
and but Not Interact in Genetic Models of Amyotrophic Lateral Sclerosis
Mutations in the SOD1 and TARDBP genes have been commonly identified in Amyotrophic Lateral Sclerosis (ALS). Recently, mutations in the Fused in sarcoma gene (FUS) were identified in familial (FALS) ALS cases and sporadic (SALS) patients. Similarly to TDP-43 (coded by TARDBP gene), FUS is an RNA binding protein. Using the zebrafish (Danio rerio), we examined the consequences of expressing human wild-type (WT) FUS and three ALS–related mutations, as well as their interactions with TARDBP and SOD1. Knockdown of zebrafish Fus yielded a motor phenotype that could be rescued upon co-expression of wild-type human FUS. In contrast, the two most frequent ALS–related FUS mutations, R521H and R521C, unlike S57Δ, failed to rescue the knockdown phenotype, indicating loss of function. The R521H mutation caused a toxic gain of function when expressed alone, similar to the phenotype observed upon knockdown of zebrafish Fus. This phenotype was not aggravated by co-expression of both mutant human TARDBP (G348C) and FUS (R521H) or by knockdown of both zebrafish Tardbp and Fus, consistent with a common pathogenic mechanism. We also observed that WT FUS rescued the Tardbp knockdown phenotype, but not vice versa, suggesting that TARDBP acts upstream of FUS in this pathway. In addition we observed that WT SOD1 failed to rescue the phenotype observed upon overexpression of mutant TARDBP or FUS or upon knockdown of Tardbp or Fus; similarly, WT TARDBP or FUS also failed to rescue the phenotype induced by mutant SOD1 (G93A). Finally, overexpression of mutant SOD1 exacerbated the motor phenotype caused by overexpression of mutant FUS. Together our results indicate that TARDBP and FUS act in a pathogenic pathway that is independent of SOD1.
Published in the journal:
. PLoS Genet 7(8): e32767. doi:10.1371/journal.pgen.1002214
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002214
Summary
Mutations in the SOD1 and TARDBP genes have been commonly identified in Amyotrophic Lateral Sclerosis (ALS). Recently, mutations in the Fused in sarcoma gene (FUS) were identified in familial (FALS) ALS cases and sporadic (SALS) patients. Similarly to TDP-43 (coded by TARDBP gene), FUS is an RNA binding protein. Using the zebrafish (Danio rerio), we examined the consequences of expressing human wild-type (WT) FUS and three ALS–related mutations, as well as their interactions with TARDBP and SOD1. Knockdown of zebrafish Fus yielded a motor phenotype that could be rescued upon co-expression of wild-type human FUS. In contrast, the two most frequent ALS–related FUS mutations, R521H and R521C, unlike S57Δ, failed to rescue the knockdown phenotype, indicating loss of function. The R521H mutation caused a toxic gain of function when expressed alone, similar to the phenotype observed upon knockdown of zebrafish Fus. This phenotype was not aggravated by co-expression of both mutant human TARDBP (G348C) and FUS (R521H) or by knockdown of both zebrafish Tardbp and Fus, consistent with a common pathogenic mechanism. We also observed that WT FUS rescued the Tardbp knockdown phenotype, but not vice versa, suggesting that TARDBP acts upstream of FUS in this pathway. In addition we observed that WT SOD1 failed to rescue the phenotype observed upon overexpression of mutant TARDBP or FUS or upon knockdown of Tardbp or Fus; similarly, WT TARDBP or FUS also failed to rescue the phenotype induced by mutant SOD1 (G93A). Finally, overexpression of mutant SOD1 exacerbated the motor phenotype caused by overexpression of mutant FUS. Together our results indicate that TARDBP and FUS act in a pathogenic pathway that is independent of SOD1.
Introduction
ALS is the most common motor neuron disorder and is characterized by loss of upper and lower motor neurons. It is the third most common neurological disorder with an incidence of 1–2 people in 100,000, a prevalence of 4–6 per 100,000 and with a lifetime risk of 1 in 1,000 [1], [2]. ALS has a devastating course with disease onset generally first detected at 50–60 years of age followed by rapid muscle weakness, atrophy and eventual paralysis resulting in death due to respiratory failure within 1–5 years. Approximately 10% of ALS patients have a familial history for this disease (FALS), whereas the majority (90%) of cases appears to be of a sporadic nature (SALS) [3], [4]. So far, three major genes have been implicated in ALS: SOD1, TARDBP and FUS. However, it is not known whether these three genes interact in a common pathway or represent distinct ALS etiologies.
Cu/Zn superoxide dismutase (SOD1) mutations were the first to be identified predominantly in FALS patients in 1993, with more than 130 mutations currently identified in approximately 20% of FALS patients [2], [4]–[6]. A toxic gain of function of mutant SOD1 causes pronounced motor deficits in vivo correlated to motor neuron degeneration in a number of animal models [2], [7], [8]. Recently, mutations in TAR DNA binding protein (TARDBP) [9]–[12] and Fused in sarcoma (FUS) [13], [14] genes were found in both FALS and SALS patients, opening novel possibilities of studying the predominant, sporadic form of this disease [11], [15]. In 2008, two concurrent reports, including ours, identified a dozen missense mutations in the TARDBP gene [9]–[11]. So far, 38 TARDBP mutations have been identified predominantly clustered in the C-terminus, glycine-rich region of the TDP-43 protein encoded by the TARDBP gene in approximately1% of SALS and 3% of FALS [11], [16]. Two concurrent publications in 2009 identified FUS mutations that occur in about 5% of FALS and less than 1% of SALS [11], [16] with 35 mutations so far identified in ALS cases. Similarly to TDP-43 (encoded by TARDBP), FUS is an RNA binding protein mainly localized in the nucleus [11]. Further comparable to the TARDBP mutations identified in ALS cases, most of the FUS mutations cluster in the C-terminus of the FUS protein, including the most common mutations, R521C present in 22 FALS and 4 SALS and R521H present in 9 FALS and 4 SALS [11], [13], [14], [17]–[28]. Interestingly, both proteins have been found to be major components of ubiquitinated inclusion bodies in autopsy tissue, not only in ALS patients, but in a number of commonly-related neurodegenerative disorders, such as frontotemporal lobar degeneration (FTLD), Alzheimer's and Parkinson's diseases [11], [16]. In rare cases, either TARDBP or FUS mutations have also been identified in FTLD cases with or without motor neuron involvement [29]–[32].
