
Cdc5-Dependent Asymmetric Localization of Bfa1 Fine-Tunes Timely Mitotic Exit
In budding yeast, the major regulator of the mitotic exit network (MEN) is Tem1, a GTPase, which is inhibited by the GTPase-activating protein (GAP), Bfa1/Bub2. Asymmetric Bfa1 localization to the bud-directed spindle pole body (SPB) during metaphase also controls mitotic exit, but the molecular mechanism and function of this localization are not well understood, particularly in unperturbed cells. We identified four novel Cdc5 target residues within the Bfa1 C-terminus: 452S, 453S, 454S, and 559S. A Bfa1 mutant in which all of these residues had been changed to alanine (Bfa14A) persisted on both SPBs at anaphase and was hypo-phosphorylated, despite retaining its GAP activity for Tem1. A Bfa1 phospho-mimetic mutant in which all of these residues were switched to aspartate (Bfa14D) always localized asymmetrically to the SPB. These observations demonstrate that asymmetric localization of Bfa1 is tightly linked to its Cdc5-dependent phosphorylation, but not to its GAP activity. Consistent with this, in kinase-defective cdc5-2 cells Bfa1 was not phosphorylated and localized to both SPBs, whereas Bfa14D was asymmetrically localized. BFA14A cells progressed through anaphase normally but displayed delayed mitotic exit in unperturbed cell cycles, while BFA14D cells underwent mitotic exit with the same kinetics as wild-type cells. We suggest that Cdc5 induces the asymmetric distribution of Bfa1 to the bud-directed SPB independently of Bfa1 GAP activity at anaphase and that Bfa1 asymmetry fine-tunes the timing of MEN activation in unperturbed cell cycles.
Published in the journal:
. PLoS Genet 8(1): e32767. doi:10.1371/journal.pgen.1002450
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002450
Summary
In budding yeast, the major regulator of the mitotic exit network (MEN) is Tem1, a GTPase, which is inhibited by the GTPase-activating protein (GAP), Bfa1/Bub2. Asymmetric Bfa1 localization to the bud-directed spindle pole body (SPB) during metaphase also controls mitotic exit, but the molecular mechanism and function of this localization are not well understood, particularly in unperturbed cells. We identified four novel Cdc5 target residues within the Bfa1 C-terminus: 452S, 453S, 454S, and 559S. A Bfa1 mutant in which all of these residues had been changed to alanine (Bfa14A) persisted on both SPBs at anaphase and was hypo-phosphorylated, despite retaining its GAP activity for Tem1. A Bfa1 phospho-mimetic mutant in which all of these residues were switched to aspartate (Bfa14D) always localized asymmetrically to the SPB. These observations demonstrate that asymmetric localization of Bfa1 is tightly linked to its Cdc5-dependent phosphorylation, but not to its GAP activity. Consistent with this, in kinase-defective cdc5-2 cells Bfa1 was not phosphorylated and localized to both SPBs, whereas Bfa14D was asymmetrically localized. BFA14A cells progressed through anaphase normally but displayed delayed mitotic exit in unperturbed cell cycles, while BFA14D cells underwent mitotic exit with the same kinetics as wild-type cells. We suggest that Cdc5 induces the asymmetric distribution of Bfa1 to the bud-directed SPB independently of Bfa1 GAP activity at anaphase and that Bfa1 asymmetry fine-tunes the timing of MEN activation in unperturbed cell cycles.
Introduction
In eukaryotes, mitotic entry is driven by a rise in cyclin-dependent kinase (Cdk) activity, which is required for the formation of a bipolar spindle and chromosome segregation (For a review, see [1]). For cells to subsequently undergo cytokinesis and enter the G1 phase of the next cell cycle, Cdk-mediated phosphorylation events are reversed and Cdk activity declines (For reviews, see [2], [3]). In budding yeast, this transition, called mitotic exit, is triggered by a signaling cascade known as the mitotic exit network (MEN). The MEN activates and releases the Cdc14 phosphatase from the nucleolus, and this phosphatase reverses the phosphorylation of Cdk substrates and inactivates the mitotic Cdks (For a review, see [4]).
The MEN must be tightly regulated for each daughter cell to receive a complete set of chromosomes. When the MEN is prematurely activated in cells undergoing mitosis, genomic instability results [5]. Therefore, the MEN is a crucial target of various checkpoints that keep mitotic Cdk activity high until the daughter chromosomes have segregated properly. The MEN coordinates spindle position and mitotic progression in asymmetrically dividing cells such as budding yeast, where the division plane is predetermined. A pathway called the spindle position checkpoint (SPOC) ensures that the MEN is activated only if the extended mitotic spindle is correctly positioned. When spindles misalign relative to the division plane, mitotic exit is delayed by preventing the MEN activation [6].
The Tem1 GTPase functions to activate the MEN [7]. The MEN signaling cascade is triggered when the two-component GTPase-activating protein (GAP) for Tem1, composed of Bfa1 and Bub2, becomes inactivated. The polo kinase Cdc5 also contributes to MEN activation by directly phosphorylating and inhibiting the GAP activity of Bfa1/Bub2 and/or disrupting its interaction with Tem1 [8], [9]. Impaired Bfa1/Bub2 GAP activity allows mitotic exit in cells that have either mitotic exit defects or activated checkpoints [10]. Consistent with this, Bfa1 remains unphosphorylated when the SPOC prevents mitotic exit [8]. Lte1, which was once suggested to promote mitotic exit as a putative guanine nucleotide exchange factor (GEF) for Tem1, has been reported to contribute to mitotic exit by controlling asymmetric Bfa1 localization and cell polarization [11], [12]. A recent study demonstrated that loading of Tem1 onto the spindle pole bodies (SPBs) is required for activation of the MEN [13]. Thus a misaligned spindle markedly delays mitotic exit in cells with low GAP activity for Tem1 [10]. These recent studies have suggested more complex ways by which MEN is regulated, including localization of MEN components to the SPB, together with the GTPase-switch model for Tem1.
The SPB acts as a scaffold for many MEN components (For a review, see [4]). The association of Tem1 with SPBs depends on Bfa1 and Bub2, which are mutually required for the other's localization to the SPB [14]. The Bfa1/Bub2 complex localizes to SPBs in an asymmetric manner: as the spindle aligns along the mother-bud axis and the dividing nucleus migrates to the bud neck, the complex is exclusively found on the bud-oriented SPB [14], [15]. Conversely, on misoriented spindles that lead to delayed mitotic exit, Bfa1/Bub2 is present on both SPBs. This suggests that the spatial distribution of Bfa1/Bub2 is directly connected to the control of mitotic exit [15], [16]. Consistent with this hypothesis, a Bub2 variant that localizes to both SPBs throughout the cell cycle prevented mitotic exit in certain MEN-impaired mutants [17]. Also a recent quantitative analysis showed that Bfa1 dynamics at the SPBs establishes asymmetry in MEN signaling and regulates MEN activity. Bfa1 associates with both SPBs in a transient fashion, but its binding to the daughter SPB (SPBd) is stabilized by cell polarity determinants and their interactions with microtubules [18]. As a consequence, Bfa1 accumulates on the SPBd during metaphase, whereas it disappears from the mother SPB (SPBm), thereby establishing Bfa1 asymmetry [18]. When the spindles are improperly positioned, Bfa1 association becomes highly dynamic on both SPBs, which is required for proper SPOC function [19].
Despite the role in the fidelity of mitosis, the molecular details governing the asymmetry of Bfa1/Bub2 positioning have yet to be fully elucidated. Furthermore, the importance of the asymmetry in the unperturbed cell cycle remains unclear. Bfa1 asymmetry is required for recruiting MEN components exclusively to the SPBd during metaphase [18]. Bfa1 reaches its maximum phosphorylation state when it associates preferentially with the SPBd, whereas Bfa1 is unphosphorylated and localizes to both SPBs in SPOC-activated cells [8], [16]. We have identified new phosphorylation sites on Bfa1 that function in directing its asymmetric distribution to SPBs. We present evidence that the phosphorylation of these sites by Cdc5 does not inhibit Bfa1 GAP activity, but induces Bfa1 asymmetry and achieves timely MEN activation during unperturbed mitotic progression.
Results
Bfa1 localizes to both SPBs in the kinase-defective cdc5-2 mutant
Bfa1 is a cell cycle-regulated phosphoprotein [8] that forms a complex with Bub2 and negatively controls the activation of Tem1, a key upstream regulator of mitotic exit [8], [9]. Cdc5 polo kinase phosphorylates Bfa1 during mitosis to down-regulate Bfa1/Bub2, and thus activates mitotic exit [8], [9]. In cdc15-2 cells, Bfa1 becomes phosphorylated by Cdc5 and Tem1 is activated, but mitotic exit is not permitted since Cdc15 acts downstream of Tem1 in the MEN [7]. Thus, we used the cdc15-2 mutant, which contains wild-type CDC5, as a control for Bfa1 phosphorylation and localization in Figure 1. In this experiment, we compared wild-type CDC5 in the cdc15-2 strain to the cdc5-1 and cdc5-2 mutants to characterize the effects of these mutations on late mitosis at the restrictive temperature [20], [21].

