
Genome Analysis Reveals Interplay between 5′UTR Introns and Nuclear mRNA Export for Secretory and Mitochondrial Genes
In higher eukaryotes, messenger RNAs (mRNAs) are exported from the nucleus to the cytoplasm via factors deposited near the 5′ end of the transcript during splicing. The signal sequence coding region (SSCR) can support an alternative mRNA export (ALREX) pathway that does not require splicing. However, most SSCR–containing genes also have introns, so the interplay between these export mechanisms remains unclear. Here we support a model in which the furthest upstream element in a given transcript, be it an intron or an ALREX–promoting SSCR, dictates the mRNA export pathway used. We also experimentally demonstrate that nuclear-encoded mitochondrial genes can use the ALREX pathway. Thus, ALREX can also be supported by nucleotide signals within mitochondrial-targeting sequence coding regions (MSCRs). Finally, we identified and experimentally verified novel motifs associated with the ALREX pathway that are shared by both SSCRs and MSCRs. Our results show strong correlation between 5′ untranslated region (5′UTR) intron presence/absence and sequence features at the beginning of the coding region. They also suggest that genes encoding secretory and mitochondrial proteins share a common regulatory mechanism at the level of mRNA export.
Published in the journal:
. PLoS Genet 7(4): e32767. doi:10.1371/journal.pgen.1001366
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001366
Summary
In higher eukaryotes, messenger RNAs (mRNAs) are exported from the nucleus to the cytoplasm via factors deposited near the 5′ end of the transcript during splicing. The signal sequence coding region (SSCR) can support an alternative mRNA export (ALREX) pathway that does not require splicing. However, most SSCR–containing genes also have introns, so the interplay between these export mechanisms remains unclear. Here we support a model in which the furthest upstream element in a given transcript, be it an intron or an ALREX–promoting SSCR, dictates the mRNA export pathway used. We also experimentally demonstrate that nuclear-encoded mitochondrial genes can use the ALREX pathway. Thus, ALREX can also be supported by nucleotide signals within mitochondrial-targeting sequence coding regions (MSCRs). Finally, we identified and experimentally verified novel motifs associated with the ALREX pathway that are shared by both SSCRs and MSCRs. Our results show strong correlation between 5′ untranslated region (5′UTR) intron presence/absence and sequence features at the beginning of the coding region. They also suggest that genes encoding secretory and mitochondrial proteins share a common regulatory mechanism at the level of mRNA export.
Introduction
In humans, ∼35% of all genes have introns in their 5′ untranslated regions (UTRs) [1]–[3]. These introns differ from those in coding regions, for example, in typical length and nucleotide composition [1]–[3]. Previously, 5′UTR introns (5UIs) were suggested to be evolving under a neutral model of random insertion and deletion events with the sole constraint of avoiding upstream open reading frames [3]. Recently, we showed that presence and length of 5UIs correlates with the level of expression across cells and tissue types [1]. More importantly, we observed an uneven distribution of 5UIs amongst genes across specific functional categories [1]. Genes with regulatory roles, including non-receptor tyrosine kinases, regulators of cytoskeleton, transcription and metabolism, were enriched in having 5UIs [1]. Our results suggested that many 5UIs are evolving under complex selective forces as opposed to a simple model of neutral evolution [1]. However, it is unclear whether there is any widely used mode of regulation that is unique to 5UIs.
In eukaryotes, splicing is coupled to key mRNA metabolic processes. During the act of splicing, several different protein complexes are deposited onto mRNA. For example, the Transcription Export (TREX) complex promotes the nuclear export of fully processed transcripts [4]. In higher eukaryotes, the TREX complex is deposited primarily onto the 5′ end of nascent transcripts by the cooperative action of the cap-binding complex and the spliceosome [5]. Given that 5UIs are necessarily proximal to 5′ ends of transcripts, an intriguing possibility is that splicing of 5UIs could have a disproportionate impact on mRNA export by promoting TREX recruitment. Although the majority of transcripts follow the splicing-dependent export pathway, alternative pathways exist. Recently, Palazzo et al. demonstrated that mRNAs that encode secreted proteins can use an alternative route for mRNA export that is mediated by a nucleotide element within the signal sequence coding region (SSCR) [6]. In contrast to the splicing-dependent pathway, this alternative RNA export (ALREX) pathway does not require splicing or a 5′ cap [6]. Vertebrate SSCRs were found to be adenine-poor and silent mutations introducing adenines into the SSCR impair its ability to promote mRNA export [6]. However, beyond adenine-depletion this element has been poorly characterized. Furthermore, it has remained unclear which SSCR-containing transcripts use ALREX and to what extent, since the vast majority of SSCR-containing transcripts are also spliced and thus could potentially use the canonical export pathway. The fact that both ALREX signals and splicing signals are found near the 5′ end of genes, suggests the interesting possibility that competition between signals at the 5′end of transcripts determines how a given mRNA is exported.
Here, we extend our computational analysis of 5UIs to identify functional groups of genes that preferentially lack these introns. We find that 5UIs are depleted in genes containing SSCRs or mitochondrial-targeting sequence coding regions (MSCRs). We demonstrate that SSCRs and MSCRs derived from 5UI-lacking (5UI−) genes contain sequence features associated with ALREX and promote export in vivo. In stark contrast, SSCRs and MSCRs derived from 5UI+ genes do not exhibit ALREX-associated features. Furthermore, we show that 5UI+ genes do not support splicing-independent mRNA export. We then characterize ALREX elements more fully by identifying and validating new ALREX-associated motifs. Taken together, our results support a model wherein the 5′-most element in a newly synthesized transcript, be it an intron or an ALREX element, dictates which pathway is employed for export. Furthermore, our results provide the first known regulatory role that is unique to 5′ UTR introns and suggest that it is widely used.
Results
SSCR – and MSCR–containing genes are depleted of introns in their 5′UTRs
Using a high quality set of 5UI definitions for human, we observed a depletion of 5UIs amongst genes with certain Gene Ontology [7] (GO) annotations (Table S1). Examples of 5UI-depleted GO terms include “MHC class II protein complex” (ratio of 5UI-containing genes to total genes annotated with particular GO term is 0/25), “aspartic endopeptidase activity” (0/23), “voltage-gated calcium channel activity” (2/35), “growth factor activity” (33/180), “electron carrier activity” (27/145), and “extracellular space” (108/497). In each case, these ratios are significantly lower than the ratio of ∼35% expected by chance (p<0.05 after adjusting for multiple hypothesis testing). More generally, we observed a depletion of 5UIs among nuclear genes encoding three protein classes.
The first class was composed of protein families encoded by mostly intronless genes. This group includes histone genes [8], olfactory receptors, G-protein coupled receptors [9], and keratins [10], [11]. Depletion of 5UIs in these gene classes does not suggest any 5UI-specific phenomena, as these genes are more generally intron-depleted.