In line with this genetic evidence, pathological reports have shown that TDP-43 and FUS antibodies co-label protein aggregates consisting of inclusion bodies observed in both SALS and FALS cases [33], distinguishing these aggregates from the SOD1 positive found in FALS patients [34]. Two recent studies in cell lines have shown that TDP-43 and FUS interact [35] with a report showing that both proteins are able to influence HDAC6 mRNA production [36]. Strong overexpression of both TARDBP and FUS (either WT or carrying ALS mutations) were shown to exacerbate a degenerative phenotype in the Drosophila eye as compared to either gene alone [37]. Further, recent studies have also demonstrated that depletion of TARDBP using RNAi in cell lines or tardbp in Drosophila leads to specific decrease in FUS mRNA levels [38], [39]. Overall, this evidence suggests that these proteins are involved in similar pathogenic mechanisms leading to motor neuron degeneration. However, the relevance of TARDBP and FUS mutations to these pathogenic mechanisms is not well understood, since none of the known functions of TDP-43 and FUS, such as binding to RNA, DNA and heterogeneous nuclear ribonucleoproteins (hnRNPs), have been shown to be perturbed by ALS-related mutations [36], [40], [41].
Recent insight in neurodegenerative diseases has revealed that several genes mutated in these disorders could participate in common molecular mechanisms, raising the possibility of a multigenic interaction at the root of the pathogenesis of neurodegeneration. One such example is the well-established genetic and functional interaction between PTEN-induced putative (mitochondrial) kinase 1 (PINK1) and the E3 ubiquitin ligase parkin (PRKN) [42], [43], two genes mutated in Parkinson's disease [44], [45]. Another recent example is the identification of interactions between TDP-43 and ataxin-2 in vivo, with ataxin-2 containing polyglutamine expansions being a potent modifier of TDP-43 toxicity [46]. This interaction was also found to have pathogenic implications since SCA1 triplet repeats are significantly increased in ALS patients as compared to controls [46].
In order to establish whether genetic interactions also exist in ALS pathogenesis we carried out a multigenic analysis of FUS, TARDBP and SOD1 in zebrafish. Zebrafish are proving to be a valuable vertebrate model to further our understanding of these multigenic interactions [47]. A number of recent studies in zebrafish with relevance to motor neuron diseases [8], [48]–[52] suggest that this vertebrate organism is ideally suited as a model to rapidly and efficiently replicate certain aspects of these disorders [53]. This allows us to better understand genetic (and eventually the cellular and molecular) mechanisms of motor neuron degeneration during a much shorter time span (in this case inside a few days). Further, these models allow us to define the crucial steps in disease development and to find ways to interfere with the development of early hallmarks of the disease, rather than to exactly replicate the (likely later) symptoms.
We have previously demonstrated that mutations of TARDBP cause a pronounced motor phenotype characterized by aberrant motor neuron morphology and motor behavior in zebrafish through both toxic gain and loss of function [50], with the toxic gain of function results being recently confirmed by another group [48]. Up to now, this represents the only vertebrate model of mutant TARDBP where a motor neuron disorder has been observed when compared to similar expression of WT TARDBP. Here we report that FUS mutations identified in ALS patients present a similar motor phenotype. Our results reveal a common genetic pathway for FUS and TARDBP in vivo, with FUS possibly acting downstream of TARDBP. These results also establish that SOD1 acts independently of FUS/TARDBP in causing a motor phenotype in these genetic models of ALS. The establishment of these new genetic models for ALS provides new tools to study the molecular mechanisms of neurodegeneration.
Results
FUS mutations characterized in this study
Three ALS-related mutations identified in our patient cohort at the University of Montréal Health Centre (CHUM; individuals from Quebec and France) were selected for this study. The R521C is the most common FUS mutation identified so far in 22 FALS and 4 SALS in a number of cohorts. The R521H mutation is also a very common mutation accounting for 9 FALS and 4 SALS [11], [13], [14], [17]–[28]. Both of these mutations have been shown to mislocalize from the nucleus to the cytosol in a number of cell line experiments [14], [54], [55]. The S57Δ mutation was identified in one SALS patient from our cohort and is one of the only variants identified in ALS cases to be located in the N-terminus region of the FUS protein [19].
Loss of function of Fus leads to a motor phenotype that can be rescued by WT FUS but not ALS–related mutations
We first confirmed by in situ hybridization that Fus mRNA was indeed expressed as early as 24 hours post-fertilization (hpf) in zebrafish embryos (Figure 1A) mainly in the hindbrain, eye and intersomitic segments as well as the spinal cord (Figure 1B). The antisense morpholino oligonucleotide (AMO) for Fus KD was designed to specifically bind near the ATG of zebrafish Fus but nowhere else in the zebrafish genome. As a control, a mismatch AMO was also designed that does not bind anywhere in the zebrafish genome. Western blot analysis using a FUS antibody demonstrated knockdown (KD) of Fus expression by approximately 60% solely in fish injected with an AMO designed to bind Fus, but not to the mismatch AMO control (Figure 1C).

Fus KD by AMO caused motor deficits consisting of a significantly abnormal motor behaviour measured as a deficient touch-evoked escape response (TEER) (Figure 2Ai) as well as reduced outgrowth of hyperbranched axons from motor neurons or unbranched axonal length (UAL) (Figure 2Bi) that we subsequently refer to as the motor phenotype. The TEER was deficient in 57% of fish injected but was negligible in zebrafish non-injected (2%), sham injected (3%) or injected with mismatch AMO (7%). Only 26% of larvae injected with Fus AMO displayed a normal touch-evoked escape response (Figure 2Ci) as compared to 95% in non-injected, 87% in sham-injected and 82% in larvae injected with the mismatch AMO. Similarly, the length of the motor axons to the point of the first branch (UAL) was significantly reduced in larvae injected with Fus AMO when compared to non-injected zebrafish from 99 to 76 µm (Figure 2B, 2Ci). These results are summarized in Table 1 row 1–4.

To determine if the human FUS and the zebrafish Fus genes are functionally similar, we co-injected human WT FUS mRNA alongside Fus AMO. The motor phenotype was rescued in these sets of injections when compared to injections of Fus AMO alone with significant increases of the percentages of zebrafish displaying a normal TEER from 26 to 70% as well as augmentation of the UAL of motor neurons from 76 to 91 µm (Figure 2A, 2B, 2Ci versus 2A, 2B, 2Cii and Table 1 rows 4–5).