When α-factor-synchronized cdc15-2 cells were released at restrictive temperature (35°C), Bfa1 was detected as a sharp band in G1 phase; the band accumulated as slower-migrating forms during mitosis, and finally attained its maximal phosphorylation states (Figure 1A left). When treated with phosphatase, the slower-migrating forms collapsed into a sharp band (Figure 1A right). The two cdc5 mutations, cdc5-1 and cdc5-2, affected Bfa1 differently. Bfa1 was phosphorylated as usual in the cdc5-1 mutant, whereas the cdc5-2 mutant was severely defective in phosphorylating Bfa1 at the restrictive temperature (Figure 1A; [8]).
We hypothesized that phosphorylation of Bfa1 by Cdc5 might influence its subcellular location. Before examining this possibility we investigated cell cycle progression and the arrest points in cdc5-1 and cdc5-2 cells. We shifted G1-synchronized cells to 35°C, and counted every 10 min the number of large budded cells with elongated nuclei stretching along the neck or two segregated nuclei (Figure 1B). Compared to the cdc15-2 mutant, both cdc5 mutants exhibited a delay during nuclear elongation (more markedly in cdc5-2 than in cdc5-1), but eventually arrested as large budded cells with separated nuclei, a phenotype similar to that of the cdc15-2 mutant (Figure 1B). Quantitative analysis of pole-to-pole distances in cells with segregated nuclei revealed that the spindle length relative to cell length was shorter in cdc5-1 and cdc5-2 cells than in cdc15-2 cells; mean spindle length was approximately 83% of that of cdc15-2 for cdc5-1, and 53% for the cdc5-2 mutant (Figure 1C). These results demonstrate that the cdc5-1 and cdc5-2 mutants have defects in nuclear spindle elongation but eventually undergo nuclear division with spindles that are not fully elongated; thereafter they arrest as large budded cells with separate nuclei, as does the cdc15-2 mutant. Hereafter, for simplicity, we refer to the arrest point of cdc5-1, cdc5-2, and cdc15-2 as late anaphase.
We then examined the localization of Bfa1 in cdc5-1, cdc5-2 and cdc15-2 cells, where Bfa1 and Spc42 were fused to GFP and RFP, respectively. Bfa1-GFP was found on both SPBs immediately after their separation and before the nucleus moved to the bud neck [14], [18]. During metaphase of the cdc15-2 cells, when the spindle was oriented along the division axis and the nucleus was positioned at the bud neck, Bfa1-GFP was predominantly localized to the SPB closest to the bud neck (Figure 1D; [18]). Bfa1 continued to be selectively localized in cdc15-2 cells, with elongated dividing nuclei or segregated nuclei (Figure 1D). The cdc5-1 mutant displayed a Bfa1-GFP localization pattern similar to the cdc15-2 mutant, whereas Bfa1-GFP remained associated with both SPBs in the cdc5-2 mutant, even after nuclear segregation (Figure 1D). In anaphase-arrested cells, Bfa1-GFP was present on both SPBs in 90.9±3.0% of cdc5-2 cells, whereas it was asymmetrically localized on the SPBd in 88.0±1.2% of cdc15-2 and 92.3±1.5% of cdc5-1 cells (Figure 1E).
We also observed that the fluorescence intensity of Bfa1-GFP on the SPBs in cdc5-2 cells was only about 25–28% of that in cdc15-2 cells (Figure 1F). Recently, Monje-Casas and Amon [18] showed that the intensity of Bfa1-GFP fluorescence is a good measure of the affinity of Bfa1 for the SPB, and its dynamics. We therefore suggest that the phosphorylation of Bfa1 by Cdc5 regulates the dynamics of its behavior, and leads to its asymmetric distribution on the two SPBs.
A decrease in Bfa1/Bub2 GAP activity is not required for Bfa1 asymmetry
Since Cdc5 inhibits Bfa1/Bub2 GAP activity toward Tem1 by phosphorylating Bfa1 [9], the presence of Bfa1 on both SPBs in cdc5-2 cells (Figure 1) could be due to uninhibited Bfa1/Bub2 GAP activity or to the absence of Bfa1 phosphorylation. To distinguish between these possibilities, we examined the localization of GAP activity-defective variants of Bfa1. In in vitro Tem1 GTPase assays with Bub2, GAP activity was almost completely absent in Bfa1W422A, markedly decreased in both Bfa1M413I and Bfa1D416A, and slightly decreased in Bfa1G411E [10]. The cdc15-2 mutant was used to analyze the localization of each Bfa1 variant at anaphase. We integrated GFP-fused BFA1, BFA1G411E, BFA1M413I, BFA1D416A, and BFA1W422A into cdc15-2SPC42-RFPΔbfa1 cells, and released these cells from G1 arrest at 35°C. Southern and Western blots verified that the Bfa1 mutants were integrated as single copies and were expressed at similar levels to wild-type Bfa1 (Figure S1). We reasoned that if Bfa1 asymmetry was promoted by inhibition of its GAP activity, the mutant forms of Bfa1 would establish Bfa1 asymmetry prematurely and localize to only one SPB throughout the cell cycle. In fact, however, like wild-type Bfa1, they associated with both SPBs immediately after SPB separation and before nuclear migration to the bud neck (data not shown). In addition, most of the Bfa1M413I, Bfa1D416A, and Bfa1W422A forms were present on both SPBs even after chromosome segregation, despite their low GAP activities (Figure 2A and 2B). Bfa1M413I, Bfa1D416A, and Bfa1W422A bound to both SPBs in 78.4±1.7, 68.7±0.6, and 70.5±10.1% of anaphase-arrested cells, respectively, while most wild-type Bfa1 (88.0±1.2%) and Bfa1G411E (83.2±4.5%) was asymmetrically localized to the SPBd.

To exclude the possibility that residual GAP activity of Bfa1M413I, Bfa1D416A, and Bfa1W422A was responsible for their association with both SPBs at anaphase, we constructed another mutant, Bfa1DDR2 (Deletion of Direct Repeat 2; Bfa1G411E M413I D416A W422A), which was predicted to have no GAP activity; indeed, in vitro assays revealed that the Bfa1DDR2/Bub2 complex completely failed to stimulate Tem1 GTPase activity (Figure 2C). Consistent with this, Bfa1DDR2 was utterly unable to prevent mitotic exit in vivo (Figure S2). After confirming single copy integration and normal expression levels (Figure S1), we observed that Bfa1DDR2 also persisted at both SPBs in anaphase (76.0±5.3%; Figure 2A and 2B), clearly demonstrating that inhibition of GAP activity does not induce Bfa1 asymmetry. These results suggest that the persistence of Bfa1 on both SPBs in the cdc5-2 mutant is not due to failure to inhibit GAP activity.
Bfa1 asymmetry is tightly linked to its phosphorylation by Cdc5
We then asked if phosphorylation by Cdc5 is required for the asymmetric distribution of Bfa1 on SPBs. We examined phosphorylation of the Bfa1 mutants shown in Figure 2 using SDS-PAGE mobility shift assays in the cdc15-2 background. When α-factor - synchronized cells were released at 35°C, Bfa1G411E became phosphorylated with similar kinetics to wild-type Bfa1 (Figure 3A). In contrast, slower-migrating forms of Bfa1M413I, Bfa1D416A, Bfa1W422A, and Bfa1DDR2 were not detected after the release from G1 arrest (Figure 3A). We did not observe any mobility shift of these Bfa1 mutants even when Cdc5 was overexpressed (Figure S3). Using yeast two-hybrid assays we showed that these Bfa1 mutants interacted with Cdc5 like wild-type Bfa1, demonstrating that the lack of Bfa1 phosphorylation by Cdc5 in these mutants is not due to reduced interaction with Cdc5 (Figure S6B).

To examine the extents of phosphorylation of the Bfa1 mutants, we purified GST-Cdc5 and GST-Cdc5KD (a kinase-dead control) from S. cerevisiae and incubated them with MBP-Bfa1 proteins (MBP-Bfa1M413I, -Bfa1D416A, -Bfa1W422A, and -Bfa1DDR2) expressed in E. coli and purified. As the amount of GST-Cdc5 was increased, wild-type Bfa1 began to appear as multiple, slower migrating forms and eventually appeared as the slowest migrating form, while the Bfa1 mutants remained as multiple, less slowly-migrating forms (Figure 3B). The results of these in vitro kinase assays differed slightly from the in vivo results in which slower migrating forms of the Bfa1 mutants were rarely seen (Figure 3A; Figure S3). This difference is probably due to either non-specific phosphorylation by the excessive Cdc5 activity used, or the presence of Cdc5 sites that are not easily phosphorylated in vivo. In either case, these Bfa1 mutants were obviously resistant to phosphorylation by Cdc5. Note that Bfa1G411E localized asymmetrically to SPBd and was phosphorylated by Cdc5 like wild-type Bfa1, whereas the other Bfa1 variants (Bfa1M413I, Bfa1D416A, Bfa1W422A, and Bfa1DDR2) were distributed to both SPBs and were not phosphorylated as efficiently as wild-type Bfa1 by Cdc5. We thus conclude that the asymmetric distribution of Bfa1 is probably linked to its phosphorylation by Cdc5.