The second class was composed of secreted or membrane-bound proteins that are trafficked through the endoplasmic reticulum (ER). We compiled a list of all genes with signal sequence coding regions (SSCRs), encoding N-terminal cleavable signal sequence peptides that target newly synthesized proteins to the ER [12] (see Materials and Methods). We observed that 5UIs were generally depleted among SSCR-containing genes (Figure 1; Fisher's Exact Test p = 8×10-8, odds ratio 0.84).

The last class included proteins localized to mitochondria. Nuclear-encoded mitochondrial genes are translated in the cytoplasm and are targeted via an N-terminal leader peptide sequence to mitochondria [13], [14]. We compiled a list of genes with mitochondrial-targeting sequence coding regions (MSCRs), and observed that 5UIs were depleted in MSCR-containing genes (Figure 1; Fisher's Exact Test p = 8×10-6; odds ratio 0.59). This depletion is even stronger than that observed for SSCR-containing genes. Thus, our results showed a general depletion of 5UIs among genes encoding either ER-targeted or mitochondrial proteins.
Next, we tested whether 5UI depletion in SSCR or MSCR-containing genes is a secondary effect of these genes having short 5′UTRs. Although 5UIs are more likely amongst genes with long 5′UTRs (Figure S1A, Wilcoxon Rank Sum Test p <2×10-16; a 99 nt greater median 5′UTR length in 5UI+ than 5UI− genes), we observed that genes encoding secreted and mitochondrial proteins have 5′UTRs that are only slightly shorter than other genes (Figure S1B, Wilcoxon Rank Sum Test p = 2×10-15, p = 9×10-9; a 25 nt and 51 nt difference in median 5′UTR length for SSCR - and MSCR-containing genes, respectively). Even after correcting for the differences in 5′UTR length, SSCR - and MSCR-containing genes were significantly depleted of 5UIs (see Text S1). Similarly, the depletion of 5UIs did not reflect an overall decrease in intronic content, as the total number of bases in non-5′UTR introns did not differ between genes containing or lacking SSCRs (Welch Two Sample t-test, p = 0.34; Figure S2).
SSCRs from 5UI− genes, but not 5UI+ genes, promote ALREX
A possible link between splicing and genes encoding secretory proteins is the nuclear export of mRNA. Several studies have indicated that export factors are loaded near the 5′ cap co-transcriptionally during the splicing of the more 5′-proximal intron [5], [15]. SSCRs, which similarly promote mRNA export via ALREX [6], are located at the 5′end of the open reading frame (ORF) and could also potentially be recognized by factors co-transcriptionally. Hence, we hypothesized that the 5′-most element in a given transcript, be it an intron or an SSCR, dictates the pathway by which that transcript is exported.
Signal peptide sequences contain a hydrophobic core with amino acids that are naturally encoded by codons with low adenine content. In addition, for pairs of biochemically similar amino acids that differ in the adenine content of their corresponding codons, SSCRs tend to prefer the amino acid with low adenine content codons [6]. We previously showed that adenine depletion in SSCRs is functionally linked to ALREX as silent adenine mutations partially inhibit ALREX [6]. Our hypothesis of a competition between export pathways, driven by whether the 5′-most element is a 5UI or an ALREX signal, predicts that the selection pressure to maintain sequence features important for ALREX-dependent mRNA export would be relaxed in transcripts with 5UIs. We therefore tested whether adenine depletion in SSCRs is attenuated in genes containing 5UIs. Remarkably, we found that SSCRs from genes lacking 5UIs contain 18.2% fewer adenines when compared to SSCRs from genes carrying 5UIs (Figure 2A; Wilcoxon Rank Sum Test p = 4×10-49). Next, we analyzed the amino acid preference of SSCR-containing genes for pairs of biochemically similar amino acids. Specifically, we observed that SSCRs of 5UI− genes have a significantly increased ratio of leucine (which has adenine-poor codons) to isoleucine (which has at least one adenine in all of its three codons) and of arginine (with relatively adenine-poor codons) relative to lysine as compared to SSCRs of 5UI+ genes (Figure 2B–2C; Fisher's Exact Test, p = 3×10-27 and 3×10-40, 95% confidence interval of odds ratio 1.4–1.7 and 1.9–2.4 respectively). SSCRs also exhibit a bias towards synonymous codons that lack adenine [6]. Importantly, this bias diminishes for 5UI+ genes (Figure 2D). This was true for codons for any given single amino acid, such as leucine or serine (Figure S3), or when all synonymous codons were aggregated (Figure 2D; Fisher's Exact Test p = 2×10-42; 95% CI of odds ratio 1.3-1.4). Taken together, our computational analysis indicates that the bias of SSCRs against adenines is relaxed in 5UI+ genes. Furthermore, this reduced bias appears to be due to a relaxation of nucleotide-level constraints, supporting the idea that the presence of 5UIs relieves selection maintaining ALREX signals.