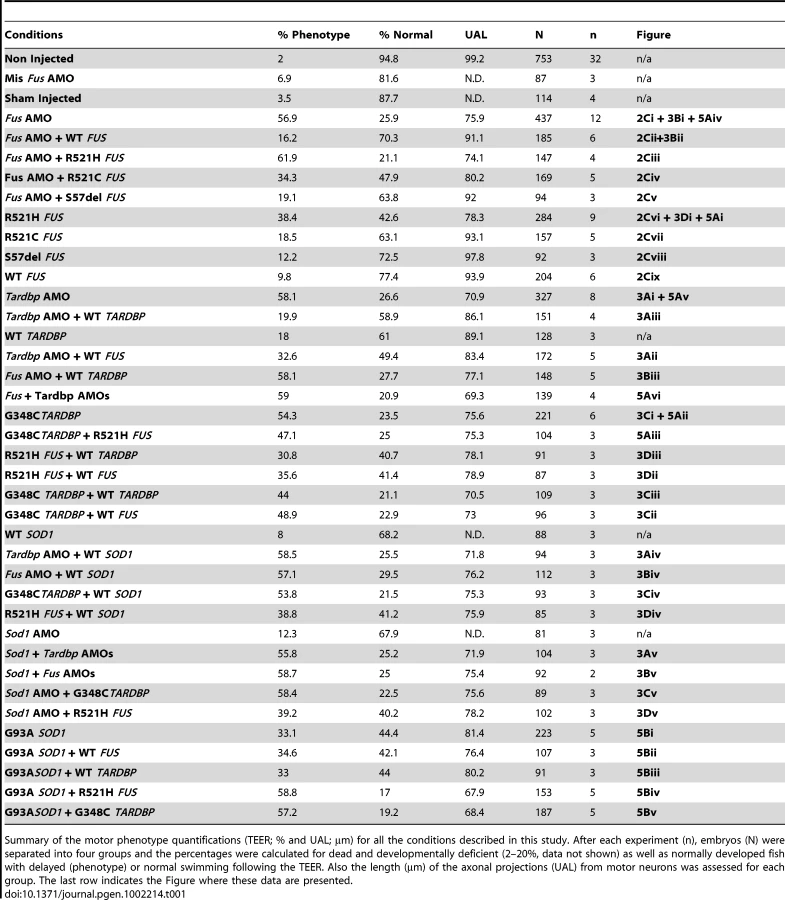
Next, we examined whether mutations in the FUS gene can cause loss of function and result in a motor phenotype similar to what we previously demonstrated for TARDBP mutations [50]. Contrary to WT FUS mRNA, co-injection of R521H FUS mRNA and Fus AMO (Figure 2Ciii) did not rescue the motor phenotype (21% of zebrafish with normal TEER and the UAL from motor neurons measured at 74 µm). Co-injection of the R521C FUS mRNA partially rescued this phenotype, from 26% with AMO (see above) to 48% of zebrafish displaying a normal TEER and UAL of 80 µm (Figure 2Civ and Figure S1A, S1Biii) since it was also found to be significantly different from the rescue of Fus KD by WT FUS mRNA injection with 70% of fish with normal TEER and UAL of 91 µm (Figure 2A, 2B, 2Cii). However, co-injection of S57Δ FUS mRNA completely rescued the phenotype induced by Fus AMO (Figure 2Cv and Figure S1A, S1Biv) to a similar degree as the co-injection WT FUS mRNA injection (Figure 2A, 2B, 2Cii) with 64% versus 70% of zebrafish displaying a normal TEER and UAL at 92 versus 91 µm. These results are summarized in Table 1 rows 4–8.
Toxic gain of function of mutant FUS
Furthermore, we determined whether expression of WT and ALS-related mutant FUS mRNAs by themselves caused a motor deficit in zebrafish. Upon overexpression of R521H FUS mRNA in zebrafish embryos we observed a motor phenotype with only 43% of zebrafish larvae displaying a normal TEER (Figure 2Aiv, 2Cvi) as compared to 77% upon WT FUS injection (Figure 2Aiii, 2Cix). Similarly, the UAL of motor neurons overexpressing R521H FUS (Figure 2Biv, 2Cvi) was reduced to 78 µm as compared to 94 µm upon WT FUS injection (Figure 2Biii, 2Cix). Surprisingly, expression at similar levels of the R521C FUS mRNA (Figure S1A, S1Bi and Figure 2Cvii) and the S57Δ FUS mutations (Figure S1A, S1Bii and Figure 2Cviii) did not elicit a motor phenotype with similar percentages of embryos displaying a normal TEER (R521C: 63% and S57Δ: 72% versus WT: 77%; Figure 2Cvii,viii and Figure S1Ai,ii) as described above for WT FUS mRNA (Figure 2Aiii, 2Cix). Similarly, the length of the UAL of motor neurons was not significantly altered when compared to WT FUS (Figure 2Cix) (R521C: 93 µm and S57Δ: 98 µm versus WT: 91 µm; Figure 2Cvii,viii and Figure S1Bi,ii). These and subsequent sets of injections presented below did not significantly affect percentages of developmentally deficient (∼10%) and of dead embryos (∼10%) within our conditions (data not shown). The differences observed upon mRNA injections were not due to different levels of protein expression, since Western blot analysis revealed no changes in expression between the WT protein and the three ALS-related FUS mutants described here (Figure 1D). The results from this section are summarized in Table 1 rows 9–12.These data indicate that the ALS-related R521H mutation of FUS can cause motor neuron deficits leading to a toxic gain of function.
These in vivo results suggest that both toxic gain and loss of function (R521H) or solely loss of function (R521C) can render FUS mutations pathogenic. A lack of phenotype for the S57Δ FUS could indicate that this variant may be a rare polymorphism not causing disease. In accordance with this functional characterization performed in our zebrafish model, the R521H and R521C FUS mutations have been identified in a large number of FALS and SALS cases with both these mutations segregating with disease in large families, whereas the S57Δ variant was identified only in one SALS case [11], [13], [14], [17]–[28].
Genetic interactions between FUS and TARDBP
We hypothesized that FUS and TARDBP operate through a common genetic pathway. To examine this possibility, we tested whether FUS or TARDBP could rescue the loss of function phenotype caused by knockdown of either of these genes in zebrafish. WT FUS mRNA was able to rescue the motor phenotype (49% of zebrafish with normal TEER and UAL of 83 µm) (Figure 3Aii and Figure 4A, 4Biii) caused by Tardbp KD alone (27% of zebrafish with normal TEER and UAL of 71 µm) (Figure 3Ai), similarly to the rescue observed with WT TARDBP (59% of zebrafish with normal TEER and UAL of 86 µm) (Figure 3Aiii and Figure 4A, 4Bii). In contrast, co-injection of WT TARDBP mRNA with Fus AMO alone (28% of zebrafish with normal TEER and UAL of 77 µm) (Figure 3Biii and Figure 4A, 4Biv) was unable to rescue the phenotype obtained by KD of Fus alone (26% of zebrafish with normal TEER and UAL of 76 µm) (Figure 2A, 2Bi and Figure 3Bi). As mentioned previously, co-injection of WT FUS mRNA with Fus AMO (Figure 2A, 2Bii and Figure 3Bii) was able to properly rescue the motor phenotype caused by Fus KD alone (Figure 2A, 2Bi and Figure 3Bi). The results from this section are summarized in Table 1 row 4, 13–14 and 16–17. These results demonstrate that a genetic interaction between FUS and TARDBP exists, with FUS overexpression being able to rescue the Tardbp KD phenotype as a downstream effector.