Novel Cdc5 phosphosites are involved in the asymmetric distribution of Bfa1
Eleven Cdc5 phosphosites have been mapped previously, and substituted with Ala in the Bfa1-11A mutant [8]. To confirm the relationship between Bfa1 localization and phosphorylation, we examined the distribution of Bfa1-11A on SPBs. Since Bfa1-11A mobility is not greatly retarded under conditions that normally produce hyperphosphorylated wild-type Bfa1 (Figure 4A), we expected this mutant to be present on both SPBs in anaphase cells. However, most of the Bfa1-11A (90.1±3.4%) was only associated with SPBd, like wild-type Bfa1 (88.0±1.25%) (Figure 4B). We therefore examined whether Bfa1-11A was further phosphorylated by Cdc5. When MBP-fused Bfa1-11A was incubated with GST-Cdc5 and γ-[32P] ATP, 32P incorporation was observed, whereas no 32P incorporation was detected in reactions with an equivalent amount of GST-Cdc5KD (Figure 4C). These observations are consistent with a previous report that mutation of these 11 residues reduces in vitro phosphorylation of Bfa1 by only 75% [8], and demonstrate that not all Cdc5 phosphorylation sites are mutated in Bfa1-11A.
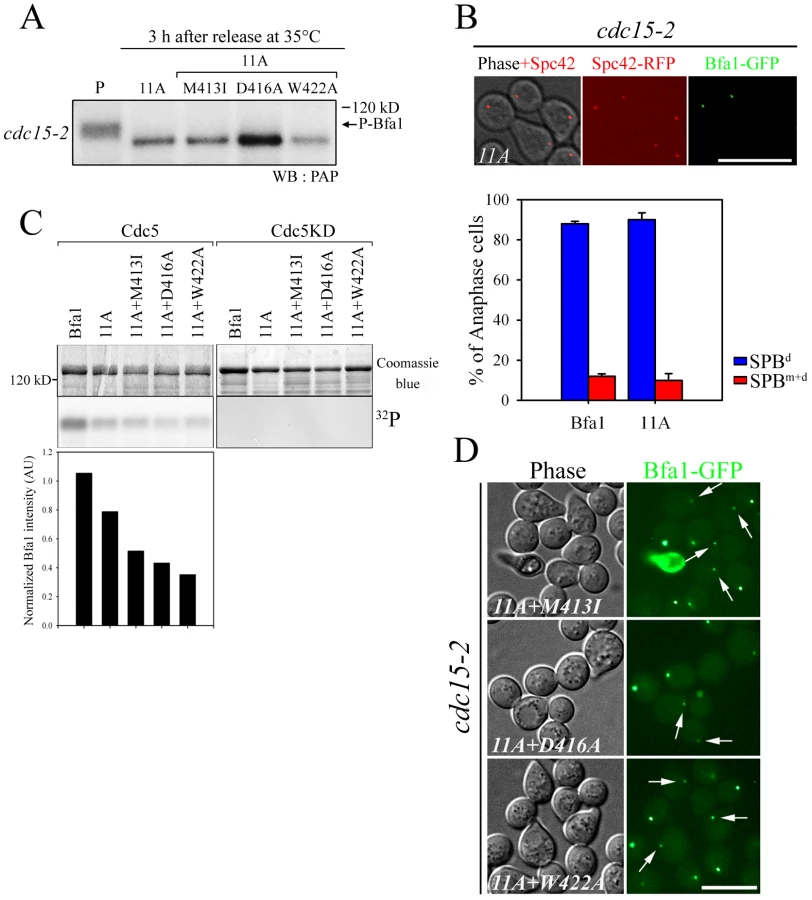
Based on these results, we reasoned that there are unidentified Cdc5 phosphosites and that these could be responsible for the asymmetric distribution of Bfa1-11A. We further hypothesized that these novel sites are not efficiently phosphorylated on Bfa1M413I, Bfa1D416A, and Bfa1W422A, thereby causing these Bfa1 variants to persist at both SPBs in anaphase. If that were the case, the introduction of the M413I, D416A, or W422A mutation into Bfa1-11A should impair Bfa1-11A asymmetry and reduce its phosphorylation. Indeed, 32P incorporation into Bfa1M413I-11A, Bfa1D416A-11A, and Bfa1W422A-11A was less efficient than into Bfa1-11A (Figure 4C), and Bfa1M413I-11A, Bfa1D416A-11A, and Bfa1W422A-11A were each symmetrically distributed to both SPBs (Figure 4D). Consistent with this, when Cdc5 was overexpressed to force phosphorylation of Bfa1 by Cdc5, we detected a mobility shift in Bfa1-11A but not in Bfa1M413I-11A, Bfa1D416A-11A, or Bfa1W422A-11A (Figure S4A). However, in the anaphase-arrested cdc15-2 background, the mobilities of Bfa1M413I-11A, Bfa1D416A-11A, and Bfa1W422A-11A were nearly the same as that of Bfa1-11A (Figure 4A), suggesting that phosphorylation of the residue(s) responsible for asymmetry is not efficient in cells expressing endogenous Cdc5 levels. Together, these results support the presence of unidentified Cdc5 target residues required for establishing Bfa1 asymmetry.
Mutation of 452S, 453S, 454S, and 559S disrupts Bfa1 asymmetry
We previously showed that the C-terminal 184 residues of Bfa1 (Bfa1-D8391–574) sufficiently inhibit the MEN and localize predominantly to the SPBd (Figure 5A and 5B; [22]). As expected, GFP-fused Bfa1-D8M413I, Bfa1-D8D416A, and Bfa1-D8W422A were found at both SPBs (Figure 5B). In addition, the Bfa1-D8 mutants were less efficiently phosphorylated by Cdc5; Bfa1-D8 was more intensely labeled by 32P and detected in a slower-migrating form than Bfa1-D8M413I, Bfa1-D8D416A, and Bfa1-D8W422A (Figure 5C). This indicated that the putative Cdc5 target site(s) responsible for Bfa1 asymmetry was probably located within the C-terminal 184 residues of Bfa1.

We next searched for possible kinase targets within the C-terminal 184 residues of Bfa1. Cdc5 is a Ser/Thr protein kinase, and Bfa1-D8 contains 23 Ser and 12 Thr residues (Figure 5A). We first systematically mutated 21 of the 23 Ser residues to Ala in Bfa1-D8 and constructed 16 different Ser mutants as GFP fusion proteins: Bfa1S395A, Bfa1S404A, Bfa1S426A, Bfa1SSS452AAA, Bfa1SKS459AAA, Bfa1S469A, Bfa1S478A, Bfa1S490A, Bfa1SS504AA, Bfa1SS510AA, Bfa1S530A, Bfa1S541A, Bfa1S551A, Bfa1S555A, Bfa1S559A, and Bfa1S571A (Figure 5A, Table 1). Both 424S and 447S were excluded because they were included in Bfa1-11A. We integrated each of the Ser mutants into cdc15-2Δbfa1 cells, and examined their localization at anaphase. The results are summarized in Table 1. Most of the GFP-fused Ser mutants exhibited the localization pattern of wild-type Bfa1-GFP (Table 1; and data not shown). However, the percentage of anaphase-arrested cells with Bfa1-GFP on both SPBs was significantly increased in cdc15-2BFA1SSS452AAA (for simplicity, cdc15-2BFA13A) and cdc15-2BFA1S559A cells; Bfa1-GFP was present on both SPBs in 12.0±1.2% of cdc15-2BFA1 cells, 67.3±6.5% of cdc15-2BFA13A cells, and 29±2.6% of cdc15-2BFA1S559A cells (Figure 5D and 5E, Table 1). Cells expressing the substitutions of each Ser in Bfa13A, GFP-tagged Bfa1S452A, Bfa1S453A, or Bfa1S454A, also had larger numbers of anaphase cells with Bfa1-GFP on both SPBs (Table 1), demonstrating that all three Ser residues contribute to Bfa1 asymmetry. In contrast, mutation of some of the Thr residues to Ala around the newly identified 452S, 453S, 454S, and 559S sites (465T, 497TT, 500T, 537T, 546T, 552T, and 572T; Figure 5A) had little effect on Bfa1 asymmetry (Table 1; data not shown).