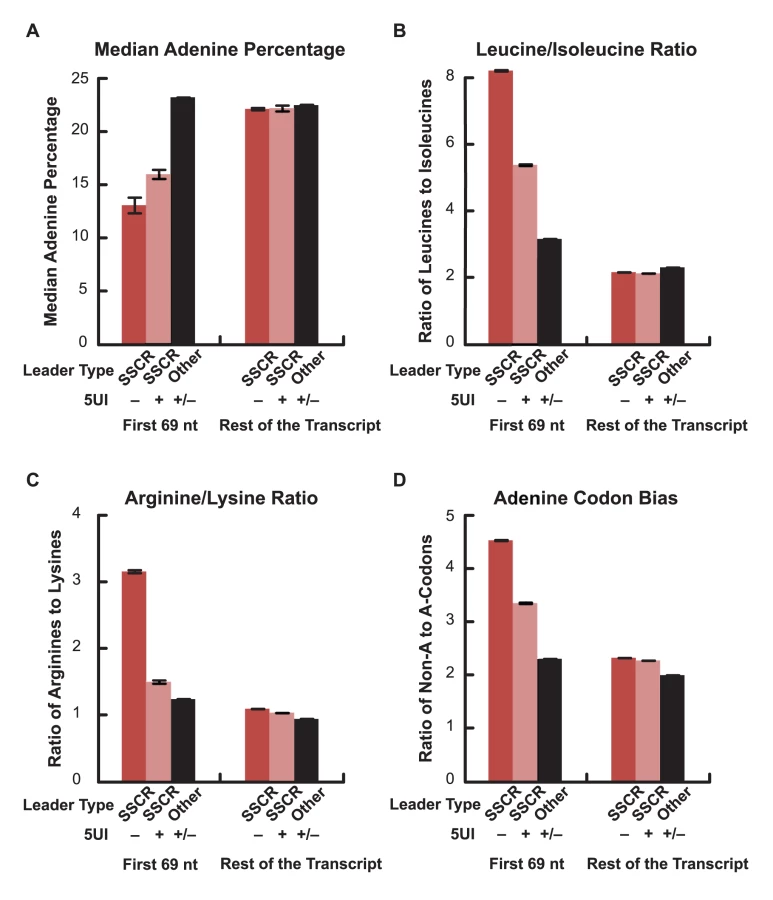
To experimentally investigate this intriguing connection between sequence features in the coding region and the presence or absence of 5UIs, we tested whether SSCRs derived from genes with 5UIs are defective in promoting mRNA export. We inserted SSCR elements into a fragment of the fushi tarazu (ftz), just downstream of the start codon. Furthermore we generated versions of ftz that either contained (ftz-i) or lacked (ftz-Δi) its endogenous intron. Modified forms of these transcripts were previously used to study splicing - and SSCR-dependent mRNA nuclear export [6], [16]. Polyadenylated forms of the ftz mRNA were microinjected into the nuclei of NIH 3T3 mouse fibroblasts. After incubating the cells for one hour, mRNA export was visually monitored by fluorescence in situ hybridization (FISH, Figure 3A) and the amount of mRNA nuclear export was quantified (Figure 3B). Nuclear injection was confirmed by co-injecting fluorescently labeled 70 kD dextran, which is too large to passively diffuse through nuclear pores (see insets, Figure 3A). As demonstrated by several groups, we found that a version of the ftz mRNA that encodes a cytoplasmic protein, but contains neither an intron nor an SSCR (c-ftz-Δi), was not efficiently exported [6], [16] (Figure 3). Nuclear export could be rescued if an intron was incorporated (c-ftz-i). As reported previously, SSCRs from the MHC class 2 gene H2-k1, which lacks a 5UI, promoted efficient export of an intronless version of ftz (Figure 3, MHC-ftz-Δi; see Palazzo et al. [6] and Figure S4 for all ftz variant sequences). We next examined the parathyroid hormone (PTH) and the prion protein (PRP) SSCRs, both derived from genes with 5UIs. Consistent with trends we observed for 5UI+ genes in general, neither PTH nor PRP SSCRs are depleted in adenine content. Furthermore, neither promoted efficient export (Figure 3, PTH-ftz-Δi and PRP-ftz-Δi). Interestingly, elimination of adenines from the PRP SSCR (PRPΔA) only marginally stimulated export (Figure 3, PRPΔA-ftz-Δi) suggesting that this SSCR lacks other features crucial for stimulating export. In summary, only SSCRs from genes lacking 5UIs promoted efficient mRNA export, experimentally demonstrating a functional relevance for the computationally-discovered connection between coding sequence features and 5UI status.

MSCRs from 5UI− genes, but not 5UI+ genes, promote ALREX
Our investigation into the relationship between 5UIs and alternative export began with the observation that 5UIs were depleted amongst secretory genes. Because 5UIs are also depleted amongst nuclear-encoded mitochondrial genes (Figure 1), we wondered whether related phenomena might be at play. Like secreted proteins, mitochondrial proteins contain a cleavable leader peptide that dictates the ultimate localization of the polypeptide chain [13], [14]. We therefore wondered whether MSCRs exhibit the same nucleotide features that had been associated with ALREX in SSCRs. Indeed MSCRs, like SSCRs, were depleted in adenines overall. Also like SSCRs, this adenine depletion was restricted to MSCRs derived from 5UI− genes (Figure 4A; Wilcoxon Rank Sum Test p = 2×10-9). We found that MSCRs, like SSCRS, tend to encode leucine relative to isoleucine (Figure 4B), and arginine relative to lysine (Figure 4C). Just as with SSCRs, this phenomenon was more pronounced when the elements were derived from 5UI− genes (Fisher's Exact Test p = 0.16 and 10−9, 95% CI of odds ratio 0.9–1.9 and 1.9–3.7 respectively). Finally, only MSCRs from 5UI− genes displayed a bias for synonymous codons that lacked adenine (Figure 4D). This was true for codons coding for any given single amino acid examined, such as leucine or serine (Figure S3), or when results for all synonymous codons were aggregated (Figure 4D; Fisher's Exact Test p = 7×10-06; 95% CI for odds ratio 1.2-1.7).

We next experimentally tested whether MSCRs from 5UI− genes promoted mRNA export in tissue culture cells. Indeed, we found that MSCRs from both the F1 ATP Synthase A (F1) and ferroredoxin reductase (FR) stimulated efficient nuclear export of the ftz transcript (Figure 5A–5B, F1-ftz-Δi, FR-ftz-Δi – see Figure S4 for all modified ftz sequences). We note that the alternative export phenotype observed for these MSCRs is at least as robust as any previously observed for SSCR-containing genes. In contrast, we found that the MSCR from the mitochondrial translation initiation factor 2a (MTIF), a 5UI+ gene, does not promote efficient export (Figure 5A–5B, MTIF-ftz-Δi). Similar to previous observations with the MHC and Insulin SSCRs [6], the introduction of seven silent adenine mutations in the FR MSCR (FR7A) partially inhibited its ability to promote export (Figure 5, FR7A-ftz-Δi).

Microinjected mRNA may behave differently from mRNA that has been endogenously transcribed. Therefore, we microinjected plasmids encoding various ftz transcripts into the nuclei of NIH 3T3 cells. After allowing the plasmids to be transcribed (20 min), further mRNA synthesis was inhibited by treating cells with the RNA Polymerase II inhibitor α-amanitin. Export of the newly synthesized transcripts was assessed two hours after treatment. We found that transcripts produced from plasmids containing FR-ftz-Δi, but not c-ftz-Δi or PTH-ftz-Δi, were efficiently exported (Figure 5C), as was previously seen for MHC-ftz-Δi [6]. Thus, we have shown that MSCR-containing transcripts are capable splicing-independent mRNA export in a manner that depends on 5UI status. This result suggests that the scope of the ALREX pathway extends from ER-trafficked genes to include nuclear mitochondrial genes.
We next wished to assess whether export was dependent on the TAP/p15 nuclear transport receptor, which is required for both SSCR - and splicing-dependent export [6]. We co-injected the viral constitutive transport element (CTE) RNA (known to inhibit TAP/p15 [17]) with the plasmid and observed that export of in vivo-transcribed FR-ftz-Δi was inhibited. Taken together, these experiments indicate that MSCRs and SSCRs from 5UI− genes promote mRNA export using a similar if not identical pathway.