TAF15 has a high structural and functional homology to FUS, since both TAF15 and FUS belong to the class of TET family of multifunctional DNA/RNA-binding proteins [56]. TAF15 has been suggested by one study as a candidate gene in ALS with few variants identified solely in FALS cases, but not in controls [57]. We thus determined the motor phenotype upon Tardbp KD with or without overexpression of TAF15 mRNA. Overexpression of TAF15 mRNA did not cause an overt motor phenotype with 65% displaying a normal TEER. Co-injecting TAF15 mRNA with Tardbp AMO (33% of zebrafish with normal TEER) failed to rescue the motor phenotype caused by Tardbp KD alone (27% of zebrafish with normal TEER). (Figure S2). These results further ascertain that the rescue of the phenotype induced by Tardbp KD by overexpressing WT FUS mRNA is specific to this gene and could be independent of the known functions of FUS and its homologue TAF15, such as DNA and/or RNA binding.
Conversely, we also determined whether the motor phenotype caused by the toxic gain of function of FUS could be rescued by overexpression of WT TARDBP and vice-versa (a dozen combinations; Figure 3A–3D, i–iii), expecting that a toxic gain of function may be irreversible. We and others have previously demonstrated that overexpression of mutant TARDBP causes a similar motor phenotype to the one described above, most pronounced upon injection of the G348C mutation [48], [50]. Overexpression of this mutant caused deficits in TEER in 54% of zebrafish with only 23% displaying a normal TEER and reduced UAL of motor axons (76 µm) (Figure 3Ci). Our results show that co-expression of mutant TARDBP with either WT FUS (23% of zebrafish with normal TEER and UAL of 73 µm) (Figure 3Cii) or WT TARDBP (21% of zebrafish with normal TEER and UAL of 70 µm) (Figure 3Ciii) failed to rescue the toxic gain of function phenotype caused by mutant TARDBP alone (23% of zebrafish with normal TEER and UAL of 76 µm) (Figure 3Ci). Similarly, we were unable to rescue the motor phenotype caused by expression of the R521H mutant FUS (43% of zebrafish with normal TEER and UAL of 78 µm) (Figure 2A, 2Biv and Figure 3Di) with either WT TARDBP (41% of zebrafish with normal TEER and UAL of 78 µm) (Figure 3Diii) or WT FUS (41% of zebrafish with normal TEER and UAL of 79 µm) (Figure 3Dii). The data from this section are summarized in Table 1 rows 9, 19–24.Thus, overexpression of WT FUS or TARDBP did not rescue the motor phenotype generated by ALS-related mutations in these two genes, consistent with their toxic gain of function.
If TARDBP and FUS interact, then overexpression of both mutant TARDBP and FUS mRNAs or simultaneous KD of both genes should yield a similar motor phenotype, whereas an exaggerated, additive phenotype could be expected if these genes act in independent pathways (see below for SOD1). As predicted for interacting genes, a similar, non-exacerbated motor phenotype was observed upon injection of the R521H mutant FUS (43% of zebrafish with normal TEER and UAL of 78 µm) (Figure 2A, 2Biv and Figure 5Ai), G348C mutant TARDBP (23% of zebrafish with normal TEER and UAL of 76 µm), (Figure 5Aii and Figure 6A, 6Biii) or both mutant TARDBP and FUS (25% of zebrafish with normal TEER and UAL of 75 µm) (Figure 5Aiii and Figure 6A, 6Biv). We also determined that co-injection of Fus and Tardbp AMOs (21% of zebrafish with normal TEER and UAL of 69 µm) (Figure 5Avi and Figure 6A, 6Bii) did not exacerbate the KD motor phenotype observed upon injection of either Fus AMO (26% of zebrafish with normal TEER and UAL of 76 µm) (Figure 5Aiv and Figure 2A, 2Bi) or Tardbp AMO (27% of zebrafish with normal TEER and UAL of 71 µm) (Figure 5Av and Figure 6A, 6Bi). These data are summarized in Table 1 rows 4, 9, 13 and 18–20. Since injections of two AMOs might harbor robust effects that would not allow the proper visualization of a possible exacerbation of the motor phenotype, we also injected suboptimal doses (see Materials and Methods) of both Fus and Tardbp AMOs and compared the motor phenotype with the one induced by single injection of Fus and/or Tardbp AMOs. The motor phenotype was not significantly altered when subdoses (half) of both Fus and Tardbp AMOs (61% of zebrafish with normal TEER versus 21% at the higher dose) was compared to single AMO injection of either Tardbp (63% of zebrafish with normal TEER versus 26% at the higher dose) and/or Fus (69% of zebrafish with normal TEER versus 27% at the higher dose) (Figure S3). Similar results were thus obtained when using either full or subdoses of both Fus and Tardbp AMOs.
Lack of genetic interactions between FUS/TARDBP and SOD1
Having provided evidence for an in vivo genetic interaction between TARDBP and FUS we sought to determine whether SOD1 interacts with these genes by performing further gain and loss of function genetic manipulations using a specific Sod1 AMO, as well as WT or mutant (G93A) SOD1 mRNAs. We then sought to determine whether SOD1 acted downstream, upstream or independently of TARDBP or FUS, comprising nineteen conditions (Figure 3A–3D iv–v grey background and Figure 4A, 4Bv and Figure 4vi). We first tested if SOD1 acts downstream of TARDBP and FUS by examining whether WT SOD1 could rescue the motor phenotypes generated by loss or toxic gain of function of TARDBP or FUS. Overexpression of WT SOD1 did not yield a motor phenotype on its own (motor phenotype consisting of 68% of zebrafish with normal TEER) and failed to rescue the motor phenotype induced by KD of Tardbp (25% of zebrafish with normal TEER and UAL of 72 µm) (Figure 3Aiv and Figure 4A, 4Bvi), KD of Fus, (29% of zebrafish with normal TEER and UAL of 76 µm) (Figure 3Biv and Figure 4A, 4Bv) as well as overexpression of the G348C mutant TARDBP (21% of zebrafish with normal TEER and UAL of 75 µm) (Figure 3Civ), or the R521H mutant FUS (41% of zebrafish with normal TEER and UAL of 76 µm) (Figure 3Div). Next, we tested whether KD of Sod1 could alleviate the motor phenotype caused by mutant TARDBP or FUS. Injection of an AMO to specifically KD Sod1 did not cause a motor phenotype on its own (consisting of 68% of zebrafish with normal TEER), consistent with the lack of phenotype observed in SOD1 knockout mice [58]. In contrast, co-injection of AMOs to Sod1 and Tardbp (25% of zebrafish with normal TEER and UAL of 72 µm) (Figure 3Av) or to Sod1 and Fus (25% of zebrafish with normal TEER and UAL of 75 µm) (Figure 3Bv) yielded a similar motor phenotype to that observed upon KD of Tardbp (motor phenotype consisting of 27% of zebrafish with normal TEER and UAL of 71 µm) (Figure 3Ai and Figure 6A, 6Bi) or Fus (26% of zebrafish with normal TEER and UAL of 76 µm) (Figure 2A, 2Bi and Figure 3Bi) alone. Co-injection of Sod1 AMO with mutant TARDBP (22% of zebrafish with normal TEER and UAL of 75 µm) (Figure 3Cv) or mutant FUS (40% of zebrafish with normal TEER and UAL of 78 µm) (Figure 3Dv) also failed to modify the motor phenotype obtained by injecting mutant TARDBP alone (21% of zebrafish with normal TEER and UAL of 75 µm) (Figure 3Ci and Figure 6A, 6Biii) and mutant FUS alone (motor phenotype consisting of 43% of zebrafish with normal TEER and UAL of 78 µm) (Figure 2A, 2Biv, Figure 3Di). From these combined results we can infer that SOD1 is not acting downstream of TARDBP or FUS. These results are summarized in Table 1 rows 4, 9, 13, 19 and 25–34.