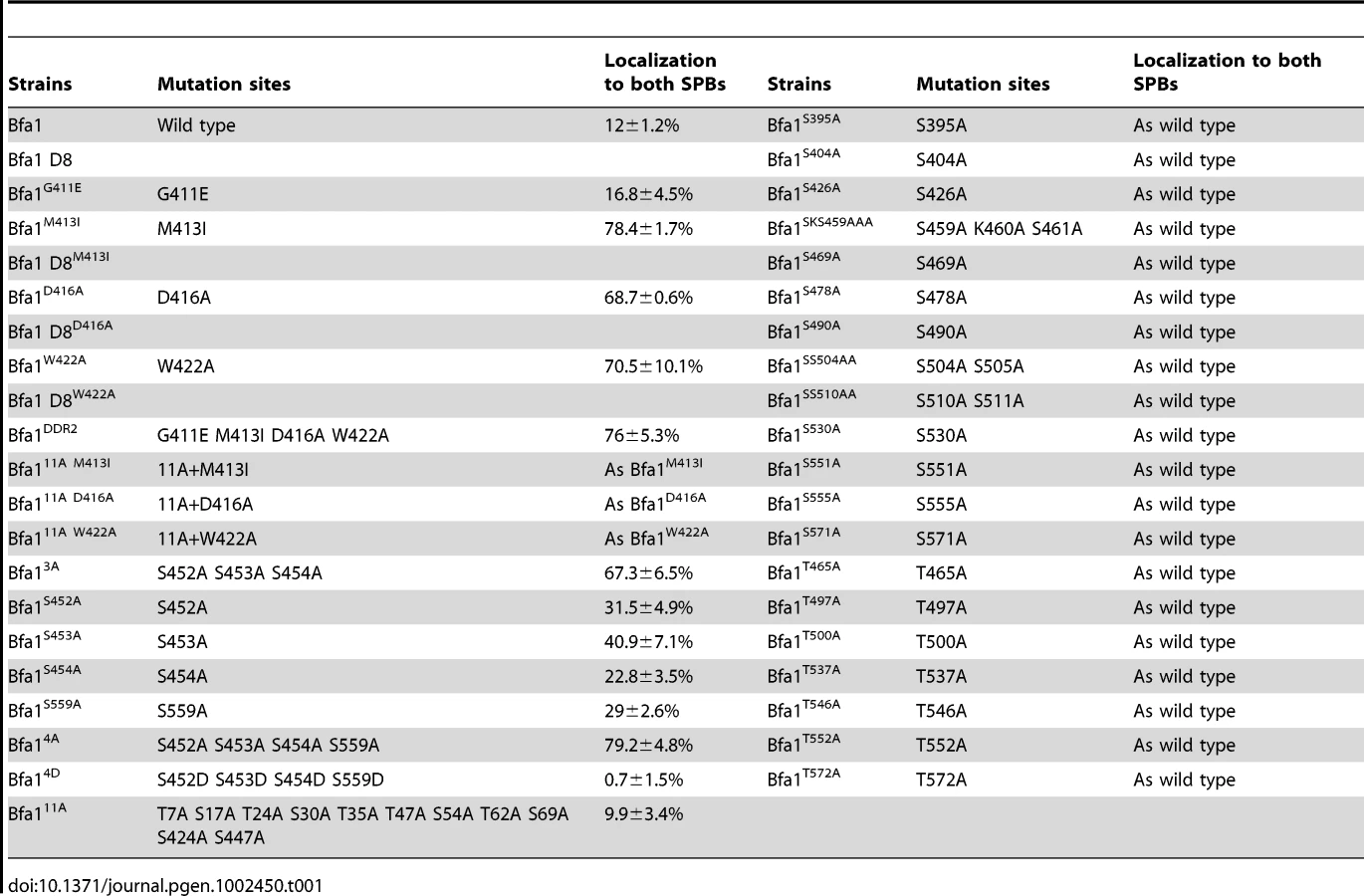
To confirm these results, we constructed Bfa14A, in which all four Ser residues were substituted with Ala (SSS452AAA S559A referred to as 4A). Bfa14A-GFP was detected on both SPBs in 79.2±4.8% of anaphase cells, compared with 67.3±6.5% for cdc15-2BFA13A and 29±2.6% for cdc15-2BFA1S559A cells, again showing that all four Ser residues play a role in establishing Bfa1 asymmetry (Figure 5D and 5E). Quantification of the GFP signal in anaphase-arrested cells showed that the fluorescence intensity of Bfa14A-GFP was nearly the same at both SPBs, weaker than that of wild-type Bfa1-GFP at the SPBd (Figure 5F) and similar to that of Bfa1-GFP in the cdc5-2 mutant (Figure 1).
In order to verify that the symmetric localization of Bfa14A was not caused by defective interaction with Cdc5, we compared the physical interactions of Bfa14A and wild-type Bfa1 with Cdc5 using yeast two-hybrid assays. As shown in Figure S6A, Bfa14A interacted as strongly with Cdc5 as wild-type Bfa1. The asymmetric localization of Tem1 to the daughter SPB in anaphase depends on Bfa1 and Bub2 [14]. Therefore we asked whether the presence of the Bfa14A mutant on both SPBs affected the asymmetric localization of Tem1. To answer this question we integrated GFP-fused BFA14A or BFA1 into cdc15-2TEM1-RFPΔbfa1 cells, arrested these cells in G1, and released them at 35°C. As expected, Tem1-RFP followed the localization pattern of Bfa1-GFP in anaphase: it was present on both SPBs in the Bfa14A background and distributed asymmetrically to the SPBd in the wild-type background (Figure 5G).
452S, 453S, 454S, and 559S can be phosphorylated by Cdc5 in vivo and in vitro
We then asked if 452S, 453S, 454S, and 559S of Bfa1 are phosphorylated by Cdc5. Measuring phosphorylation of these four Ser without phosphorylation of the 11 known targets was not feasible. Therefore, since the N-terminus of Bfa1 contains nine of the Cdc5 targets of Bfa1-11A, we used the C-terminal 184 residues of Bfa1 in in vitro kinase assays. MBP-tagged Bfa1-D8, Bfa1-D8-11A, Bfa1-D84A, and Bfa1-D84A-11A were incubated with GST-Cdc5 or GST-Cdc5KD. The extents of phosphorylation were determined from the resulting mobility shifts and phospho-Bfa1-D8 bands by Phos-tag SDS-PAGE (Figure 6A). The band representing Bfa1-D84A-11A was tighter in mobility shifts and the phospho-Bfa1-D84A-11A forms were not detected in Phos-tag SDS-PAGE (Figure 6A). The extents of phosphorylation were further confirmed by measuring γ-[32P] incorporation and Bfa1-D84A-11A less intensely labeled with 32P than that of Bfa1-D8-11A (Figure 6B), demonstrating that the 4A mutations reduce Bfa1 phosphorylation by Cdc5. In addition, we speculated that the slower-migrating forms of Bfa1-11A observed in CDC5-overexpressing cells (Figure S4A) resulted, at least in part, from the phosphorylation of residues, such as 452S, 453S, 454S, and S559, required for Bfa1 asymmetry. Indeed, the 4A mutations abolished the slower migrating forms of Bfa1-11A in cells overexpressing CDC5 (Figure S4A), consistent with the results in Figure 6A and 6B. Thus, we suggest that Cdc5 phosphorylates 452S, 453S, 454S, and 559S of Bfa1.

To confirm this we used mass spectrometry (MS) to map Bfa1 phosphorylation sites (Figure 6C). Recombinant MBP-Bfa1 was phosphorylated in vitro with GST-Cdc5 or with GST-Cdc5KD as a negative control (Figure 6C). Subsequently, MBP-Bfa1 was purified by SDS-PAGE, excised, digested with trypsin, and analyzed by phosphopeptide-selective precursor ion-scanning liquid chromatography (LC) MS. The success of this approach was assessed by seeing if we could detect the 11 previously identified Cdc5 phosphorylation sites. Although we were unable to identify eight of the 11 phosphorylation residues of Bfa1, likely due to their presence on extremely small (547S-549K; three amino acids) or large (28F-75K; 48 amino acids) trypsin peptides, we did detect phosphorylated forms of 17S, 24T, and 447S with high frequency. The same tandem LC-MS/MS analysis identified two phosphopeptide species, 452SSpSPFLR458 (pS, phosphorylated S; Figure 6C) and 452SpSpSPFLR458 (data not shown), containing 453S and 454S, two of the four Bfa1 phosphorylation residues responsible for asymmetry identified above. Phosphorylation of 452S and 559S was not detected by this approach.
To ask if the GAP activity of Bfa14A affects its association with both SPBs at anaphase, we directly measured the GAP activity of Bfa14A/Bub2. When MBP-Bfa14A was added to the reaction together with Tem1 and GST-Bub2, as in the experiment of Figure 2C, γ-Pi increased rapidly with kinetics similar to those obtained with wild-type Bfa1, indicating that the 4A substitutions had no effect on Bfa1 GAP activity (Figure 6D). We also examined the control of mitotic exit by Bfa14A in vivo: BFA14A cells arrested as large budded cells, as did wild-type BFA1 cells, in the presence of various checkpoint-activating signals (Figure S5A–S5C). Together, these results show that the Bfa14A has functional GAP activity and demonstrate that the presence of Bfa14A at both SPBs is independent of GAP activity.
Asymmetric localization of Bfa1 fine-tunes the timing of MEN activation
Since Bfa1 is found on both SPBs in cells with misaligned spindles [16], it has been proposed that symmetrically localized Bfa1, and in particular, the Bfa1 associated with the SPB in mother cells, contributes to the arrest of mitotic exit [17]. However, the BFA1M413I, BFA1D416A, BFA1W422A, and BFA14A cells grew well and did not show any apparent cell cycle delay in unperturbed conditions, despite having Bfa1 on both SPBs throughout the cell cycle. To better understand the function of Bfa1 asymmetry in mitotic exit, we examined cell cycle progression in these asymmetry-defective BFA1 mutants. Following G1 synchronization and release at room temperature, we monitored large budded cells with two divided nuclei (for simplicity, anaphase cells). In both wild-type and mutant BFA1 cultures, anaphase cells began to accumulate approximately 70 min after release (Figure 7A). In BFA1 cells, numbers of anaphase cells began to decrease about 110 min after release. Interestingly, in the BFA1M413I, BFA1D416A, BFA1W422A, and BFA14A cultures, the decrease in anaphase cells was delayed by about 10 min (Figure 7A).