Identification of motifs associated with ALREX
Although our experimental findings supported the importance of adenine-depletion for ALREX, they also indicated that other sequence features may be involved. For example, the PRP SSCR (from a 5UI+ gene) did not promote efficient export even after adenines were eliminated (Figure 3, PRPΔA-ftz-Δi). Furthermore, the incorporation of silent adenines only partially inhibited export by the FR MSCR (Figure 5, FR7A-ftz-Δi), or the MHC SSCR [6]. Therefore, we wished to search for additional ALREX-associated sequence features.
Identification of nucleotide motifs responsible for ALREX function is challenging, because enriched RNA-level motifs might arise due to recurrent patterns at the protein sequence level. Although numerous bioinformatics tools exist to search for nucleotide features (such as transcription factor binding sites) in non-coding regions, few are tailored to the problem of identifying RNA motifs within coding regions. We sought to exploit the idea that we have two collections of SSCRs that differ in the expected abundance of ALREX signals. Specifically, we compared SSCRs from genes with and without 5UIs to identify nucleotide signals exhibiting differential abundance between the sets. Although RNA-level features may be artifactually enriched relative to random RNA sequence due to protein sequence-level constraints, such an artifactual enrichment would not be expected in 5UI− relative to 5UI+ SSCR-containing genes.
We first extended codon usage analyses of the SSCR and MSCR regions to identify other representative signatures. In addition to previously noted adenine depletion, 5UI− SSCR and MSCR genes strongly preferred codons lacking thymine, with a ∼1.4 and a ∼1.7 fold enrichment relative to 5UI+ SSCR and MSCR genes (Figure 6A, Fisher's Exact Test p = 7×10-46 and 4×10-13; 95% CI for odds ratio 1.3–1.5 and 1.5–2.0, for SSCRs and MSCRs respectively).

Next, we searched for primary sequence elements using a discriminative motif finding approach. Specifically, we searched for nucleotide sequences that are significantly enriched among SSCR-containing 5UI− genes relative to 5UI+ genes using the DEME algorithm [18]. We found a likely candidate motif (Figure 6B), which can be roughly described by the consensus sequence CGSSGC (where S represents a mixture of C and G). This motif is highly depleted of adenines and thymines consistent with our analysis (Figure 2, Figure 3, and Figure 6A) and had high information content.
The motif did not show a strong preference for a particular frame of translation (Figure 6B) suggesting that this signal is relevant at the RNA as opposed to protein level. The motif not only appeared in a higher fraction of 5UI− SSCR sequences (47.5% versus 22.2% in 5UI+ SSCRs; see Materials and Methods), but also was much more likely to occur in multiple copies in the SSCRs of 5UI− genes (Figure 6C, 6D; 26.8% versus 7.14%). The CGSSGC motif also revealed a strong positional bias, occurring more frequently toward the 5′ end of coding regions from 5UI− genes (Figure 6E, Figure S5, Wilcoxon Rank Sum Test p = 0.002, median position was 39 and 45 among 5UI− and 5UI+ genes, respectively; see Materials and Methods).
We wished to further examine the question of whether the non-canonical mRNA export function of SSCRs is acting via the same mechanism as that of MSCRs. We therefore tested whether the CGSSGC motif (which was enriched among 5UI− SSCR genes) could also predict the absence of 5UIs among genes with an MSCR. We compared performance of the CGSSGC motif (discovered without use of any MSCR-containing genes) in discriminating 5UI− from 5UI+ MSCRs and found it to outperform at least 99% of 100,000 randomly generated motifs (Figure 6F; False-positive Rate range 10% to 70%; see Materials and Methods). This result indicates that MSCRs and SSCRs, despite differences in the protein sequences they encoded, each play host to a common RNA-level motif associated both with the lack of 5UIs and the ability to support non-canonical mRNA export.
To identify additional motifs, we used the AlignACE [19] algorithm on the set of SSCR sequences from 5UI− genes. This algorithm has the advantage that it can identify multiple nucleotide sequences and allows greater flexibility in motif length. We filtered the discovered sequences for their discriminative ability and found 19 motifs that were significantly enriched among 5UI− relative to 5UI+ genes (Table S2, see Materials and Methods). The discovered motifs displayed mutual similarity and included several close variants of the CGSSGC motif discovered by DEME (See Figure S6 for the PSSM logos of the most discriminative AlignACE motifs).
We next focused on the properties of the four most discriminative AlignACE motifs. All four motifs were more likely to occur in multiple copies among the 5UI− genes compared to 5UI+ genes (Figure S7). Even though these four motifs were discovered based on their ability to discriminate 5UI− from 5UI+ genes among those genes with SSCRs, these motifs were also predictive of 5UI absence for genes with MSCRs (Figure S8). All four motifs performed in the top quartile compared to 100,000 random motifs (Figure S8; see Materials and Methods). However, unlike the CGSSGC motif, three of these motifs displayed a significant bias for occurring in a particular frame of translation. These three motifs may thus be detecting protein sequence-level differences between 5UI− and 5UI+ genes (Figure S9). In fact, consensus sequences of many AlignACE motifs included CTGs that can encode leucines, which were highly enriched among SSCRs and MSCRs from 5UI− genes relative to their 5UI+ counterparts.
We next decided to test whether synthetic elements matching the discovered motifs could promote the export of ftz mRNA. We used versions of the ftz-Δi mRNA containing either three copies of an element matching the consensus CGSSGC motif (M1-ftz-Δi), a CUG repeat-containing element (M2-ftz-Δi), or a single copy of each (M3-ftz-Δi see Figure 7A for the sequences of all these constructs). We chose CUG repeats as they appeared in many of the consensus sequences of AlignACE motifs (Table S2). In addition, there are several RNA binding proteins, such as CUG-BP1 [20] and the Muscleblind family of proteins [21] that are known to recognize CUG repeats.