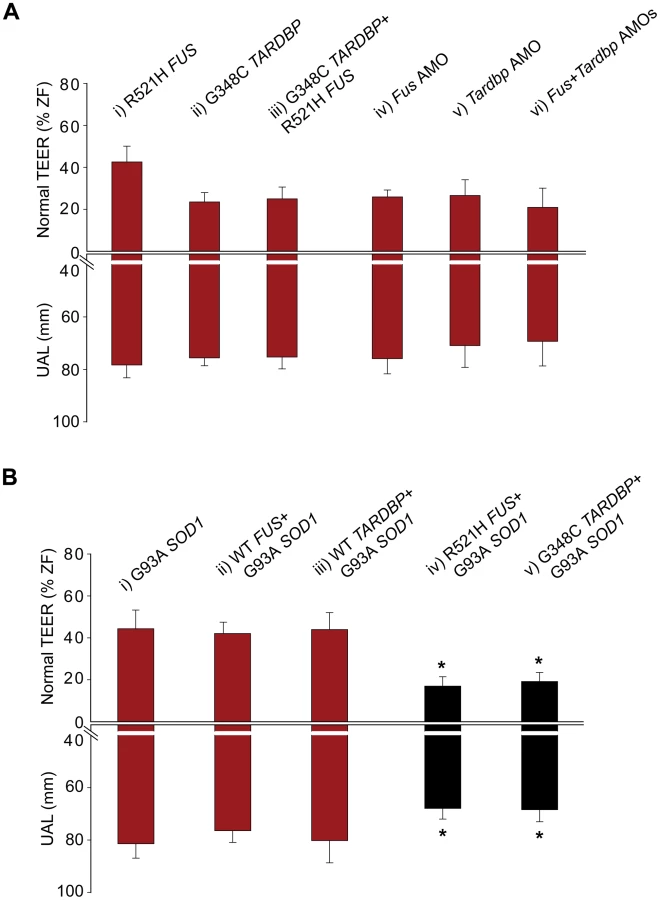
Alternatively, to determine if SOD1 acts upstream we tested for rescue of the mutant SOD1-induced motor phenotype by overexpressing WT TARDBP or FUS. Overexpression of mutant SOD1 mRNA in zebrafish embryos has been shown to cause shortening and premature branching of axonal projections from the motor neurons in the spinal cord [8] and is consistent with the toxic gain of function observed in ALS [2], [7], [8]. Consistent with these published results, we observed a motor phenotype consisting of 33% of zebrafish with swimming deficits, 44% of zebrafish with normal TEER and UAL of 81 µm when we overexpressed the G93A mutant SOD1 in our zebrafish model (Figure 5Bi and Figure 6A, 6Bv). Similarly, co-expression with mutant SOD1 of WT TARDBP (motor phenotype consisting of 44% of zebrafish with normal TEER and UAL of 80 µm) (Figure 5Biii) or FUS (42% of zebrafish with normal TEER and UAL of 76 µm) (Figure 5Bii) failed to rescue this motor deficit to any significant extent, indicating that mutant SOD1 does not act upstream of either of these two genes. These results are summarized in Table 1 rows 9, 19 and 35–37.

SOD1 acts independently of FUS/TARDBP
The foregoing results strongly suggest that SOD1 acts independently of TARDBP and FUS in our models. If this is true, then the motor phenotype yielded by either mutant SOD1 or TARDBP/FUS alone should be less severe than the “additive” phenotype of mutant SOD1 and TARDBP/FUS. Indeed, co-injection of both the R521H mutant FUS and the G93A mutant SOD1 mRNAs (motor phenotype consisting of 17% of zebrafish with normal TEER and UAL of 68 µm) (Figure 5Biv and Figure 6A, 6Bvi) yielded an exaggerated motor phenotype with a higher percentage of embryos affected as well as an exacerbated axonal shortening from motor neurons when compared to injection of mutant FUS alone (42% of zebrafish with normal TEER and UAL of 78 µm) (Figure 2A, 2Biv and Figure 5Ai) or mutant SOD1 (44% of zebrafish with normal TEER and UAL of 81 µm) (Figure 5Bi and Figure 6A, 6Bv) alone. Similarly, co-injection of mutant SOD1 and of mutant TARDBP (motor phenotype consisting of 19% of zebrafish with normal TEER and UAL of 68 µm) (Figure 5Bv) led to a significantly exacerbated motor phenotype when compared to mutant SOD1 alone (44% of zebrafish with normal TEER and UAL of 81 µm) (Figure 5Bi and Figure 6A, 6Bv). The data in this section are summarized in Table 1 rows 9, 19 and 38–39. These results indicate that mutant SOD1 may yield a motor phenotype which is additive with that generated by ALS-related FUS and TARDBP mutations, suggesting that SOD1 may act independently of TARDBP and FUS.
Discussion
Does a central pathogenic mechanism involving genetic interactions of FUS and TARDBP lead to motor neuron degeneration in ALS?
As summarized in the matrix of Figure 7A, here we show that expression of WT FUS is able to rescue the motor phenotype induced by KD of Fus (Figure 2) as well as the motor phenotype caused by KD of zebrafish Tardbp (Figure 3 and Figure 4; summarized in Figure 7A, green cell). On the other hand, expression of WT TARDBP is unable to rescue the phenotype caused by KD of Fus (Figure 3 and Figure 4; Figure 7A, red cell), whereas it does rescue the motor phenotype induced by KD of Tardbp. These results as well as in vitro reports of physical interactions [35], [36] suggest that TARDBP and FUS share a common genetic pathway, with FUS being downstream of TARDBP. Alternatively, as certain studies have previously demonstrated, FUS could be a more general transcriptional regulator, potentially capable of compensating for certain of the functions of TARDBP [15]. This possibility is less likely since overexpression of TAF15, a gene belonging to the class of TET family of multifunctional DNA/RNA-binding proteins with high functional and structural similarity to FUS was unable to rescue the motor phenotype caused by Tardbp KD (Figure S2). The lack of exacerbation of partial or more complete double knockdowns also indicates a non-additive effect of TARDBP and FUS (Figure 5 and Figure S3). Although we were unable to generate an increased phenotype upon double KD of Tardbp and Fus in this study, we cannot exclude the possibility of phenotype exacerbation at different doses of AMOs.