To further examine the delay in cell cycle progression in the Bfa1 mutant cells, we measured Pds1 and Sic1 levels. Pds1 is an anaphase inhibitor that is degraded upon sister chromatid separation [23] and Sic1 is a negative regulator of mitotic CDKs that accumulates following activation of the MEN (For a review, see [24]). Consistent with Figure 7A, the wild-type and all the BFA1 mutants exhibited a drop in Pds1 levels approximately 60 min after release (Figure 7B and 7C), indicating that Bfa1 asymmetry and its persistent association with the SPBm does not alter the timing of anaphase onset. Importantly, Sic1 accumulation in the wild-type began about 80 min after release, whereas it began about 90 min after release in BFA1M413I, BFA1D416A, BFA1W422A, and BFA14A cells (Figure 7B and 7C). BFA1G411E cells, with a normal Bfa1 distribution, displayed the Pds1 and Sic1 kinetics of the wild-type (data not shown). These results show that asymmetry-defective BFA1 cells activate the MEN approximately 10 min later than cells with normal Bfa1 localization.
We confirmed the 10 min delay of mitotic exit in BFA14A mutant cells by examining the dynamics of Cdc14 release in BFA1 and BFA14A mutant cells by live cell analysis using time-lapse confocal microscopy. Mitotic exit requires full activation of Cdc14 by releasing it from the nucleolus in late anaphase [25]. As shown in the captured images of Figure 7D, Cdc14 was fully released out of the nucleolus 95 and 105 min after BFA14A mutant cells were released from G1 arrest, while wild-type BFA1 cells released Cdc14 approximately 10 min earlier. When we examined time lapse images of several BFA14A and wild-type cells, the average time of Cdc14 release was 95.8±9.1 min after release from G1 arrest in the BFA14A mutant and 86.7±13.5 min in BFA1 wild-type (Figure 7D). These observations clearly demonstrate that in the asymmetry-defective BFA14A cells the activation of MEN is delayed by approximately 10 min compared with wild-type BFA1 cells.
We then investigated the physical interaction of Bfa14A with Tem1 and Bub2 using yeast two-hybrid assays to verify that the 10 min delay of mitotic exit was due to its symmetric localization resulting from lack of phosphorylation and not to any reduction in its interaction with Tem1 and Bub2. As shown in Figure S6C and S6D, Bfa14A interacted with Tem1 and Bub2 like wild-type Bfa1. Together, these observations demonstrate that Cdc5-dependent phosphorylation of the four identified serine residues in Bfa1 controls its displacement from the SPBm for timely mitotic exit in unperturbed mitosis.
Phospho-mimicking of 452S, 453S, 454S, and 559S results in Bfa1 asymmetry and timely mitotic exit
In order to confirm that Cdc5-dependent phosphorylation of 452S, 453S, 454S, and 559S in Bfa1 controls the asymmetric localization of Bfa1 for timely mitotic exit, we constructed Bfa14D (SSS452DDD S559D referred to as 4D) that mimics the negative charges due to phosphorylation. GFP-fused BFA14D was integrated into cdc15-2SPC42-RFPΔbfa1 cells, and as shown in Figure 8A, Bfa14D-GFP remained localized asymmetrically on the SPBs throughout the cell cycle, even in G2/M when wild-type Bfa1 is present on both SPBs. At G2/M, the cells with asymmetrically localized Bfa1 on the SPBs were significantly increased in cdc15-2BFA14D cells than in cdc15-2BFA1 cells; 73.9±6.3% of cdc15-2BFA14D and 19.7±5.8% of cdc15-2BFA1 cells (Figure 8A). When cells were arrested in late anaphase, Bfa14D-GFP was asymmetrically localized on the daughter SPB in 98.6±1.5% of the cells, compared to 20.8±4.8% in cdc15-2BFA14A cells (Figure 8A and Figure 5E). These observations demonstrate that symmetric localization of Bfa14A is caused by loss of phosphorylation, and suggest that phosphorylation of Bfa1 by Cdc5 on 452S, 453S, 454S, and 559S is necessary for establishing Bfa1 asymmetry.

In kinase-defective cdc5-2 cells, Bfa1 was unphosphorylated and localized to both SPBs (Figure 1D). The finding that Cdc5-dependent Bfa1 phosphorylation on 452S, 453S, 454S, and 559S residues regulates its asymmetric localization prompted us to ask whether the phospho-mimicking Bfa14D is asymmetrically located in cdc5-2 cells. For this, we integrated pRS304-BFA14D-GFP into the TRP1 locus of cdc5-2Δbfa1 cells. In late anaphase-arrested cdc5-2 cells, Bfa14D was asymmetrically localized in 72.7±0.1% of the cells, while Bfa1-GFP was present on both SPBs in 90.9±3.0% of cells (Figure 1E and Figure 8B). These observations further support the notion that Cdc5-dependent phosphorylation of 452S, 453S, 454S, and 559S in Bfa1 is important for its asymmetric localization.
Since Bfa1 is a target of Cdc5 phosphorylation for triggering mitotic exit, its deletion has been reported to rescue kinase-defective cdc5-2 cells arrested in late anaphase at restrictive temperatures [8]. To see whether Bfa14D can inhibit the MEN, we tested whether the viability of cdc5-2 could be restored by BFA14D. As shown in Figure S7, BFA14D as well as wild-type BFA1 suppressed the growth of cdc5-2Δbfa1 cells, while knock-out of BFA1 rescued the viability of cdc5-2 cells. These results demonstrate that Bfa14D is able to inhibit MEN like wild-type Bfa1.
Since Bfa14D-GFP is exclusively localized on one of the SPBs during mitosis (Figure 8A) but functions as a negative regulator of the MEN, we asked whether the lack of dynamic localization of phospho-mimetic Bfa14D affects cell cycle progression. cdc15-2 cells expressing wild-type BFA1 or phospho-mimetic BFA14D were synchronized in G1 and released at room temperature, and their cell cycle progression was monitored by counting metaphase and anaphase cells. As shown in Figure 8C, BFA14D cells exhibited the same kinetics of cell cycle progression as wild-type BFA1 cells. In both wild-type and BFA14D cells, metaphase cells began to accumulate at 60 min and peaked at 100 min, while anaphase cells appeared at 100 min and reached a peak at 120 min after release (Figure 8C).
To further analyze the cell cycle progression, we measured the mitotic cyclin Clb2, which is degraded upon activation of the MEN [26]. Consistent with the above result, Clb2 began to accumulate at approximately 60 min after release in both wild-type and BFA14D mutant cells, peaked at 90 min, and then declined (Figure 8D and 8E). These results showed that BFA14D cells allow timely mitotic exit like wild-type BFA1. Together they confirm that phosphorylation of 452S, 453S, 454S, and 559S regulates the asymmetric localization of Bfa1 and timely mitotic exit in unperturbed cell cycle.
In addition, the asymmetric presence of Bfa14D protein on SPBs in G2/M did not affect early mitotic progression (Figure 8). Therefore we suggest that Bfa1 asymmetry is required for timely activation of the MEN but is not necessary for mitotic progression before anaphase in the unperturbed cell cycle.
Phospho-mimetic BFA14D cells delay mitotic exit in response to spindle misalignment
Previous studies have suggested that the symmetrical distribution of Bfa1/Bub2 is directly related to the delay of mitotic exit when the spindle is not properly aligned [15], [16]. We therefore asked whether Bfa14D is able to function in the spindle position checkpoint and whether it is symmetrically localized in cells with misaligned spindles. Proper positioning of the mitotic spindle relies on two independent pathways, one involving the minus-end microtubule motor dynein, the other Bim1, a plus-end microtubule-binding protein [27], [28]. The absence of DYN1 induces anaphase spindle misalignment in the mother cell and thus triggers the spindle position checkpoint [6], [27]. We first examined the localization of Bfa14D in Δdyn1 cells by integrating pRS304-BFA14D-GFP into the TRP1 locus of Δdyn1mCherry-TUB1Δbfa1 cells, as described in Materials and Methods. Surprisingly, Bfa14D was present on both SPBs in cells with misaligned spindles like wild-type Bfa1 (Figure 9A).

