONE 3 e3337 doi:10.1371/journal.pone.0003337
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 4
Nejčtenější v tomto čísle
- Survival Motor Neuron Protein Regulates Stem Cell Division, Proliferation, and Differentiation in
- PTG Depletion Removes Lafora Bodies and Rescues the Fatal Epilepsy of Lafora Disease
- Evolution of Vertebrate Transient Receptor Potential Vanilloid 3 Channels: Opposite Temperature Sensitivity between Mammals and Western Clawed Frogs
- An Evolutionary Genomic Approach to Identify Genes Involved in Human Birth Timing