Expression of SOD1 was unable to rescue the phenotypes caused by KD of Fus and/or Tardbp suggesting independent molecular pathways (Figure 5 and Figure 6). Further, mutant SOD1 does not appear to genetically converge with the pathogenic cascades elicited by mutant TARDBP or FUS, since expression of mutant SOD1 exacerbates the motor phenotype induced by mutant FUS (as summarized in Figure 7A).
Although the pathological mechanisms through which ALS-related TARDBP mutations cause motor neuron degeneration are not understood, protein misfolding and phosphorylation, nuclear to cytosolic shuttling and RNA imbalance are presumed to be involved [11], [15], [16]. Our results suggest that certain ALS-related FUS mutations, similarly to TARDBP [50], can cause motor neuron deficits through both loss and toxic gain of function mechanisms. As illustrated in Figure 7B, the molecular mechanisms that cause motor neuron degeneration could be initiated with a loss of function of FUS or TARDBP due to mislocalization from the nucleus to the cytosol, whereas the toxic gain of function could be the result of abnormal accumulation of aggregate-prone proteins as reported for two of the FUS mutations described here, R521H and R521C [14], [54], [55], as well as the mutations R495X, R522G and P525L [59]–[61]. A similar toxic gain of function has been described for the A315T, G348C and A382T TARDBP mutations [48], [50]. In fact, several of these recent studies and others have shown that in neurons, expression of TDP-43 and/or FUS in the cytosol causes aggregation of these proteins with subsequent recruitment into stress granules, thus initiating pathogenic events [59]–[63].
While this article was under review, a study in Drosophila performed a functional characterization of several FUS mutations, including the R521H and R521C mutations described here [37]. When strongly overexpressed solely in neurons mutant FUS led to an increased severity of the “rough-eye” phenotype, widespread neurodegeneration and lethality as compared to WT FUS overexpression [37]. The authors also suggested genetic interactions between TARDBP and FUS since overexpression of both mutants together caused exacerbation of this phenotype as compared to expression of either mutant TARDBP or FUS. However, overexpression of WT TARDBP and WT FUS together caused similar increases in phenotype severity when compared to overexpression of either WT genes alone [37]. Thus, this exacerbation could be likely a result of excessive amounts of these proteins, which have been found as likely to aggregate in a number of models, including yeast [64]–[66]. On the other hand, we describe here a genetic interaction due to rescue (as opposed to exacerbation) by WT FUS of the motor phenotype upon Tardbp KD, suggesting that FUS is downstream to TARDBP in this pathway.
Consistent with a possible action downstream from TARDBP, FUS is thought to have a more critical role in regulating neuronal morphology and connectivity [15]. In cell lines, TDP-43 was shown to form complexes with hnRNPs and a fraction of TDP-43 in these complexes does interact directly with FUS, with this in vitro interaction being enhanced in cell lines from ALS patients harboring TARDBP mutations [35]. Another study using cell lines demonstrated that a common biochemical pathway exists where FUS and TDP-43 interact by binding competitively HDAC6 mRNA, with TDP-43 being upstream in this pathway. Further, since FUS antibodies have been shown to co-label TDP-43 positive protein aggregates observed in both SALS and FALS cases, a similar pathogenic function for these mutant proteins has been suggested [33]. Finally, ubiquitinated aggregates observed in FALS cases with SOD1 mutations were not immunopositive against TDP-43 or FUS antibodies [33], [34], again consistent with independent pathogenic mechanisms for SOD1 and TDP-43/FUS.
Interactors of TARDBP are central in ALS genetics and pathology
Interestingly, a number of in vivo studies have demonstrated that TARDBP has a number of genetic interactors such as Ataxin-2 [46], progranulin (GRN) [48], [67]–[69], valosin-containing protein (VCP) [70], [71] and histone deacetylase 6 (HDAC6) [36], [38]. Most of these interactors are mutated in other neurodegenerative disorders, such as dementia and expanded polyglutamine repeat disorders, whereas some may participate in generalized processes such as autophagy. Further, recent genetic studies have shown that mutations in Ataxin-2 and VCP are prevalent in ALS patients, with intermediate polyglutamine expansions significantly associated with ALS [46] and a study finding VCP mutations in 1–2% of FALS patients [72]. These combined results suggest that TARDBP plays a pivotal role in the pathogenic pathways leading to motor neuron degeneration culminating in ALS. These results also suggest that a multigenic pathway shared in a number of neurodegenerative disorders may exist. Unraveling the molecular and genetic components of this network of neurodegenerative interactions could have major implications in our understanding of the pathophysiology of these neurological disorders and could accelerate the discovery of future treatments for these increasingly prevalent diseases.
Materials and Methods
Ethics statement
Zebrafish were raised from a colony maintained according to established procedures. All procedures described here were carried out in compliance with the Canadian Council for Animal Care.
Embryonic RNA manipulation
Injections were performed in 1–4 cell stage blastulae. FUS WT and mutants (R521H, R521C, S57Δ), TARDBP WT and mutant (G348C), SOD1 WT and mutant (G93A) mRNAs were transcribed from NotI-linearized pCS2+ using SP6 polymerase with the mMESSAGE Machine Kit (Ambion). This was followed by a phenol-chloroform purification and ethanol precipitation, and diluted in nuclease-free water (Ambion). The mRNAs were diluted in nuclease free water (Ambion) with 0.05% Fast Green vital dye (Sigma) at a concentration of 60 ng/µl (FUS), 25 ng/µl (TARDBP) and 100 ng/µl (SOD1) and were pulse-injected into early embryos using a Picospritzer III (General Valve) pressure ejector. The zebrafish TARDBP, FUS and SOD1 gene orthologues, Tardbp, Fus, and Sod1 (NM_201476; NM_201083.2; and NM_131294.1 respectively) were identified using the Ensembl's gene homology prediction program (http://www.ensembl.org). AMOs were designed to bind and inhibit specifically the ATG of the following genes (and no other genomic sequence): Fus (GGCCATAATCATTTGACGCCATGTT), Tardbp (GTACATCTCGGCCATCTTTCCTCAG) and Sod1 (GCACACAAACGGCCTTGTTCACCAT) mRNA translation (Gene Tools) were designed complimentary to the region of translational initiation of the Fus CGAAGGCGACTGTACGTATAACACCTCAGAAATTGTTATTCTGCATCATTTCTAAAAGGATTTTAAGCCCAAAC [(ATG)GCGTCAAATGATTATGGCC]AAA, Tardbp (GGAAACAGTTAGCACAGCTCGCGCATTCGGTGTAATC [(ATG)ACGGAGTGCTATATTCGTGTGG]), and Sod1 TCTTATCAAACACAGTCGGTTTCTTTCACTCTCTCACAACTTCTCAGTTTGCATAATCTACAGTCAGC [(ATG)GTGAACAAGGCCGTTTGTGTGC] mRNAs to inhibit protein translation. These AMOs were designed to bind solely to the 1st ATG of the appropriate region of translational initiation for each gene and were confirmed by BLAST searches to not recognize any other sequences in the zebrafish genome (or any human transcripts). An AMO having nucleotides that were mismatched represented as lowercase to disrupt specificity were also designed for Fus (GGCgAaAATgATTTcACcCCATGTT). Dose-dependence curves of AMO and mRNA toxicity were performed and AMOs were injected at a concentration of 0.6 (Fus), 0.1 (Tardbp) and 0.5 (Sod1) mM to minimize morpholino-induced developmental delay and toxicity and to yield a consistent motor phenotype. Suboptimal doses were established for Fus (0.3 mM) and Tardbp (0.05 mM).