When the anaphase spindle is misaligned in the parent of Δdyn1 cells, BFA1 deletion induces improper mitotic exit, as a result of which both multinucleate and anucleate cells accumulate [14], [29]. To assess the spindle position checkpoint functioning of Bfa14D, we monitored multinucleate and anucleate cells in Δdyn1BFA14D and compared them with Δdyn1 cells with the wild-type BFA1 (Δdyn1BFA1) after arrest with α-factor and release at 16°C, when the spindle orientation defect is most pronounced. As shown in Figure 9B, the improper mitotic exit seen in Δdyn1Δbfa1 cells was significantly decreased in both Δdyn1BFA1 and Δdyn1BFA14D cells; 5.7±0.1% in Δdyn1BFA1 and 4.7±0.3% in Δdyn1BFA14D cells. We also examined the spindle position checkpoint function of phospho-mimetic Bfa14D in Δbim1Δbfa1 cells. Consistent with the above result, BFA14D rescued the viability of Δbim1Δbfa1 cells like wild-type BFA1 (Figure 9C).
These results demonstrate that BFA14D cells contain SPOC activity like wild-type BFA1 cells. The symmetric localization of Bfa14D is consistent with the SPOC activity of Bfa14D as well as previous evidence that the symmetrical localization of Bfa1 in cells with misaligned spindles is directly connected to the activation of SPOC [15], [16]. Based on these observations, we suggest that the newly identified Cdc5-dependent phosphorylation residues in Bfa1, 452S, 453S, 454S, and 559S, are only important for its asymmetrical localization and the timing of mitotic exit in unperturbed cells.
Discussion
In budding yeast, Kar9 and dynein preferentially associate with the bud-directed SPB, from which astral microtubules emanate [30], [31]. If these proteins distribute symmetrically to both SPBs, the mitotic spindle does not align properly, showing that SPB asymmetry is essential for mitosis [30], [31]. Konig et al. showed that cyclin-dependent kinase 1 (Cdk1) is asymmetrically recruited to the SPBm in early anaphase and negatively regulates MEN activity at the SPBm [32]. Caydasi and Pereira [19] reported that forced targeting of Bfa1 and Bub2 to both SPBs compromised SPOC function and Valerio-Santiago et al. demonstrated that control of Tem1 localization is essential for the proper functioning of the MEN and SPOC [13]. Recently, Bertazzi et al. showed that Lte1-promoted exclusion of Kin4 from the SPBd is essential for proper mitotic exit [33]. These previous studies mainly focused on the biological function of SPB asymmetry in cells with misaligned spindles. Here, we have demonstrated that Cdc5-dependent phosphorylation of Bfa1 contributes to its asymmetric distribution at SPBd, which is required for timely mitotic exit and, therefore, is required for the fidelity of cell division in unperturbed cells without misaligned spindles.
Previously, GAP activity-defective Bub2-Myc was reported to lead to localization of the Bfa1/Bub2 complex to both SPBs throughout the cell cycle. This complex inhibits mitotic exit, but only in mutant backgrounds in which the MEN is partially impaired [17]. However, it was not clear whether the inhibition of mitotic exit was due to lack of asymmetry or to the absence of GAP activity. Therefore, the importance of Bfa1 asymmetry and its specific function in normal cell cycle progression were not well understood. In this study, we identified various asymmetry-defective Bfa1 mutants that persist on both SPBs throughout the cell cycle. In particular, unlike Bub2-Myc, the Bfa14A mutant stimulated the Tem1 GTPase (Figure 6D), and activated checkpoints for mitotic exit control (Figure S5A–S5C). These results demonstrate that wild-type Bfa1 and Bfa14A differ only in their localization patterns. Bfa14A delayed Sic1 accumulation and Cdc14 release by approximately 10 min relative to cells with normal localization of Bfa1 (Figure 7B–7D). On the other hand, the phospho-mimetic Bfa14D mutant allowed timely mitotic exit like wild-type Bfa1 (Figure 8C–8E). Therefore, we suggest that Bfa1 asymmetry and its disappearance from the SPBm regulate the timing of MEN activation in unperturbed cell division cycles.
However in cells with misaligned spindles, Bfa14D was located on both SPBs and there was full SPOC activity (Figure 9). We therefore consider that the newly identified Cdc5-dependent phosphorylation residues in Bfa1, 452S, 453S, 454S, and 559S, are only important for its asymmetrical localization and the timing of mitotic exit in unperturbed cells. The symmetric localization as well as the SPOC activity of Bfa14D is consistent with previous studies that showed that the symmetrical localization of Bfa1 in cells with misaligned spindles is directly connected with activation of the SPOC [15], [16]. We speculate that cells override the Cdc5-depedent asymmetric localization of Bfa1 in the presence of a spindle orientation defect. Thus, the previously reported mechanisms that account for the symmetric localization of Bfa1 and the arrest of mitotic exit in response to misaligned spindles may apply to Bfa14D.
It has been suggested that a 10 min delay in the cell division cycle is not biologically significant in controlling the cell division cycle. However, considering that the entire cell cycle of budding yeast is about 90 min and mitosis takes approximately 30 min [34], a 10 min delay is not negligible. In fact, mitotic exit is only delayed by 15 minutes in the presence of constant peak levels of Clb2, which blocks spindle disassembly [35].
Valerio-Santiago et al. recently showed that localization of Tem1 to the SPBs is essential for activation of the MEN [13]. As we showed in Figure 5G, Tem1-RFP localized to both SPBs in BFA14A cells. We also showed that Bfa14A binds to Tem1 like wild-type Bfa1 (Figure S6C). These observations suggest that the delay of mitotic exit in BFA14A cells is a consequence of disrupting the asymmetric localization of Tem1.
What molecular details underlie Bfa1 asymmetry in unperturbed mitosis? One significant contribution may come from cell polarity determinants [18]. Monje-Casas and Amon reported that the correct interaction of astral microtubules with the bud cortex alters the affinity of Bfa1 for SPBs and affects its asymmetry [18]. Consistent with this observation, Geymonat et al. showed that the activity of Lte1 in mitotic regulation depends on its localization to the bud cortex and contributes to the asymmetric localization of Bfa1 to the daughter SPB [12]. How can information in the cortex control the distribution of Bfa1 at SPBs, and how is Bfa1 able to bind to the SPBs with different affinities? When spindles misalign, Kin4 kinase activity and its localization to SPBs are reported to regulate the residence time of Bfa1 at SPBs, as well as SPOC activity [19]. However, if the spindle is correctly aligned, Kin4 begins to associate with the SPBm in mid-anaphase at a time when Bfa1 asymmetry has already been established [36]. In addition, in Δkin4 cells with proper spindle positioning, Bfa1 has a normal localization pattern [19]. Furthermore, symmetric localization of Bfa14A is not caused by its defective interaction with Kin4, since Bfa14A interacted with Kin4 like wild-type Bfa1 (Figure S5D). Thus, other factors must regulate Bfa1 asymmetry, particularly in unperturbed cells. Fraschini et al. [17] proposed that the disappearance of Bfa1/Bub2 from the mother-directed SPB requires Bfa1/Bub2 GAP activity. We also observed that various GAP activity-defective Bfa1 mutants persisted at the SPBm during anaphase. However, we showed that Bfa1 asymmetry was not dependent on GAP activity. Because MEN activation requires inhibition of Bfa1 GAP activity and Bfa1 asymmetry, if Bfa1 asymmetry is regulated by its GAP activity, only a decline in GAP activity could promote Bfa1 loss from the SPBm. Nevertheless, this is probably not the case, as is shown by the symmetric localization of the GAP-defective Bub2-Myc and Bfa1DDR2 proteins. Bfa1 also localized on both SPBs in BFA14A cells with normal GAP activity, and in cdc5-2 cells where Bfa1 GAP activity is expected to be high due to lack of phosphorylation.
Although we have shown in this study that Bfa1 asymmetry is not dependent on its GAP activity, we should consider the possibility that its GAP activity influences its localization indirectly, by affecting its phosphorylation. However, Bfa1D416A, which retains approximately 50% of the GAP activity of wild-type Bfa1, is also defective in phosphorylation by Cdc5, like Bfa1 mutants completely lacking GAP activity (Figure 3). Thus, it is unlikely that the GAP activity of Bfa1 influences its localization indirectly by affecting its phosphorylation. It may still be possible that asymmetric localization requires a certain threshold level of Bfa1 GAP activity (which must be higher than the level in Bfa1D416A) as a prerequisite for phosphorylation of the four serine residues that we have identified.
Our observations that mutation of the four Cdc5-depedent phosphorylation residues, 452S, 453S, 454S, and 559S to Ala in Bfa14A significantly reduced its phosphorylation by Cdc5, as well as affecting its localization, strongly support the role of these residues in directing asymmetric localization in unperturbed mitosis. This notion was further supported by phospho-mimetic Bfa14D, which was asymmetrically localized to the SPBs in cdc15-2-dependent arrested cells (Figure 8A) and even in kinase-defective cdc5-2 (Figure 8B). However, Bfa14D were not asymmetrically localized in 100% of the cdc5-2 cells (Figure 8B) and phosphorylation of Bfa1-D811A+4A by Cdc5 was only reduced by approximately 25% compared with Bfa1-D8 (Figure 6B). Therefore, we cannot exclude the possibility that Bfa1 contains some additional residue(s) that is/are also phosphorylated by Cdc5 and is/are involved in the asymmetric localization of Bfa1.