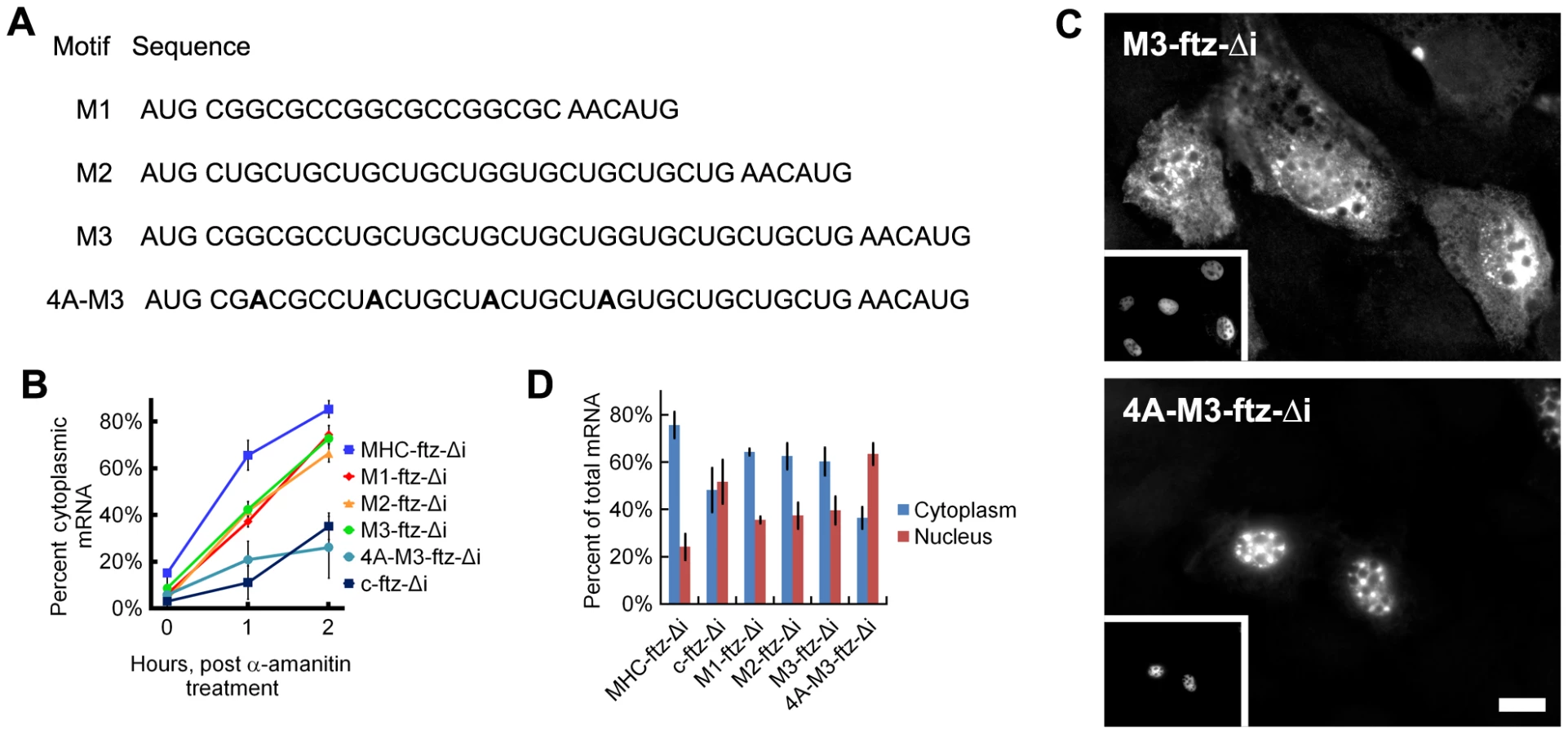
To assay for export activity we microinjected plasmids that contained versions of the ftz gene fused to segments containing elements matching ALREX-enriched motifs and their combinations (Figure 7A) into the nuclei of NIH 3T3 cells. After allowing the plasmids to be transcribed (20 min), further mRNA synthesis was inhibited by treating cells with α-amanitin. We found that all three motif-containing ftz constructs (M1-, M2-, M3-ftz-Δi) were exported more efficiently than c-ftz-Δi but substantially less efficiently than MHC-ftz-Δi mRNA (Figure 7B). Adenine depletion was required for export, as mRNA generated from plasmid containing a mutant form of M3-ftz-Δi bearing four silent adenine mutations (4A-M3-ftz-Δi, see Figure 7A) collectively disrupting each of the two component elements was not efficiently exported (Figure 7B–7C). To further validate these results, we transfected plasmids encoding the motif-containing ftz genes with elements corresponding to these motifs into COS-7 cells and measured the steady state distribution of mRNA. In agreement with our microinjection experiments, we found that the three motif-containing ftz constructs were exported to a level that was clearly higher than c-ftz-Δi but lower than MHC-ftz-Δi (Figure 7D). As observed for microinjected NIH3T3 cells (Figure 5), mRNA generated from a plasmid containing the 4A-M3-ftz-Δi construct was not efficiently exported from transfected COS-7 cells (Figure 7D).
Discussion
The function and evolution of introns has been intensely studied since their discovery (reviewed in [22], [23]). Despite the presence of a large number of introns in untranslated regions, especially in the 5′ untranslated regions of transcripts, these studies have been largely focused on introns in coding regions [3]. We established that the distribution of 5UIs in the human genome is non-random, with specific functionally related groups of genes being enriched [1] or depleted (this study) for 5UIs. Here we show that, in both secreted and mitochondrial genes, the presence or absence of 5UIs correlates with sequence features at the beginning of the coding region. Minimally, our results further support the conclusion that complex selective forces govern the evolution of 5′UTR introns. Moreover, our results are best explained by the existence of a regulatory mechanism that is both special to 5UIs and has relevance to thousands of genes across the genome.
Our results show that nuclear transcripts encoding both secretory and mitochondrial proteins share RNA-level signals capable of directing mRNA export, even for an intronless message. It has frequently been observed that mRNAs of functionally related genes are co-regulated at the post-transcriptional level (‘the regulon hypothesis’ [24]). Our results suggest that, consistent with this phenomenon, the ALREX pathway can facilitate coordinated expression of functionally related genes at the level of mRNA export. Moreover, our analyses support a model whereby the first transcript element emerging from RNA Polymerase II during transcription—be it an intron or an ALREX-promoting element—determines which RNA export pathway is predominantly followed (Figure 8). Under this model, presence of a 5UI would supersede downstream SSCR or MSCR export signals and relax selection pressures that maintain ALREX-promoting sequence features.

Although we have made progress in defining some sequence features that mediate the ALREX function (see Figure 6 and Figure 7), it is clear that a more extensive description of ALREX features individually and in combination will be quite useful. We found specific nucleotide-level motifs in the 5′ end of coding regions which discriminate between genes with and without 5UIs. Substantial future efforts will be required to combine information about 5UI absence with the presence and placement of ALREX signals within a unified framework that can predict ALREX activity. This information could be used to compile a full list of transcripts using the ALREX pathway. It will be interesting to determine whether other genes, such as those that encode membrane-bound proteins but lack a signal sequence (and hence an SSCR), can use this alternative export pathway.
The most fundamental challenge for future studies will be to understand the biological role or roles of the ALREX pathway. Why is its selection maintained even in transcripts that contain coding region introns and are therefore enabled to use the canonical mRNA export pathway?
Although the functional downstream consequences of using either the splicing-dependent or ALREX-pathway remain unknown, silent mutations within the SSCR not only impair mRNA export but also disrupt proper ER-targeting of the transcripts [6]. This suggests that multiple post-transcriptional events, such as mRNA export, mRNA transport in the cytoplasm and mRNA translation, are coupled [25].