Touch-evoked escape response (TEER)
Morphology and behavioral touch responses were assessed with a stereomicroscope (Zeiss, Oberkochen, Germany) and only fish with no obvious developmental deficits were selected to determine the TEER. For escape swimming at 48 hpf, embryos were touched lightly at the level of the tail or head with a pair of blunt forceps. Fish that were unable to escape were touched several times (3–4 times) in order to ascertain their failure to respond. Thus, for each injection set, larvae were separated in four groups; dead and developmentally deficient, fish with deficits in TEER and fish displaying a normal TEER. The percentages for the two last groups are described in Table 1 for each condition. Their responses were also recorded using a Photron (San Diego, CA) Fastcam PCI high-speed video camera at a rate of 125 frames/s.
Immunohistochemistry
For immunohistochemical analysis of axonal projections of motor neurons, monoclonal antibodies anti-SV2 (Developmental Studies Hybridoma) were used to assess the motor neuron morphology at 48 and 72 hpf. Fluorescent images of fixed embryos were taken using a Quorum Technologies spinning-disk confocal microscope mounted on an upright Olympus BX61W1 fluorescence microscope equipped with an Hamamatsu ORCA-ER camera. Image acquisition was performed with Volocity software (PerkinElmer). As previously described [50], axonal projections from primary and secondary motor neurons at a defined location in the intersomitic segments were determined. Analysis of Z-stacks by confocal microscopy was performed in three to four axonal projections per animal. The axonal length to the first branching (UAL) was determined by tracing the labeled axon from the spinal cord to the point where it branches using Image J. These values were averaged for each of the animal analyzed (10–30 zebrafish per condition) for the various conditions in our study.
In situ hybridization
Sense and antisense probes of 500 bp length against FUS mRNA were designed. 24 hpf zebrafish embryos were processed for in situ hybridization using fluorescent FastRed as previously described [73], with minor modifications.
Western blotting
Zebrafish embryos were lysed in ice using cold SDS sample buffer (63 mM Tris-HCl pH 6.8, 10% glycerol, 5% ß-mercaptoethanol, 3.5% sodium dodecyl sulfate) and were maintained on ice and homogenized using a hand-held pestle. The lysates were centrifuged for 15 minutes at 13000 rpm and separated into soluble and insoluble fractions. SDS/PAGE Western blotting of both fractions were carried out as previously described [9], using monoclonal antibodies against myc (Invitrogen), actin (Clone C4; ICN BIOMEDICALS, Inc.), a polyclonal antibodies against TDP-43 (ProteinTech) and a polyclonal antibody (Bethyl Laboratories) as well as a monoclonal antibody against FUS (BD Transduction Laboratories).
Statistics
Statistical significance was determined by anova and post hoc analysis by Tukey's multiple comparison test using Prism software (Prism Software Ltd.) as well as a two-tailed distribution, two-sample equal variance t-test using Sigma Plot software (Systat Software Inc., San Jose, CA, USA). Significance was established at p<0.05.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. NelsonLM 1995 Epidemiology of ALS. Clin Neurosci 3 327 331
2. BoilleeSVande VeldeCClevelandDW 2006 ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52 39 59
3. Gros-LouisFGasparCRouleauGA 2006 Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta
4. PasinelliPBrownRH 2006 Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7 710 723
5. RosenDR 1993 Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364 362
6. KabashiEValdmanisPNDionPRouleauGA 2007 Oxidized/misfolded superoxide dismutase-1: the cause of all amyotrophic lateral sclerosis? Ann Neurol 62 553 559
7. BendottiCCarriMT 2004 Lessons from models of SOD1-linked familial ALS. Trends Mol Med 10 393 400
8. LemmensRVan HoeckeAHersmusNGeelenVD'HollanderI 2007 Overexpression of mutant superoxide dismutase 1 causes a motor axonopathy in the zebrafish. Hum Mol Genet 16 2359 2365
9. KabashiEValdmanisPNDionPSpiegelmanDMcConkeyBJ 2008 TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40 572 574
10. SreedharanJBlairIPTripathiVBHuXVanceC 2008 TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319 1668 1672
11. Lagier-TourenneCClevelandDW 2009 Rethinking ALS: the FUS about TDP-43. Cell 136 1001 1004
12. GitchoMABalohRHChakravertySMayoKNortonJB 2008 TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol 63 535 538
13. VanceCRogeljBHortobagyiTDe VosKJNishimuraAL 2009 Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323 1208 1211
14. KwiatkowskiTJJrBoscoDALeclercALTamrazianEVanderburgCR 2009 Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323 1205 1208
15. Lagier-TourenneCPolymenidouMClevelandDW TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19 R46 64
16. MackenzieIRRademakersRNeumannM TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9 995 1007
17. BlairIPWilliamsKLWarraichSTDurnallJCThoengAD 2009 FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J Neurol Neurosurg Psychiatry
18. CorradoLDel BoRCastellottiBRattiACeredaC 2009 Mutations of FUS Gene in Sporadic Amyotrophic Lateral Sclerosis. J Med Genet
19. BelzilVVValdmanisPNDionPADaoudHKabashiE 2009 Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology 73 1176 1179
20. ChioARestagnoGBrunettiMOssolaICalvoA 2009 Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol Aging 30 1272 1275
21. TicozziNSilaniVLeClercALKeaglePGelleraC 2009 Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 73 1180 1185
22. ChioACalvoAMogliaCOssolaIBrunettiM 2010 A de novo missense mutation of the FUS gene in a “true” sporadic ALS case. Neurobiol Aging
23. YanJDengHXSiddiqueNFectoFChenW 2010 Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 75 807 814
24. MillecampsSSalachasFCazeneuveCGordonPBrickaB 2010 SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J Med Genet 47 554 560
25. RademakersRStewartHDejesus-HernandezMKriegerCGraff-RadfordN 2010 Fus gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve 42 170 176
26. TateishiTHokonoharaTYamasakiRMiuraSKikuchiH 2010 Multiple system degeneration with basophilic inclusions in Japanese ALS patients with FUS mutation. Acta Neuropathol 119 355 364