Bfa14A bound to both SPBs of anaphase-arrested cdc15-2 cells with properly segregated nuclei, whereas the 11 previously identified Cdc5 target sites (Bfa1-11A) had little or no effect on Bfa1 localization. However, the association of Bfa14A with the two SPBs at anaphase was not as stable as that of wild-type Bfa1 for the SPBd. We also found that the Bfa1DDR2 mutant formed a stronger association with the SPBs than wild-type Bfa1 (Figure S8). Thus, further characterization of this mutant may help uncover the molecular mechanisms underlying Bfa1 dynamics. One possibility is that all Bfa1 becomes phosphorylated, and phospho-Bfa1 has different affinities for the two SPBs. Alternatively, Cdc5 may differentially phosphorylate Bfa1 at one of the two SPBs. Another possibility raised by Monje-Casas and Amon [18], is that some proteins mediating the association of Bfa1 with SPBs may control the affinity of Bfa1 for the SPBs by introducing various modifications.
When we mapped the phosphosites of Bfa1 by mass spectrometry, phosphorylation of several characterized sites, such as 7T and 424S, was detected with higher efficiency than phosphorylation of the novel residues we identified as required for Bfa1 asymmetry (Figure S9). It is tempting to speculate that the Cdc5 target sites regulating Bfa1 asymmetry in unperturbed mitosis are phosphorylated with higher fidelity, and/or that other factor(s) are involved in modulating the efficiency of phosphorylation to control Bfa1 dynamics. Due to low phosphorylation efficiency, only 453S and 454S were detected as phosphor forms by MS. The low phosphorylation efficiency of these residues is consistent with the proposed biological role of their phosphorylation in controlling the timing of mitotic exit during unperturbed cell division cycles. Although phosphorylation of the newly identified phosphosites of Bfa1 by Cdc5 was not very efficient, a similar SSS934FL sequence in Claspin had been identified as the target of phosphorylation by Plx1, a frog ortholog of budding yeast Cdc5 [37].
While mapping the Cdc5 target sites, we identified p150S and p180S, which were reported to be phosphorylated by Kin4 and to prevent further modification of Bfa1 by Cdc5 (10; [38]). Although we cannot exclude the possibility that p150S and p180S were phosphorylated nonspecifically due to the extremely high Cdc5 kinase activity, the phosphorylation efficiency at these sites was comparable to that of other Cdc5 targets responsible for GAP activity, such as 7T and 424S (Figure S9). Furthermore, despite the phosphorylation of these sites, other sites were phosphorylated by Cdc5. Thus, we suspect that Bfa1 phosphorylation by Cdc5 and Kin4, and the biological functions of these modifications are far more complicated than we currently understand, and will require further study.
We found that the 452S, 453S, 454S in budding yeast Bfa1 are conserved as 589S, 590T, 591S in its fission yeast homologue byr4 (Figure S11), which is also localized asymmetrically to SPBs in anaphase [39]. It would be interesting to examine whether these conserved residues of byr4 are also phosphorylated by a polo-like kinase, contribute to the asymmetric localization of byr4 and regulate the timing of SIN in fission yeast.
In summary, we have shown that Cdc5-mediated phosphorylation of the newly identified residues on Bfa1 modulates the affinity of Bfa1 for SPBs, and as a consequence contributes to the asymmetric distribution of Bfa1 at anaphase. The asymmetric Bfa1 distribution is required for timely mitotic exit, thus probably ensuring tight coupling of MEN activation and chromosome segregation during normal cell cycle progression. We have also uncovered a novel function of the polo kinase, Cdc5, in the control of mitotic exit. Further studies are needed to identify factors that control the Cdc5-dependent Bfa1 phosphorylation responsible for asymmetric localization in unperturbed mitosis, and how it is overridden in the presence of a misaligned spindle. These studies promise to provide crucial insights into how centrosome asymmetry is generated, and its biological importance in the asymmetric division of eukaryotic cells.
Materials and Methods
Yeast strains, culture, and cell cycle synchronization
All yeast cultures and genetic techniques were carried out as described by Kim et al. [22]. The S. cerevisiae strains used in this study are described in Table S1. Strains were generated by PCR-based methods and verified by PCR, and Southern and western blot analysis [40], [41]. The integrating plasmid, pRS304-BFA1-GFP, was linearized with EcoRV and integrated into the TRP1 locus, as described by Kim et al. [10]. Bfa1 mutants were constructed by PCR-based site-directed mutagenesis, as described by Kim et al. [10]. Cells were synchronized in G1 by adding 10 µg/ml or 50 ng/ml α-factor (Sigma-Aldrich) to BAR1 or bar1 cells, respectively, and at S phase with 0.2 M hydroxyurea (Sigma-Aldrich) for 2–3 h. CDC5 expression was driven by the GAL promoter.
Microscopy and imaging analysis
Fluorescence microscopy was performed essentially as described by Kim et al. [22]. Cellular labeling was visualized on an Axioplan2 (Zeiss) microscope with a Zeiss 100× Plan Neofluar oil immersion objective. Images were acquired using an Axiocam CCD (Zeiss) camera and AxioVision software (Zeiss). The fluorescence intensity of GFP and RFP-fused proteins was quantitatively analyzed by confocal microscopy. For confocal images, we used a Nipkov disk-based UltraVIEW RS confocal system (PerkinElmer) equipped with a Nikon microscope (TE2000-PFS). The 100× NA 1.4 oil immersion objective lens was controlled by a piezoelectric z stepper. In each experiment, 10 to 15 z sections were acquired at 0.5 µm steps with 2×2 binning, the same laser power and exposure time, and projected in UltraVIEW RS software (PerkinElmer). Fluorescence intensity of selected regions of interest was quantified using UltraVIEW RS and Image J software (Version 1.38u, NIH), and the background fluorescence was subtracted by placing the same measurement circle in nearby intracellular regions without a Bfa1-GFP or Tem1-RFP signal. Since the fluorescence intensity of Bfa1-GFP generally increased with Spc42-RFP intensity, we normalized the intensity of Bfa1-GFP to Spc42-RFP to precisely measure the amount of Bfa1 associated with the SPB. For time-lapse experiment, a Nipkov disk-based UltraVIEW RS confocal system (PerkinElmer) equipped with a Nikon microscope (TE2000-PFS) was used. Images for cells on agar plugs were taken every 5 min and processed with Adobe Photoshop 7.0. No manipulations were added other than adjustments in brightness and contrast.
Protein analyses and preparation of recombinant proteins
For phosphatase treatment, TAP-tagged Bfa1 was precipitated with IgG Sepharose beads (Amersham) from total cellular lysates (1 mg in 700 µl modified H-buffer containing 1% NP-40) as described by Kim et al. [10] and treated with Calf Intestinal Alkaline Phosphatase (CIP, New England Biolabs) for 30 min at 37°C. For co-immunoprecipitation of TAP-tagged Bfa14A and 3XHA-tagged Kin4, Kin4 was purified with anti-HA (Roche) followed by protein A-agarose (sigma) and co-precipitates were blotted with peroxidase anti-peroxidase (PAP, Sigma) for Bfa1 or Bfa14A. For western blot analysis, peroxidase anti-peroxidase (PAP, Sigma), monoclonal anti-HA (Roche), monoclonal anti-Myc (Roche), polyclonal anti-GFP (Santa Cruz Biotechnology), Clb2 (produced in the lab), monoclonal anti-α-Tubulin (Sigma), and polyclonal anti-Actin (Santa Cruz Biotechnology) were used. Band intensity was quantified and analyzed using the LAS-3000 image analyzer (Fujifilm) and Image J software (Version 1.38u, NIH).
GST-Cdc5 (Δ70N-Cdc5 harboring S165D and T238D) and GST-Cdc5KD (Δ70N-Cdc5 harboring an N209A) were expressed in S. cerevisiae, as described by Geymonat et al. [9]. Tem1, GST-Bub2, MBP-Bfa1, and MBP-D8-Bfa1 were prepared from E. coli, as described by Kim et al. [10].
In vitro Tem1 GTPase assay and yeast two-hybrid assay
The intrinsic or GAP-stimulated GTPase activity of Tem1 was quantified, as described by Kim et al. [10] with an EnzCheck Phosphate Assay Kit (E-6646; Molecular Probes). The amount of γ-Pi released from Tem1-GTP was monitored by measuring the absorbance at 360 nm. Yeast two-hybrid assays were performed as previously described Kim et al. [22].
In vitro kinase assay
In vitro kinase assays were performed, as described by Geymonat et al. [9]. For radioactive kinase assays, 100 ng of substrate was mixed with 10–50 ng of either GST-Cdc5 or GST-Cdc5KD in 15 µl kinase buffer (50 mM Tris-Cl, pH 7.5, 10 mM MgCl2, 1 mM dithiothreitol) with 50 µM ATP and 0.1 µl γ-[32P]ATP (Amersham Biosciences, 370 MBq/ml, 3000 Ci/mmol). After incubation at 30°C for either 15 min for full-length Bfa1 or 30 min for Bfa1-D8, Laemli buffer was added to stop the phosphorylation reaction. γ-[32P]-labeling was visualized by autoradiography. To better detect the phosphorylation of Bfa1-D8 by Cdc5, 1–10 µg substrates were incubated with 1–3 µg of either GST-Cdc5 or GST-Cdc5KD in the same kinase buffer with 0.5 mM non-radioactive ATP, separated on 7.5% SDS-PAGE containing 100 µM Phos-tag acrylamide (MANAC Incorporated) [42] and 200 µM MnCl2, and stained with Coomassie brilliant blue.