Here, we have discovered and validated two motifs that promote mRNA export, suggesting that ALREX may recruit more than one nuclear factor. Such factors could not only dictate RNA export but perhaps also dictate how the mRNA is distributed and translated once in the cytoplasm. Investigation of these questions awaits identification of ALREX factors, and of mRNA localization or other phenotypes associated with disrupted ALREX function. One of the motifs we discovered is a long CUG repeat that could potentially bind to CUG binding proteins. However, MHC-ftz-Δi mRNA is exported from HeLa cells that were depleted of both MBNL1 and MBNL2, two members of the muscleblind family of CUG-repeat binding proteins (unpublished findings), suggesting that these are not the responsible factors. Identification of the ALREX-element binding protein(s) will shed light onto how ALREX operates and provide insight into the biological role of this pathway.
The question of biological role is particularly intriguing in the case of nuclear-encoded mitochondrial genes. The textbook description of nuclear-encoded mitochondrial genes has translation of these genes occurring within the general pool of cytoplasmic proteins, with subsequent protein localization due solely to the mitochondrial targeting peptide sequence. However, there is evidence that nuclear-encoded mitochondrial transcripts can localize to the vicinity of mitochondria prior to translation [26], [27]. Although we do not detect any mitochondrial targeting of MSCR-bearing transcripts (Figure 5), it is possible that a fraction of these mRNAs are indeed localized. It will be interesting to learn what role ALREX could play in the localization and translation of nuclear-encoded mitochondrial genes.
Substantial future studies will be required to further explore mechanisms of the ALREX pathway. For example, it is unclear whether ALREX signals are inhibited by other complexes deposited on the transcript in a splicing dependent manner. One example is the Exon Junction Complex (EJC), which potentiates the translation of properly spliced mRNA [28], [29] and the nonsense-mediated degradation of improperly spliced transcripts [30], [31]. Some mRNAs, such as those of PrP and PTH genes, encode secreted proteins but lack any ALREX-promoting element. For such mRNAs, it is possible that the proper ER - targeting and efficient translation of these transcripts requires the recruitment of the EJC or TREX components to the 5′UTR. Identification of the nuclear proteins that associate with ALREX elements, and how these factors are coupled to other processes, will yield significant insight into the role of ALREX in mediating gene expression, and localization of both mRNAs and proteins.
Materials and Methods
Assembly of a collection of genes with 5UIs and analysis of total exonic and intronic length
NCBI's human Reference Gene Collection (RefSeq) [32] and the associated annotation table, retrieved from the UCSC genome browser genome assembly May 2004 (http://hgdownload.cse.ucsc.edu/downloads.html), were used to extract a high confidence set of 5UIs. The lengths of 5′UTR-associated genomic features were determined using RefSeq intron-exon definitions (downloaded June 2007). Out of a total ∼24.5 k RefSeq transcripts, ∼8.5 k contained at least one intron. Genomic coordinates of 5UIs examined were as previously described [1]. When multiple splice variants involving a given 5′UTR exhibited identical splicing patterns within that 5′UTR region, a single identifier was selected randomly as the representative for that 5′UTR.
For the remaining transcripts, total lengths of coding region introns were determined from the RefSeq Annotation (downloaded from UCSC genome browser, May 2004 genome assembly on May 15th 2009 http://hgdownload.cse.ucsc.edu/downloads.html).
Microinjection and mRNA imaging
DNA constructs encoding ftz isoforms were assembled by first digesting the pBR322 plasmid containing c-ftz-Δi [6] with Nco I and ligating oligonucleotides encoding various SSCRs and MSCRs (see Figure S4) so that the extra sequences were all inserted just downstream of the start codon. The constructs were then transcribed into mRNA, which was then polyadenylated, purified and then microinjected into NIH 3T3 fibroblast nuclei at 200 µg/ml with Alexa488 conjugated 70 kD dextran (1 mg/ml) as previously described [6], [33]. DNA microinjections were performed as previously described [6]. Briefly, ftz isoforms were subcloned into pCDNA3 using Hind III and Xho I and microinjected at 50 µg/ml with Alexa488-conjugated 70 kD dextran (1 mg/ml) into NIH 3T3 fibroblast nuclei. After allowing the RNA to be transcribed for 20 min, the cells were treated with α-amanitin (50 ng/ml) to prevent further transcription. CTE RNA was synthesized as previously described [6] and microinjected at a concentration of 200 µg/ml along with DNA and Alexa488-conjugated 70 kD dextran. All microinjected cells were incubated for the indicated time to allow for mRNA export, then fixed with 4% paraformaldehyde in phosphate buffered saline (PBS). DNA transfections into COS-7 cells were performed as described previously [6]. Transfected cells were incubated for 12–18 hrs, then fixed with 4% paraformaldehyde in PBS. The ftz mRNA was stained by fluorescence in situ hybridization followed by imaging and quantification of RNA nuclear export as previously described [6]. Cell imaging and mRNA quantification were also performed as previously described [33].
Functional enrichment of Gene Ontology categories
FuncAssociate [34], [35] beta version was used for Gene Ontology (GO) analysis, and Synergizer [36] was used for mapping RefSeq IDs into the ‘namespace’ of GO association files using Ensembl as the synonym authority. We restricted the space of genes in which GO correlations were sought to RefSeq because our 5UI genes were drawn only from this set. To quantify the effect size of GO correlations, the results in Table S1 were sorted according to their log10 odds ratio, with significance calculated by Fisher's Exact Test as previously described [35]. Multiple hypothesis correction was achieved via a resampling approach that preserves the dependency structure between the tested hypotheses [35]. Adjusted p-values were calculated using 10000 resampling simulations.
Analysis of SSCR and MSCR sequences
We retrieved the complete set of transcripts with signal peptide annotations from the Ensembl 50 database using Biomart [37] (downloaded on February 2009 http://www.ensembl.org/biomart/martview). Of the 38396 transcripts in this database, 4953 were annotated as having a signal peptide, and 4704 of these were in our set of RefSeq genes. The coding region sequences for all the genes in our set were downloaded from NCBI Refseq Collection release 33 (ftp://ftp.ncbi.nih.gov/refseq/H_sapiens/mRNA_Prot). The ratio of the amino acids, total adenine counts and the codon usage bias were calculated for the first 69 nt and the rest of the sequences. There were 135 coding region sequences that had a length that was not a multiple of three. These sequences in addition to those with total length less than 150 nt were removed from further analysis.
The list of mitochondrial genes was retrieved from the Organelle DB [38] website (downloaded on February 2009 from http://organelledb.lsi.umich.edu/). Identifiers were translated to RefSeq ID using Synergizer [36]. Nine genes were removed from this list as they were encoded by the mitochondrial genome. For some genes, Synergizer could not find a RefSeq ID corresponding to the “standard name”. These genes were manually inspected and the synonyms provided by Organelle DB website were used to find corresponding RefSeq IDs. When multiple splice variants were exact duplicates with respect to the first 69 nts of their coding region, a single identifier was selected as the representative. This procedure yielded 364 RefSeq transcripts out of ∼25 k transcripts having an MSCR. The manually edited list of mitochondrial genes is available in Dataset S1. The software package R 2.6.0 was used for all the statistical analyses, except where otherwise noted.