27. SuzukiNAokiMWaritaHKatoMMizunoH 2010 FALS with FUS mutation in Japan, with early onset, rapid progress and basophilic inclusion. J Hum Genet 55 252 254
28. MackenzieIRRademakersRNeumannM 2010 TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9 995 1007
29. BenajibaLLe BerICamuzatALacosteMThomas-AnterionC 2009 TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol 65 470 473
30. BorroniBBonviciniCAlbericiABurattiEAgostiC 2009 Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum Mutat 30 E974 983
31. SleegersKCrutsMVan BroeckhovenC 2010 Molecular pathways of frontotemporal lobar degeneration. Annu Rev Neurosci 33 71 88
32. Van LangenhoveTvan der ZeeJSleegersKEngelborghsSVandenbergheR 2010 Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 74 366 371
33. DengHXZhaiHBigioEHYanJFectoF FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol 67 739 748
34. MackenzieIRBigioEHIncePGGeserFNeumannM 2007 Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61 427 434
35. LingSCAlbuquerqueCPHanJSLagier-TourenneCTokunagaS ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci U S A
36. KimSHShanwareNPBowlerMJTibbettsRS 2010 Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J Biol Chem 285 34097 34105
37. LansonNAJrMaltareAKingHSmithRKimJH 2011 A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum Mol Genet
38. FieselFCVoigtAWeberSSVan den HauteCWaldenmaierA 2010 Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J 29 209 221
39. PolymenidouMLagier-TourenneCHuttKRHuelgaSCMoranJ 2011 Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14 459 468
40. AyalaYMZagoPD'AmbrogioAXuYFPetrucelliL 2008 Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 121 3778 3785
41. BurattiEBaralleFE 2010 The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol 7 420 429
42. ParkJLeeSBLeeSKimYSongS 2006 Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441 1157 1161
43. PallanckLGreenamyreJT 2006 Neurodegenerative disease: pink, parkin and the brain. Nature 441 1058
44. ValenteEMAbou-SleimanPMCaputoVMuqitMMHarveyK 2004 Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304 1158 1160
45. KitadaTAsakawaSHattoriNMatsumineHYamamuraY 1998 Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392 605 608
46. EldenACKimHJHartMPChen-PlotkinASJohnsonBS Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466 1069 1075
47. ValdmanisPNMeijerIAReynoldsALeiAMacLeodP 2007 Mutations in the KIAA0196 gene at the SPG8 locus cause hereditary spastic paraplegia. Am J Hum Genet 80 152 161
48. LairdASVan HoeckeADe MuynckLTimmersMVan den BoschL Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLoS ONE 5 e13368 doi:10.1371/journal.pone.0013368
49. Gros-LouisFKrizJKabashiEMcDearmidJMillecampsS 2008 Als2 mRNA splicing variants detected in KO mice rescue severe motor dysfunction phenotype in Als2 knock-down zebrafish. Hum Mol Genet 17 2691 2702
50. KabashiELinLTradewellMLDionPABercierV 2010 Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet 19 671 683
51. SimpsonCLLemmensRMiskiewiczKBroomWJHansenVK 2009 Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum Mol Genet 18 472 481
52. RameshTLyonANPinedaRHWangCJanssenPM A genetic model of amyotrophic lateral sclerosis in zebrafish displays phenotypic hallmarks of motoneuron disease. Dis Model Mech 3 652 662
53. KabashiEChampagneNBrusteinEDrapeauP In the swim of things: recent insights to neurogenetic disorders from zebrafish. Trends Genet
54. ItoDSekiMTsunodaYUchiyamaHSuzukiN 2011 Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol 69 152 162
55. GalJZhangJKwinterDMZhaiJJiaH 2010 Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging
56. TanAYManleyJL 2009 The TET family of proteins: functions and roles in disease. J Mol Cell Biol 1 82 92
57. TicozziNVanceCLeclercALKeaglePGlassJD 2011 Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am J Med Genet B Neuropsychiatr Genet
58. ReaumeAGElliottJLHoffmanEKKowallNWFerranteRJ 1996 Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 13 43 47
59. BoscoDALemayNKoHKZhouHBurkeC Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet 19 4160 4175
60. BarmadaSJSkibinskiGKorbERaoEJWuJY Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 30 639 649
61. DormannDRoddeREdbauerDBentmannEFischerI ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29 2841 2857
62. Liu-YesucevitzLBilgutayAZhangYJVanderwydeTCitroA Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS ONE 5 e3250 doi:10.1371/journal.pone.0013250
63. ColombritaCZennaroEFalliniCWeberMSommacalA 2009 TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem 111 1051 1061
64. KryndushkinDWicknerRBShewmakerF FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast model of amyotrophic lateral sclerosis. Protein Cell 2 223 236
65. FushimiKLongCJayaramNChenXLiL Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy. Protein Cell 2 141 149
66. Lagier-TourenneCPolymenidouMClevelandDW 2010 TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19 R46 64
67. ZhangYJXuYFDickeyCABurattiEBaralleF 2007 Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci 27 10530 10534
68. RademakersREriksenJLBakerMRobinsonTAhmedZ 2008 Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet 17 3631 3642
69. KleinbergerGWilsHPonsaertsPJorisGTimmermansJP 2010 Increased caspase activation and decreased TDP-43 solubility in progranulin knockout cortical cultures. J Neurochem 115 735 747
70. RitsonGPCusterSKFreibaumBDGuintoJBGeffelD TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci 30 7729 7739
71. GitchoMAStriderJCarterDTaylor-ReinwaldLFormanMS 2009 VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J Biol Chem 284 12384 12398
72. JohnsonJOMandrioliJBenatarMAbramzonYVan DeerlinVM 2010 Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 68 857 864
73. JowettT 2001 Double in situ hybridization techniques in zebrafish. Methods 23 345 358
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 8
Nejčtenější v tomto čísle
- An EMT–Driven Alternative Splicing Program Occurs in Human Breast Cancer and Modulates Cellular Phenotype
- Chromosome Painting Reveals Asynaptic Full Alignment of Homologs and HIM-8–Dependent Remodeling of Chromosome Territories during Meiosis
- A Pre-mRNA–Associating Factor Links Endogenous siRNAs to Chromatin Regulation
- Discovery of Sexual Dimorphisms in Metabolic and Genetic Biomarkers