Determination of Cdc5-dependent phosphorylation by mass spectrometry
After in vitro phosphorylation of Bfa1 was performed with purified Cdc5 kinase and the product was digested with trypsin, liquid chromatography was carried out on a Dionex LC Packings nano HPLC system (LC-Packings) coupled to the QSTAR Pulsar ESI-hybrid Q-TOF tandem mass spectrometer (Applied Biosystems), as described in Lee et al [43]. The column outlet was coupled directly to the high voltage ESI source (typically 2.3 kV) and peptides eluting from the column were sprayed directly into the orifice of the mass spectrometer. Information-Dependent Acquisition (IDA) mode was performed to acquire MS/MS spectra based on an inclusion mass list and dynamic assessment of relative ion intensity. For MS/MS, a full mass scan range mode was m/z = 100–2000 Da. After determining the charge states of an ion on zoom scans, product ion spectra were acquired in MS/MS mode with relative collision energy of 55%. The individual spectra from MS/MS were processed using the Analyst QS software (v1.1, Applied Biosystems) and searched against a limited database containing only the protein of interest, Bfa1, which was performed with mass tolerance 0.1 Da and with a confidence value no less.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. NiggEA 2001 Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2 21 32
2. MorganDO 1999 Regulation of the APC and the exit from mitosis. Nat Cell Biol 1 E47 53
3. QueraltEUhlmannF 2008 Cdk-counteracting phosphatases unlock mitotic exit. Curr Opin Cell Biol 20 661 668
4. BardinAJAmonA 2001 Men and sin: what's the difference? Nat Rev Mol Cell Biol 2 815 826
5. WangYHuFElledgeSJ 2000 The Bfa1/Bub2 GAP complex comprises a universal checkpoint required to prevent mitotic exit. Curr Biol 10 1379 1382
6. BloecherAVenturiGMTatchellK 2000 Anaphase spindle position is monitored by the BUB2 checkpoint. Nat Cell Biol 2 556 558
7. LeeSEFrenzLMWellsNJJohnsonALJohnstonLH 2001 Order of function of the budding-yeast mitotic exit-network proteins Tem1, Cdc15, Mob1, Dbf2, and Cdc5. Curr Biol 11 784 788
8. HuFWangYLiuDLiYQinJ 2001 Regulation of the Bub2/Bfa1 GAP complex by Cdc5 and cell cycle checkpoints. Cell 107 655 665
9. GeymonatMSpanosAWalkerPAJohnstonLHSedgwickSG 2003 In vitro regulation of budding yeast Bfa1/Bub2 GAP activity by Cdc5. J Biol Chem 278 14591 14594
10. KimJJangSSSongK 2008 Different levels of Bfa1/Bub2 GAP activity are required to prevent mitotic exit of budding yeast depending on the type of perturbations. Mol Biol Cell 19 4328 4340
11. ShirayamaMMatsuiYTohEA 1994 The yeast TEM1 gene, which encodes a GTP-binding protein, is involved in termination of M phase. Mol Cell Biol 14 7476 7482
12. GeymonatMSpanosAde BettigniesGSedgwickSG 2009 Lte1 contributes to Bfa1 localization rather than stimulating nucleotide exchange by Tem1. J Cell Biol 187 497 511
13. Valerio-SantiagoMMonje-CasasF 2011 Tem1 localization to the spindle pole bodies is essential for mitotic exit and impairs spindle checkpoint function. J Cell Biol 192 599 614
14. PereiraGHofkenTGrindlayJMansonCSchiebelE 2000 The Bub2p spindle checkpoint links nuclear migration with mitotic exit. Mol Cell 6 1 10
15. MolkJNSchuylerSCLiuJYEvansJGSalmonED 2004 The differential roles of budding yeast Tem1p, Cdc15p, and Bub2p protein dynamics in mitotic exit. Mol Biol Cell 15 1519 1532
16. PereiraGTanakaTUNasmythKSchiebelE 2001 Modes of spindle pole body inheritance and segregation of the Bfa1p-Bub2p checkpoint protein complex. EMBO J 20 6359 6370
17. FraschiniRD'AmbrosioCVenturettiMLucchiniGPiattiS 2006 Disappearance of the budding yeast Bub2-Bfa1 complex from the mother-bound spindle pole contributes to mitotic exit. J Cell Biol 172 335 346
18. Monje-CasasFAmonA 2009 Cell polarity determinants establish asymmetry in MEN signaling. Dev Cell 16 132 145
19. CaydasiAKPereiraG 2009 Spindle alignment regulates the dynamic association of checkpoint proteins with yeast spindle pole bodies. Dev Cell 16 146 156
20. CharlesJFJaspersenSLTinker-KulbergRLHwangLSzidonA 1998 The Polo-related kinase Cdc5 activates and is destroyed by the mitotic cyclin destruction machinery in S. cerevisiae. Curr Biol 8 497 507
21. ShirayamaMZachariaeWCioskRNasmythK 1998 The Polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. EMBO J 17 1336 1349
22. KimJJeongJSongK 2004 The C-terminus of Bfa1p in budding yeast is essential to induce mitotic arrest in response to diverse checkpoint-activating signals. Genes Cells 9 399 418
23. Cohen-FixOPetersJMKirschnerMWKoshlandD 1996 Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev 10 3081 3093
24. ZachariaeWNasmythK 1999 Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev 13 2039 2058
25. ShouWSeolJHShevchenkoABaskervilleCMoazedD 1999 Exit from mitosis is triggered by Tem1-dependent release of the phosphatase Cdc14 from nucleolar RENT complex. Cell 97 233 244
26. WäschRCrossFR 2002 APC-dependent proteolysis of the mitotic cyclin Clb2 is essential for mitotic exit. Nature 418 495 6
27. LiYYYehEHaysTBloomK 1993 Disruption of mitotic spindle orientation in a yeast dynein mutant. Proc Natl Acad Sci U S A 90 10096 10100
28. LeeLTirnauerJSLiJSchuylerSCLiuJY 2000 Positioning of the mitotic spindle by a cortical-microtubule capture mechanism. Science 287 2260 2262
29. AdamesNROberleJRCooperJA 2001 The surveillance mechanism of the spindle position checkpoint in yeast. J Cell Biol 153 159 168
30. LiakopoulosDKuschJGravaSVogelJBarralY 2003 Asymmetric loading of Kar9 onto spindle poles and microtubules ensures proper spindle alignment. Cell 112 561 574
31. GravaSSchaererFFatyMPhilippsenPBarralY 2006 Asymmetric recruitment of dynein to spindle poles and microtubules promotes proper spindle orientation in yeast. Dev Cell 10 425 439
32. KonigCMaekawaHSchiebelE 2010 Mutual regulation of cyclin-dependent kinase and the mitotic exit network. J Cell Biol 188 351 368
33. BertazziDTKurtulmusBPereiraG 2011 The cortical protein Lte1 promotes mitotic exit by inhibiting the spindle position checkpoint kinase Kin4. J Cell Biol 193 1033 1048
34. MorganD 2007 The Cell Cycle: Principle of control University of Oxford press 6
35. DrapkinBJLuYProckoALTimneyBLCrossFR 2009 Analysis of the mitotic exit control system using locked levels of stable mitotic cyclin. Mol Syst Biol 5 328
36. PereiraGSchiebelE 2005 Kin4 kinase delays mitotic exit in response to spindle alignment defects. Mol Cell 19 209 221
37. LoweryDMLimDYaffeMB 2005 Structure and function of Polo-like kinases. Oncogene 24 248 259
38. MaekawaHPriestCLechnerJPereiraGSchiebelE 2007 The yeast centrosome translates the positional information of the anaphase spindle into a cell cycle signal. J Cell Biol 179 423 436
39. CeruttiLSimanisV 1999 Asymmetry of the spindle pole bodies and spg1p GAP segregation during mitosis in fission yeast. J Cell Sci 112 2313 2321
40. LongtineMSMcKenzieA3rdDemariniDJShahNGWachA 1998 Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14 953 961
41. JankeCMagieraMMRathfelderNTaxisCReberS 2004 A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21 947 962
42. KinoshitaEKinoshita-KikutaETakiyamaKKoikeT 2006 Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5 749 757
43. LeeJMKimSLeeJYYooEYChoMC 2006 A differentially expressed proteomic analysis in placental tissues in relation to pungency during the pepper fruit development. Proteomics 6 5248 5259
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 1
Nejčtenější v tomto čísle
- Poly(ADP-Ribose) Polymerase 1 (PARP-1) Regulates Ribosomal Biogenesis in Nucleoli
- Microenvironmental Regulation by Fibrillin-1
- Parallel Mapping and Simultaneous Sequencing Reveals Deletions in and Associated with Discrete Inherited Disorders in a Domestic Dog Breed
- Two-Component Elements Mediate Interactions between Cytokinin and Salicylic Acid in Plant Immunity