Motif discovery, scoring, and disambiguation of overlaps
For motif discovery, the first 99 nt of SSCR-containing genes were used to ensure that all signal peptides were included in their entirety. Highly similar sequences were removed to avoid overweighting closely related sequences. Specifically, the first 99 nt from each sequence was aligned to all others using blastn [39]. A threshold (E-value <10−25) was used to group similar sequences, and one randomly selected representative from each such set was used after this filter.
We used the DEME [18] software to search for a motif that is highly enriched in the 5UI− set of sequences relative to the 5UI+ set. We also used the AlignACE software [19] to search for a set of highly enriched motifs in the 5UI− set. AlignACE searches for frequently occurring motifs in both the forward and complementary strands of DNA sequences. Choosing to focus on RNA motifs, we discarded 2 of the 20 motifs reported that were constructed from less than 10 representative forward-strand sites.
Positional Specific Scoring Matrices (PSSM) of the discovered motifs were extracted from the forward-strand sites of each motif. For a given sequence s and a motif with length m, all windows of size m within the first 99 bases were scored using the PSSM of the motif. To avoid calling multiple overlapping motifs, only the highest scoring window in a contiguous series of overlapping windows was selected. For each motif, an initial PSSM score threshold (t*) was selected such that t* yields the highest enrichment of motif-containing sequences among the SSCR-containing and 5UI− genes on the p-value generated from Fisher's Exact Test using the 2×2 contingency table (Table 1).

Given the total number of genes N, the number of 5UI− genes m, and the number of motif-containing sequences n, this test estimates the probability that k or more genes would be found to overlap between the 5UI− genes and the motif-containing sequences under the null hypothesis of independence:where the probability of observing exactly i overlaps given N, m and n follows from the hypergeometric distribution:
Among the 18 AlignACE motifs, we focused on the four that were most enriched among 5UI− genes compared to 5UI+ genes based on the resulting p-value. Further analyses on motif occurrences and positional distributions were performed on these four AlignACE motifs and the DEME motif.
PSSM score threshold selection
While PSSM threshold selection using Fisher's Exact Test provided a quick way identify discriminative AlignACE motifs, the selection of thresholds did not take into account the likelihood that such discrimination may have occurred by chance.
To account for this possibility, we randomly generated four sets of PSSMs matching the discovered motifs' lengths (6, 10, 14, and 16 nt). We modeled each position of the PSSM as an independent sample from a Dirichlet distribution with parameters (αi) equal to the background nucleotide frequency such that Σαi = 1. The background nucleotide frequency was calculated among the first 99 nts of either SSCR-containing or MSCR-containing 5UI− genes. For each given motif length, we generated 40,000 random PSSMs for SSCR set and 100,000 random PSSMs for the MSCR set. We generated receiver operating characteristic (ROC) plots to compare the discriminative performance of these randomly generated PSSMs with that of the discovered motifs. First, we scanned each sequence to find the maximum score for each PSSM. We classified a sequence as motif-containing if its maximum PSSM score was greater than a given threshold t*. For all random and discovered motifs, we calculated the true positive rate (TPR) as the fraction of motif-containing 5UI− genes, and the false positive rate (FPR) as the fraction of motif-containing 5UI+ genes as a function t*. Therefore, each point on an ROC plot corresponds to (TPR, FPR) of a particular PSSM at some threshold t*. These ROC plots are informative about the analyzed motif's power to discriminate 5UI− from 5UI+ genes.
For each discovered motif, we used the ROC plots generated from SSCR-containing genes (Figure S8) to choose the PSSM score threshold value (t′) for subsequent analysis. The threshold t′ was chosen such that it maximizes the difference between the discovered motif's TPR and the median TPR of the random motifs was the most at the FPR value corresponding to t′. Since we discovered motifs using the SSCR-containing set only, the ROC plots for the MSCR set were not subject to any overfitting that might have occurred during motif discovery.
Positional distribution of sequence elements that match the motifs
To assess whether there is any significant deviation in the positional distribution of motifs in the 5UI− set from that in the 5UI+ set, we performed the Wilcoxon Rank Sum test. We examined differences in distributions for the positions of all motif occurrences in each sequence. We also generated histograms for the reading frame at which motifs occur in the coding region to look for differences between the 5UI− and 5UI+ sets.
Supporting Information
Zdroje
1. CenikC
DertiA
MellorJ
BerrizG
RothF
2010 Genome-wide functional analysis of human 5′ untranslated region introns. Genome Biology 11 R29 doi:10.1186/gb-2010-11-3-r29
2. PesoleG
MignoneF
GissiC
GrilloG
LicciulliF
2001 Structural and functional features of eukaryotic mRNA untranslated regions. Gene 276 73 81 doi:10.1016/S0378-1119(01)00674-6
3. HongX
ScofieldDG
LynchM
2006 Intron Size, Abundance, and Distribution within Untranslated Regions of Genes. Mol Biol Evol 23 2392 2404 doi:10.1093/molbev/msl111
4. SträβerK
MasudaS
MasonP
PfannstielJ
OppizziM
2002 TREX is a conserved complex coupling transcription with messenger RNA export. Nature 417 304 308 doi:10.1038/nature746
5. ChengH
DufuK
LeeC
HsuJL
DiasA
2006 Human mRNA Export Machinery Recruited to the 5′ End of mRNA. Cell 127 1389 1400 doi:10.1016/j.cell.2006.10.044
6. PalazzoAF
SpringerM
ShibataY
LeeC
DiasAP
2007 The Signal Sequence Coding Region Promotes Nuclear Export of mRNA. Plos Biol 5 e322 doi:10.1371/journal.pbio.0050322
7. BarrellD
DimmerE
HuntleyRP
BinnsD
O'DonovanC
2009 The GOA database in 2009—an integrated Gene Ontology Annotation resource. Nucleic Acids Res 37 D396 D403 doi:10.1093/nar/gkn803
8. HentschelCC
BirnstielML
1981 The organization and expression of histone gene families. Cell 25 301 313 doi:10.1016/0092-8674(81)90048-9
9. NiimuraY
NeiM
2006 Evolutionary dynamics of olfactory and other chemosensory receptor genes in vertebrates. Journal of Human Genetics 51 505 517 doi:10.1007/s10038-006-0391-8
10. HesseM
ZimekA
WeberK
MaginTM
2004 Comprehensive analysis of keratin gene clusters in humans and rodents. European Journal of Cell Biology 83 19 26 doi:10.1078/0171-9335-00354
11. WuD
IrwinD
ZhangY
2008 Molecular evolution of the keratin associated protein gene family in mammals, role in the evolution of mammalian hair. BMC Evolutionary Biology 8 241 doi:10.1186/1471-2148-8-241
12. BlobelG
DobbersteinB
1975 Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J Cell Biol 67 835 851
13. RoiseD
HorvathSJ
TomichJM
RichardsJH
SchatzG
1986 A chemically synthesized pre-sequence of an imported mitochondrial protein can form an amphiphilic helix and perturb natural and artificial phospholipid bilayers. EMBO J 5 1327 1334
14. HeijneGV
1986 Mitochondrial targeting sequences may form amphiphilic helices. EMBO J 5 1335 1342
15. VisaN
IzaurraldeE
FerreiraJ
DaneholtB
MattajI
1996 A nuclear cap-binding complex binds Balbiani ring pre-mRNA cotranscriptionally and accompanies the ribonucleoprotein particle during nuclear export. J Cell Biol 133 5 14 doi:10.1083/jcb.133.1.5
16. LuoM
ReedR
1999 Splicing is required for rapid and efficient mRNA export in metazoans. Proceedings of the National Academy of Sciences of the United States of America 96 14937 14942 doi:VL-96
17. GrüterP
TaberneroC
von KobbeC
SchmittC
SaavedraC
1998 TAP, the Human Homolog of Mex67p, Mediates CTE-Dependent RNA Export from the Nucleus. Molecular Cell 1 649 659 doi:10.1016/S1097-2765(00)80065-9
18. RedheadE
BaileyTL
2007 Discriminative motif discovery in DNA and protein sequences using the DEME algorithm. BMC Bioinformatics 8 385 doi:10.1186/1471-2105-8-385
19. RothFP
HughesJD
EstepPW
ChurchGM
1998 Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotech 16 939 945 doi:10.1038/nbt1098-939
20. TimchenkoL
MillerJ
TimchenkoN
DeVoreD
DatarK
1996 Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucl Acids Res 24 4407 4414 doi:10.1093/nar/24.22.4407
21. KinoY
MoriD
OmaY
TakeshitaY
SasagawaN
2004 Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Hum Mol Genet 13 495 507 doi:10.1093/hmg/ddh056
22. Rodríguez-TrellesF
TarríoR
AyalaFJ
2006 Origins and Evolution of Spliceosomal Introns. Annu Rev Genet 40 47 76 doi:10.1146/annurev.genet.40.110405.090625
23. William RoyS
GilbertW
2006 The evolution of spliceosomal introns: patterns, puzzles and progress. Nat Rev Genet 7 211 221 doi:10.1038/nrg1807
24. KeeneJD
2007 RNA regulons: coordination of post-transcriptional events. Nat Rev Genet 8 533 543 doi:10.1038/nrg2111
25. MooreMJ
ProudfootNJ
2009 Pre-mRNA Processing Reaches Back toTranscription and Ahead to Translation. Cell 136 688 700 doi:10.1016/j.cell.2009.02.001
26. GarciaM
DelaveauT
GoussardS
JacqC
2010 Mitochondrial presequence and open reading frame mediate asymmetric localization of messenger RNA. EMBO Rep 11 285 291 doi:10.1038/embor.2010.17
27. SylvestreJ
VialetteS
Corral DebrinskiM
JacqC
2003 Long mRNAs coding for yeast mitochondrial proteins of prokaryotic origin preferentially localize to the vicinity of mitochondria. Genome Biology 4 R44 doi:10.1186/gb-2003-4-7-r44
28. NottA
Le HirH
MooreMJ
2004 Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes & Development 18 210 222 doi:10.1101/gad.1163204
29. MaXM
YoonS
RichardsonCJ
JülichK
BlenisJ
2008 SKAR Links Pre-mRNA Splicing to mTOR/S6K1-Mediated Enhanced Translation Efficiency of Spliced mRNAs. Cell 133 303 313 doi:10.1016/j.cell.2008.02.031
30. KimVN
KataokaN
DreyfussG
2001 Role of the Nonsense-Mediated Decay Factor hUpf3 in the Splicing-Dependent Exon-Exon Junction Complex. Science 293 1832 1836 doi:10.1126/science.1062829
31. Le HirH
GatfieldD
IzaurraldeE
MooreMJ
2001 The exon–exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J 20 4987 4997 doi:10.1093/emboj/20.17.4987
32. PruittKD
TatusovaT
MaglottDR
2005 NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 33 D501 D504 doi:10.1093/nar/gki025
33. GueroussovS
TarnawskySP
CuiXA
MahadevanK
PalazzoAF
2010 Analysis of mRNA Nuclear Export Kinetics in Mammalian Cells by Microinjection. J Vis Exp 46 doi:10.3791/2387
34. BerrizGF
BeaverJE
CenikC
TasanM
RothFP
2009 Next generation software for functional trend analysis. Bioinformatics doi:10.1093/bioinformatics/btp498 btp498
35. BerrizGF
KingOD
BryantB
SanderC
RothFP
2003 Characterizing gene sets with FuncAssociate. Bioinformatics 19 2502 2504 doi:10.1093/bioinformatics/btg363
36. BerrizGF
RothFP
2008 The Synergizer service for translating gene, protein and other biological identifiers. Bioinformatics 24 2272 2273 doi:10.1093/bioinformatics/btn424
37. HaiderS
BallesterB
SmedleyD
ZhangJ
RiceP
2009 BioMart Central Portal–unified access to biological data. Nucl Acids Res 37 W23 27 doi:10.1093/nar/gkp265
38. WiwatwattanaN
KumarA
2005 Organelle DB: a cross-species database of protein localization and function. Nucl Acids Res 33 D598 604 doi:10.1093/nar/gki071
39. AltschulSF
GishW
MillerW
MyersEW
LipmanDJ
1990 Basic local alignment search tool. Journal of Molecular Biology 215 403 410 doi:10.1016/S0022-2836(05)80360-2
40. CrooksGE
HonG
ChandoniaJ
BrennerSE
2004 WebLogo: A Sequence Logo Generator. Genome Res 14 1188 1190 doi:10.1101/gr.849004
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 4
Nejčtenější v tomto čísle
- Survival Motor Neuron Protein Regulates Stem Cell Division, Proliferation, and Differentiation in
- PTG Depletion Removes Lafora Bodies and Rescues the Fatal Epilepsy of Lafora Disease
- Evolution of Vertebrate Transient Receptor Potential Vanilloid 3 Channels: Opposite Temperature Sensitivity between Mammals and Western Clawed Frogs
- An Evolutionary Genomic Approach to Identify Genes Involved in Human Birth Timing